
ABSTRACT
Introduction
Human prion diseases are heterogeneous, and often rapidly progressive, transmissible neurodegenerative disorders associated with misfolded prion protein (PrP) aggregation and self-propagation. Despite their rarity, prion diseases comprise a broad spectrum of phenotypic variants determined at the molecular level by different conformers of misfolded PrP and host genotype variability. Moreover, they uniquely occur in idiopathic, genetically determined, and acquired forms with distinct etiologies.
Area covered
This review provides an up-to-date overview of potential therapeutic targets in prion diseases and the main results obtained in cell and animal models and human trials. The open issues and challenges associated with developing effective therapies and informative clinical trials are also discussed.
Expert opinion
Currently tested therapeutic strategies target the cellular PrP to prevent the formation of misfolded PrP or to favor its elimination. Among them, passive immunization and gene therapy with antisense oligonucleotides against prion protein mRNA are the most promising. However, the disease’s rarity, heterogeneity, and rapid progression profoundly frustrate the successful undertaking of well-powered therapeutic trials and patient identification in the asymptomatic or early stage before the development of significant brain damage. Thus, the most promising therapeutic goal to date is preventing or delaying phenoconversion in carriers of pathogenic mutations by lowering prion protein expression.
1. Introduction
Prion diseases are a group of typically rapidly progressive and transmissible neurodegenerative disorders affecting humans and other mammalian species. The pathogenic hallmark is the deposition of pathological conformers of the prion protein (PrP), mainly in the central and less frequently in the peripheral nervous system and organs such as spleen, tonsils, appendix, etc. In humans, prion diseases are rare, with an annual mortality rate of about 2 cases per 1 million population [Citation1].
Current terminology and classification of human prion diseases use historical eponyms (Creutzfeldt – Jakob disease, CJD, and Gerstmann – Sträussler – Scheinker disease, GSS) and clinicopathological or protein chemistry descriptions (Fatal Familial Insomnia, FFI, and Variably Protease-Sensitive Prionopathy, VPSPr). These terms are combined with the designation of the putative etiology (i.e. sporadic, genetic, or acquired). Finally, a terminology mainly referring to molecular features is used to classify CJD, the most common human prion disease, in several distinct subtypes (see below).
Based on pathological features, two main groups of human prion disease are distinguished. The first, the most prevalent, including CJD and most FFI cases, is characterized by widespread spongiform change involving multiple brain areas. The second comprises the PrP-amyloidoses, including GSS and other rare genetic prion diseases characterized by parenchymal and vessel-related cerebral amyloid angiopathy (CAA), with or without systemic PrP deposits [Citation2]. Notably, this significant difference in the distinctive histopathology of the two groups (spongiform change vs. amyloid plaques) strongly correlates with the physicochemical properties of abnormal PrP (PrPSc) aggregates. In spongiform encephalopathy, proteinase K (PK) treatment of brain homogenates generates N-terminally truncated PrPSc fragments retaining the glycophosphatidylinositol (GPI) anchor and the glycosylation sites referred to as PrP27–30. In contrast, in the PrP-amyloidoses, PK-resistant PrPSc comprises unglycosylated anchorless fragments ragged at both N- and C- termini [Citation2]. As an exception to this general rule, a subgroup of rare prion disorders, including VPSPr and some GSS cases linked to the PRPN P102L-129M haplotype are characterized by the coexistence of both PrPSc fragments correlating with a mixed phenotype that includes both spongiform change and PrPSc amyloid deposits.
Sporadic prion disease, which accounts for more than 80% of all human cases, is thought to originate spontaneously from unknown stochastic cellular events leading to the conversion of the cellular prion protein (PrPC) into PrPSc. The genetic form is causally linked to autosomal-dominant pathogenic mutations in the prion protein gene (PRNP) and contributes to about 10–15% of cases. Finally, the acquired form originates either from the accidental human-to-human transmission though surgical/medical procedures as in iatrogenic CJD (iCJD), blood transfusions as in variant CJD (vCJD), and ritual cannibalism as in kuru, or from the ingestion of bovine spongiform encephalopathy (BSE)-derived prions as zoonosis (vCJD).
At the molecular level, two types of PrP27–30 with distinct physicochemical properties (i.e. type 1 and type 2), in combination with the PRNP genotype at the polymorphic codon 129 (coding for methionine/M or valine/V), largely determine the CJD phenotype, allowing a histo-molecular disease classification [Citation3,Citation4]. Six molecular groups, MM1, MV1, VV1, MM2, MV2, and VV2, correlating with the clinical and neuropathological phenotypic heterogeneity of sporadic CJD (sCJD), were initially defined. However, given that two of these molecular combinations were found to be associated with two distinct clinicopathological subtypes rather than one, ‘histopathological’ hallmarks were also introduced. Accordingly, the MM2 group includes a ‘cortical’ (MM2C) and a ‘thalamic’ variant (MM2T), and the MV2 group comprises a prevalent subtype with cerebellar kuru-type amyloid plaques (MV2K subtype) and a rare variant phenotypically indistinguishable from the MM2C group (MV2C subtype). Interestingly, the classification also applies to the prevalent genetic forms of CJD (gCJD), where the histotypes mainly depend on the codon 129 polymorphism in the mutated allele/PrPSc type 1 or 2 combinations rather than on the mutation per se [Citation5]. The classification of human prion diseases, including the CJD subtypes, is summarized in .
Table 1. Summary of human prion disorders, their prevalence and main histo-molecular features.
1.1. Molecular pathogenesis and potential therapeutic targets
The primary mechanism governing prion disease pathogenesis in all disease forms is the misfolding of physiologically expressed PrPC into the abnormal isoform PrPSc that is partially and variably resistant to protease digestion and insoluble in detergents [Citation6]. Prion protein misfolding prompts PrPSc accumulation in highly ordered aggregates that mediate neurotoxicity [Citation7].
PrPC expression is the absolute requirement for prion formation and replication since mice devoid of PrPC do not develop the disease after experimental injection of prion-infected tissue homogenates [Citation8]. Human PrPC is encoded by the PRNP located in the short arm of chromosome 20 and in mature form includes 208 amino acids (PrP23–231). After synthesis as a 253 amino acid precursor, N- and C-terminal signal peptides are removed, and chaperones guide the nascent PrPC into the endoplasmic reticulum (ER) and Golgi apparatus to fold correctly and undergo post-translational modifications, including the variable attachment of glycans at residues Asn181 and Asn197, and the addition of a GPI anchor to the C-terminus. Following these steps, the PrPC fold in absence of Cu2+ comprises an intrinsically disordered N-terminal domain and a structured, predominantly α-helical C-terminal domain [Citation9].
There is incomplete knowledge about the key events triggering PrPSc formation. Pathogenic PRNP mutations and/or external factors such as oxidative stress, age, and inflammatory insults may all play a role [Citation10]. Failure of cellular quality control mechanisms involving molecular chaperones, such as heat shock proteins, that normally favor the elimination of misfolded species by refolding or degradation through the ubiquitin-proteasome quality control (UPS) [Citation11,Citation12], may also promote the stabilization of soluble oligomers, leading to the formation of higher-ordered structures [Citation10].
There is growing convergence on seeded nucleation being the mechanism underlying prion aggregation. During this process, PrPSc oligomers incorporate and convert PrPC monomers, growing in size and eventually generating protofibrils and fibrils [Citation13]. Then, fibril fragmentation creates new nucleation sites that amplify the reaction by incorporating new PrPC monomers [Citation7]. Using animal-derived prions (i.e. 263K and anchorless RML), recent cryo-EM studies demonstrated the assembly of PrPSc fibrils with parallel in-register intermolecular β sheets, in which each misfolded PrPSc monomer represents one rung of the fibril core and templating surface for incoming monomers at fibril ends, where prion growth occurs [Citation14].
In physiologic conditions, PrPC is localized primarily on the cell surface, where it is GPI-anchored in the lipid rafts of the membrane. Evidence suggests that this site is critically involved in converting PrPC into PrPSc [Citation15]. As part of this process, PrPC could act as a toxicity-inducing receptor or trap and concentrate PrPSc molecules on the cell surface that induce toxic effects by compromising the integrity of cell membranes [Citation16]. Additionally, given the attachment to the membrane through the GPI moiety, during the conversion phase, PrPC twists along one side of the fibril while binding to membranes, causing its distortion and promoting cellular damage [Citation14]. Besides the cell surface, PrPC conversion can also occur shortly after internalization during an endocytic process. In such conditions, the internalized PrPSc aggregates can foster the seeded conversion by blocking the UPS system and activating the unfolded protein response (UPR) signaling pathways related to ER stress [Citation17]. As a result, the impairment of protein synthesis leads to synaptic dysfunction and neuronal loss, which may also depend on the activation of the apoptotic cascade stimulated by the saturation of the UPS system [Citation18] (). Additionally, protracted UPR response leads to deacylation and degradation of PIKfyve, reducing phosphoinositide diphosphate PI(3,5)P2 levels. This, in turn, alters endosome maturation, resulting in enlarged endolysosomes that eventually become intracellular vacuoles reminiscent of prion-induced spongiosis [Citation19].
