
Abstract
Objectives
Schizophrenia is a psychiatric disorder affecting 1% of the population. Accumulating evidence indicates that neuroinflammation is involved in the pathology of these disorders by altering neurodevelopmental processes and specifically affecting glutamatergic signalling and astrocytic functioning. The aim of this study was to curate interactive biological pathways involved in schizophrenia for the identification of novel pharmacological targets implementing pathway, gene ontology, and network analysis.
Methods
Neuroinflammatory pathways were created using PathVisio and published in WikiPathways. A transcriptomics dataset, originally created by Narla et al. was selected for data visualisation and analysis. Transcriptomics data was visualised within pathways and networks, extended with transcription factors, pathways, and drugs. Network hubs were determined based on degrees of connectivity.
Results
Glutamatergic, immune, and astrocytic signalling as well as extracellular matrix reorganisation were altered in schizophrenia while we did not find an effect on the complement system. Pharmacological agents that target the glutamate receptor subunits, inflammatory mediators, and metabolic enzymes were identified.
Conclusions
New neuroinflammatory pathways incorporating the extracellular matrix, glutamatergic neurons, and astrocytes in the aetiology of schizophrenia were established. Transcriptomics based network analysis provided novel targets, including extra-synaptic glutamate receptors, glutamate transporters and extracellular matrix molecules that can be evaluated for therapeutic strategies.
Introduction
Schizophrenia is a psychiatric disorder with a prevalence of one percent in the general population (Tamminga and Holcomb Citation2005). Individuals affected by the illness endure periods of psychosis, accompanied by a large individual and social burden. The disorder can be characterised by a variety of symptoms, including hallucinations, delusions, thought disorder, social withdrawal, apathy, and cognitive deficits (Patel et al. Citation2014). The onset of these symptoms generally occurs between late adolescence and early 30s.
Historically, the pathophysiology of schizophrenia was hypothesised to involve deficiencies in neurodevelopment (Murray et al. Citation2017). Neurodevelopment in general has a growth stage in early development and a pruning stage. The former is predominant in early development whereas the latter occurs throughout lifetime, but especially during puberty. The neurodevelopmental hypothesis has been substantiated by reports of abnormalities in processes typically associated with development, these being neuronal proliferation, migration, myelination, and synaptic plasticity (Birnbaum and Weinberger Citation2017). The multi-hit model postulates that certain gene-environmental interactions affect these processes at pivotal stages during prenatal, early, and adolescent neurodevelopment, thereby predisposing a person to develop schizophrenia (Müller et al. Citation2015; Liu et al. Citation2017). Correspondingly, genetic epidemiology has revealed various environmental stressors, such as childhood trauma and maternal infection, and genetic risks associated with the disorder (Birnbaum and Weinberger Citation2017).
With respect to the underlying pathogenic mechanisms, accumulating evidence indicates that neuroinflammation, specifically chronic inflammation of the central nervous system (CNS), may play a central role in the aetiology of schizophrenia (Miller and Goldsmith Citation2017; Comer et al. Citation2020). The aforementioned gene-environmental interactions, like childhood trauma, potentially cause hyper activation of the peripheral immune system and subsequent permeability of the blood brain barrier (Hanson and Gottesman Citation2005). Several studies reported indeed an increase in pro-inflammatory cytokines in individuals with schizophrenia (Müller et al. Citation2015). Consequently, pass over of peripheral immune cells and/or molecules into the CNS might occur, thereby inducing inflammation. Prolonged CNS inflammation damages the brain parenchyma and sensitises the residing microglia to future inflammatory events (DiSabato et al. Citation2016). Moreover, inflammatory factors may directly interfere with neural signalling (Haroon et al. Citation2017; Miller and Goldsmith Citation2017).
Genome-wide association studies support the notion that in the pathophysiology of schizophrenia there are alterations in neuronal and immune signalling (Miller and Goldsmith Citation2017; Mei et al. Citation2018; Comer et al. Citation2020). The Psychiatric Genomics Consortiums study indicated that both inflammatory and glutamatergic signalling genes are associated with schizophrenia (Trubetskoy et al. Citation2022). Glutamate is the most prominent excitatory neurotransmitter and facilitates synaptic plasticity by acting on receptors such as the ionotropic N-methyl-D-aspartate (NMDA) receptor (Hardingham and Bading Citation2010; Haroon et al. Citation2017). Additionally, NMDAR containing parvalbumin inhibitory interneurons in the prefrontal cortex modulate dopaminergic activity in the mesocortical and mesolimbic systems. Dysregulation of dopamine levels in these areas has been associated with negative and positive symptoms (Schwartz et al. Citation2012; Haroon et al. Citation2017; Comer et al. Citation2020). Astrocytes are responsible for glutamate uptake from the synaptic cleft as well as glutamate conversion into glutamine, which can be transported to presynaptic glutamatergic neurons and be converted back into glutamate (Mei et al. Citation2018). Pro-inflammatory cytokines, however, can alter astrocytic functioning. In combination with abnormal expression of glutamatergic genes, this might dysregulate the glutamate system. Consequently, excitotoxicity, NMDA-receptor hypo function, and reduced synaptic plasticity may be physiological consequence (Haroon et al. Citation2017).
