
ABSTRACT
Introduction
Complement-based drug discovery is undergoing a renaissance, empowered by new advances in structural biology, complement biology and drug development. Certain components of the complement pathway, particularly C1q and C3, have been extensively studied in the context of neurodegenerative disease, and established as key therapeutic targets. C5 also has huge therapeutic potential in this arena, with its druggability clearly demonstrated by the success of C5-inhibitor eculizumab.
Areas covered
We will discuss the evidence supporting C5 as a target in neurodegenerative disease, along with the current progress in developing different classes of C5 inhibitors and the gaps in knowledge that will help progress in the field.
Expert opinion
Validation of C5 as a therapeutic target for neurodegenerative disease would represent a major step forward for complement therapeutics research and has the potential to furnish disease-modifying drugs for millions of patients suffering worldwide. Key hurdles that need to be overcome for this to be achieved are understanding how C5a and C5b should be targeted to bring therapeutic benefit and demonstrating the ability to target C5 without creating vulnerability to infection in patients. This requires greater biological elucidation of its precise role in disease pathogenesis, supported by better chemical/biological tools.
1. Introduction
The complement cascade is a system of approximately 30 different proteins that circulate in the plasma and tissue fluids or are found on cell membranes [Citation1]. There are numerous excellent overviews covering the intricacies of complement activation, its role in the pathogenesis of numerous diseases, and the promise of therapies targeting the components within the pathway [Citation2–22], and so we will provide a relatively short description within this review. Upon recognition of a foreign pathogen or damage-associated molecular patterns on tissues within the body, the complement system is activated in a sequence via a chain of protein–protein interactions driven by conformational changes arising from proteolytic cleavage. The activation of one protein via enzymatic cleavage enables binding and activation of the next protein in the cascade, giving rise to the rapid innate immune responses of cell lysis, opsonization, chemotaxis, and inflammation () [Citation1]. The last enzymatic step in the cascade is the cleavage of C5 to C5a and C5b. The former, C5a, is a chemotactic and anaphylactic peptide, whereas C5b binds C6 marking the start of the terminal pathway. All the interactions within the terminal pathway are non-enzymatic, driven purely by conformational changes that reveal binding sites for the next protein in the pathway. The end point of the terminal pathway is the formation of the lytic pore, the membrane attack complex (MAC). Nucleated cells are rarely lysed by MAC because ion pumps counteract osmotic lysis and active mechanisms of ectocytosis and endocytosis remove MAC complexes from the cell surface. In nucleated cells, MAC lesions are transient and cells usually survive, however, MAC attack is not without consequence and downstream events are frequently inflammatory [Citation23]. Influx of calcium ions through the pore and subsequent release of additional calcium from the ER activates calmodulin resulting in activation of multiple downstream proteases including Akt and PI3k (phosphatidylinositol-3-kinase), triggering numerous downstream inflammatory processes. Increased calcium also supports assembly and activation of the pyrin domain-containing protein 3 (NLRP3) inflammasome leading to release of active IL18 and IL1 beta [Citation24]. Thus, cleavage and activation of C5 can trigger numerous processes which might play a role in complement-mediated degenerative diseases.
Figure 1. Overview of the complement cascade and its activation. Three different activation pathways (classical, alternative and lectin) mediate the immune response through a series of protein–protein interactions in a cascade that leads to cleavage of C3. The cleaved components go on to perform multiple roles; C3a acts as an anaphylatoxin, effecting inflammation and chemotaxin, while C3b can either cause phagocytosis of foreign species through opsonization or combine with other complement components to form C5 convertase, which cleaves C5 into C5a and C5b, initiating the terminal complement pathway. C5a, like C3a, is an anaphylatoxin, causing amplification of the inflammatory signal. C5b undergoes a series of interactions with further complement components to form the membrane attack complex (MAC), a cytolytic pore which then destroys pathogens.
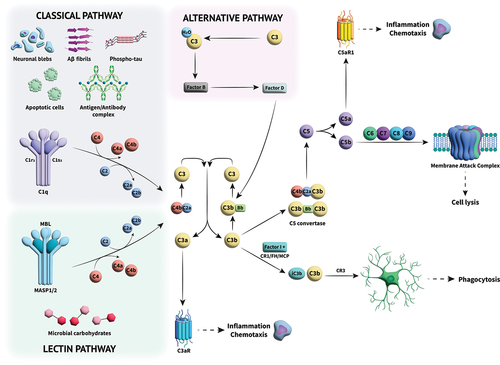
Complement kills and removes infected cells and bacteria either through cell lysis with the MAC, or more commonly, because of opsonization where the cell/bacteria are coated with the opsonins C3b, iC3b, and C4b, making them susceptible to phagocytosis triggered by receptors such as CR1 or CR3 (complement receptor 3; CD11b/CD18) on infiltrating neutrophils and macrophage. The complement cascade also plays a role within the adaptive immune response helping with immunologic memory, and cross-talking with T-cells, B-cells and antigen presenting cells [Citation10,Citation25].
1.1. Complement activation & control
There are three main pathways to activate complement: the classical pathway, lectin pathway, and the alternative pathway, all of which can be seen in . All three of these pathways lead to the cleavage of the complement 3 protein (C3) into complement component 3a (C3a) and complement component 3b (C3b). The classical pathway involves complement components C1, C2, and C4. C1 has three subcomponents: C1q, C1r, and C1s. C1q binds to an antibody-antigen complex and undergoes a conformational change resulting in the autoactivation of C1r that subsequently cleaves and activates C1s [Citation26]. The C1s subunit cleaves C4 to give C4a and C4b, the latter can bind covalently to the target surface where it can bind C2 in a magnesium-dependent manner. Once bound to C4b, C2 is cleaved by C1s, releasing the small fragment, C2b, and retaining the large enzymatic subunit, C2a. This bimolecular complex is termed the C3 convertase. The C2a subunit within the C3 convertase then cleaves C3 into C3a and C3b, the latter of which can covalently bind to the target surface [Citation1]. The lectin pathway occurs when mannose-binding lectin (MBL) binds to mannose residues on the surface of bacteria or damaged brain cells. Binding activates MBL-associated serine proteases (MASPs) to cleave C2 and C4 as described above to form the C3 convertase [Citation1]. The alternative pathway is triggered by the magnesium-dependent binding of factor B (FB) to C3b to form the proenzyme, C3bB. The bound FB is a substrate for the serine protease factor D (FD) that cleaves FB to Ba and Bb; Ba is released while the enzymatic subunit, Bb, is retained. The resulting C3 convertase, C3bBb, can bind a stabilizing factor called properdin that lengthens its otherwise very short half-life. The alternative pathway is always ticking over in the body, primed through the hydrolysis of an internal thioester bond, producing C3(H2O), a C3b-like molecule, or by cleavage of C3 to C3b by proteases present at sites of infection or tissue damage [Citation27]. The C3 convertase of any of the three activating pathways cleaves multiple molecules of C3 to C3b that bind covalently in the vicinity of the activating nidus, each forming a C3 convertase in turn, causing amplification of the system and generating clusters of C3b (or C4b and C3b). These oligomers form C5 convertases within which the Bb or C2a enzymatic domains cleave C5 to C5a and C5b.
As the convertase enzymes are proinflammatory and easily amplified, it is essential that the components are tightly controlled and inactivated when they deposit on self surfaces. When this happens there is the potential for ‘bystander lysis’ of normal self-cells, hence these express an array of complement regulatory proteins including CD35 (complement receptor 1, CR1), CD46 (MCP, membrane cofactor protein), CD55 (DAF, decay accelerating factor) and CD59 that prevent complement activation (CD35, CD46), amplification (CD35, CD55) and MAC formation (CD59). There are also proteins in the fluid phase that recognize structures on self-cells and bind to deposited C3b (factor H, FH) or C4b (C4b binding protein, C4BP), causing rapid inactivation by acting as cofactors for factor I (FI), a proteolytic enzyme that cleaves and inactivates C3b (by forming iC3b) and C4b (forming C4c and C4d). Many of these protective proteins also possess an activity termed decay accelerating activity (FH, CD55, CD35, C4BP), whereby binding to a multimolecular convertases causes rapid dissociation of the enzymatic subunit, C2a or Bb. The lack of these protective proteins on pathogen surfaces facilitates amplification of complement and opsonization or lysis of foreign cells.