Figure 1. Schematic representation of the putative mechanisms of PrP aggregate formation and clearance and mechanism of action of different anti-prion compounds. After synthesis, the nascent PrPC enters the lumen of the ER, where the N-terminal signal peptide is removed. Then, the protein moves to the Golgi apparatus to undergo post-translational modifications. Once fully folded, PrPC moves along the secretory pathway to the outer leaflet of the plasma membrane, where it anchors via the C-terminal GPI moiety to lipid rafts. Both cell surface and endosomal pathways are putative sites of PrPC misfolding and oligomerization. Endocytosed PrPC and misfolded PrP are rapidly recycled into the plasma membrane or removed through macroautophagy following the extracellular release of exosomes. Internalized aggregates promote the ER stress resulting in the activation of UPR and ERAD response, which, in turn, induce the degradation of misfolded PrP by the UPS system. Red solid lines indicate potential therapeutic intervention points, including ASOs targeting PrP mRNA, prion clearance (antibody therapy), prion replication (PPS and Quinacrine), and oligomers stabilization (Doxycycline). Blue arrows and dotted lines represent new potential sites of intervention (UPR and ERAD response enhancement; exosome blockade). Black solid arrows indicate molecular steps (endocytosis of PrPC; conversion of PrPC to PrPSc; macroautophagy pathways). Red solid arrows indicate the cascade triggered by the accumulation of PrP aggregates.
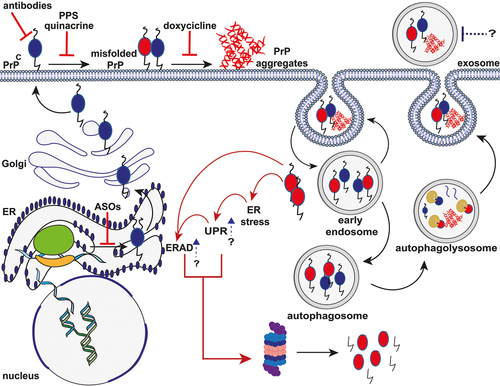
All evidence above indicates that PrPC and PrPSc are both potential therapeutic candidates for prion disease because of their pivotal role in disease pathogenesis. Strategies to deplete the PrPC substrate or prevent recruitment and contact between PrPC and PrPSc should hinder the conversion mechanism. Additionally, an intervention targeting cell-surface PrPC should prevent its binding with the neurotoxic pathologic aggregates [Citation7]. Given the current modeling of prion strain selection [Citation20], removing PrPC as substrate, compared to targeting PrPSc, has the potential additional advantage of avoiding the risk of selecting a resistant PrPSc conformer generating a novel dominant strain.
Another critical point in the pathogenesis of prion disease is the ability of protein aggregates to propagate within the central nervous system (CNS), spreading from cell to cell and between different brain areas [Citation21]. Different mechanisms may explain how PrPSc aggregates enter and spread between cells, including exosomes [Citation22], nanotubes [Citation23], and receptor-mediated internalization [Citation21]. All these mechanisms are potential targets of therapeutic interventions aiming to slow the diffusion of the disease to different brain regions. In this regard, it is noteworthy that the susceptibility of cellular populations and brain regions to prion strains is variable and leads to distinguishable disorders with unique phenotypic features [Citation24]. At the cellular level, there is an involvement of both neurons and glial cells. Evidence from transcriptomic studies indicated that microglial and astrocytic activation occurs since the early phase of the disease, and changes in these cell groups precede neuronal damage. Microglial cells are involved in either neuroprotection by clearing PrPSc or neurodegeneration by prompting the release of inflammatory cytokines that induce neuronal apoptosis [Citation25]. Therefore, modulating the expression of cell-specific cofactors related to neuroinflammatory pathways, such as receptors and chaperones, could be an additional therapeutic strategy [Citation26].
1.2. History of treatment failures
In the last 30 years, scientists tested several chemical compounds for their potential anti-prion effect in cell cultures and animal models of prion disease. However, only a few eventually reached a trial in patients affected by human prion disease. Moreover, most of these studies included a limited number of individuals and were non-comparable in their experimental design since they comprised case reports, observational and case-control studies, as well as randomized placebo-controlled clinical trials ().
Table 2. Summary of anti-prion molecules tested by clinical studies on human prion diseases.
Pursuing the ‘slow virus’ hypothesis of CJD etiology, initial attempts to treat CJD involved the antiviral Amantadine, a drug initially developed against the influenza virus. In anecdotal CJD case reports, the drug positively affected survival time [Citation27], or clinical symptoms [Citation28,Citation29]. However, other single-case observations did not confirm the positive effect [Citation30,Citation31]. In a study by Terzano and colleagues, Amantadine treatment did not increase survival in 4 CJD patients compared with 5 CJD individuals undergoing only palliative/supportive care [Citation32]. As the only distinguishing feature between groups, subjects treated with Amantadine showed a transient improvement in wakefulness [Citation32]. A subsequent study confirmed the lack of a positive effect in 8 CJD patients [Citation33]. Additionally, approximately 35 other CJD patients were treated unsuccessfully with Amantadine [Citation34]. Similarly, antiviral drugs such as Acyclovir and Interferon failed to show a positive clinical effect in CJD case reports [Citation35–37].
Flupirtine, a pyridine derivative with analgesic and anti-apoptotic properties [Citation38], reduced cell death in neuronal cells treated in vitro with the neurotoxic PrP(106–126) fragment [Citation39]. The drug was then evaluated in a double-blind prospective clinical trial including 28 sCJD patients (13 treatment arm, 15 placebo arm). The treatment demonstrated some beneficial effects on cognition but failed to prolong survival [Citation40].
In vitro and animal studies demonstrated that pentosan polysulphate (PPS), a polyanion compound, reduced PrPSc formation [Citation41,Citation42]. Intra-cerebroventricular administration of PPS in transgenic mice intracerebrally infected with different prion strains (263K, RML, Fukuoka-1) prolonged incubation time. Consistently, the drug reduced abnormal PrP deposition, the extent of neuropathologic change (atrophy, neuronal loss), and the associated infectivity [Citation42]. Two different observational studies in the United Kingdom (UK) and Japan evaluated PSS in 18 individuals affected by prion disorders, including 6 sCJD (all from the Japanese study), 4 iCJD (2 from each study), 3 GSS-P102L (2 from the UK, one from Japan) and 3 vCJD patients (all from the British study) [Citation43,Citation44]. Both studies reported a likely extended survival in treated patients. Moreover, the treatment decreased the PrPSc/total PrP and the oligomeric PrP/total PrP ratios in some CJD cases in one study [Citation45]. However, PPS did not significantly affect neurological functions, and brain postmortem pathology [Citation43,Citation44].
One of the most studied anti-prion agents is Quinacrine, an antiprotozoal drug used in the past as an antimalarial. The drug targets the conversion of PrPC into PrPSc [Citation46]. In early 2000s, initial evidence demonstrated its efficacy in inhibiting PrPSc accumulation in scrapie-infected neuroblastoma cells [Citation47,Citation48]. In subsequent animal studies, Quinacrine had mixed results. Some reported no effect on incubation time and disease duration [Citation49,Citation50], while others showed an extension of both incubation time and survival after prion injection [Citation51]. A first study involving 4 CJD patients documented minimal and transient improvement of arousal and integrative functions [Citation52]. In the three following independent studies with case-control [Citation53], observational [Citation54], and randomized trial designs [Citation55], which included 95 individuals undergoing active Quinacrine treatment, there were no significant clinical effects. Of note, the study populations were significantly different among studies: one included sCJD patients (n = 23) [Citation55], another both sCJD (n = 30) and vCJD (n = 2) cases [Citation53], whereas the last included patients of various etiologies (sporadic, iatrogenic, inherited, and vCJD) [Citation54]. Some authors attributed the inefficacy of Quinacrine in vivo to the selection of drug-resistant prions after continuous treatment exposure [Citation56].