Alternatively, several immune-related genes and loci can help to maintain neurological processes involved in schizophrenia, examples include TLR4, IL-6, TGFβ, and CRP (Haroon et al. Citation2017; Comer et al. Citation2020). The most well-known locus is the major histocompatibility complex located on chromosome 6 region 6p22.1–6p21.3 (Woo et al. Citation2020; Hogenaar and van Bokhoven Citation2021). In particular, genes encoding for proteins of the complement system have been associated in the pathogenesis (Magdalon et al. Citation2020; Woo et al. Citation2020). The complement system comprises a comprehensive set of proteins essential for the host defence against invading microorganisms and clearance of injured cells. Within the CNS, however, this system is also involved in synaptic pruning, neuronal migration, and proliferation (Coulthard et al. Citation2018; Magdalon et al. Citation2020; Hogenaar and van Bokhoven Citation2021).
The knowledge about all these molecular processes can be used for data analysis using molecular, machine-readable pathways. These pathways provide an interactive representation of biological pathways, including interactions between genes, metabolites, and proteins as well as proper annotation and literature references (Kelder et al. Citation2012). WikiPathways allows the creation and publication of community created, but expert curated pathways in its repository (Martens et al. Citation2021). Pathway analysis positions the omics data in the context of these pathways and associated biological functions. Additionally, it facilitates the assessment of how pathway components are expressed in different samples.
Molecular pathways allow comprehensive and dynamic visualisation of the emerging neurodevelopmental roles of the complement system in neuroinflammation. In combination with Gene Ontology (GO) and network analysis, it indicates the contributions of specific risk genes, biological processes, and transcription factors to development of schizophrenia. Furthermore, this could help reveal potential targets for new therapeutic strategies. Hence, the aim of the current study was to create and publish interactive biological pathways involved in schizophrenia (with focus on immunology and glutamate) and use them for the identification of novel pharmacological targets based on transcriptomics analysis consisting of pathway, GO, and network analysis.
Materials and methods
Pathway construction
A literature search was conducted using PubMed, Google Scholar, and Online Mendelian Inheritance in Man (OMIM). Medical Subject Headings terms were used to identify relevant articles about the processes, pathways, and gene interactions contributing to the dysfunctional glutamatergic neurotransmission and complement system in schizophrenia (Supplementary information file 1 for an overview of search terms). The pathways were created using PathVisio (version 3.3.0) (Kutmon et al. Citation2015). The genes, proteins, and metabolites identified in the literature were inserted as new nodes. Concerning the annotations of these nodes, the identifier mapping database BridgeDb Hs_Derby_Ensembl_91, metabolites_20210109.bridge and interactions 20210109.bridge identifier mapping databases (van Iersel et al. Citation2010). NCBI Gene (Sayers et al. Citation2021), UniProt (UniProt Citation2021), and ChEBI (Hastings et al. Citation2016) identifiers were used for genes, proteins and metabolites, respectively. Pathway and process nodes were added to the pathways and annotated with WikiPathways and Wikidata identifiers (Waagmeester et al. Citation2020). The curation guidelines described on the WikiPathways website and the advice for pathway drawing given in the 10 simple rules paper by Hanspers et al. (Hanspers et al. Citation2021) were applied. Molecular Interaction Map interactions (Kohn et al. Citation2006) were used for standard interaction types like ‘stimulation’ and ‘conversion’, interactions not captured in these such as exocytosis and transportation were drawn using unspecified basic interaction types. Literature references were added to the interactions and nodes.
Transcriptomics dataset
To investigate gene expression in the pathways a dataset originally created by Narla et al. (GSE92874) (Narla et al. Citation2017) and published on Gene Expression Omnibus (Barrett et al. Citation2013) was selected. The dataset contains transcriptomics (mRNA) data, created by RNA sequencing of neural committed cells. These were acquired from human induced pluripotent stem cell (hiPSC)-derived NPCs of healthy control (n = 4) and schizophrenia (n = 4) patients that were genotyped by Brennand et al. (Brennand et al. Citation2011). Characteristics of the patient population are summarised in . Additional information about the procedure and subjects is described in the original publications.
Table 1. Clinical and demographic characteristics of GSE92874 subjects.
Quality control and differential expression analysis
For quality control i.a. principal component analysis and hierarchical cluster analysis was done (supplementary information file 2). We used the data of the differential expression analysis as described by the authors. The data included the log2 fold change (log2FC), p-value, and the Benjamini–Hochberg false discovery rate adjusted p-value (q-value) based on fragments per kilobase of transcript per million mapped reads (FPKM). in the current study, missing values and least significant duplicates were omitted. Genes encompassing at least one non-expression value (FPKM = 0) across the samples were removed from the dataset. Genes were considered differentially expressed (genes, DEGs) in case the corresponding q-value was < 0.05 and the log2FC was either < −1.0 or > 1.0.
Overrepresentation analysis
GO-based overrepresentation analysis, alias gene set enrichment analysis, was performed using the web service of g:Profiler (Raudvere et al. Citation2019) and the list of DEGs obtained as described above. This allowed for the identification of GO biological processes, cellular components, and molecular functions associated with the DEGs. A q-value below 0.05 for the resulting GO terms indicated statistical significance.
Pathway enrichment analysis was performed using the statistics function of PathVisio (version 3.3.0) based on log2FC and q-values (Kutmon et al. Citation2015). The analysis was performed using the Human Pathway Collection of WikiPathways (version 20210510) and additionally the two pathways created in the current study (Martens et al. Citation2021). Pathways were ranked based on their calculated Z-score, we used a cut-off value of 1.96 (Z > 1.96) to select affected pathways.