The background tick over of complement, producing C3b, is continuous in plasma and body fluids, enabling rapid response in the face of infection. As explained above, this low-level activation is tightly controlled to prevent damage to self, only amplifying in response to a foreign surface. However, several mechanisms cause dysregulation of the complement cascade, potentially causing overt activation on self-cells and leading to tissue damage, inflammation, and disease. These mechanisms include common polymorphisms or rare mutations in complement proteins that produce gain-of-function of activating proteins or loss-of-function in control proteins [Citation28,Citation29]. Often an array of ‘risk’ common polymorphisms occurs in combination with other disease-causing triggers (including environmental factors) that cause over-activation of the complement system whereas rare mutations may in themselves be highly penetrant and strongly associated with disease risk. Autoantibodies are also associated with disease, including those that directly bind tissue antigens causing inappropriate activation and overwhelming the innate control mechanisms (for example, in myasthenia gravis or neuromyelitis optica)[Citation30,Citation31] or autoantibodies that bind to activated complement proteins or complexes, preventing the action of regulators (such as nephritic factors, associated with renal disease) [Citation32]. Environmental factors vary from disease to disease but might include infections, pregnancy, damaged tissue, and accumulation of debris, such as sub-retinal drusen in age-related macular degeneration or plaques in Alzheimer’s disease (AD) [Citation32].
1.2. The anaphylatoxins
The by-products of complement activation include the anaphylactic and chemotactic peptides, C3a and C5a. C3a is produced at high amounts at sites of complement activation due to the actions of the amplification loop that propagates C3 cleavage; in most circumstances C3a initiates and promotes inflammation, although it is postulated to be anti-inflammatory in certain situations and diseases [Citation33,Citation34]. C3a is a small fragment (9kDa) which binds to a C3a-specific G-protein-coupled receptor (C3aR) on infiltrating cells and tissue cells; due to the actions of widespread inhibitors, carboxy peptidases, that remove the C-terminal arginine residue inactivating C3a (forming C3a desArg), its actions tend to be localized to the area where the complement cascade was activated.
C3 and C5 have arisen through gene duplication, hence their structures are highly similar, and they undergo similar structural changes following cleavage and activation. As with C3, C5 is a large 196 kDa molecule that comprises two chains, alpha and beta, and is cleaved by C5 convertases in the alpha chain generating C5a and C5b () [Citation35,Citation36]. Unlike C3, C5 does not contain an internal cysteine-glutamine thioester bond that enables covalent binding and opsonization of target surfaces [Citation35,Citation37]. Instead, C5 contains the amino acids serine (Ser1007) and alanine (Ala1010) in the corresponding positions. Due to the lack of an internal thioester bond, C5 has a higher tolerance for the presence of water and is less hydrophobic than C3. C5a is produced by cleavage at the peptide/amide bond between amino acids arginine (Arg751) and leucine (Leu752) by C5 convertase, located toward the C-terminal end of the alpha chain [Citation38]. Like C3a, C5a is an anaphylactic and chemotactic peptide that promotes inflammation by binding to C5-specific transmembrane receptors (C5aR1 and 2) on leukocytes, recruiting them to sites of infection and inflammation and triggering activation phenomena, such as the oxidative burst [Citation33,Citation39,Citation40]. C5a is far more potent than C3a and its actions are more wide-ranging. Although C5a is also a substrate for carboxypeptidases, C5a desArg retains significant activity and can still bind C5aRs [Citation41]. Due to the highly inflammatory activity of C5a, the native protein C5a, the anaphylatoxin C5a or the better-characterized receptor, C5aR1, have dominated as drug targets for a number of inflammatory diseases.
Figure 2. Structure of C5 with C5a residues highlighted in blue, in complex with a small molecule ligand (PDB:6I2X) [Citation36].
![Figure 2. Structure of C5 with C5a residues highlighted in blue, in complex with a small molecule ligand (PDB:6I2X) [Citation36].](/cms/asset/429d313d-2a38-41b3-9453-a40f6de00085/iett_a_2177532_f0002_oc.jpg)
2. C5 and neurodegenerative disease
C5 has emerged as an important target with a growing body of evidence from clinical trials and drug approvals that blockade of C5 brings therapeutic benefit in diseases such as paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome [Citation42–44]. Despite a wealth of evidence placing the complement system and the terminal pathway as a key player in neurodegenerative (ND) pathologies, clinical trials targeting C5 in ND disease are limited to amyotrophic lateral sclerosis (ALS; terminated -NCT04248465, ongoing -NCT04436497) [Citation45]. Evidence of a role of complement in human ND disease is restricted to post-mortem analyses of central nervous system (CNS) tissue, limited biomarker analyses of cerebrospinal fluid (CSF) and peripheral biomarkers in-life, genetic associations, and animal models of disease [Citation30,Citation46–48]. Limitation of tissue analyses to post-mortem in humans makes it difficult to understand causality and restricts the ability to understand the role of complement in the progression of disease, knowledge of which may be critical for targeting the right drugs at the right time to delay onset or slow progression.
AD is particularly well-researched, with animal models providing evidence to place dysregulated complement at the forefront of ND, specifically in the context of neuroinflammation. Use of mouse knock-out strains and judicious tissue staining puts C1q, C3 and the C5 activation products in the spotlight [Citation30,Citation46–63]. Compelling evidence comes from a study where DBA/2 J mice, which are deficient in C5, and C57Bl6 mice transgenic for the human APP gene were compared, with a lower amyloid burden and milder pathology observed in DBA/2 J mice [Citation64]. Recent investigations using C6-deficient mice and a C7-blocking antibody strongly suggest that MAC, rather than C5a, may be a critical player in synapse loss in several murine AD models [Citation65]. Importantly, animal model data are backed by genetic associations [Citation66,Citation67] between complement and AD include clusterin and CD35 (complement receptor 1, CR1), suggesting a role for complement control in disease etiology [Citation68–70]. The role of clusterin is unclear- while it does inhibit the MAC, it also binds debris and likely accumulates on amyloid plaques. Critically, however, CR1 controls the complement cascade and is one of the few control proteins that is capable of directly inhibiting C5 convertase, potentially indicating a role for C5 in AD [Citation30,Citation68]. Despite the accumulating evidence for a role of the complement terminal pathway in AD, challenges of early diagnosis, understanding of the stage of disease in which complement plays a role and access of drugs to the CNS tissue continues to hamper progression into clinical trials.
Although there are no genetic associations of complement with ALS, analysis of post-mortem tissue demonstrates abundant complement deposition in affected tissues, and complement proteins are upregulated in affected tissue [Citation71–74]. Plasma levels of C5a and C5b-C9 are also found to be significantly elevated in ALS patients, suggesting upregulation of the terminal complement pathway [Citation66]. The picture is similar in Huntington’s Disease (HD) and Parkinson’s Disease (PD), with complement deposition in tissues being observed post-mortem and complement proteins being expressed at higher levels in diseased tissue [Citation75–78]. Although it is difficult to distinguish cause or effect in these human analyses, animal models of ALS and HD do illustrate a pathogenic role of complement with knockout or blockade of C5aR1 having a beneficial effect [Citation79–81].
The studies described above indicate that complement is activated in neurodegenerative disease (ND) and that activation progresses through the terminal pathway. It remains unclear whether C5 inhibition will provide therapeutic benefit in these diseases as stage of disease and access of drugs to the right place remains critical, the outcome of trials utilizing systemically administered zilucoplan in ALS may throw some light on therapeutic potential.
2.1. C5 in neuroinflammation
Several ND diseases have been associated with a hallmark of chronic and/or aberrant neuroinflammation, with this area becoming increasingly attractive as an avenue for treating diseases such as AD, ALS, PD, and HD, though this list is by no means exhaustive [Citation82–92]. There are numerous excellent reviews covering the intricacies of complement in neuroinflammation, with some highlighted by the authors here [Citation27,Citation45,Citation93–100]. Neuroinflammation presents a complex target for drug discovery; the balance of pro-inflammatory and pro-healing actions by the immune system has profound and differing effects on factors such as neuroprotection, neurogenesis, phagocytosis, and cell recruitment dependent on the indication, the severity and the timing of intervention [Citation84,Citation86,Citation92,Citation93]. It is crucial, therefore, that the ability to precisely control immune cell phenotype in the brain is realized, and the impact of this fully interrogated, to maximize translation to patients.
The innate immune cells of the brain, microglia, adopt a range of highly dynamic roles spanning surveillance, cytokine release, cell recruitment, phagocytosis, and synapse pruning, with their activation state shown to be pivotal in the neuroinflammatory health of the brain [Citation89]. In the healthy brain microglia produce little to no complement components, but expression rapidly increases on activation or insult [Citation27,Citation101]. This inflammatory response is a key part of the immune response to injury and pathogenic species, including viral or bacterial infections and misfolded proteins, but it is crucial that this response is appropriately resolved once the threat is eliminated in order to promote healing [Citation86]. Dysregulated inflammation can quickly spiral, activating other inflammatory pathways and causing toxicity to the surrounding tissue; this collateral damage is considered to be a key driver of neuroinflammatory degradation [Citation86]. Complement activation has been linked with inflammasome activity [Citation24,Citation102], interferon signaling [Citation103], and NfκB signaling [Citation27].
