
Abstract
Short-rib thoracic dysplasia 3 with or without polydactyly (OMIM # 613091) represents a clinical spectrum encompassing a heterogeneous group of skeletal dysplasias associated with homozygous or compound heterozygous mutations of DYNC2H1. We describe the case of a couple with two consecutive therapeutic abortions due to a diagnosis of short-rib thoracic dysplasia mutations. In the first pregnancy, the diagnosis has been made at 21 weeks. In the second one, an accurate and early ultrasound examination allowed a diagnosis at 12 weeks. DYNC2H1 mutations were confirmed in both cases. In this report, we underline the importance of an ultrasound evaluation at the end of the first trimester of pregnancy in the detection of early signs of skeletal dysplasias. An early prenatal diagnosis of a short-rib skeletal dysplasia, such as for other severe skeletal dysplasias, is critical to offer a couple the chance of a weighted, informed, and less traumatic decision about the continuation of the pregnancy.
Introduction
DYNC2H1 encodes the 4307 amino acids heavy chain of the cytoplasmic dynein 2 complex. This complex is involved in intraflagellar transport and includes an intermediate chain (D2IC), a light intermediate chain (D2LIC), and a light chain (LC) [Citation1].
Intraflagellar transport is essential for the development of several mammalian organs and tissues. Cilia and flagella consist of an internal microtubule structure called an axoneme. The axoneme is assembled progressively adding tubulin and other proteins to the ciliary or flagellar tip, which becomes further and further away from the site of protein synthesis. For this reason, a mechanism of anterograde intraflagellar transport, usually driven by kinesin proteins, become necessary to deliver proteins to the growing tip. On the other hand, cells need a form of retrograde intraflagellar transport, in order to modulate the flagellar length and carry back particles of the intraflagellar transport complex. Dynein-2 is the main motor of retrograde intraflagellar transport. DYNC2H1 has a key role in the translocation along the microtubules of proteins necessary for retrograde transport. A disruption of DYNC2H1 leads to the accumulation, on the flagellar tip, of particles of the retrograde transport complex, which can’t be recycled, causing a disruption of the flagellar development [Citation2,Citation3].
Based on their motility, cilia can be classified as motile, for instance, those involved in mucus clearance and in primary ciliary dyskinesia, and non-motile. Non-motile cilia have sensory functions since they transduce chemical signals headed to a cell and trigger intracellular responses. There are several signaling pathways involving cilia, including Hedgehog, Wnt, Notch, Hippo, GPCR, PDGF, mTOR, and TGF-beta. All of them are crucial for normal embryonic development [Citation4,Citation5]. Mutations in DYNC2H1 cause an abnormal morphology and function of the cilia and disrupt Sonic Hedgehog signaling pathway by leading to the accumulation of hedgehog pathway proteins [Citation6].
Homozygous or compound heterozygous mutations in DYNC2H1 are associated with a spectrum of skeletal disorders belonging to the family of ciliopathies. Currently, we use the name short-rib thoracic dysplasia 3 with or without polydactyly, referring to this spectrum of autosomal recessive skeletal ciliopathies characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a “trident” appearance of the acetabular roof. This group of short-rib polydactyly skeletal dysplasias includes several disorders such as Jeune syndrome, short rib-polydactyly syndrome, and Mainzer-Saldino syndrome [Citation7,Citation8].
Dagoneau and colleagues first reported DYNC2H1 in three families with Jeune asphyxiating thoracic dystrophy and in two families with short-rib polydactyly syndrome [Citation9]. Jeune asphyxiating thoracic dystrophy is characterized by genetic heterogeneity and its main features are short ribs, narrow thorax with secondary, often lethal, respiratory insufficiency, short long bones, “trident” aspect of the acetabula, and metaphyseal dysplasia [Citation10]. Short-rib thoracic dysplasia with polydactyly is characterized by a more severe phenotype and a higher prevalence of polydactyly and heart, brain, and urogenital malformations [Citation11]. Also, the Saldino phenotype has been observed in association with DYNC2H1 mutations; this variant typically presents with extreme micromelia, very short, poorly mineralized long bones, and multiple organ system anomalies [Citation12].