Network analysis
Pathway based networks: The curated pathways were converted into networks within Cytoscape software (version 3.8.2) (Shannon et al. Citation2003) using the WikiPathways app (Kutmon et al. Citation2014). The values from the list of DEGs were imported as additional columns to the data nodes. The network analysis function of Cytoscape was used to determine the degree of each node for the identification of hubs (nodes with the most connections). Subnetworks were created from the network of the pathway ‘Neuroinflammation and glutamatergic signalling’ (WikiPathways:WP5083) (Supplementary information file 3).
Drug-target and pathway-gene analysis: The CyTargetLinker app was used to extend the networks with drug-target and pathway-gene interactions (Kutmon et al. Citation2018). For drug targets the linkset drugbank4-2.xgmml originally created from DrugBank content was used (Law et al. Citation2014). Genes that were not targeted by any drug were removed from the network, and drugs categorised as approved were selected for further analysis. In another, broader, approach, the complete drug networks were extended with the ChEMBL linkset (chembl_23_hsa_20180126.xgmml) which contains chemical compounds (Gaulton et al. Citation2017), investigating the identification of potential genetic targets for repurposable as well as novel pharmacological agents. For extension with pathways, two WikiPathways pathway-gene link sets, WikiPathways-20190610-hsa.xgmml and a smaller linkset that included the two new neuroinflammation pathways from this study were used (Martens et al. Citation2021). The latter was created using the Linkset-Creator software, which can be found on https://github.com/CyTargetLinker/linksetCreator.
In order to investigate the hierarchy of the overrepresented GO biological processes, the BiNGO app in Cytoscape was used (Maere et al. Citation2005). Similar to g:Profiler (Raudvere et al. Citation2019), this app implements a functional over representation analysis to visualise the hierarchy of enriched GO terms within a directed network. A subnetwork was created including GO terms with a q-value < 0.01. For DEGs-based networks a STRING network was generated from the list of DEGs using Cytoscape’s STRING app (Doncheva et al. Citation2019). The chosen evidence scores for protein-protein interaction were general confidence, nervous system tissue, and text mining scores of 0.9, 0.375 and 0.2, respectively.
Results
Differential expression analysis
The dataset originally created by Narla et al. consisted of 24,331 transcripts and differential expression analysis data of four healthy controls and four schizophrenia hiPSC-derived neural committed cell lines. Mean ages were 16.3 ± 9.6 and 24.5 ± 2.1 years, respectively (). All subjects were Caucasian, and the majority were male (M/F = 5/3). Boxplots of the log2 transformed FPKM values indicated no particular differences between the samples. Principal component analyses and heatmaps showed two clear clusters of healthy controls and schizophrenia samples (Supplementary information file 2). After quality control and normalisation, data of 15,268 genes was selected for further analysis. 1030 genes were determined to be DEGs in schizophrenia, with 361 being downregulated and 611 being upregulated (Supplementary information file 2).
Gene ontology-based overrepresentation analysis
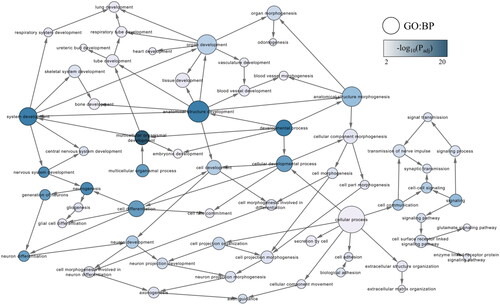
GO-based overrepresentation analysis by g:Profiler and BiNGO indicated a wide range of biological processes (n = 2193) associated with the DEGs (, and the tables in the Supplementary information file 3). Regarding biological processes, the strongest associations were identified for developmental processes, including ‘nervous system development’. Visualisation of the GO biological processes in Cytoscape indicated that several neuronal processes such as ‘neurogenesis’, ‘neuron differentiation’ and ‘neuron development’ were particularly overrepresented (). Likewise, regulatory processes underlying cellular and biological development were also overrepresented (Supplementary information file 4).
Figure 1. Gene ontology (GO) subnetwork of developmental biological processes. The subnetwork was created using BiNGO functional overrepresentation analysis, and filtering for GO terms with a false discovery rate adjusted p-value (q-value) below .01. The blue colour gradient indicates the significance of the enrichment, with darker colours corresponding to smaller q-values. Node sizes indicate the number of direct interactions (degree) as determined in the original network.
Pathway analysis
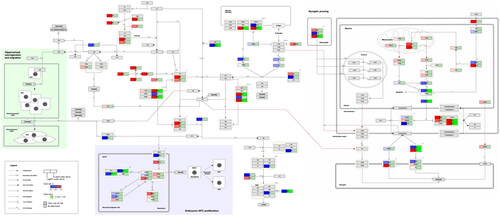
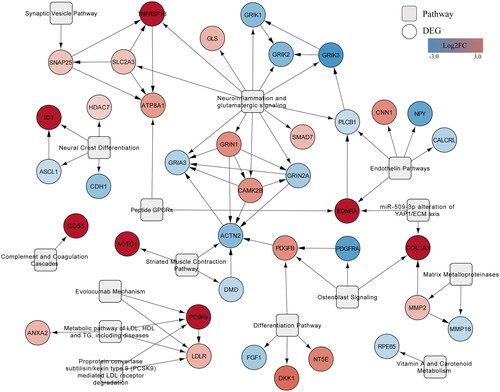
and show the curated pathways, ‘Neuroinflammation and glutamatergic signalling (WikiPathways:WP5083)’ and ‘Complement system in neuronal development and plasticity (WikiPathways:WP5090)’.