Generation of C5a likely drives inflammation and microglial activation, whereas formation of the MAC causes the cell activation events described above and induction of NLRP3 inflammasome activity [Citation67]. The vast majority of studies related specifically to C5 and its effect on microglial phenotype have focused on C5a, either by altering its binding with one of the receptors, or by treating microglia with C5a and assessing the change in activation state [Citation58,Citation104–109]. Such studies have also demonstrated the ability of C5a to recruit microglia to the site of an injury [Citation58]. The MAC has also been shown to cause a more inflammatory microglial activation state [Citation24,Citation67]. Collectively, these observations suggest that further investigation into the effect of C5 blockade on microglial activation state and neuroinflammation is well warranted.
3. C5 inhibitors
All the clinically available C5 inhibitors are biologics, such as the anti-C5 antibody eculizumab, but there has also been progress toward small molecule and peptide modulators for the CNS. shows the binding sites of these different types of modulators, and their location relative to the C5 cleavage site. Very interestingly, the reported biologics, small molecules, and peptides appear to interact with different sites of the protein, respectively. The availability of structural data for each of these classes of molecules paves the way for computational modeling to facilitate the development of new modulators of each of these distinct sites in C5, allowing us to probe the biological effects of interacting with different parts of the protein. This could prove powerful in the development of new drugs, giving greater precision if these sites result in different effects (e.g. reduced cleavage, conformational changes preventing C5b binding to the next proteins in the cascade). A challenge to be overcome regardless of the type of inhibitor chosen is the abundance of complement proteins in plasma, along with their rapid turnover rates, necessitating high, frequent doses that are orders of magnitude larger than therapies targeting other immune system proteins [Citation1]. This has knock-on effects on cost, ease-of-use and quality of life for patients, particularly with first-generation drugs which, for example, are given intravenously at short intervals. The advent of next-generation drugs, such as recycling antibodies, orally bioavailable drugs or gene therapies, are gradually changing the landscape, potentially overcoming some of these pitfalls [Citation1].
Figure 3. Diagram of C5 with the residues forming C5a on cleavage highlighted in light blue, the biologic eculizumab (PDB ID: 5I5K) in Orange [Citation110], small molecule inhibitor H1H (PDB ID: 6I2X) in grey [Citation36], and allosteric modulator knob peptide (PDB ID: 7AD6) in green [Citation154]. The interaction site for cobra venom factor (CVF) is also indicated in light grey.
![Figure 3. Diagram of C5 with the residues forming C5a on cleavage highlighted in light blue, the biologic eculizumab (PDB ID: 5I5K) in Orange [Citation110], small molecule inhibitor H1H (PDB ID: 6I2X) in grey [Citation36], and allosteric modulator knob peptide (PDB ID: 7AD6) in green [Citation154]. The interaction site for cobra venom factor (CVF) is also indicated in light grey.](/cms/asset/7593edbc-6913-473a-bae6-71b046cc0cd8/iett_a_2177532_f0003_oc.jpg)
3.1. Biologics
The strongest evidence for the therapeutic potential of complement 5 inhibition in neurodegenerative diseases can be found in the success of various C5-inhibitory biologics to treat related disorders. The biggest success in this area is the monoclonal recombinant antibody eculizumab, which was developed by Alexion Pharmaceuticals and prevents terminal complement activation by inhibiting the cleavage of C5 to C5a and C5b [Citation111]. Eculizumab was originally developed and approved for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) and has since been approved for atypical hemolytic uremic syndrome (aHUS), as well as other diseases which affect the nervous system, namely anti–acetylcholine receptor–positive (AChR+) refractory generalized myasthenia gravis (gMG) and anti-aquaporin-4 (AQP4) antibody-positive neuromyelitis optica spectrum disorder (NMOSD) [Citation43,Citation44,Citation112–119]. Although both gMG and NMOSD are driven by antibodies, the downstream pathogenic effects are mediated by MAC that damages the neuromuscular endplate (in the case of gMG) or the astrocytes that support neuronal function (in the case of NMOSD). In phase 3 clinical trials, blockade of C5 activation dramatically reduced relapses in NMOSD (NCT01892345) and improved clinical responsiveness and symptoms that affected daily activities in gMG patients (NCT01997229) [Citation44,Citation120]. Yan et al. also developed a C5-specific single-chain variable fragment which, through intraparenchymal injection, reduced inflammatory infiltration, MAC formation, neuromyelitis optica spectrum disorder (NMOSD)-like lesions and cell loss in the brain in a mouse model [Citation121]. Other biologics developed to inhibit C5 cleavage include the recycling antibody, ravulizumab, a modified version of eculizumab with a longer half-life to allow for less frequent maintenance dosing. Other antibodies being developed for C5 include tesidolumab (Novartis), pozelimab (Regeneron Pharmaceuticals) and another recycling antibody and crovalimab (Roche). An interesting problem with anti-C5 therapy using eculizumab/ravulizumab is their inability to bind a C5 variant with an arginine-to-histidine change at position 885 making these drugs ineffective in a small subset of patients [Citation122]. Development of other antibodies or biologics with differing epitopes can overcome this issue.
Several other biologics are in development for C5 inhibition that are not based on antibodies. Nomacopan (rVA576; coversin) is a small biologic (~17kDa) that binds C5, including the R885H variant, and prevents its interaction with the C5 convertase [Citation35,Citation123]. Nomacopan was originally isolated from the saliva of ticks as an anti-inflammatory molecule that prevented inflammation that might result from repeated blood feeds from a variety of host species; it was termed Ornithodoros moubata complement inhibitor (OmCI) and inhibits C5 across a wide spectrum of species [Citation124,Citation125]. More recently, it has been shown to have a dual activity with the ability to sequester and bind leukotriene B4 with high affinity in addition to C5 [Citation123]. It has shown promise in many different animal models of disease although its small size conveys a short half-life [Citation123,Citation126–128]; PASylated forms of the molecule that enable retention at localized sites have been used with success in animal models [Citation128]. Nomacopan is in phase 3 clinical development for transplant-associated thrombotic microangiopathies (NCT04784455) and phase 2 for bullous pemphigoid (NCT04563923) [Citation129].
One drawback of using anti-C5 drugs may be a resultant susceptibility to meningococcal infection amongst treated patients [Citation117,Citation130,Citation131]. Protocols for treatment with eculizumab/ravulizumab always include meningococcal vaccination 2 weeks prior to commencement of drug administration although this does not completely eliminate the risk and patients require close monitoring [Citation117,Citation130–136]. Whilst biologics have undoubtedly been game changing in the field of complement modulation, they have other limitations that hamper their widespread use and application to neurodegenerative disease. First is their high cost, with the price of eculizumab being the subject of a number of publications assessing the relative cost-benefit of its use; eculizumab costs approximately £340,000 per annum per patient. The cost of biologics is a consistent issue across medical care [Citation137–139].
Another challenge for treatment of neurodegenerative disease is the pharmacokinetic properties of biologics. While the long half-life of antibodies can be beneficial for the treatment of systemic disease or other organs, their inability to penetrate the blood-brain barrier (BBB) limits their utility for the treatment of CNS disease. In addition, biologics currently cannot be dosed orally due to their poor bioavailability and their susceptibility to breakdown by the body’s gastrointestinal system [Citation140,Citation141]. As a result, biologics require intravenous (IV) or subcutaneous (SC) administration, which can hugely increase cost if hospital IV administration is required and can create potential issues with compliance amongst patients. Oral dosing could be preferable in terms of cost and patient uptake, particularly for chronic conditions such as neurodegenerative disease. The converse side of this argument is the requirement for cognitively impaired patients to remember to take an oral drug; the rapid turnover of complement species means that activity would be restored rapidly if a dose were missed, making consistency key. Biologics’ large size and generally polar nature also limit their cell permeability and particularly their BBB penetrance, where generally only small, lipophilic molecules are able to easily pass unless they are able to utilize an active transport mechanism [Citation142]. This restrictive barrier is predicted to be inaccessible to almost all biologics and up to 98% of small molecules [Citation143]. Research is currently investigating novel delivery mechanisms to reach the brain, enter cells, and allow for oral dosing but, until this is realized, biologics have limited utility for CNS treatment despite their promise for diseases with systemic delivery regimes [Citation140,Citation141,Citation143–145].