Afterward, more than 100 variants have been reported, including both miss-sense and non-sense DYNC2H1 mutations, although no patient with truncating mutations affecting both alleles has been described. Short ribs, small thorax, brachydactyly, and shortened long bones are the typical features. Polydactyly, gastrointestinal, urogenital, and heart malformations are rather rare [Citation3,Citation13], such as situs viscerum inversus [Citation14].
The most recent classification of genetic skeletal disorders [Citation15] includes all the DYNC2H1-related disorders in group 9: Ciliopathies with major skeletal involvement.
We describe two consecutive therapeutic abortions for a Short-rib thoracic dysplasia 3, associated with DYNC2H1 variants.
Clinical report
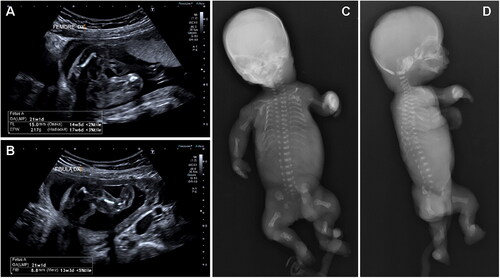
A 27-year-old woman and a 26-year-old man requested genetic counseling because of fetal anomalies. They are healthy, non-consanguineous, and in their family history there are no congenital defects. Ultrasound at 21 weeks showed: fetal biometry corresponding to 18 weeks; mild cerebellar hypoplasia; narrow, bell-shaped, thorax with hypoplastic lungs; ventricular septal defect; severe, generalized micromelia (). Considering the lack of time necessary for a genetic test, the couple chose to terminate the pregnancy.
Figure 1. Instrumental examination at the first pregnancy. Ultrasounds at 21 weeks (A, B) show severe, generalized micromelia. X-rays after abortion (C, D) show rhizo-mesomelic micromelia with reduced mineralization of the long bones and spurred metaphyses, short ribs, and platyspondyly.
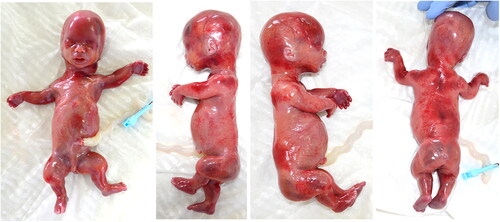
At our dysmorphological examination, the fetus, female, presented hypertelorism, low-set ears, long philtrum, narrow thorax, severely shortened upper and lower limbs, short and stubby fingers, single palmar crease on the left ().
Figure 2. 21 weeks fetus from the first pregnancy of the couple. The fetus, female, presented with hypertelorism, low-set ears, long philtrum, narrow thorax, severely shortened upper and lower limbs, short and stubby fingers, and a single palmar crease on the left.
The anatomopathological examination reported rhizo-mesomelic dwarfism with dysplasia of the epiphyseal plates and irregular intramembranous ossification, bell-shaped thorax, intestinal malrotation, and incomplete lung fissures.
X-ray showed marked rhizo-mesomelic micromelia with reduced mineralization of the long bones and spurred metaphyses, short ribs with the absence of the 11th and 12th pairs, platyspondyly, trident acetabulum ().
A fetal skin biopsy was performed for cytogenetic and molecular testing. Karyotype was normal female (46,XX). Clinical exome sequencing detected two heterozygous variants in the DYNC2H1 gene: c.6077C > T, of maternal origin, and c.12610T > G, of paternal origin. The test also identified a heterozygous variant of maternal origin in the CEP120 gene: c.832 C > G.