After the Quinacrine trial failures, Doxycycline, a tetracycline-class antibiotic commonly used to treat bacteria and parasite infections, received rising scientific attention. Doxycycline acts as an anti-prion drug by binding PrPSc conformers, hindering their assembly into amyloid fibrils, and sensitizing their proteolytic degradation [Citation57,Citation58]. In scrapie-infected (263K) transgenic mice, Doxycycline was effective in prolonging the incubation time independently from the route of administration [Citation59]. In humans, a randomized controlled trial conducted in France and Italy evaluated the use of Doxycycline in 121 individuals affected by sCJD, iCJD, vCJD, and gCJD (62 included in the treatment arm, and 59 in the placebo group) [Citation60]. The trial was interrupted ahead of schedule after an interim analysis revealed no superiority of Doxycycline compared to the placebo. In 2018, a combined analysis including a group of 12 patients (7 under treatment and 5 controls) undergoing a randomized, double-blind controlled trial and a separate group of 88 sCJD cases (55 treated with Doxycycline and 33 controls) revealed a slightly statistically significant prolongation of survival [Citation61]. This effect was more pronounced in subjects carrying the genotype MM at PRNP codon 129 [Citation61]. A clinical trial with Doxycycline (DOXIFF) involving asymptomatic individuals that bear the PRNP D178N-129M haplotype linked to FFI is ongoing in Italy [Citation62]. The first results are expected by the end of 2023. Mutation carriers will receive doxycycline 100 mg/day (200 mg/day since May 2019) over a 10 year-period [Citation63]. The trial represents the first attempt to assess the feasibility of preventive treatment in asymptomatic individuals with a high risk of developing prion disease.
1.3. Promising treatments
1.3.1. Immunotherapy and the development of anti-PrP antibodies
Immunotherapy represents a promising approach for the treatment of a variety of neurological disorders, including neurodegenerative diseases. Accordingly, in the last decade, enormous efforts have been made to develop immunotherapies for Alzheimer’s (AD) and Parkinson’s (PD) related proteinopathies [Citation64,Citation65].
In the prion field, the phenomenon of self-tolerance, determined by the widespread PrPC expression in tissues and organs, hindered the application of active immunotherapy (i.e. the production of anti-PrP antibodies by the host following vaccination) [Citation66]. Nonetheless, a wide range of small molecules, including truncated or modified recombinant PrP peptides, PrP dimers, and heterologous PrP peptides, have been tested in vitro for their ability to elicit an anti-PrPC immunity [Citation67], with contrasting results [Citation68–70]. Even if some studies documented a delayed symptom onset in vaccinated mice, the overall effect of active immunization was marginal.
Given the difficulty of developing vaccination strategies breaking the self-tolerance efficiently, researchers pursued passive immunization (i.e. administering a pre-made antibody able to block or modulate a specific target). However, the development of antibodies that selectively bind PrPSc proved challenging. Therefore, most researchers focused on targeting PrPC. Indeed, PrPSc requires PrPC for its propagation [Citation71], and limiting the conversion of PrPC into PrPSc prevents neurotoxicity [Citation72].
In the first study of this kind, conducted in the mid-80s, a polyclonal rabbit PrP antiserum raised against PrP27–30 reduced prion infectivity in vitro [Citation73,Citation74]. Later, the development of several monoclonal antibodies by immunization of homozygous PrPC knock-out mice [Citation8] allowed the targeting of various epitopes and the search for the most favorable efficacy-side effect profile. Transgenic expression of the Mu heavy chain of anti-PrPC antibody 6H4 provided the first evidence of neuroprotection against prions in vivo [Citation75]. The analysis of the efficacy-side effect profile of the anti-prion antibodies in prion-infected cellular models and, to a lesser extent, in prion-infected mice showed a significant association with the target epitope [Citation76]. Antibodies targeting the globular domain of PrPC (POM1, D18, ICSM18) reduced PrPSc formation [Citation75,Citation77], but also induced intracellular toxicity [Citation78,Citation79], likely by disrupting the intramolecular docking between N- and C-terminal domains of PrPC [Citation80]. Specifically, a recent study revealed that the formation of an intramolecular R208-H140 hydrogen bond (‘H-latch’) that alters the flexibility of the α2–α3 and β2–α2 loops of PrPC mediates the neurotoxic effect of these antibodies [Citation81]. Accordingly, abolishing the H-latch formation confers resistance to POM1 toxicity [Citation81].
In contrast, antibodies raised against the flexible tail of PrPC, including those targeting the octapeptide repeat region, did not show significant toxicity. Yet, most of these antibodies conferred neuroprotection against prions [Citation66,Citation79]. As a significant exception, 4H11, an anti-PrPC antibody targeting the octapeptide repeat region, caused neuronal and glial toxicity associated with behavioral deficits in BSE-infected mice with no therapeutic effects [Citation82]. Moreover, the safety profile of the ICSM18 antibody, targeting the PrPC α1 region in the globular domain, thus expected to be neurotoxic, has been the object of intense debate. The antibody effectively reduced PrPSc levels and prion infectivity in a mouse model [Citation83]. However, while the intracerebral stereotactic injection of 2 μg of ICSM18 was well tolerated [Citation84], a dose escalation up to 6 μg showed a dose-dependent neurotoxic effect with an estimated 3.1 μg for the upper limit of the ICSM18 intracerebrally injected safe dose [Citation79]. These data illustrate that anti-prion antibodies’ efficacy and toxicity profiles are complex and likely depend on multiple intrinsic (e.g. targeted epitope) and extrinsic (e.g. route of administration, dosage) factors.
Nonetheless, the impressive therapeutic effect of ICSM18 in mice infected by intraperitoneal injection of RML prions with a survival time >500 days with no clinical sign of disease (N.B, when the administration started after 7 or 30 days after injection) [Citation83], prompted the first human trial using the humanized version of ICSM-18, PRN100. The study included six patients (5 sCJD and one iCJD) who received PRN100 through repeated intravenous administrations [Citation85]. PRN100 was not effective in modifying the clinical trajectory of the disease. Still, in 1 out of 2 cases undergoing postmortem brain examination (i.e. the only iCJD case), neuropathologic examination suggested that treatment could clear disease-associated PrP. Remarkably, no treatment-related clinical side effects were reported, and no evidence of subclinical neurotoxicity was noted at the neuropathologic examination, although PRN100 reached therapeutic levels only in 4 out of 6 individuals. Considering the small number of patients included, these are encouraging results and highlight the feasibility of antibody-based clinical trials for CJD. However, it should be noted that in mice, ICSM18 was ineffective in blocking prion disease when the administration started after clinical onset [Citation84]: this could at least partially explain the preliminary observations in CJD patients. Given these findings, the late administration during the symptomatic phase should also be considered a possible cause of the negative outcome of previous trials with other drugs (e.g. Doxycycline, Quinacrine, etc.).
1.3.2. Therapeutic gene modulation
Several lines of evidence suggest that lowering the PrPC levels could represent an effective disease-modifying therapy. Indeed, the conversion of PrPC into PrPSc is the critical molecular event for disease development and propagation after infection, as demonstrated by conditional and constitutive knock-out models [Citation72,Citation86,Citation87].
Gene therapy in prion disease may be especially suitable for patients or presymptomatic individuals carrying heterozygous pathogenic PRNP mutations with variable penetrance associated with gCJD, FFI, GSS, and PrP-CAA with or without systemic amyloidosis phenotypes, which is supported by the successful development of genetic modulation for other genetic neurodegenerative disorders [Citation88,Citation89]. Although no trials adopting therapeutic gene modulation for prion disease are ongoing, a wealth of evidence has been collected to answer the following preliminary questions: 1) can humans tolerate PrPC lowering without relevant side effects? 2) What is the best modality to lower PrPC expression?
Abolishing PrPC expression in mice, goats, and cattle did not induce CNS pathology or a clinical phenotype indicating abnormalities of CNS functions [Citation90–92] but showed an effect on peripheral myelin maintenance [Citation93,Citation94]. The reduction of PrPC was linked to a late onset peripheral neuropathy, demyelinating type, in homozygous knock-out mice (Prnp−/−) but not in heterozygous (Prnp+/-) [Citation93]. In humans, research has focused on loss-of-function genetic variants. Interestingly, the finding in apparently healthy mid-age adults of heterozygous loss of function truncating mutation localized in the N-terminal region (before codon 131) indicates that a 50% reduction of PrPC expression might be well-tolerated [Citation95].