3.2. Small molecules
Small molecule drugs have the considerable advantage of being far more readily developed for good BBB penetrance and bioavailability, so they would appear a natural choice for complement therapeutics. However, the development of a small molecule able to interrupt protein–protein interactions is notoriously challenging [Citation146], and so there has been much less success in the development of small molecule C5 modulators than there has been in biologics. In 2012, however, Zhang and co-workers reported two related small molecules, (S)-1-(4-methoxy-3-((4-methylpentyl)oxy)phenyl)-3-(1-phenylethyl)urea () and (S)-1-(3-(hexyloxy)-4-methoxyphenyl)-3-(1-phenylethyl)urea (), which were able to block deposition of C9, but not C3 and C4 [Citation147].
In 2019, Jendza and co-workers identified the target of those small molecules [Citation36]. In their study, they activated complement in serum with and without the small molecule present and investigated the generation of complement breakdown products. They found that the cascade was inhibited at the point of C5 cleavage, since neither the C5 breakdown products C5a or C5b nor any other component downstream of C5 could be detected [Citation36]. To investigate the binding mode of these compounds and their structure-activity relationships (SAR), a series of analogues has been synthesized and assessed by the group. They reported that substitution at the 6-position of the phenyl core was tolerated (). However, the substitution at the 2-position () led to a complete loss of activity. They subsequently used molecular modeling techniques to predict bioactive conformations of those compounds and complemented their study by solving a cryo-EM structure at 3.35 Å resolution of C5 bound to cobra venom factor (CVF) and the small molecule H1H shown in . An illustration of this is shown in (PDB code: 6I2X). This structure suggested the mechanism by which these small molecules prevent C5 cleavage [Citation36]. It also revealed that the 2-methoxy group of this compound responsible for the 30-fold boost in potency interacted with a loop from the MG8 domain of C5 (), although the precise atomic coordinates are not clearly defined from the observed density in this region. This group is additionally stabilizing the active conformation through an intramolecular hydrogen bond with the urea NH moiety (). Publication of these data will likely serve as a stepping-stone toward structure-guided design of novel small molecular inhibitors of C5 in the conceivable future. The effect of these molecules on inflammatory phenotype or neuroprotection in the brain is yet to be studied.
Figure 4. Development of small molecule C5 modulators [Citation36].
![Figure 4. Development of small molecule C5 modulators [Citation36].](/cms/asset/58a6f3f5-dd15-4850-b7c5-09f977caa781/iett_a_2177532_f0004_oc.jpg)
In 2019, a study by Mishra & Rana was published, showing four approved non-steroidal anti-inflammatory drugs: sulindac, raloxifene, oxaprozin, and carprofen to directly bind to a recombinant human C5a fragment [Citation148]. A combination of cheminformatic and structure-based in silico approaches, all-atom molecular dynamics simulations, in addition to cell-free biophysical assays such as circular dichroism (CD), fluorescence spectroscopy, and isothermal titrational calorimetry (ITC) were employed to demonstrate this. They showed that all four ligands bound to C5a with sub-micromolar affinity (Kd ranging from 0.57 μM for sulindac to 0.71 μM for raloxifene). Upon addition of the ligands over 1–100 μM range, substantial conformational alterations in C5a were observed. At low concentrations (≤1 μM), moderate conformational changes occurred in the protein, whereas increasing the concentrations beyond 10 μM completely rearranged the native structure of C5a [Citation148]. However, none of the compounds demonstrated molecular aggregation over 1–100 μM range. Based on these results, it can be speculated that at low concentration, specific binding of the drugs at their distinct high affinity sites may induce an intrinsically disordered state within C5a, which acts as a transition state in triggering subsequent unfolding of the protein and thus might inhibit subsequent C5a-C5aR interactions. However, this speculation requires further corroboration from follow-up studies. It is also unknown whether these ligands may bind uncleaved C5, or whether such binding may prevent cleavage.
3.3. Other inhibitor classes
3.3.1. Therapeutic peptides
To date, more than 80 peptide-based therapeutics have gained approval from regulatory bodies and hundreds more are in active development worldwide [Citation149]. Short synthetic peptides offer an attractive therapeutic strategy for inhibiting proteins of interest and have advantages over larger biopharmaceuticals, including more facile administration and unique binding modes, which may be advantageous to those patients carrying mutations in target proteins that confer resistance to the treatment with biologics.
Zilucoplan, also known as RA 101495, is a small (15 residue) macrocyclic peptidomimetic inhibiting C5, which entered clinical trials several years ago [Citation150]. Zilucoplan works by binding to C5 with high affinity (Kd = 2.6 nM), inhibiting cleavage of C5 and subsequent formation of the MAC [Citation151]. Zilucoplan also binds to cleaved C5b and inhibits the interaction between C5b and C6, thereby further blocking MAC assembly [Citation152]. It has shown anti-inflammatory and cell protective activities in preclinical studies [Citation151]. Zilucoplan is currently in phase III clinical trials for gMG and ALS [Citation153]. Future work may expand the label for Zilucoplan, assessing its efficacy in the diseases that are driven by neuroinflammation beyond the neuromuscular junction.
In 2021, Elsen and co-workers reported a compelling structural biology and pharmacology study on allosteric modulation of C5 by knob domain peptides, the protrusions from bovine antibodies that have been credited for cows’ strong protection against microbes [Citation154]. This study was the first to investigate the molecular mechanisms of this novel disulfide-rich class of peptides (). Previously, the group showed that knob domain peptides derived from bovine antibodies have great potential for therapeutic applications [Citation155]. Using this approach, the group obtained four knob domain peptides: K8, K57, K92, and K149, which display high affinity binding to human C5. Reported equilibrium dissociation constants were of 17.8 nM for K8, 1.4 nM for K57, <0.6 nM for K92, and 15.5 nM for K149. For functional characterization of these peptides, complement assays for classical pathway and alternative pathway activation in human serum were performed, assessing C5b-9 neo-epitope formation and C5a release, in combination with orthogonal ELISAs measuring C3b and C9 deposition. These studies showed that K57 was a potent and fully efficacious inhibitor of C5 activation, preventing release of C5a, and deposition of C5b and C9, while showing no effect on C3b, which is upstream of C5. Other peptides displayed more nuanced mechanisms of action, ranging from high affinity ‘silent’ binding with no discernible effect on C5a release, formation of C5b neo-epitope or C9 deposition (K149), to allosteric effects exerted on C5 (K8 and K92). To elucidate the structural basis for the allosteric modulation of C5, the crystal structure of the C5-K8 complex was determined at a resolution of 2.3 Å (PDB code: 7AD7) [Citation154]. The solved structure shows the K8 peptide binding to a previously unreported regulatory site on C5 (). The peptide bound adopted a cysteine knot-like configuration, with its 3D structure constrained by three disulfide bonds. A crystal structure of the C5-K92 complex was also reported (PDB code: 7AD6). K92 adopted a topology highly like K8, but it was stapled with only two disulfide bonds. This work introduced knob domains as a new peptide modality, with yet unexplored therapeutic potential for the modulation of complement C5. It represents the first application of knob domain peptides as allosteric modulators of C5, expanding its scope for complement-targeted therapies. Peptides bearing knob domains can offer a range of advantages over the current macromolecular C5 inhibitors, and their development into therapeutics will hopefully come to fruition over the next years.
3.3.2. RNAi
A different approach to anti-C5 therapy is to prevent the molecule being expressed in the first place, rather than to block the protein or its activation fragments/receptors. Alnylam Pharmaceuticals have pioneered C5 RNAi (ribonucleic acid interference) therapy with the development of cemdisiran [Citation156]. This modality targets liver biosynthesis of C5 and in phase 1 clinical trials was reported to reduce serum hemolytic activity by 98% [Alnylam press release 6 December 2015, Alnylam Reports that ALN-CC5 Achieves Up to 99 Percent Knockdown of Serum C5 and Up to 98 Percent Inhibition of Serum Hemolytic Activity with Durability Supportive of Once Monthly and Possibly Once Quarterly Subcutaneous Dose Regimen]. The molecule was originally trialed in the hematological disease, paroxysmal nocturnal hemoglobinuria. Despite effective knockdown of C5, very low levels of the circulating molecule were sufficient to cause some lysis of erythrocytes. Generation of C5 at extra-hepatic sites might be a consideration for some diseases and is very pertinent to treatment of diseases of the CNS where C5 is known to be made locally, particularly in the context of inflammation. It remains to be seen whether RNAi therapies can be delivered locally to the CNS. Cemdisiran is currently in phase 3 clinical development as a combination with pozelimab for the treatment of generalized myasthenia gravis (NCT05070858); the dual combination has potential to drastically reduce the dose of anti-C5 antibody (pozelimab).