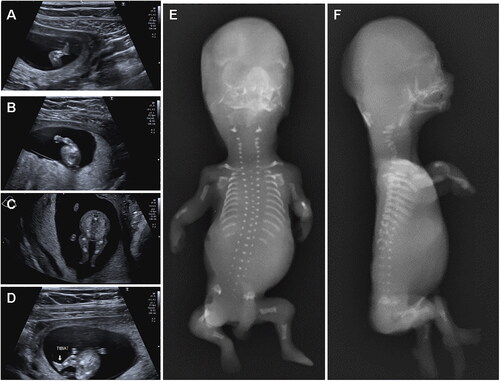
One year later another natural pregnancy started. Ultrasound at 12 + 1 weeks showed: mesomelic and rhizomelic shortening of both upper and lower limbs; redundant subcutaneous tissue of the limbs; small hands without polydactyly (). The couple chose to terminate the pregnancy.
Figure 3. Instrumental examination at the second pregnancy. Ultrasounds at 12 + 1 weeks (A, B, C, D) show: mesomelic and rhizomelic shortening of both upper and lower limbs; redundant subcutaneous tissue of the limbs; small hands without polydactyly. X-rays after abortion (E, F) show shortened, but not bowed, long bones of the limbs, short ribs, absence of the 12th pair of ribs, small vertebral bodies with platyspondyly, reduced mineralization of the cervical vertebral bodies.
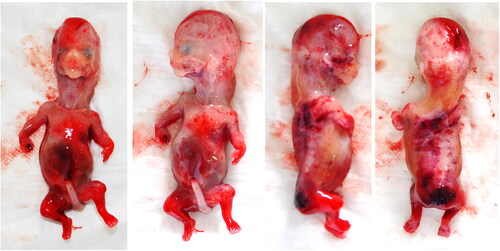
At our dysmorphological examination, the fetus, female, presented low-set and small ears, flattened nasal bridge, micrognathia, long neck, narrow thorax, mesomelic, and rhizomelic shortening of the limbs ().
Figure 4. 14 weeks fetus from the second pregnancy of the couple. The fetus, female, presented with facial edema, long neck, low-set and small ears, micrognathia, narrow thorax, mesomelic and rhizomelic shortening of the limbs.
Fetal autopsy reported nuchal edema and the absence of evident defects of thoracic and abdominal organs. No lung or kidney anomalies were observed, but we have to consider that the pregnancy had been interrupted at an early gestational age, with congestive phenomena occurring.
X-ray showed reduced mineralization of the cervical vertebral bodies, small vertebral bodies with platyspondyly, short ribs, absence of the 12th pair of ribs, and markedly shortened, but not bowed, long bones of the limbs ().
NGS analysis on DNA from fetal fibroblasts confirmed both the DYNC2H1 variants observed in the first pregnancy.
The couple had a third, normal pregnancy. All the ultrasound evaluations didn’t show anomalies. NGS analysis on amniocytes excluded both parental mutations. A healthy female was born by natural birth.
Materials and methods
After obtaining informed consent for the genetic analyses, clinical exome enrichment and parallel sequencing were performed on genomic DNA extracted from fetal tissue and circulating leukocytes of the parents. Library preparation and clinical exome capture were performed by using the Twist Human Core Exome Kit (Twist Bioscience) according to the manufacturer’s protocol and sequenced on the Illumina NovaSeq 6000 platform. The GenomeUp software (GenomeUp) was used for the variant calling and annotating variants. The sequencing data were aligned to the hg19 human reference genome. The functional impact of the variants was analyzed by Combined Annotation Dependent Depletion (CADD) V.1.3, Sorting Intolerant from Tolerant (SIFT), and Polymorphism Phenotyping v2 (PolyPhen-2). Rare variants (MAF < 0.1%) were filtered based on the gnomAD database. Based on the guidelines of the American College of Medical Genetics and Genomics (S1), a minimum depth coverage of 30X was considered suitable for analysis. Variants were examined for coverage and Qscore (minimum threshold of 30), and visualized by the Integrative Genome Viewer (IGV).
Ultrasound imaging was performed using Toshiba Aplio i700, with convex transabdominal, transvaginal, and linear probes.