Additionally, inhibitory drugs could successfully target loss-of-function variants involving essential genes [Citation96], indicating that lowering PrPC expression represents a potential therapeutic approach even if PRNP loss-of-function has harmful effects.
Remarkably, although the minimal effective PrPC lowering necessary to accomplish clinical outcomes remains elusive, in vitro findings of dose-dependent efficacy were recently replicated in vivo. Indeed, a study reported that a transient pharmacologic 21% PrPC knockdown (i.e. treatment administered twice at −14 and 76 days post-inoculation during the presymptomatic phase) is sufficient to extend survival in RML prion-infected mice [Citation97]. This finding aligns with evidence that a 50% reduction of PrPC expression in Prnp+/- mice are beneficial against prion infection.
Currently, there is no univocal therapeutic method to lower the PrPC levels. The approaches investigated include RNA interference [Citation98], the use of small molecules identified on the surface of human glioblastoma (T98G) and neuroblastoma (IMR32) cells [Citation99], adeno-associated virus vector type 2 encoding a short hairpin RNA targeting Prnp mRNA (AAV2-PrP-shRNA) [Citation100], and antisense oligonucleotides (ASOs) targeting PrP RNA [Citation101]. In recent years, different ASOs have been designed and extensively studied [Citation97,Citation102,Citation103]. Researchers found that intracerebroventricular administration of PrP-lowering ASOs extends survival by 61–98% in RML prion-infected mice [Citation102]. Treatment efficacy was strictly dependent on the ability of specific ASOs to lower PrPC expression (i.e. aptameric interaction between ASOs and PrP was ineffective) as evaluated by CSF PrP concentrations. Even a single administered dose close to the clinical disease onset, or even after the appearance of neuropathologic change, showed a benefit [Citation102]. A subsequent study reported the efficacy of ASOs against four additional strains at different time points, even during the symptomatic phase of the disease [Citation97]. However, the administration of some ASOs induced a subacute decline requiring euthanasia in a subgroup of mice with established prion neuropathology [Citation97,Citation101,Citation102]. The finding is apparently uncoupled from PrP-lowering since it was also observed with ASOs not targeting PrP RNA [Citation103]. Before evaluating the translation of ASO-based therapy in human trials, the mechanisms underlying this phenomenon must be clarified and prevented.
1.4. Open issues, challenges, and possible solutions
1.4.1. Low disease incidence and the design of well-powered clinical trials
An annual incidence of approximately 2 cases per million people makes prion disease an exceedingly rare condition [Citation95,Citation104]. Besides, the prevalence differs according to the etiology and phenotype. Finally, prion disease is a phenotypically heterogeneous disorder, including several disease subtypes. All these features prevent the inclusion of sizable homogeneous patient cohorts in clinical trials [Citation76]. To date, all therapeutic trials, mainly designed as single-center studies and including small patient cohorts (<50 individuals), have focused on symptomatic individuals diagnosed with prion disease. In such a context, an early and accurate diagnosis would be pivotal since diagnostic delay could be primarily responsible for the failure of anti-prion therapies. Indeed, several studies demonstrated that the effectiveness of therapies increases with early treatment. However, despite the significant advances in the development and validation of diagnostic tests, the diagnosis of prion disease is still reached with significant delay attended by severe brain damage. To overcome this limit, identifying groups at increased risk of developing the disease and eventually close to the clinical onset would be an option for future trials. However, no specific factors are known to predict with a high level of certainty the age of disease onset [Citation105,Citation106], although etiology and genetic host factors are known to play a role in the age distribution of clinical onset [Citation3,Citation5,Citation107], and lifetime disease risk [Citation95]. The recent development of gene modulation therapeutic strategies has raised interest in asymptomatic PRNP mutation carriers. A recent international collaborative study including 1094 individuals with highly penetrant PRNP mutations (i.e. E200K, D178N, P102L) found a broad and not predictable variability in age at onset [Citation106]. Based on the age-dependent hazards, it has been estimated that randomized preventive trials would require hundreds (or even thousands) of at-risk individuals to be statistically powered for an endpoint of clinical onset [Citation106]. Given the exceedingly high number of required individuals for a feasible preventive trial, post-marketing trials possibly using historical controls appear to be the best option [Citation106].
1.4.2. Heterogeneity of disease (strains, subtypes, and mixed phenotypes)
Prion disease includes a highly heterogeneous spectrum of phenotypes determined at the molecular level by different PrPSc conformers and host genotype variability. Focusing on sCJD, the most common prion disorder, the current classification recognizes six clinicopathological subtypes (MM1/MV1, VV2, MV2K, MM2C, MM2T, VV1) [Citation3,Citation24]. Five of them behaved as prion strains after transmission to syngeneic hosts, producing a distinctive phenotype [Citation108]. Evidence suggests that the same human prion strains are also responsible for genetic and iatrogenic CJD forms [Citation2]. Interestingly, the combination of two or more subtypes co-occurs within the same brain in about one-third of sCJD cases [Citation4]. However, most demonstrate a predominant subtype since the non-dominant one usually shows a focal (regional) distribution [Citation109].
Identifying these subtypes accurately in vivo has multiple implications. Firstly, they are biologically distinct disorders that accumulate structurally different isoforms of PrPSc, and consequently, treatments selectively targeting PrPSc (e.g. anti-PrPSc antibodies) should account for this conformational heterogeneity. Secondly, sCJD subtypes have different clinical trajectories, including a remarkable variability of disease duration that largely depends on PrPSc regional distribution, speed of replication and spreading, and neurotoxicity of aggregates. Similar considerations can be extended to other rarer prion disease (i.e. VPSPr, GSS) that are also characterized by significant clinicopathological heterogeneity. Therefore, future clinical trials should consider phenotypic diversity for patient selection and outcome assessments.
1.4.3. Identification of the presymptomatic phase, prediction of clinical conversion, and early diagnosis
Two possible recruitment scenarios are predictable for patient enrollment into future clinical trials: the inclusion of symptomatic or presymptomatic (at-risk) individuals.
Given the rapid clinical progression of prion disorders, a requisite for clinical trials including symptomatic individuals is the possibility of reaching an early and accurate diagnosis. In this regard, the development and the introduction in clinical practice of ultrasensitive seeding assays, such as the real-time quaking-induced conversion (RT-QuIC) assay and the protein misfolded amplification assay (PMCA), represented a breakthrough for the diagnostic work-up of these disorders [Citation110]. RT-QuIC is particularly useful for diagnosing sCJD and PMCA of vCJD, while genetic testing remains the gold standard for inherited forms. A recent meta-analysis including cases with definite, probable, and possible CJD reported a pooled 94% accuracy (91% sensitivity and 97% specificity) of prion RT-QuIC [Citation111]. However, the meta-analysis has the limitation of not considering factors such as the CJD subtype, the reaction protocol adopted, and the tissue/biofluid analyzed that substantially influence RT-QuIC sensitivity [Citation112]. Recent studies involving surveillance centers from Europe and the US demonstrated that the introduction of RT-QuIC in the diagnostic criteria for sCJD significantly improved diagnostic accuracy [Citation113–115], even at the time of the first neurological evaluation [Citation116].
In two different studies, cerebrospinal fluid (CSF) RT-QuIC has demonstrated effective detection of prion seeding activity in a few presymptomatic E200K carriers (3 out of 28) and a single carrier of the P102L mutation (out of 23 tested) [Citation117,Citation118]. By contrast, there was no seeding activity in 6-OPRI, A117V, and D178N carriers. Interestingly, two of the three E200K carriers remained asymptomatic after two and three years of follow-up, revealing that CSF RT-QuIC could show prion seeding activity long before conversion to the symptomatic stage. Together, these findings confirmed the effectiveness of CSF RT-QuIC in gCJD [Citation114,Citation119–125], and in a lesser extent in GSS, and provided further insights for its potential uses in trials for presymptomatic individuals. In summary, the assay showed an overall low and variable sensitivity during the presymptomatic stage, being PRNP mutations at least in part responsible for such variability. Secondly, CSF RT-QuIC potentially detects prion seeding activity in the presymptomatic phase but within a large time frame before conversion to the symptomatic stage.