4. To cleave, or not to cleave?
Whilst the case for targeting C5 in ND is clear, the mechanism of action that will provide the best outcome for a given disease under the ND umbrella is yet to be determined. One of the key questions that remains is whether to develop a drug preventing C5 cleavage, thus removing both C5a and C5b, or merely block the activity of one of the cleaved components. The familiar story of the delicate balance between beneficial and deleterious effects of the immune system in resolving disease is very applicable here, and this is especially the case for C5a. The most potent anaphylatoxin in the complement system has a crucial role to play in clearing inflammatory deposits and pathogens, so it is little surprise that there is a fine line between the benefit of clearing pathogenic species and the harm of the inflammation caused by C5a itself.
Well-characterized and potent peptide antagonists for C5aR1 specifically were developed in 1999 and have been used as tools to study C5a signaling in many diseases, including neurodegenerative disease [Citation157]. In fact, one of these peptides, PMX205, has recently completed Phase I clinical trials for ALS and is in preclinical development for Parkinson’s Disease and Huntington’s Disease [Citation158,Citation159]. In an ALS rat model, treatment with the C5aR1 signaling inhibitor PMX205 resulted in longer survival and better end-stage motor-deficit scores [Citation62]. Reduced astroglia proliferation was also found in areas where motor neurons were degenerating, suggesting a role for C5a in disease pathogenesis.
There is also evidence of a pathogenic role for C5a in AD. Within in vitro studies, C5a was shown to exacerbate Aβ-induced neuroinflammation in BV-2 microglia, as inhibition of the receptor rescued cell proliferation and increased expression of inflammatory cytokines [Citation105]. This effect was ameliorated by PMX205 administration. Treatment with PMX53, a structurally related C5aR1 antagonist, also reduced Aβ-induced neurotoxicity in primary mouse neurons. Multiple in vivo studies from the Tenner group have demonstrated the beneficial effect of inhibiting C5a signaling in AD mouse models [Citation52,Citation53,Citation58,Citation62,Citation160].
In contrast to this, other studies demonstrate the importance of maintaining C5a signaling in other cases [Citation161–163]. Oral administration of C5a in a mouse model of transthyretin V30M (ATTR V30M) amyloidosis increased phagocytosis and clearance of amyloid deposits, whilst PMX53 increased amyloid deposition [Citation161]. This study, however, did not investigate the sensory-motor or autonomic impact of this reduced amyloid burden in the mice. C5a was shown to be detrimental to healing when administered either before or immediately after spinal cord injury but was able to mediate caspase-3-induced apoptosis and increase neurite outgrowth when administered 24 hours after the injury [Citation162].
Development of further specific and selective probes, along with thorough understanding of how current C5 inhibitors work, will play a huge role in elucidating exactly how we need to target C5, and when, in different neurodegenerative diseases.
5. Conclusion
Within this review, we present the evidence supporting C5 as a target for neurodegenerative disease, in addition to the known inhibitors either used clinically or in development. We also discuss the complex scenario surrounding C5a and its potential beneficial/negative role in the development and progression of neurodegenerative disease. Following on from this, we briefly discuss the current methods for measuring complement modulation and identify the next technological advances that would enable us to probe the therapeutic benefit of C5 inhibition further, increasing the translation of future drug discovery programs. We hope that this accelerates momentum in C5 drug development, particularly for neurodegenerative disease, and leads to new medicines providing lifesaving benefit for millions of patients globally.
6. Expert opinion
There is a compelling case for C5 as a target in a number of ND diseases, along with evidence that C5 blockade can also be a successful clinical strategy in disease, as exemplified by biologics such as eculizumab. Multiple hurdles exist along the path to validation however, including the risks associated with reduced infection response, the rapid turnover of complement proteins, the need for BBB penetrance and the challenge in developing drugs for such a complex protein, especially in the context of small molecule drug discovery.
Even if these problems can be addressed, there still remains the challenge of balancing beneficial and damaging effects of complement 5 and its cleaved components, a consideration that will surely be different across ND. The diverse and complex biology behind ND makes the development of one C5 targeting drug for all indications a likely impossibility. Understanding the right way, time and method of intervention at C5 in various ND is crucial to bringing real benefit to millions of patients worldwide. A crucial advance toward this goal would be the development of well-characterized, specific and BBB penetrant tools for C5 cleavage inhibition and MAC formation, adding to the peptides already available to study C5a receptor binding.
A strategy may well be to turn some of the above challenges to our advantage. The ready alteration of small molecules to adjust turnover could be harnessed to rapidly restore anti-infective function if it was found that constant C5 inhibition was not required to improve neurodegeneration for some indications. The longer half-life of biologics may well address challenges with compliance by allowing for less frequent dosing than a once or twice daily pill that may be forgotten by cognitively impaired patients if agents could be successfully targeted to the brain.
The underlying theme throughout this review is one of personalized medicine, a goal that the medical community is striving for across the whole of health care. There is likely no area where this could be more powerful than in ND. The ability to move away from the classical labels such as AD, PD etc., used to describe a collection of symptoms brought about by complex pathology, and accurately assess the needs of the degenerative microenvironment presented by an individual patient supported by a range of therapies with precise immunological and neurological effects on a molecular level, will revolutionize this field and may well be the key to effectively treating ND. To achieve this, a thorough understanding of inflammatory state linked to the degenerative microenvironment and accurate testing technologies associated with well-defined biomarkers is also needed. In a sense, a back-to-basics approach to ND research may be the key to achieving this, with a focus on unraveling the pathology behind ND in order to move forward in drug discovery.
Applying this approach to C5 as a target requires the kind of toolkit detailed above; a versatile, but well-characterized suite of biological and chemical tools that can be used to interrogate the role of C5 and its cleaved components within the brain milieu of diseased and aging patients. Appropriate cellular models may be able to expedite this, though this is also a grand challenge in neuroscience research. Interrogation of C5 as an ND target will likely benefit from new technologies such as co-cultures iPSC-derived brain cells and 3D cultures of mixed cell types (e.g. neurons, astrocytes, and microglia), where communication within the delicately balanced environment of the brain can be better approximated. For some indications elsewhere in the body, such as microbial infections and selected cancers where translation has successfully been achieved, these technologies certainly can bolster our understanding and provide confidence, but for neurological diseases such as ND or brain cancer they may prove crucial to bringing new drugs through the clinic.
Article highlights
The complement system is a crucial regulator of the body’s immune response
Neurodegenerative disease is one of the world’s greatest unmet clinical needs, with drug development efforts hampered by failure
Complement inhibition has huge promise as a therapeutic approach to neurodegenerative disease due to its role in neuroinflammation
There is a wealth of evidence supporting complement component 5 as a therapeutic target in neurodegenerative disease, along with demonstrations of its druggability
Challenges with targeting C5 include its abundance throughout the body, high turnover of complement proteins and access to C5 beyond the blood-brain barrier
Practically, a small molecule inhibitor may be more suitable for CNS diseases if the challenges associated with drugging complement and protein–protein interaction disruption can be overcome
The molecular mechanism by which C5 should be targeted is as yet undetermined and requires more investigation.
This box summarizes key points contained in the article.
Declaration of interests
Claire L. Harris has consulted for and collaborated with companies developing anti-complement therapeutics. CLH is also an employee of Gyroscope Therapeutics, a Novartis company; the opinions in this manuscript are entirely those of the authors and not necessarily those of Gyroscope Therapeutics nor Novartis.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov. 2015;14(12):857–877.
- Ricklin D, Hajishengallis G, Yang K, et al. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797.
- Bajic G, Degn SE, Thiel S, et al. Complement activation, regulation, and molecular basis for complement‐related diseases. EMBO J. 2015;34:2735–2757.
- Carroll MC, Fischer MB. Complement and the immune response. Curr Opin Immunol. 1997;9:64–69.
- Wagner E. Therapeutic potential of complement modulation. Nat Rev Drug Discov. 2010;9:43–56.
- Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. 2007;171:715–727.
- Hajishengallis G, Reis ES, Mastellos DC, et al. Novel mechanisms and functions of complement. Nat Immunol. 2017;18:1288–1298.
- Mollnes TE, Kirschfink M. Strategies of therapeutic complement inhibition. Mol Immunol. 2006;43:107–121.
- Dunkelberger JR. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20:34–50.
- Freeley S, Kemper C, Le FG. The “ins and outs” of complement-driven immune responses. Immunol Rev. 2016;274:16–32.
- Harris CL. Tailoring anti-complement therapeutics. Biochem Soc Trans. 2002;30:1019–1026.