Figure 2. Neuroinflammation and glutamatergic signalling. (www.WikiPathways.org/instance/WP5083) The colours indicate the differential expression of mRNA SCZ vs. healthy control according to the dataset by Narla et al. (Citation2017). Log2 fold change values are reported, with red and blue representing upregulation and downregulation, respectively. Green indicates a false discovery rate adjusted p-value (q-value) < .05.
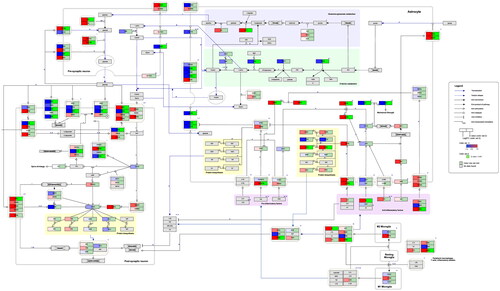
Figure 3. Complement system in neuronal development and plasticity. (http://www.WikiPathways.org/instance/WP5090). The colours indicate the differential expression of mRNA SCZ vs. healthy control according to the dataset by Narla et al. (Citation2017). Log2 fold change values are reported, with red and blue representing upregulation and downregulation, respectively. Green indicates a false discovery rate adjusted p-value (q-value) < .05.
Glutamatergic signalling, astrocytes, and neuroinflammation
Presynaptic neurons release glutamate, which acts on various receptors, thereby regulating long-term potentiation and depression, neuronal survival, and proliferation. Glutamate retro-signalling negatively regulates its own exocytotic release. The remainder is taken up by astrocytes through excitatory amino acid transporters. Glutamine is generated from blood-derived glucose, transported to presynaptic neurons, and converted into glutamate. Alternatively, glucose is converted into D-serine, which functions as a co-agonist for N-methyl-D-aspartate receptors and is broken down by D-amino acid oxidase. CNS inflammation induces M1 and M2 phenotype in microglia, which can modulate neuronal and astrocytic function (www.wikipathways.org/instance/WP5083).
In this pathway several alterations in the expression of genes involved in glutamatergic signalling, astrocytic functioning and neuroinflammation were found (). For instance, there was an increase in expression of genes promoting glutamine-glutamate synthesis (GOT1, GLS), glutamine transport to presynaptic neurons (SLC38A1), glutamate transport into vesicles (SLC17A6), and astrocytic glutamate reuptake (SLC1A2/3). In contrast, the metabotropic glutamate receptor 7 (GRM7)-mediated inhibition of presynaptic glutamate release as well as astrocytic glutamate clearance (SLC1A2/3) were attenuated. Concerning post-synaptic glutamate signalling, NMDA-receptor subunits encoded by GRIN1 and GRIN2A were upregulated and downregulated, respectively. In addition, genes encoding for other glutamate receptors, for example, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor and kainate receptor subunits were downregulated. Similarly, with the exception of CAM2KB, the expression of genes producing enzymes involved in downstream glutamate signalling and phosphorylation of AMPA (PLCB1, PRKCA, PP1CB) were reduced. There were, however, no significant changes in genes implicated in protein biosynthesis, neuroprotection, and cell survival (CREB, BDNF, FOS, BCL2, ARC).
Astrocytic glucose transporters (SL2A1/3) were upregulated in samples from schizophrenia patients. Furthermore, there was a reduction in the expression of genes responsible for glycine uptake (SLC6A9), D-serine release (LRC8B/D) and metabolism (PSPH, PHGDH). GFAP, which is required for mechanical strength in astrocytes, was also downregulated. Immune signalling was affected as indicated by upregulation of both anti-inflammatory (TGFB3, LIF, IL13RA1) and pro-inflammatory (TNFRSF1B) transcription factors and receptors. Additionally, a number of transcription factors downstream of the receptors described above were enriched, with significant upregulation in the case of SMAD7 and SOCS3.
Complement system in neural and immune signalling
The complement system can be activated by ligands displayed on microbes or spontaneous hydrolysis. All pathways terminate in opsonisation, cell lysis or chemotaxis. As shown in the green box C3d-CR2 signalling inhibits adult NPC proliferation and anaphylatoxins and lectin components enhance migration. C5a-C5aR1 interactions contribute to NPC polarity and proliferation (blue box). TGFβ, C1q, C3 and CR3 mediate selective synaptic pruning of weak and apoptotic synapses (http://www.WikiPathways.org/instance/WP5090). Regarding the complement factors and receptors that were present in the dataset, no significant alterations in expression levels were found. Several complement control proteins that inhibit C3 cleavage, generation of C5, and cell lysis were differentially expressed. The results visualised in the pathway indicates upregulation of CSMD1 and downregulation of CD55 and CD46.
Concerning embryonic NPC proliferation, the expression of apical proteins CRB1 and MPP5 was reduced. However, PRKCZ and the C5a receptor (C5AR1and C5AR2) were not significantly enriched. In contrast, various genes and proteins that mediate synaptic pruning were significantly altered in schizophrenia. For example, the C1q-inducing factor TGFB3 was upregulated. The pro-apoptotic CASP3 was downregulated and the anti-apoptotic ATPIIA and ATPIIC upregulated. PROS1 and GAS6 were differentially expressed as well. These factors interact with the TAM family of tyrosine kinases receptors present on the surface of microglia (Nonaka and Nakanishi Citation2019). This promotes microglial pruning of the synapses marked for degradation, a process that is hypothesised to be overactive in schizophrenia (Woo et al. Citation2020; Hogenaar and van Bokhoven Citation2021). Among them GAS6 was found upregulated and PROS1 downregulated, thus leaving an ambiguous effect of these factors on synaptic pruning in schizophrenia.