Other fluid biomarkers, including total-tau, neurofilament light chain (NfL), glial fibrillar acid protein (GFAP), and ubiquitin C-terminal hydrolase L1, have also been investigated in both CSF and plasma for their ability to predict the conversion from the presymptomatic to the symptomatic stage [Citation113,Citation114]. Among them, plasma NfL levels increase abruptly close to clinical conversion, especially in genetic prion disorders showing a fast disease progression (gCJD-E200K and FFI). In contrast, a more progressive (linear) elevation of both NfL and GFAP levels starting more than two years before phenoconversion has been observed in P102L-associated disease [Citation118]. The finding of increased levels of NfL in prion-inoculated Prnp wild-type mice as early as 60 days post-inoculation also supports its use since the presymptomatic phase [Citation97].
Evidence also supports the use of neuroimaging (i.e. brain magnetic resonance imaging, MRI, cerebral positron emission tomography with 18F-fluorodeoxyglucose) [Citation126–128], or neuropsychologic testing [Citation129] as proximity markers heralding the conversion to the symptomatic stage. However, they could be more helpful in identifying the early symptomatic stage rather than predicting conversion.
1.4.4. Definition of standardized outcomes
Defining and accurately measuring the outcomes is critical to any clinical trial. Identifying reliable and standardized evaluation strategies is mandatory to determine the efficacy of a specific treatment and promote reliability and comparison between trials. To date, the use of different outcome measures between studies, except for survival, has significantly prevented comparing drug compounds for CJD treatment in historical trials.
As a general rule, given the heterogeneity of prion disease, composite outcomes should be preferred to a single measure.
Acknowledging the disease’s lethality, all trials investigated the survival time as the primary outcome. In this regard, researchers should be aware that the sCJD subtype influences disease duration (e.g. the natural disease duration is three months in sCJD MM1 vs. 18 months in sCJD MV2K) [Citation3]. Therefore, survival assessment must be coupled with the subtype (neuropathological) definition. Additionally, trials should consider factors that may influence survival, such as enteral feeding [Citation130]. In this regard, to consider the time from the clinical onset to akinetic mutism could be an alternative option. Although less standardized and relevant for individuals’ prognoses, the severity of neuropathologic change compared to that described in historical cohorts could be an additional measure of treatment efficacy. Similarly, postmortem brain tissues can be analyzed for the amount of PrPSc accumulation by either neuropathology or western blot.
Quantifying disease-associated molecules in biofluid could help clinicians monitor treatment in vivo. CSF PrP concentration is stable in presymptomatic PRNP mutation carriers [Citation117], and therefore represents a candidate biomarker for PrP lowering therapies, such as ASOs. Drug trials in other neurodegenerative diseases, such as Tofersen in amyotrophic lateral sclerosis [Citation131], evaluated plasma NfL as a secondary endpoint to assess treatment efficacy. Similarly, monitoring NfL levels in plasma could be helpful in trials involving CJD patients since they are associated with survival [Citation132,Citation133].
Putaminal diffusion tensor MRI has also been proposed as a biomarker of disease severity. The decrease in putamen radial diffusivity predicted clinical worsening as defined by the Medical Research Council (MRC) scale [Citation134]. However, compared with blood biomarkers, performing serial brain MRI has limitations related to availability in rural hospitals, costs, administration in poorly cooperative patients, and greater complexity in analyzing the results that could prevent its application in large patient cohorts.
Finally, standardized scales and questionnaires are broadly used to evaluate trial outcomes. In the past, studies included a variety of scores on cognitive and functional tests, such as the Barthel index, the Mini-Mental State Examination, the modified Rankin score, and so on, that are tools commonly used in neurologic clinical practice but are not specific for the assessment of CJD patients. In 2013, Thompson and colleagues developed and validated the MRC prion disease rating scale, a particular outcome measure for prion disease therapeutic trials [Citation135]. The scale has been drawn in English and currently needs translation and validation in non-English speaking countries. A further criticism of the MRC prion disease rating scale is that it measures functional impairment without focusing on the ‘type’ of deficits contributing to it. To overcome this limit, the same research group recently developed two novel scales, the sCJD Motor and Cognitive scales, allowing them to quantify how these functional domains are selectively impaired [Citation136].
2. Conclusion
Human prion diseases are a heterogeneous group of disorders related to PrP misfolding, representing the prototype of neurodegenerative diseases caused by protein misfolding, amyloidogenic aggregation, and transcellular propagation by seeding-induced conversion. Currently, without effective pharmacotherapies, PrPC represents the main potential target of therapeutic interventions to slow PrPC conversion into PrPSc and the diffusion of the disease to different brain regions. The strategies to target PrPC include passive immunization with anti-PrP antibodies and lowering PrPC expression by ASOs administration. Both approaches have shown promising results in animal models by prolonging survival. In a recent clinical trial involving six symptomatic CJD patients, an anti-PrP antibody was well-tolerated and showed no toxicity signs. The treatment was ineffective in modifying the disease’s clinical trajectory, although neuropathologic examination suggested a possible positive effect on PrPSc clearing from the brain in one case. The main therapeutic challenge for most patients with prion disease is initiating treatment as soon as possible after the onset of symptoms in sporadic human prion disease before developing severe, irreversible brain damage. Thus, the most promising realistic scenario currently involves presymptomatic individuals carrying heterozygous pathogenic PRNP mutations in whom proximity biomarkers identify the disease onset in the pre-clinical stage.
3. Expert opinion
The study of prion diseases has provided fundamental insight into the pathogenesis of neurodegenerative disorders. The rapid propagation, the development of infectivity in most cases, and the prominent neurotoxicity of misfolded PrP have made the prion models more informative than those involving other proteinopathies. However, when it comes to therapy, many of these peculiar features of prion diseases represent challenges rather than advantages. Therefore, it is likely that the development of effective treatments for these disorders will follow the success obtained with other more prevalent and slowly progressive conditions such as AD and PD.
Given their pivotal and isolated role in disease pathogenesis, PrPC and PrPSc represent the most logical and promising currently identified potential therapeutic target candidates for prion disease. The gene STX6, a risk variant recently identified in a genome-wide association study of sCJD [Citation137], is the only possible exception. Indeed, the results of a study showing a modest positive effect on survival in experimentally infected mice with knockout of Stx6 expression seem to support further exploration of STX6 as a potential therapeutic target for prion disease and, possibly, other neurodegenerative disorders [Citation138].
In the last 30 years, scientists tested several chemical compounds for their possible anti-prion effect in cell cultures and animal models of prion disease. However, only a few reached a trial in patients affected by prion disease, and all of them eventually failed to demonstrate a significant clinical effect. As for other neurodegenerative disorders, current strategies to deplete the PrPC substrate or prevent recruitment and contact between PrPC and PrPSc include immunization and gene therapy with ASOs against PrP mRNA. Regarding immunotherapy, developing vaccination strategies that break the self-tolerance efficiently or antibodies that selectively bind PrPSc proved challenging. Following the demonstration of the efficacy of anti-PrPC antibodies in reducing PrPSc levels in prion-infected cellular and mice models and extensive efforts to understand the structural basis of the neurotoxicity shown by some of them, anti-PrPC antibodies have been recently administered to humans for the first time.
Several lines of evidence suggest that lowering PrPC expression levels through the administration of ASOs targeting PrP mRNA could also represent an effective disease-modifying therapy. Gene therapy to lower PrPC expression may be especially suitable for patients or presymptomatic individuals carrying heterozygous pathogenic PRNP mutations with variable penetrance. The results of preliminary studies in genetically determined neurodegenerative disorders, such as amyotrophic lateral sclerosis, spinal muscular atrophy and Huntington’s disease, are promising for developing further ASOs, or other PrP-lowering therapies and the eventual translation to trials treating human prion disease.
Unfortunately, the low incidence of disease and the scarcity of factors predicting the clinical onset limit the design of randomized controlled (pre-approval) trials for both sporadic and genetic prion disorders. The inclusion of highly penetrant PRNP mutation carriers in post-marketing studies is, to date, the most promising approach to reduce the sample size and develop well-powered clinical trials in the field. Nonetheless, multicentric and long-lasting recruitments would be necessary to reach the required sample size. Monitoring blood markers of neuroaxonal damage (NfL), astroglial activation (GFAP), and CSF prion seeds by RT-QuIC are feasible and promising strategies to identify the conversion to the symptomatic stage in presymptomatic PRNP mutation carriers.
Future clinical trials dealing with the sporadic disease must also face the challenge of disease heterogeneity for patient selection and outcome assessments. Along this line, models are needed to predict disease duration and subtype in vivo based on demographic, laboratory, genetic, and clinical findings [Citation139,Citation140]. The application of these models will help physicians decide on patient eligibility and stratification. Future clinical trials should also include, besides survival time, composite outcomes accounting for disease progression and severity through functional (clinical) and biological (laboratory) measures. Clinical assessment must consist of standardized evaluation scales specifically developed for CJD, at least for the prevalent subtypes. At present, blood NfL is the most promising candidate biomarker for monitoring drug effects and disease progression.