- Reis ES, Mastellos D C, Yancopoulou D, et al. Applying complement therapeutics to rare diseases. Clin Imm. 2015;161(2):225–240.
- Kirschfink M. Targeting complement in therapy. Immunol Rev. 2001;180:177–189.
- Sjöberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol. 2009;30:83–90.
- Thurman JM, Holers VM. The Central Role of the Alternative Complement Pathway in Human Disease. J Immunol. 2006;176:1305–1310.
- Carroll M. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5:981–986.
- Hawksworth OA, Coulthard LG, Woodruff TM. Complement in the fundamental processes of the cell. Mol Immunol. 2017;84:17–25.
- Ricklin D. Complement in immune and inflammatory disorders: therapeutic interventions. J Immunol. 2013;190:3839–3847.
- McGeer P, Lee M, McGeer E. A review of human diseases caused or exacerbated by aberrant complement activation. Neurobiol Aging. 2016;52:12–22.
- Harris CL, Pouw RB, Kavanagh D, et al. Developments in anti-complement therapy; from disease to clinical trial. Mol Immunol. 2018;102:89–119.
- Mastellos DC, Ricklin D, Lambris JD. Clinical promise of next-generation complement therapeutics. Nat Rev Drug Discov. 2019;18:707–729.
- Ricklin D, Lambris JD. New milestones ahead in complement-targeted therapy. Semin Immunol. 2016;28:208–222.
- Morgan BP. The membrane attack complex as an inflammatory trigger. Immunobiology. 2016;221:747–751.
- Triantafilou K, Hughes TR, Triantafilou M, et al. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci. 2013;126:2903–2913.
- Fearon DT, Carter RH. The CD19/CR2/TAPA-1 Complex of B Lymphocytes: linking Natural to Acquired Immunity. Annu Rev Immunol. 1995;13:127–149.
- Taylor BES, Appelboom G, Zilinyi R, et al. Role of the complement cascade in cerebral aneurysm formation, growth, and rupture. Neuroimmunol Neuroinflamm. 2015;2:93–101.
- Schartz ND, Tenner AJ. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J Neuroinflammation. 2020;17:354.
- Harris CL, Heurich M, de Cordoba SR, et al. The complotype: dictating risk for inflammation and infection. Trends Immunol. 2012;33:513–521.
- de Córdoba SR, Harris CL, Morgan BP, et al. Lessons from functional and structural analyses of disease-associated genetic variants in the complement alternative pathway. Biochim Biophys Acta (BBA) - Mol Basis Dis. 2011;1812:12–22.
- Carpanini SM, Torvell M, Morgan BP. Therapeutic inhibition of the complement system in diseases of the central nervous system. Front Immunol. 2019;10:362.
- Chamberlain JL, Huda S, Whittam DH, et al. Role of complement and potential of complement inhibitors in myasthenia gravis and neuromyelitis optica spectrum disorders: a brief review. J Neurol. 2021;268:1643–1664.
- Iatropoulos P, Daina E, Curreri M, et al. Cluster analysis identifies distinct pathogenetic patterns in c3 glomerulopathies/immune complex–Mediated membranoproliferative GN. J Am Soc Nephrol. 2018;29:283–294.
- Wadhwa M, Prabhakar A, Anand JP, et al. Complement activation sustains neuroinflammation and deteriorates adult neurogenesis and spatial memory impairment in rat hippocampus following sleep deprivation. Brain Behav Immun. 2019;82:129–144.
- Coulthard LG, Woodruff TM. Is the Complement Activation Product C3a a Proinflammatory Molecule? Re-evaluating the Evidence and the Myth. J Immunol. 2015;194:3542–3548.
- Fredslund F, Laursen NS, Roversi P, et al. Structure of and influence of a tick complement inhibitor on human complement component 5. Nat Immunol. 2008;9:753–760.
- Jendza K, Kato M, Salcius M, et al. A small-molecule inhibitor of C5 complement protein. Nat Chem Biol. 2019;15:666–668.
- Law SKA, Dodds AW. The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein Sci. 1997;6:263–274.
- Jore MM, Johnson S, Sheppard D, et al. Structural basis for therapeutic inhibition of complement C5. Nat Struct Mol Biol. 2016;23:378–386.
- Li XX, Lee JD, Kemper C, et al. The complement receptor C5aR2: a powerful modulator of innate and adaptive immunity. J Immunol. 2019;202:3339–3348.
- Pandey S, Maharana J, Li XX, et al. Emerging Insights into the structure and function of complement C5a receptors. Trends Biochem Sci. 2020;45:693–705.
- Reis ES, Chen H, Sfyroera G, et al. C5a receptor-dependent cell activation by physiological concentrations of desarginated C5a: insights from a novel label-free cellular assay. J Immunol. 2012;189:4797–4805.
- Dmytrijuk A, Robie-Suh K, Cohen MH, et al. FDA report: eculizumab (Soliris®) for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Oncologist. 2008;13:993–1000.
- Licht C, Greenbaum LA, Muus P, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. 2015;87:1061–1073.
- Howard JF, Utsugisawa K, Benatar M, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol Internet]. 2017 [cited 2022 Jul 14];16:976–986. Available from http://www.thelancet.com/article/S1474442217303691/fulltext
- Bonifati DM, Kishore U. Role of complement in neurodegeneration and neuroinflammation. Mol Immunol. 2007;44:999–1010.
- Morgan BP. Complement in the pathogenesis of Alzheimer’s disease. Semin Immunopathol Springer Verlag; Jan. 2018;1:113–124.
- Hakobyan S, Harding K, Aiyaz M, et al. Complement biomarkers as predictors of disease progression in Alzheimer’s disease. J Alzheimers Dis. 2016;54:707–716.
- Krance SH, Wu CY, Zou Y, et al. The complement cascade in Alzheimer’s disease: a systematic review and meta-analysis. Mol Psychiatry. 2019;26:5532–5541.
- Benoit ME, Tenner AJ. Complement protein C1q-mediated neuroprotection is correlated with regulation of neuronal gene and MicroRNA expression. J Neurosci. 2011;31:3459–3469.
- Zhou J, Fonseca MI, Pisalyaput K, et al. Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer’s disease. J Neurochem. 2008;106:2080–2092.
- Stephan AH, Madison DV, Mateos JM, et al. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. 2013;33:13460–13474.
- Woodruff TM, Tenner Yazan AJ, Sanderson SD, et al. Alzheimer’s disease performance in murine models of pathology and enhances behavioral treatment with a C5aR antagonist decreases. J Immunol. 2009;183:1375–1383. Available from: http://www.jimmunol.org/content/183/2/1375
- Carvalho K, Schartz ND, Balderrama-Gutierrez G, et al. Modulation of C5a–C5aR1 signaling alters the dynamics of AD progression. J Neuroinflammation. 2022;19:1–21. Available from: https://jneuroinflammation.biomedcentral.com/articles/10.1186/s12974-022-02539-2
- Matsuoka Y, Picciano M, Malester B, et al. Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer’s disease. Am J Pathol. 2001;158:1345–1354.
- Fonseca MI, Chu SH, Berci AM, et al. Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer’s disease. J Neuroinflammation. 2011;8:1–12.
- Benoit ME, Clarke EV, Morgado P, et al. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J Immunol. 2012;188:5682–5693.
- Fonseca M, McGuire S, Counts S, et al. Complement activation fragment C5a receptors, CD88 and C5L2, are associated with neurofibrillary pathology. J Neuroinflammation. 2013;10:25.
- Ager RR, Fonseca MI, Chu SH, et al. Microglial C5aR (CD88) expression correlates with amyloid-β deposition in murine models of Alzheimer’s disease. J Neurochem. 2010;113:389–401.
- Linnartz B, Kopatz J, Tenner AJ, et al. Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J Neurosci. 2012;32:946–952.
- Fonseca MI, Chu SH, Hernandez MX, et al. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation. 2017;14:1–15.
- Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. 2010;112:733–743.
- Fonseca MI, Ager RR, Chu S-H, et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J Immunol. 2009;183:1375–1383.
- Bohlson SS, Fraser DA, Tenner AJ. Complement proteins C1q and MBL are pattern recognition molecules that signal immediate and long-term protective immune functions. Mol Immunol. 2007;44:33–43.
- Ryman D, Gao Y, Lamb BT. Genetic loci modulating amyloid-beta levels in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2008;29:1190–1198.
- Carpanini SM, Torvell M, Bevan RJ, et al. Terminal complement pathway activation drives synaptic loss in Alzheimer’s disease models. Acta Neuropathol Commun. 2022;10:1–16. Available from: https://actaneurocomms.biomedcentral.com/articles/10.1186/s40478-022-01404-w
- Mantovani S, Gordon R, Macmaw JK, et al. Elevation of the terminal complement activation products C5a and C5b-9 in ALS patient blood. J Neuroimmunol. 2014;276:213–218.