Results
Clinical exome analysis revealed two heterozygous variants in the DYNC2H1 gene, NM_001377.2: c.6077C > T (p.Thr2026Ile) and c.12610T > G (p.Trp4204Gly), inherited from the mother and the father, respectively. The missense change p.Thr2026Ile is present in the GnomAD database (https://gnomad.broadinstitute.org/) with an allele frequency of 0.000004029, is localized close to the Hydrolytic ATP binding site of dynein motor region D1, involving a very highly conserved residue, is predicted as deleterious and probably damaging by SIFT, Mutation Taster and PolyPhen 2.0, has a CADD score of 26.20 and can be classified as Likely Pathogenic according to the ACMG criteria (PM1 + PM2 + PP1 + PP3). The missense variant p.Trp4204Gly is not present in the GnomAD database, is localized in the dynein heavy chain C-terminal domain of the protein, is predicted as deleterious and probably damaging by SIFT, Mutation Taster, and PolyPhen 2.0, has a CADD score of 33 and can be classified as Likely Pathogenic according to the ACMG criteria (PM1 + PM2 + PP1 + PP3).
Discussion
Skeletal dysplasias are a wide and heterogeneous group including more than 300 disorders affecting the development of the bones. Although most of these disorders are very rare, taken together they have an incidence at the birth of 2/10000–3,2/10000 [Citation16,Citation17,Citation18]. The most frequent skeletal dysplasias, at birth, are achondroplasia, with an incidence of 1/10000–1/20000, and osteogenesis imperfecta (1–1,5/20000). Among them, about 70% are diagnosed prenatally [Citation17]. These data also consider stillbirths and early neonatal deaths, which can compose up to 50% of these numbers.
Incidence at birth doesn’t take into account the most severe skeletal dysplasias, which are lethal in utero or lead to therapeutic abortion. Their frequency is harder to estimate.
Ultrasound scans, other instrumental evaluations, and genetic tests contribute to formulating a clinical suspicion and hypothesizing the severity of a condition in a pregnancy complicated by clinical signs of skeletal dysplasia. Establishing a correct diagnosis is a key point to guiding a couple to an informed decision about continuing or not a pregnancy.
The introduction of Next-Generation Sequencing methods, mainly exome sequencing, has deeply innovated prenatal diagnostics and has extended the diagnostic yield by 8.5–50% [Citation19], significantly increasing the chances to obtain a molecular diagnosis in a timely manner.
Along with the increased genetic detection rate, prenatal sequencing techniques can also reveal new associations between fetal phenotypes and genetic variations, considerably extending the spectrum of prenatal manifestations of disease-causing variants [Citation19].
At the same time, the detection and the interpretation of variants of uncertain significance represent a crucial issue for a definite diagnosis and a proper estimate of the recurrence risk. In this case, NGS-based sequencing allowed the identification of two Likely Pathogenic (class 4) variants in DYNC2H1. A meticulous process of Reverse Phenotyping became necessary to confirm the diagnosis of short-rib thoracic dysplasia 3 with or without polydactyly, considering the high clinical heterogeneity of this skeletal dysplasia. The fetal ribs were moderately shortened and there wasn’t polydactyly, differently from the Saldino-Noonan type. The absence of heart, palate, gastrointestinal and urogenital malformations differs from the typical phenotype of short rib-polydactyly syndrome. On the other hand, the presence of narrow thorax, micromelia, “trident” appearance of the acetabular roof, and metaphyseal abnormalities, in absence of polydactyly, fits with the original definition of Jeune syndrome, also encompassed in the clinical spectrum associated with DYNC2H1 mutations.
A second, consecutive, malformed fetus, in association with the same DYNC2H1 variants, further strengthened the attribution of pathogenicity of the variants.
In the second pregnancy, the echographic suspicion of skeletal dysplasia was fixed at a very earlier stage, as the clinician scheduled a specific checkup at 12 weeks, due to the course of the first pregnancy. A diagnosis by molecular analysis of DYNC2H1 on amniocytes/chorionic villus sampling or by a second-trimester ultrasound would have led to an unnecessarily delayed termination of pregnancy (in our case we should have considered a 12 week chorionic villus sampling and two weeks for the genetic test). This case suggests that, in couples with an assessed state of carriers of skeletal dysplasia, and even in couples with a family history of skeletal dysplasia, with or without a molecular definition, a dedicated ultrasound at the end of the first trimester of pregnancy can detect the early signs of the disorder and lead to a precocious and, possibly, less traumatic decision.