Pathway overrepresentation analysis
The pathway overrepresentation analysis identified the pathways ‘Striated Muscle Contraction (WikiPathways:WP3795)’, ‘Differentiation pathway (WikiPathways:WP2848)’, ‘Neural crest differentiation (WikiPathways:WP2064)’, and ‘Oligodendrocytes specification and differentiation (WikiPathways:WP4304)’ as the most enriched (highest Z-score). The newly created pathways, describing glutamatergic signalling, astrocytes, and neuroinflammation (WikiPathways:WP5083) and the complement system (WikiPathways:WP5090) also appear in overrepresentation analysis with Z-scores of 3.5 and 1.9, respectively.
Network analysis
Merged curated pathways
A merged network was generated from the pathways neuroinflammation (WikiPathways:WP5083) and complement (WikiPathways:WP5090) pathways (Supplementary information file 3). The resulting network indicated two hub nodes, glutamate (degree = 16) and C3b (degree = 22). Additionally, NGF, CFBb, CREB1, STAT6, and TGFB2/3 and D-serine exhibited degrees between 9 and 11. The genes connecting the two pathways were TGFB1-3 and IFNG.
Drug-target and protein-chemical compound interactions
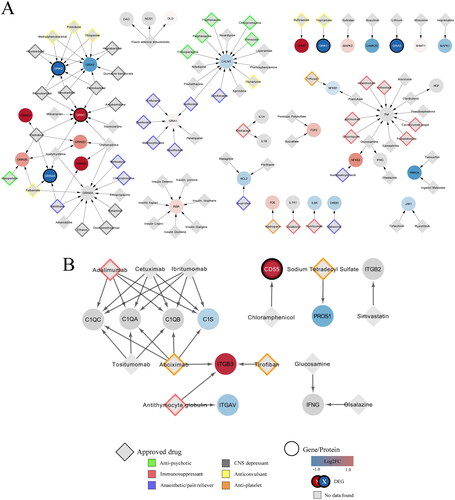
Food and Drug Administration (FDA) approved drugs from DrugBank targeted 37 proteins in the neuroinflammation (WikiPathways:WP5083) and 10 in the complement (WikiPathways:WP5090) pathway (). The full list with all identified drugs, also non-approved and experimental, is given in the supplementary tables. Barbiturate and excitatory amino acid-targeting drugs, which both function as CNS depressants, were among the identified drugs as well as anaesthetics, pain reducers, anticonvulsants, and immunosuppressants (). Antipsychotic drugs target CALM1 and GRIN2B. In contrast, none of the genes in WikiPathways:WP5090 were targeted by any approved antipsychotic drugs (). The identified drugs were generally immunosuppressive or antiplatelet in nature. The differentially expressed CD55, for instance, is targeted by the antibiotic chloramphenicol. After extension of the network with the chemical compounds from ChEMBL more chemical components which can be future drugs were added (Supplementary information file 4). Many drugs are known to target the glutamate receptors, respectively of their subunits as can be seen in .
Figure 4. Network of interactions between FDA approved drugs and their targets in the combined neuroinflammation and complement system network. (A) Interactions derived from WikiPathways:WP5083 network. (B) Interactions derived from WikiPathways:WP5090 network. Log2 fold change values of the genes (ellipse) are represented by a colour gradient, with red and blue indicating upregulation and downregulation, respectively. DEGs are signified by a white label and black border. FDA approved drugs (grey diamonds) targeting the genes are categorised into antipsychotics (green), immunosuppressants (red), anaesthetics (purple), CNS depressants (grey), anticonvulsants (yellow) and antiplatelet (orange).
Gene-pathway interactions
Extension of the network, created from STRING with WikiPathways pathways based on the list of DEGs, included all pathways found by the pathway statistical analysis with one exception (). Regarding the types of pathways, the network incorporated various neuronal pathways, including the ‘Neural crest differentiation (WikiPathways:WP2064)’, ‘Synaptic Vesicles pathway (WikiPathways:WP2267)’, and ‘Neuroinflammation and glutamatergic signalling (WikiPathways:WP5083)’ (). Several metabolic pathways, involving vitamin A and carotenoids (WikiPathways:WP716), and cholesterol (WikiPathways:WP4522, WikiPathways:WP2846), were among the most enriched pathways. Another category encompassed the extracellular matrix pathways ‘Matrix metalloproteinases (WikiPathways:WP129)’, ‘MiR509-3p alteration of YAPI/ECM axis (WikiPathways:WP3967)’, and ‘Endothelin pathways (WikiPathways:WP2197)’.
Figure 5. Network of interactions between DEGs and the 15 most significantly enriched WikiPathways pathways. Pathways (rounded rectangles) with the highest pathway statistics-determined Z-scores and their first neighbour DEGs that were upregulated (red) or downregulated (blue).