Article highlights
Human prion diseases are rare, often rapidly progressive, transmissible neurodegenerative disorders caused by prion protein misfolding, characterized by broad clinical, histopathological, and molecular heterogeneity.
No pharmacologic treatment has shown a positive effect on survival in patients with prion disease, but novel promising approaches, including immunotherapy and genetic modulation, are now available.
A first observational trial using an anti-prion protein antibody (PRN100) documented a possible brain clearance of the abnormal prion protein in one Creutzfeldt-Jakob disease-affected brain and no adverse events in six individuals with clinical disease.
Antisense oligonucleotides effectively reduced the levels of physiologic cellular prion protein in animal models and currently represent the most promising approach for inherited prion disease, mainly to prevent phenoconversion in PRNP mutation carriers.
The rarity of disease limits the enrollment of large patient cohorts, impairing the ability to successfully undertake statistically meaningful trials.
Future clinical trials should consider the molecular heterogeneity of the disease related to different prion strains, validate novel biomarkers of phenoconversion in at-risk individuals, and further standardize clinical and/or biological outcomes.
List of abbreviations
CJD Creutzfeldt-Jakob diseasePrP prion proteinsCJD sporadic Creutzfeldt-Jakob diseaseGSS Gerstmann – Sträussler – Scheinker disease FFI Fatal Familial InsomniaVPSPr Variably Protease-Sensitive PrionopathyCAA cerebral amyloid angiopathyPK proteinase K BSE bovine spongiform encephalopathyGPI glycophosphatidylinositolPrPC cellular prion proteiniCJD iatrogenic CJDvCJD variant CJDER endoplasmic reticulumUPS ubiquitin-proteasome quality controlUPR unfolded protein responseCNS central nervous systemPPS pentosan polysulphateAD Alzheimer’s diseasePD Parkinson’s diseaseASOs antisense oligonucleotidesRT-QuIC real-time quaking-induced conversionPMCA protein misfolding cyclic amplificationCSF Cerebrospinal fluidNfL neurofilament light chainGFAP glial fibrillar acid proteinMRI magnetic imagingMRC Medical Research Council
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Watson N, Brandel J-P, Green A, et al. The importance of ongoing international surveillance for Creutzfeldt-Jakob disease. Nat Rev Neurol. 2021;17:362–379.
- Baiardi S, Rossi M, Capellari S, et al. Recent advances in the histo-molecular pathology of human prion disease. Brain Pathol. 2019;29:278–300.
- Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233.
- Parchi P, Strammiello R, Notari S, et al. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol. 2009;118:659–671.
- Baiardi S, Rossi M, Mammana A, et al. Phenotypic diversity of genetic Creutzfeldt-Jakob disease: a histo-molecular-based classification. Acta Neuropathol. 2021;142:707–728.
- Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383.
- Scheckel C, Aguzzi A. Prions, prionoids and protein misfolding disorders. Nat Rev Genet. 2018;19:405–418. doi: 10.1038/s41576-018-0011-4.
- Büeler H, Aguzzi A, Sailer A, et al. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347.
- Watts JC, Bourkas MEC, Arshad H. The function of the cellular prion protein in health and disease. Acta Neuropathol. 2018;135:159–178.
- Sweeney P, Park H, Baumann M, et al. Protein misfolding in neurodegenerative diseases: implications and strategies. Transl Neurodegener. 2017;6:6.
- Yedidia Y, Horonchik L, Tzaban S, et al. Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. Embo J. 2001;20:5383–5391.
- Ma J, Lindquist S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci U S A. 2001;98:14955–14960.
- Cox B, Ness F, Tuite M. Analysis of the generation and segregation of propagons: entities that propagate the [PSI+] prion in yeast. Genetics. 2003;165:23–33.
- Kraus A, Hoyt F, Schwartz CL, et al. High-resolution structure and strain comparison of infectious mammalian prions. Mol Cell. 2021;81:4540–4551.e6.
- Rouvinski A, Karniely S, Kounin M, et al. Live imaging of prions reveals nascent PrPSc in cell-surface, raft-associated amyloid strings and webs. J Cell Biol. 2014;204:423–441.
- Ntt L, Wu B, Harris DA. Prion neurotoxicity. Brain Pathol. 2019;29:263–277.
- Thellung S, Corsaro A, Dellacasagrande I, et al. Proteostasis unbalance in prion diseases: mechanisms of neurodegeneration and therapeutic targets. Front Neurosci. 2022;16:966019.
- O’donovan CN, Tobin D, Cotter TG. Prion protein fragment PrP-(106-126) induces apoptosis via mitochondrial disruption in human neuronal SH-SY5Y cells. J Biol Chem. 2001;276:43516–43523.
- Lakkaraju AKK, Frontzek K, Lemes E, et al. Loss of PIKfyve drives the spongiform degeneration in prion diseases. EMBO Mol Med. 2021;13:e14714.
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318(5852):930–936.
- Aguzzi A, Akk L. Cell biology of prions and prionoids: a status report. Trends Cell Biol. 2016;26:40–51.
- Fevrier B, Vilette D, Archer F, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A. 2004;101:9683–9688.
- Gousset K, Schiff E, Langevin C, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol. 2009;11:328–336.
- Parchi P, de Boni L, Saverioni D, et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol. 2012;124:517–529.
- Poggiolini I, Saverioni D, Parchi P. Prion protein misfolding, strains, and neurotoxicity: an update from studies on Mammalian prions. Int J Cell Biol. 2013;2013:910314.
- de Melo ASLF, Lima JLD, Malta MCS, et al. The role of microglia in prion diseases and possible therapeutic targets: a literature review. Prion. 2021;15:191–206.
- Sanders WL. Creutzfeldt-Jakob disease treated with amantadine. J Neurol Neurosurg Psychiatry. 1979;42:960–961.
- Braham J. Jakob-Creutzfeldt disease: treatment by amantadine. Br Med J. 1971;4:212–213.
- Sanders WL, Dunn TL. Creutzfeldt-Jakob disease treated with amantidine. A report of two cases. J Neurol Neurosurg Psychiatry. 1973;36:581–584.
- Scully R, Galdabini J, McNeely B. Case records of the massachusetts general hospital. N Engl J Med. 1980;303:1162–1171.
- Ratcliffe J, Rittman A, Wolf S, et al. Creutzfeldt-Jakob disease with focal onset unsuccessfully treated with amantadine. Bull Los Angeles Neurol Soc. 1975;40:18–20.
- Terzano MG, Montanari E, Calzetti S, et al. The effect of amantadine on arousal and EEG patterns in Creutzfeldt-Jakob disease. Arch Neurol. 1983;40:555–559.
- Neri G, Figà-Talamanca L, Di Battista GC, et al. Amantadine in Creutzfeldt-Jakob disease. Review of the literature and case contribution. Riv Neurobiol. 1984;30:47–56.
- Stewart LA, Rydzewska LHM, Keogh GF, et al. Systematic review of therapeutic interventions in human prion disease. Neurology. 2008;70:1272–1281.
- David AS, Grant R, Ballantyne JP. Unsuccessful treatment of Creutzfeldt-Jakob disease with acyclovir. Lancet. 1984;1:512–513.
- Newman PK. Acyclovir in Creutzfeldt-Jakob disease. Lancet. 1984;1:793.
- Kovanen J, Haltia M, Cantell K. Failure of interferon to modify Creutzfeldt-Jakob disease. Br Med J. 1980;280:902.
- Dhar S, Bitting RL, Rylova SN, et al. Flupirtine blocks apoptosis in batten patient lymphoblasts and in human postmitotic CLN3- and CLN2-deficient neurons. Ann Neurol. 2002;51:448–466.
- Perovic S, Schröder HC, Pergande G, et al. Effect of flupirtine on Bcl-2 and glutathione level in neuronal cells treated in vitro with the prion protein fragment (PrP106-126). Exp Neurol. 1997;147:518–524.
- Otto M, Cepek L, Ratzka P, et al. Efficacy of flupirtine on cognitive function in patients with CJD: a double-blind study. Neurology. 2004;62:714–718.
- Caughey B, Raymond GJ. Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J Virol. 1993;67:643–650.
- Doh-Ura K, Ishikawa K, Murakami-Kubo I, et al. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol. 2004;78:4999–5006.