- Xie CB, Qin L, Li G, et al. Complement membrane attack complexes assemble NLRP3 inflammasomes triggering IL-1 activation of IFN-γ-primed human endothelium. Circ Res. 2019;124:1747–1759.
- Carpanini SM, Harwood JC, Baker E, et al. The impact of complement genes on the risk of late-onset Alzheimer’s disease. Genes (Basel). 2021;12:443.
- Jun G, Naj AC, Beecham GW, et al. Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol. 2010;67:1473–1484.
- Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099.
- Grewal RP, Morgan TE, Finch CE. C1qB and clusterin mRNA increase in association with neurodegeneration in sporadic amyotrophic lateral sclerosis. Neurosci Lett. 1999;271:65–67.
- Kawamata T, Akiyama H, Yamada T, et al. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol. 1992;140:691.
- Donnenfeld H, Kascsak RJ, Bartfeld H. Deposits of IgG and C3 in the spinal cord and motor cortex of ALS patients. J Neuroimmunol. 1984;6:51–57.
- Sta M, Rmr S-S, Casula M, et al. Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of complement activation. Neurobiol Dis. 2011;42:211–220.
- Yamada T, Mcgeer PL, Mcgeer EG. Lewy bodies in Parkinson’s disease are recognized by antibodies to complement proteins. Acta Neuropathol. 1992;84:100–104.
- McGeer PL, McGeer EG. Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism Relat Disord. 2004;10:S3–S7.
- Singhrao SK, Neal JW, Morgan BP, et al. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp Neurol. 1999;159:362–376.
- Hodges A, Strand AD, Aragaki AK, et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet. 2006;15:965–977. Available from: https://academic.oup.com/hmg/article/15/6/965/582386
- Woodruff TMTM, Costantini KJ, Crane JW, et al. The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J Immunol. 2008;181:8727–8734.
- Lee JD, Kumar V, Fung JNT, et al. Pharmacological inhibition of complement C5a-C5a1 receptor signalling ameliorates disease pathology in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Br J Pharmacol. 2017;174:689–699.
- Woodruff T, Woodruff TM, Crane JW, et al. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J. 2006;20:1407–1417. Available from: https://onlinelibrary.wiley.com/doi/full/10.1096/fj.05-5814com
- Moller T. Neuroinflammation in Huntington’s disease. J Neural Transm. 2010;117:1001–1008.
- Heneka MT, Carson MJ, Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. Available from: http://www.sciencedirect.com/science/article/pii/S1474442215700165
- Frank-Cannon TC, Alto LT, McAlpine FE, et al. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener. 2009;4:47.
- Hooten KG, Beers DR, Zhao W, et al. Protective and toxic neuroinflammation in amyotrophic lateral sclerosis. Neurotherapeutics. 2015;12:364–375. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4404435/pdf/13311_2014_Article_329.pdf
- DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139:136–153.
- Kölliker-Frers R, Udovin L, Otero-Losada M, et al. Neuroinflammation: an integrating overview of reactive-neuroimmune cell interactions in health and disease. Mediators Inflamm. 2021;2021:1–20.
- Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8: 382–397. Available from: http://www.sciencedirect.com/science/article/pii/S1474442209700626
- Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014;11:98.
- Harry GJ, Kraft AD. Neuroinflammation and microglia: considerations and approaches for neurotoxicity assessment. Expert Opin Drug Metab Toxicol. 2008;4: 1265–1277. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18798697
- Hong H, Kim BS, Im HI. Pathophysiological role of neuroinflammation in neurodegenerative diseases and psychiatric disorders. Int Neurourol J. 2016;20:S2–S7.
- Russo I, Barlati S, Bosetti F. Effects of neuroinflammation on the regenerative capacity of brain stem cells. J Neurochem. 2011;116:947–956.
- Alexander JJ. The complement cascade: yin-Yang in neuroinflammation–neuro-protection and -degeneration. J Neurochem. 2008;107:1169–1187.
- Orsini F, de Blasio D, Zangari R, et al. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front Cell Neurosci. 2014;0:380.
- Warwick CA, Keyes AL, Woodruff TM, et al. The complement cascade in the regulation of neuroinflammation, nociceptive sensitization, and pain. J Biol Chem. 2021;297:101085.
- Alawieh A, Langley EF, Weber S, et al. Identifying the role of complement in triggering neuroinflammation after traumatic brain injury. J Neurosci. 2018;38:2519–2532.
- Alexander JJ, Anderson AJ, Barnum SR, et al. The complement cascade: yin–Yang in neuroinflammation – neuro-protection and -degeneration. J Neurochem. 2008;107:1169–1187. Available from: https://onlinelibrary.wiley.com/doi/full/10.1111/j.1471-4159.2008.05668.x
- Mallah K, Couch C, Alshareef M, et al. Complement mediates neuroinflammation and cognitive decline at extended chronic time points after traumatic brain injury. Acta Neuropathol Commun. 2021;9:72.
- Hammad A, Westacott L, Zaben M. The role of the complement system in traumatic brain injury: a review. J Neuroinflammation. 2018;15:24.
- Brennan FH, Anderson AJ, Taylor SM, et al. Complement activation in the injured central nervous system: another dual-edged sword? J Neuroinflammation. 2012;9:1–13. Available from: https://jneuroinflammation.biomedcentral.com/articles/10.1186/1742-2094-9-137
- Crehan H, Hardy J, Pocock J. Microglia, Alzheimer‘s disease, and complement. Int J Alzheimers Dis. 2012;2012:983640.
- Laudisi F, Spreafico R, Evrard M, et al. Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1β release. J Immunol. 2013;191:1006–1010.
- Roy ER, Wang B, Wan YW, et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. 2020;130:1912–1930. Available from. DOI:10.1172/JCI133737DS1
- Möller T, Nolte C, Burger R, et al. Mechanisms of C5a and C3a complement fragment-induced [Ca2+](i) signaling in mouse microglia. J Neurosci. 1997;17:615–624.
- An XQ, Xi W, Gu CY, et al. Complement protein C5a enhances the β-amyloid-induced neuro-inflammatory response in microglia in Alzheimer’s disease. médecine/sciences. 2018;34:116–120.
- Hschner CNHK S. Complement factor C5a and epidermal growth factor trigger the activation of outward potassium currents in cultured murine microglia. Neuroscience. 1996;73:1109–1120.
- M Persson MPEHLR. The complement-derived anaphylatoxin C5a increases microglial GLT-1 expression and glutamate uptake in a TNF-alpha-independent manner. Eur J Neurosci. 2009;29:267–274.
- Miller AM, Stella N. Microglial cell migration stimulated by ATP and C5a involve distinct molecular mechanisms: quantification of migration by a novel near-infrared method. Glia. 2009;57:875–883.
- Yang C, Yang L, Liu Y. Soluble complement complex C5b-9 promotes microglia activation. J Neuroimmunol. 2014;267:16–19.
- Schatz-Jakobsen JA, Zhang Y, Johnson K, et al. Structural basis for eculizumab-mediated inhibition of the complement terminal pathway. J Immunol. 2016;197:337–344.
- Rother RP, Rollins SA, Mojcik CF, et al. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25:1256–1264.
- Muppidi S, Utsugisawa K, Benatar M, et al. Long-term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve. 2019;60:14–24.
- Pittock SJ, Lennon VA, McKeon A, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. 2013;12:554–562.
- Mantegazza R, O’Brien FL, Yountz M, et al. Consistent improvement with eculizumab across muscle groups in myasthenia gravis. Ann Clin Transl Neurol. 2020;7:1327–1339.
- Siddiqi ZA, Nowak RJ, Mozaffar T, et al. Eculizumab in refractory generalized myasthenia gravis previously treated with rituximab: subgroup analysis of REGAIN and its extension study. Muscle Nerve. 2021;64:662–669.
- Howard JF, Karam C, Yountz M, et al. Long-term efficacy of eculizumab in refractory generalized myasthenia gravis: responder analyses. Ann Clin Transl Neurol. 2021;8:1398–1407.
- Socié G, Caby-Tosi MP, Marantz JL, et al. Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome: 10-year pharmacovigilance analysis. Br J Haematol. 2019;185:297–310.
- Zuber J, Le Quintrec M, Krid S, et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant. 2012;12:3337–3354.
- Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368:2169–2181.
- Palace J, Wingerchuk DM, Fujihara K, et al. Benefits of eculizumab in AQP4+ neuromyelitis optica spectrum disorder: subgroup analyses of the randomized controlled phase 3 PREVENT trial. Mult Scler Relat Disord. 2021;47:102641.