Discussion
In the present study, two new interactive neuroinflammatory pathways describing processes that are suspected to contribute to schizophrenia development were created. They were investigated using a transcriptomics dataset, originally created by Narla et al. (Citation2017) from iPSC samples of schizophrenia patients and healthy controls. In contrast to the original study, which focused on microRNA, nuclear FGFR1, and gene correlations, we used the transcriptomics dataset to investigate the special role of inflammation processes in schizophrenia development. While results indicating a direct connection between the transcriptomics alterations in schizophrenia and the complement system remained inconclusive in this study; pathway, GO and network analyses of the transcriptomics data indicated alterations in neurodevelopment and the crosstalk between glutamatergic, astrocytic, and immune signalling. A notable limitation to this study is that the dataset from four patients and healthy controls is relatively small and the study should be repeated on a larger scale.
The present findings are in accordance with the glutamate hypothesis, which claims that glutamatergic dysfunction, in particular downregulation of the glutamate activated NMDA-receptor, is involved in the aetiology of schizophrenia (Mei et al. Citation2018). In this study, most synaptic glutamate receptors, including the NMDAR subunit GRIN2A, were downregulated, suggesting a decrease in normal functioning and aligns with the hypothesis mentioned. In contrast, although non-significant, extra-synaptic NMDA-receptor subunits were upregulated, which has been associated with neurotoxicity and abnormal signalling (Hardingham and Bading Citation2010; Mei et al. Citation2018).
The glutamatergic system modulates the activity of dopaminergic neurons in the mesocortical and mesolimbic systems (Schwartz et al. Citation2012; Haroon et al. Citation2017). The latter is normally involved in reward. Glutamate inhibits the mesolimbic system through acting on GABA inhibitory neurons such as parvalbumin interneurons (Schwartz et al. Citation2012). This prevents the mesolimbic system from becoming hyper-activated, a phenomenon, which has been associated with positive schizophrenia symptoms. The synaptic NMDA-receptor downregulation in these interneurons that was observed in previous studies fails to achieve that as it does not properly contribute to the inhibition of these dopaminergic neurons (Mei et al. Citation2018). Within the cognition-associated mesocortical system, glutamate directly stimulates dopamine-releasing neurons (Schwartz et al. Citation2012), so downregulation of the system’s glutamate receptors would impair normal functioning, contributing to cognitive symptoms.
According to previous studies, the reported alterations in glutamatergic gene expression might be due to excessive extracellular glutamate levels (Miller and Goldsmith Citation2017; Mei et al. Citation2018; Comer et al. Citation2020). The current study suggests that there are changes in astrocytic signalling pathways as well as in associated inflammatory markers.
Astrocytic changes, found in the respective part of the relevant pathway (WikiPathways:WP5083) included a decrease in glutamate reuptake and D-serine synthesis as well as an increase in glutamate synthesis. This could result in excessive extracellular glutamate levels, whereupon glutamate spill over would occur, thus activating neighbouring as well as more distant synapses (Hardingham and Bading Citation2010; Mei et al. Citation2018). The subsequent hyper activation may result in a decrease in synapse independence leading to neurotoxicity and could account for the neuronal loss and impairment of learning and memory associated with schizophrenia (Haroon et al. Citation2017). Moreover, strong over activation of the intra-synaptic NMDA- and AMPA-receptors would induce rapid calcium influx, causing cytotoxicity as well as receptor desensitisation and internalisation (Hardingham and Bading Citation2010; Mei et al. Citation2018). This would contribute to the aforementioned synaptic NMDA-receptor hypo activation found in previous studies.
In contrast to some previous studies (Müller et al. Citation2015; Miller and Goldsmith Citation2017), the present findings do not support a generalised pro-inflammatory state in schizophrenia as the majority of both pro- and anti-inflammatory factors were upregulated. Previous studies incorporated different types of samples, these being post-mortem and blood serum samples, which are challenging systems to draw firm conclusions from. Cultured hiPSC and in vivo cells differ in tissue interactions, adjacent cell presence and the microenvironment. It is well-accepted that this may influence a variety of inflammatory factors (Haroon et al. Citation2017). If the opposing actions of anti and pro-inflammatory signalling in hiPSCs are indicative of what happens in vivo then that would diminish the overall effect on astrocytic functioning. Despite the complexity and ambiguity of the crosstalk between the immune factors and astrocytes, the findings indicate that astrocytes alter the functioning of glutamatergic neurons.
Additionally, the extracellular matrix and its relation to astrocytes and neuroinflammation might explain the changes in glutamatergic signalling (Berretta Citation2012). In the current study, various processes involved in the degradation and organisation of the extracellular matrix were found to be altered in the schizophrenia model used, which is in agreement with the conclusions reached in a different way by Narla et al. (Narla et al. Citation2017) who produced the dataset we used. The extracellular matrix is also the environment in which factors, cytokines, and inflammatory mediators distribute and interact with different structures and cells (Pantazopoulos et al. Citation2021). Extracellular matrix changes, potentially also induced by neuroinflammation and astrocytic alterations (Berretta Citation2012), might contribute to impaired neural signalling, development, and maintenance.