- Bone I, Belton L, Walker AS, et al. Intraventricular pentosan polysulphate in human prion diseases: an observational study in the UK. Eur J Neurol. 2008;15:458–464.
- Tsuboi Y, Doh-Ura K, Yamada T. Continuous intraventricular infusion of pentosan polysulfate: clinical trial against prion diseases. Neuropathology. 2009;29:632–636.
- Honda H, Sasaki K, Minaki H, et al. Protease-resistant PrP and PrP oligomers in the brain in human prion diseases after intraventricular pentosan polysulfate infusion. Neuropathology. 2012;32:124–132.
- Kamatari YO, Hayano Y, Yamaguchi K, et al. Characterizing antiprion compounds based on their binding properties to prion proteins: implications as medical chaperones. Protein Sci. 2013;22:22–34.
- Doh-Ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol. 2000;74:4894–4897.
- Korth C, May BC, Cohen FE, et al. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci U S A. 2001;98:9836–9841.
- Collins SJ, Lewis V, Brazier M, et al. Quinacrine does not prolong survival in a murine Creutzfeldt-Jakob disease model. Ann Neurol. 2002;52:503–506.
- Barret A, Tagliavini F, Forloni G, et al. Evaluation of quinacrine treatment for prion diseases. J Virol. 2003;77:8462–8469.
- Murakami-Kubo I, Doh-Ura K, Ishikawa K, et al. Quinoline derivatives are therapeutic candidates for transmissible spongiform encephalopathies. J Virol. 2004;78:1281–1288.
- Nakajima M, Yamada T, Kusuhara T, et al. Results of quinacrine administration to patients with Creutzfeldt-Jakob disease. Dement Geriatr Cognit Disord. 2004;17:158–163.
- Haïk S, Brandel JP, Salomon D, et al. Compassionate use of quinacrine in Creutzfeldt-Jakob disease fails to show significant effects. Neurology. 2004;63:2413–2415.
- Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 2009;8:334–344.
- Geschwind MD, Kuo AL, Wong KS, et al. Quinacrine treatment trial for sporadic Creutzfeldt-Jakob disease. Neurology. 2013;81:2015–2023.
- Ghaemmaghami S, Ahn M, Lessard P, et al. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLOS Pathog. 2009;5:e1000673.
- Tagliavini F, Forloni G, Colombo L, et al. Tetracycline affects abnormal properties of synthetic PrP peptides and PrP(Sc) in vitro. J Mol Biol. 2000;300:1309–1322.
- Forloni G, Iussich S, Awan T, et al. Tetracyclines affect prion infectivity. Proc Natl Acad Sci U S A. 2002;99:10849–10854.
- De Luigi A, Colombo L, Diomede L, et al. The efficacy of tetracyclines in peripheral and intracerebral prion infection. PLoS ONE. 2008;3:e1888.
- Haïk S, Marcon G, Mallet A, et al. Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13:150–158.
- Varges D, Manthey H, Heinemann U, et al. Doxycycline in early CJD: a double-blinded randomised phase II and observational study. J Neurol Neurosurg Psychiatry. 2017;88:119–125.
- Forloni G, Tettamanti M, Lucca U, et al. Preventive study in subjects at risk of fatal familial insomnia: innovative approach to rare diseases. Prion. 2015;9:75–79.
- Forloni G, Roiter I, Artuso V, et al. Preventive pharmacological treatment in subjects at risk for fatal familial insomnia: science and public engagement. Prion. 2022;16:66–77.
- van Dyck CH. Anti-amyloid-β monoclonal antibodies for alzheimer’s disease: pitfalls and promise. Biol Psychiatry. 2018;83:311–319.
- Antonini A, Bravi D, Sandre M, et al. Immunization therapies for Parkinson’s disease: state of the art and considerations for future clinical trials. Expert Opin Investig Drugs. 2020;29:685–695.
- Aguzzi A, Lakkaraju AKK, Frontzek K. Toward therapy of human prion diseases. Annu Rev Pharmacol Toxicol. 2018;58:331–351.
- Ma Y, Ma J. Immunotherapy against Prion Disease. Pathogens. 2020;9:216.
- Schwarz A, Krätke O, Burwinkel M, et al. Immunisation with a synthetic prion protein-derived peptide prolongs survival times of mice orally exposed to the scrapie agent. Neurosci Lett. 2003;350:187–189.
- Petsch B, Müller-Schiffmann A, Lehle A, et al. Biological effects and use of PrPSc- and PrP-specific antibodies generated by immunization with purified full-length native mouse prions. J Virol. 2011;85:4538–4546.
- Nitschke C, Flechsig E, van den Brandt J, et al. Immunisation strategies against prion diseases: prime-boost immunisation with a PrP DNA vaccine containing foreign helper T-cell epitopes does not prevent mouse scrapie. Vet Microbiol. 2007;123:367–376.
- Weissmann C. A “unified theory” of prion propagation. Nature. 1991;352:679–683.
- Mallucci G, Dickinson A, Linehan J, et al. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874.
- Bendheim PE, Barry RA, DeArmond SJ, et al. Antibodies to a scrapie prion protein. Nature. 1984;310:418–421.
- Gabizon R, McKinley MP, Groth D, et al. Immunoaffinity purification and neutralization of scrapie prions. Prog clin biol res. 1989;317:583–600.
- Heppner FL, Musahl C, Arrighi I, et al. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science. 2001;294:178–182.
- Frontzek K, Aguzzi A. Recent developments in antibody therapeutics against prion disease. Emerg Top Life Sci. 2020;4:169–173.
- Peretz D, Williamson RA, Kaneko K, et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001;412:739–743.
- Sonati T, Reimann RR, Falsig J, et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature. 2013;501:102–106.
- Reimann RR, Sonati T, Hornemann S, et al. Differential toxicity of antibodies to the prion protein. PLOS Pathog. 2016;12:e1005401.
- Wu B, McDonald AJ, Markham K, et al. The N-terminus of the prion protein is a toxic effector regulated by the C-terminus. Elife. 2017;6:e23473.
- Frontzek K, Bardelli M, Senatore A, et al. A conformational switch controlling the toxicity of the prion protein. Nat Struct Mol Biol. 2022;29:831–840.
- Lefebvre-Roque M, Kremmer E, Gilch S, et al. Toxic effects of intracerebral PrP antibody administration during the course of BSE infection in mice. Prion. 2007;1:198–206.
- White AR, Enever P, Tayebi M, et al. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003;422:80–83.
- Klöhn P-C, Farmer M, Linehan JM, et al. PrP antibodies do not trigger mouse hippocampal neuron apoptosis. Science. 2012;335:52.
- Mead S, Khalili-Shirazi A, Potter C, et al. Prion protein monoclonal antibody (PRN100) therapy for Creutzfeldt-Jakob disease: evaluation of a first-in-human treatment programme. Lancet Neurol. 2022;21:342–354.
- Büeler H, Raeber A, Sailer A, et al. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med. 1994;1:19–30.
- Safar JG, DeArmond SJ, Kociuba K, et al. Prion clearance in bigenic mice. J Gen Virol. 2005;86:2913–2923.
- Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377:1723–1732.
- Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, et al. Targeting huntingtin expression in patients with huntington’s disease. N Engl J Med. 2019;380:2307–2316.
- Büeler H, Fischer M, Lang Y, et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582.
- Richt JA, Kasinathan P, Hamir AN, et al. Production of cattle lacking prion protein. Nat Biotechnol. 2007;25:132–138.
- Benestad SL, Austbø L, Tranulis MA, et al. Healthy goats naturally devoid of prion protein. Vet Res. 2012;43:87.
- Bremer J, Baumann F, Tiberi C, et al. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010;13:310–318.
- Küffer A, Lakkaraju AKK, Mogha A, et al. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature. 2016;536:464–468.
- Minikel EV, Vallabh SM, Lek M, et al. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med. 2016;8:322ra9.
- Minikel EV, Karczewski KJ, Martin HC, et al. Evaluating drug targets through human loss-of-function genetic variation. Nature. 2020;581:459–464.
- Minikel EV, Zhao HT, Le J, et al. Prion protein lowering is a disease-modifying therapy across prion disease stages, strains and endpoints. Nucleic Acids Res. 2020;48:10615–10631.
- White MD, Farmer M, Mirabile I, et al. Single treatment with RNAi against prion protein rescues early neuronal dysfunction and prolongs survival in mice with prion disease. Proc Natl Acad Sci U S A. 2008;105:10238–10243.