The case also emphasizes the impact of the diagnosis by NGS in presence of a fetus with skeletal dysplasia. Molecular diagnosis can guide, when possible, a more informed decision about the current pregnancy and, in all the cases, allows a correct assessment of the recurrence risk and offers the couple the option of a pre-implantation genetic diagnosis.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Vaughan KT, Mikami A, Paschal BM, et al. Multiple mouse chromosomal loci for dynein-based motility. Genomics. 1996;36(1):29–38.
- Taschner M, Lorentzen E. The intraflagellar transport machinery. Cold Spring Harb Perspect Biol. 2016;8(10):a028092.
- Schmidts M, Arts HH, Bongers EMHF, et al. Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. J Med Genet. 2013;50(5):309–323.
- Pala R, Alomari N, Nauli SM. Primary cilium-dependent signaling mechanisms. IJMS. 2017; 18(11):2272.
- Wheway G, Nazlamova L, Hancock JT. Signaling through the primary cilium. Front Cell Dev Biol. 2018; 6:8.
- Ocbina PJR, Eggenschwiler JT, Moskowitz I, et al. Complex interactions between genes controlling trafficking in primary cilia. Nat Genet. 2011;43(6):547–553.
- Huber C, Cormier-Daire V. Ciliary disorder of the skeleton. Am J Med Genet C Semin Med Genet. 2012;160C(3):165–174.
- Handa A, Voss U, Hammarsjö A, et al. Skeletal ciliopathies: a pattern recognition approach. Jpn J Radiol. 2020;38(3):193–206.
- Dagoneau N, Goulet M, Geneviève D, et al. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am J Hum Genet. 2009;84(5):706–711.
- Keppler-Noreuil KM, Adam MP, Welch J, et al. Clinical insights gained from eight new cases and review of reported cases with Jeune syndrome (asphyxiating thoracic dystrophy). Am J Med Genet A. 2011;155A(5):1021–1032.
- Naki MM, Gür D, Zemheri E, et al. Short rib-polydactyly syndrome. Arch Gynecol Obstet. 2005;272(2):173–175.
- Badiner N, Taylor SP, Forlenza K, et al. Mutations in DYNC2H1, the cytoplasmic dynein 2, heavy chain 1 motor protein gene, cause short-rib polydactyly type I, Saldino-Noonan type. Clin Genet. 2017;92(2):158–165.
- El Hokayem J, Huber C, Couve A, et al. NEK1 and DYNC2H1 are both involved in short rib polydactyly Majewski type but not in beemer langer cases. J Med Genet. 2012;49(4):227–233.
- Cheng C, Li X, Zhao S, et al. Compound heterozygous variants in DYNC2H1 in a foetus with type III short rib-polydactyly syndrome and situs inversus totalis. BMC Med Genomics. 2022;15(1):55.
- Mortier GR, Cohn DH, Cormier-Daire V, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019; 179(12):2393–2419.
- Krakow D, Rimoin DL. The skeletal dysplasias. Genet Med. 2010;12(6):327–341.
- Barbosa-Buck CO, Orioli IM, da Graça Dutra M, et al. Clinical epidemiology of skeletal dysplasias in South America. Am J Med Genet A. 2012 May;158A(5):1038–1045.
- Cavalcanti DP, Fano V, Mellado C, et al. Skeletal dysplasias in Latin America. Am J Med Genet C Semin Med Genet. 2020;184(4):986–995.
- Smogavec M, Gerykova Bujalkova M, Lehner R, et al. Singleton exome sequencing of 90 fetuses with ultrasound anomalies revealing novel disease-causing variants and genotype-phenotype correlations. Eur J Hum Genet. 2022;30(4):428–438.