Regarding the complement pathway (WikiPathways:WP5090), the complement system was dysregulated on a regulatory but not on an effector level. There were significant alterations in various complement system regulators, which is in accordance with previous studies (Magdalon et al. Citation2020; Woo et al. Citation2020). These experimental and genetic studies reported downregulation of the complement regulator CSMD1 as well. It is hypothesised that the resulting loss of negative regulation might lead to over activation of the complement system, thereby increasing the rate of synaptic pruning and causing excessive loss of synapses (Woo et al. Citation2020). The increase in the expression of negative complement system regulators CD55 and CD46, observed in the present study, however, might induce the opposite. Furthermore, adenosine triphosphatase-dependent phospholipid transporters limiting the exposure of phosphatidylserine and integrating apoptotic signalling were upregulated. This potentially results in a decrease in interactions between the neuron and phagocytic microglia (Hogenaar and van Bokhoven Citation2021). Since synaptic pruning relies on these interactions, the changes in the aforementioned molecules suggest a decrease in synaptic pruning. This is in disagreement with the neurodevelopmental hypothesis of schizophrenia, which posits that this process is increased in schizophrenia (Murray et al. Citation2017). The discordance might be due to synaptic pruning occurring at a later developmental stage, and hence, pruning-associated changes in the expression of these molecules would not be observed in neural progenitor or neural committed cells (Coulthard et al. Citation2018).
Other alterations included the downregulation of several apical proteins, suggesting dysregulated proliferation (Coulthard et al. Citation2018). MABL and TGFB3 were upregulated, which might contribute to over activation of the complement system as a whole (Magdalon et al. Citation2020; Hogenaar and van Bokhoven Citation2021). However, it was not possible to verify this in the present study due to expression data for several complement factors being filtered out because of not reaching the required significance levels. Assessment of the cleavage products of the complement factors would require analysis of proteomics or peptidomics data as well as proper annotation for machine readability. Moreover, there were no changes in the expression level of mannan-binding lectin-associated serine proteases complexes, C4A nor C4B, which is in disagreement with previous studies (Woo et al. Citation2020).
The network extended with DrugBank and ChEMBL indicated various targets within the dysregulated processes, which might have potential for future clinical application. Drugs targeting glutamate receptor subunits have been shown to be effective in modulating glutamatergic transmission (Patel et al. Citation2014). However, their use has been associated with detrimental side effects, addiction, and resistance. Looking at the potential targets within this study, it might be more beneficial to indirectly reverse NMDA-receptor hypo activation and glutamate dysfunction by targeting the DEGs encoding extra-synaptic NMDA-receptors, glutamate transporters or extracellular matrix molecules. We also found some drugs that might modulate the complement system, but because the exact roles of the complement regulators are not fully established, conclusions for these are not yet possible.
The current study contributes to the growing body of evidence for impaired neurodevelopment and maintenance induced by neuroinflammation in schizophrenia. The existing knowledge around glutamate toxicity and the complement system in the brain was captured in molecular pathways and made available for data analysis. The pathways are now part of the regular WikiPathways releases. Visualising and analysing a transcriptomics dataset from a previously published study in these new pathways showed alterations in inflammatory and neurotrophic mediators induced a wide range of effects on glutamatergic and astrocytic signalling. These changes may contribute to neurotoxicity, the inhibition of synaptic plasticity, and neuronal survival. Astrocytes and the immune system were associated with alterations in matrix constituents, thereby impairing their roles in neuronal migration, proliferation, and synaptogenesis.
Statement of interest
None to declare.
Supplementary information file 4.docx
Download MS Word (1.3 MB)Supplementary information file 1.docx
Download MS Word (12.3 KB)Supplementary Tables.xlsx
Download MS Excel (30.5 KB)Supplementary information file 2.docx
Download MS Word (711.5 KB)Supplementary information file 3.docx
Download MS Word (14.9 KB)Acknowledgements
None.
Additional information
Funding
References
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. 2013. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 41:D991–D995. doi: 10.1093/nar/gks1193.
- Berretta S. 2012. Extracellular matrix abnormalities in schizophrenia. Neuropharmacology. 62(3):1584–1597. doi: 10.1016/j.neuropharm.2011.08.010.
- Birnbaum R, Weinberger DR. 2017. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat Rev Neurosci. 18(12):727–740. doi: 10.1038/nrn.2017.125.
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, et al. 2011. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 473(7346):221–225. doi: 10.1038/nature09915.
- Comer AL, Carrier M, Tremblay M-È, Cruz-Martín A. 2020. The inflamed brain in schizophrenia: the convergence of genetic and environmental risk factors that lead to uncontrolled neuroinflammation. Front Cell Neurosci. 14:274. doi: 10.3389/fncel.2020.00274.
- Coulthard LG, Hawksworth OA, Woodruff TM. 2018. Complement: the emerging architect of the developing brain. Trends Neurosci. 41(6):373–384. doi: 10.1016/j.tins.2018.03.009.
- DiSabato DJ, Quan N, Godbout JP. 2016. Neuroinflammation: the devil is in the details. J Neurochem. 139(Suppl 2):136–153. doi: 10.1111/jnc.13607.
- Doncheva NT, Morris JH, Gorodkin J, Jensen LJ. 2019. Cytoscape StringApp: network analysis and visualization of proteomics data. J Proteome Res. 18(2):623–632. doi: 10.1021/acs.jproteome.8b00702.
- Gaulton A, Hersey A, Nowotka M, Bento AP, Chambers J, Mendez D, Mutowo P, Atkinson F, Bellis LJ, Cibrián-Uhalte E, et al. 2017. The ChEMBL database in 2017. Nucleic Acids Res. 45(D1):D945–D954. doi: 10.1093/nar/gkw1074.
- Hanson DR, Gottesman II. 2005. Theories of schizophrenia: a genetic-inflammatory-vascular synthesis. BMC Med Genet. 6(1):7. doi: 10.1186/1471-2350-6-7.