- Silber BM, Gever JR, Rao S, et al. Novel compounds lowering the cellular isoform of the human prion protein in cultured human cells. Bioorg Med Chem. 2014;22:1960–1972.
- Ahn M, Bajsarowicz K, Oehler A, et al. Convection-enhanced delivery of AAV2-PrPshRNA in prion-infected mice. PLoS ONE. 2014;9:e98496.
- Nazor Friberg K, Hung G, Wancewicz E, et al. Intracerebral infusion of antisense oligonucleotides into prion-infected mice. Mol Ther Nucleic Acids. 2012;1:e9.
- Raymond GJ, Zhao HT, Race B, et al. Antisense oligonucleotides extend survival of prion-infected mice. JCI Insight. 2019;5:e131175.
- Reidenbach AG, Minikel EV, Zhao HT, et al. Characterization of the prion protein binding properties of antisense oligonucleotides. Biomolecules. 2019;10:1.
- Klug GMJA, Wand H, Simpson M, et al. Intensity of human prion disease surveillance predicts observed disease incidence. J Neurol Neurosurg Psychiatry. 2013;84:1372–1377.
- Goldman JS, Vallabh SM. Genetic counseling for prion disease: updates and best practices. Genet Med. 2022;24:1993–2003.
- Minikel EV, Vallabh SM, Orseth MC, et al. Age at onset in genetic prion disease and the design of preventive clinical trials. Neurology. 2019;93:e125–134.
- Heinemann U, Krasnianski A, Meissner B, et al. Creutzfeldt-Jakob disease in Germany: a prospective 12-year surveillance. Brain. 2007;130:1350–1359.
- Bishop MT, Will RG, Manson JC. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A. 2010;107:12005–12010.
- Rossi M, Baiardi S, Parchi P. Understanding prion strains: evidence from studies of the disease forms affecting humans. Viruses. 2019;11:309.
- Candelise N, Baiardi S, Franceschini A, et al. Towards an improved early diagnosis of neurodegenerative diseases: the emerging role of in vitro conversion assays for protein amyloids. Acta Neuropathol Commun. 2020;8:117.
- Rübsamen N, Pape S, Konigorski S, et al. Diagnostic accuracy of cerebrospinal fluid biomarkers for the differential diagnosis of sporadic Creutzfeldt-Jakob disease: a (network) meta-analysis. Eur J Neurol. 2022;29:1366–1376.
- Poleggi A, Baiardi S, Ladogana A, et al. The use of real-time quaking-induced conversion for the diagnosis of human prion diseases. Front Aging Neurosci. 2022;14:874734.
- Hermann P, Laux M, Glatzel M, et al. Validation and utilization of amended diagnostic criteria in Creutzfeldt-Jakob disease surveillance. Neurology. 2018;91:e331–338.
- Rhoads DD, Wrona A, Foutz A, et al. Diagnosis of prion diseases by RT-Quic results in improved surveillance. Neurology. 2020;95:e1017–1026.
- Watson N, Hermann P, Ladogana A, et al. Validation of revised international creutzfeldt-jakob disease surveillance network diagnostic criteria for sporadic creutzfeldt-jakob disease. JAMA Netw Open. 2022;5:e2146319.
- Mastrangelo A, Mammana A, Baiardi S, et al. Evaluation of the impact of CSF prion RT-Quic and amended criteria on the clinical diagnosis of Creutzfeldt-Jakob disease: a 10-year study in Italy. J Neurol Neurosurg Psychiatry. 2022;94(2):121–129. DOI:10.1136/jnnp-2022-330153
- Vallabh SM, Minikel EV, Williams VJ, et al. Cerebrospinal fluid and plasma biomarkers in individuals at risk for genetic prion disease. BMC Med. 2020;18:140.
- Mok TH, Nihat A, Majbour N, et al. Trajectories of neurodegeneration and seed amplification biomarkers prior to disease onset in individuals at risk of prion disease. Brain. 2022:awad101. DOI:10.1093/brain/awad101
- Sano K, Satoh K, Atarashi R, et al. Early detection of abnormal prion protein in genetic human prion diseases now possible using real-time QUIC assay. PLoS ONE. 2013;8:e54915.
- Lattanzio F, Abu-Rumeileh S, Franceschini A, et al. Prion-specific and surrogate CSF biomarkers in Creutzfeldt-Jakob disease: diagnostic accuracy in relation to molecular subtypes and analysis of neuropathological correlates of p-tau and Aβ42 levels. Acta Neuropathol. 2017;133:559–578.
- Franceschini A, Baiardi S, Hughson AG, et al. High diagnostic value of second generation CSF RT-Quic across the wide spectrum of CJD prions. Sci Rep. 2017;7:10655.
- Bongianni M, Orrù C, Groveman BR, et al. Diagnosis of human prion disease using real-time quaking-induced conversion testing of olfactory mucosa and cerebrospinal fluid samples. JAMA Neurol. 2017;74:155–162.
- Foutz A, Appleby BS, Hamlin C, et al. Diagnostic and prognostic value of human prion detection in cerebrospinal fluid. Ann Neurol. 2017;81:79–92.
- Mok TH, Nihat A, Luk C, et al. Bank vole prion protein extends the use of RT-Quic assays to detect prions in a range of inherited prion diseases. Sci Rep. 2021;11:5231.
- Cramm M, Schmitz M, Karch A, et al. Stability and reproducibility underscore utility of RT-Quic for diagnosis of creutzfeldt-jakob disease. Mol Neurobiol. 2016;53:1896–1904.
- Cortelli P, Perani D, Montagna P, et al. Presymptomatic diagnosis in fatal familial insomnia: serial neurophysiological and 18FDG-PET studies. Brain. 2006;129:668–675.
- Cohen OS, Chapman J, Korczyn AD, et al. Familial Creutzfeldt-Jakob disease with the E200K mutation: longitudinal neuroimaging from asymptomatic to symptomatic CJD. J Neurol. 2015;262:604–613.
- Novi G, Canosa A, Nobili F, et al. Longitudinal brain magnetic resonance imaging and real-time quaking induced conversion analysis in presymptomatic Creutzfeldt-Jakob disease. Eur J Neurol. 2018;25:e127–128.
- Mole J, Mead S, Rudge P, et al. Cognitive decline heralds onset of symptomatic inherited prion disease. Brain. 2021;144:989–998.
- McNiven K, Nihat A, Mok TH, et al. Enteral feeding is associated with longer survival in the advanced stages of prion disease. Brain Commun. 2019;1:fcz012.
- Miller TM, Cudkowicz ME, Genge A, et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. 2022;387:1099–1110.
- Staffaroni AM, Kramer AO, Casey M, et al. Association of blood and cerebrospinal fluid tau level and other biomarkers with survival time in sporadic creutzfeldt-jakob disease. JAMA Neurol. 2019;76:969–977.
- Abu-Rumeileh S, Baiardi S, Ladogana A, et al. Comparison between plasma and cerebrospinal fluid biomarkers for the early diagnosis and association with survival in prion disease. J Neurol Neurosurg Psychiatry. 2020;91:1181–1188.
- Hyare H, De Vita E, Porter M-C, et al. Putaminal diffusion tensor imaging measures predict disease severity across human prion diseases. Brain Commun. 2020;2:fcaa032.
- Thompson AGB, Lowe J, Fox Z, et al. The Medical Research Council prion disease rating scale: a new outcome measure for prion disease therapeutic trials developed and validated using systematic observational studies. Brain. 2013;136:1116–1127.
- Nihat A, Mok TH, Odd H, et al. Development of novel clinical examination scales for the measurement of disease severity in Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2022;93:404–412.
- Jones E, Hummerich H, Viré E, et al. Identification of novel risk loci and causal insights for sporadic Creutzfeldt-Jakob disease: a genome-wide association study. Lancet Neurol. 2020;19(10):840–848. DOI:10.1016/S1474-4422(20)30273-8
- Jones E, Hill E, Linehan J, et al. Knockout of sporadic creutzfeldt-jakob disease risk gene Stx6 in mice extends prion disease incubation time BioRxiv; 2023. DOI: 10.1101/2023.01.10.523281.
- Nihat A, Ranson JM, Harris D, et al. Development of prognostic models for survival and care status in sporadic Creutzfeldt-Jakob disease. Brain Commun. 2022;4:fcac201.
- Llorens F, Rübsamen N, Hermann P, et al. A prognostic model for overall survival in sporadic Creutzfeldt-Jakob disease. Alzheimers Dement. 2020;16:1438–1447.