- Zhu W, Wang Z, Hu S, et al. Human C5-specific single-chain variable fragment ameliorates brain injury in a model of NMOSD. Neurol Neuroimmunol Neuroinflammat. 2019;6:561.
- Nishimura JI, Ando K, Masuko M, et al. Tesidolumab (LFG316) for treatment of C5-variant patients with paroxysmal nocturnal hemoglobinuria. Haematologica. 2022;107:1483–1488.
- Roversi P, Ryffel B, Togbe D, et al. Bifunctional lipocalin ameliorates murine immune complex-induced acute lung injury. J Biol Chem. 2013;288:18789–18802.
- Nunn MA, Sharma A, Paesen GC, et al. Complement inhibitor of C5 activation from the soft tick ornithodoros moubata. J Immunol. 2005;174:2084–2091.
- Barratt-Due A, Thorgersen EB, Lindstad JK, et al. Ornithodoros moubata complement inhibitor is an equally effective C5 inhibitor in pigs and humans. J Immunol. 2011;187:4913–4919.
- Hepburn NNJ, Williams AS, Nunn MA, et al. In vivo characterization and therapeutic efficacy of a C5-specific inhibitor from the soft tick Ornithodoros moubata. J of Bio Chemistry. 2007;282:8292–8299.
- Sezin T, Murthy S, Attah C, et al. Dual inhibition of complement factor 5 and leukotriene B4 synergistically suppresses murine pemphigoid disease. JCI Insight. 2019;4:15.
- Eskandarpour M, Chen YH, Nunn MA, et al. Leukotriene B4 and its receptor in experimental autoimmune uveitis and in human retinal tissues: clinical severity and LTB4 dependence of retinal Th17 cells. Am J Pathol. 2021;191:320–334.
- Sadik CD, Rashid H, Hammers CM, et al. Evaluation of nomacopan for treatment of bullous pemphigoid: a phase 2a nonrandomized controlled trial. JAMA Dermatol. 2022;158:641–649.
- McNamara LA, Topaz N, Wang X, et al. High risk for invasive meningococcal disease among patients receiving eculizumab (Soliris) despite receipt of meningococcal vaccine. MMWR Morb Mortal Wkly Rep. 2019;66:734–737.
- Konar M, Granoff DM. Eculizumab treatment and impaired opsonophagocytic killing of meningococci by whole blood from immunized adults. Blood. 2017;130:891–899.
- Gäckler A, Kaulfuß M, Rohn H, et al. Failure of first meningococcal vaccination in patients with atypical haemolytic uraemic syndrome treated with eculizumab. Nephrol Dialysis Transplantation. 2020;35:298–303.
- Granoff DM, Kim H, Topaz N, et al. Differential effects of therapeutic complement inhibitors on serum bactericidal activity against non-groupable meningococcal isolates recovered from patients treated with eculizumab. Haematologica. 2019;104:e340–e344.
- Lewis LA, Ram S. Meningococcal disease and the complement system. Virulence. 2014;5:98–126.
- Muri L, Ispasanie E, Schubart A, et al. Alternative complement pathway inhibition abrogates pneumococcal opsonophagocytosis in vaccine-naïve, but not in vaccinated individuals. Front Immunol. 2021;12:732146.
- Ispasanie E, Muri L, Schubart A, et al. Alternative complement pathway inhibition does not abrogate meningococcal killing by serum of vaccinated individuals. Front Immunol. 2021;12:4198.
- Makurvet FD. Biologics vs. Small Molecules: Drug Costs and Patient Access. Med Drug Discov. 2021;9:100075.
- Levy AR, Chen P, Johnston K, et al. Quantifying the economic effects of ravulizumab versus eculizumab treatment in patients with atypical hemolytic uremic syndrome. J Med Econ. 2022;25:249–259.
- Wang Y, Johnston K, Popoff E, et al. A US cost-minimization model comparing ravulizumab versus eculizumab for the treatment of atypical hemolytic uremic syndrome. J Med Econ. 2020;23:1503–1515.
- Biologics: SS. An update and challenge of their pharmacokinetics. Curr Drug Metab. 2014;15:271–290.
- Mantaj J, Vllasaliu D. Recent advances in the oral delivery of biologics. Pharm J. 2020;304:7933.
- Oo C, Kalbag SS. Leveraging the attributes of biologics and small molecules, and releasing the bottlenecks: anew wave of revolution in drug development. Expert Review of Clinical Pharmacology. 2016;9:747–749.
- Smith AJ. New horizons in therapeutic antibody discovery: opportunities and challenges versus small-molecule therapeutics. SLAS Discovery. 2015;20:437–453.
- Adami G, Saag KG, Chapurlat RD, et al. Balancing benefits and risks in the era of biologics. Ther Adv Musculoskelet Dis. 2019;11.
- Zelek WM, Morgan BP. Targeting complement in neurodegeneration: challenges, risks, and strategies. Trends Pharmacol Sci. 2022;43:615–628.
- Scott DE, Bayly AR, Abell C, et al. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov. 2016;15:533–550.
- Zhang M, Yang XY, Tang W, et al. Discovery and structural modification of 1-phenyl-3-(1-phenylethyl)urea derivatives as inhibitors of complement. ACS Med Chem Lett. 2012;3:317–321.
- Mishra R, Rana S. A rational search for discovering potential neutraligands of human complement fragment 5a (hC5a). Bioorg Med Chem. 2019;27:115052.
- Wang L, Wang N, Zhang W, et al. Therapeutic peptides: current applications and future directions. Signal Transduction and Targeted Therapy. 2022;7:48.
- Albazli K, Kaminski HJ, Howard J. Complement inhibitor therapy for myasthenia gravis. Front Immunol. 2020;11:917.
- Ricardo A, Arata M, DeMarco S, et al. Preclinical evaluation of RA101495, a potent cyclic peptide inhibitor of C5 for the treatment of paroxysmal nocturnal hemoglobinuria. Blood. 2015;126:939–939.
- Ricardo A, Arata M, DeMarco SJ, et al. Development of RA101348, a potent cyclic peptide inhibitor of C5 for complement-mediated diseases. Blood. 2014;124:2936–2936.
- Howard JF, Nowak RJ, Wolfe GI, et al. Clinical effects of the self-administered subcutaneous complement inhibitor zilucoplan in patients with moderate to severe generalized myasthenia gravis: results of a phase 2 randomized, double-blind, placebo-controlled, multicenter clinical trial. JAMA Neurol. 2020;77:582–592.
- Macpherson A, Laabei M, Ahdash Z, et al. The allosteric modulation of complement c5 by knob domain peptides. Elife. 2021;10:1–49.
- Macpherson A, Scott-Tucker A, Spiliotopoulos A, et al. Isolation of antigen-specific, disulphide-rich knob domain peptides from bovine antibodies. PLoS Biol. 2020;18:e3000821.
- Kusner LL, Yucius K, Sengupta M, et al. Investigational RNAi therapeutic targeting C5 is efficacious in pre-clinical models of myasthenia gravis. Mol Ther Methods Clin Dev. 2019;13:484–492.
- Finch AM, Wong AK, Paczkowski NJ, et al. Low-molecular-weight peptidic and cyclic antagonists of the receptor for the complement factor C5a. J Med Chem. 1999;42:1965–1974.
- MND | brisbane, QLD | alsonex [Internet]. [cited 2022 Oct 11]. Available from: https://www.alsonex.com.au/The-technology.
- Taylor N. ANZCTR - Registration. Australia New Zealand Clinical Trial Registry. 2019. https://www.anzctr.org.au/Trial/Registration/Trial.
- Hernandez MXM, Namiranian P, Nguyen E, et al. C5a increases the injury to primary neurons elicited by fibrillar amyloid beta. ASN Neuro. 2017;9:1–10. Available from: https://us.sagepub.com/en-us/nam/open-access-at-sage
- Fella E, Sokratous K, Papacharalambous R, et al. Pharmacological stimulation of phagocytosis enhances amyloid plaque clearance; evidence from a transgenic mouse model of ATTR neuropathy. Front Mol Neurosci. 2017;10:138.
- Guo Q, Cheng J, Zhang J, et al. Delayed post-injury administration of C5a improves regeneration and functional recovery after spinal cord injury in mice. Clin Exp Immunol. 2013;174:318–325.
- Mukherjee P, Thomas S, Pasinetti GM. Complement anaphylatoxin C5a neuroprotects through regulation of glutamate receptor subunit 2 in vitro and in vivo. J Neuroinflammation. 2008;5:1–7.