- Hanspers K, Kutmon M, Coort SL, Digles D, Dupuis LJ, Ehrhart F, Hu F, Lopes EN, Martens M, Pham N, et al. 2021. Ten simple rules for creating reusable pathway models for computational analysis and visualization. PLOS Comput Biol. 17(8):e1009226. doi: 10.1371/journal.pcbi.1009226.
- Hardingham GE, Bading H. 2010. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 11(10):682–696. doi: 10.1038/nrn2911.
- Haroon E, Miller AH, Sanacora G. 2017. Inflammation, glutamate, and glia: a trio of trouble in mood disorders. Neuropsychopharmacology. 42(1):193–215. doi: 10.1038/npp.2016.199.
- Hastings J, Owen G, Dekker A, Ennis M, Kale N, Muthukrishnan V, Turner S, Swainston N, Mendes P, Steinbeck C. 2016. ChEBI in 2016: improved services and an expanding collection of metabolites. Nucleic Acids Res. 44(D1):D1214–1219. doi: 10.1093/nar/gkv1031.
- Hogenaar JTT, van Bokhoven H. 2021. Schizophrenia: complement cleaning or killing. Genes (Basel). 12(2):259. doi: 10.3390/genes12020259.
- Kelder T, Van Iersel MP, Hanspers K, Kutmon M, Conklin BR, Evelo CT, Pico AR. 2012. WikiPathways: building research communities on biological pathways. Nucleic Acids Res. 40:D1301–D1307. doi: 10.1093/nar/gkr1074.
- Kohn KW, Aladjem MI, Weinstein JN, Pommier Y. 2006. Molecular interaction maps of bioregulatory networks: a general rubric for systems biology. Mol Biol Cell. 17(1):1–13. doi: 10.1091/mbc.e05-09-0824.
- Kutmon M, Ehrhart F, Willighagen EL, Evelo CT, Coort SL. 2018. CyTargetLinker app update: a flexible solution for network extension in cytoscape. F1000Res. 7:743. doi: 10.12688/f1000research.14613.1.
- Kutmon M, Lotia S, Evelo CT, Pico AR. 2014. WikiPathways app for cytoscape: making biological pathways amenable to network analysis and visualization. F1000Res. 3:152. doi: 10.12688/f1000research.4254.2.
- Kutmon M, van Iersel MP, Bohler A, Kelder T, Nunes N, Pico AR, Evelo CT. 2015. PathVisio 3: an extendable pathway analysis toolbox. PLOS Comput Biol. 11(2):e1004085. doi: 10.1371/journal.pcbi.1004085.
- Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, et al. 2014. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 42:D1091–D1097. doi: 10.1093/nar/gkt1068.
- Liu X, Li Z, Fan C, Zhang D, Chen J. 2017. Genetics implicate common mechanisms in autism and schizophrenia: synaptic activity and immunity. J Med Genet. 54(8):511–520. doi: 10.1136/jmedgenet-2016-104487.
- Maere S, Heymans K, Kuiper M. 2005. BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 21(16):3448–3449. doi: 10.1093/bioinformatics/bti551.
- Magdalon J, Mansur F, Teles E Silva AL, de Goes VA, Reiner O, Sertié AL. 2020. Complement system in brain architecture and neurodevelopmental disorders. Front Neurosci. 14:23. doi: 10.3389/fnins.2020.00023.
- Martens M, Ammar A, Riutta A, Waagmeester A, Slenter DN, Hanspers K, A Miller R, Digles D, Lopes EN, Ehrhart F, et al. 2021. WikiPathways: connecting communities. Nucleic Acids Res. 49(D1):D613–D621. doi: 10.1093/nar/gkaa1024.
- Mei Y-Y, Wu DC, Zhou N. 2018. Astrocytic regulation of glutamate transmission in schizophrenia. Front Psychiatry. 9:544. doi: 10.3389/fpsyt.2018.00544.
- Miller BJ, Goldsmith DR. 2017. Towards an immunophenotype of schizophrenia: progress, potential mechanisms, and future directions. Neuropsychopharmacology. 42(1):299–317. doi: 10.1038/npp.2016.211.
- Müller N, Weidinger E, Leitner B, Schwarz MJ. 2015. The role of inflammation in schizophrenia. Front Neurosci. 9:372. doi: 10.3389/fnins.2015.00372.
- Murray RM, Bhavsar V, Tripoli G, Howes O. 2017. 30 Years on: how the neurodevelopmental hypothesis of schizophrenia morphed into the developmental risk factor model of psychosis. Schizophr Bull. 43(6):1190–1196. doi: 10.1093/schbul/sbx121.
- Narla ST, Lee YW, Benson CA, Sarder P, Brennand KJ, Stachowiak EK, Stachowiak MK. 2017. Common developmental genome deprogramming in schizophrenia – Role of integrative nuclear FGFR1 signaling (INFS). Schizophr Res. 185:17–32. doi: 10.1016/j.schres.2016.12.012.
- Nonaka S, Nakanishi H. 2019. Microglial clearance of focal apoptotic synapses. Neurosci Lett. 707:134317. doi: 10.1016/j.neulet.2019.134317.
- Pantazopoulos H, Katsel P, Haroutunian V, Chelini G, Klengel T, Berretta S. 2021. Molecular signature of extracellular matrix pathology in schizophrenia. Eur J Neurosci. 53(12):3960–3987. doi: 10.1111/ejn.15009.
- Patel KR, Cherian J, Gohil K, Atkinson D. 2014. Schizophrenia: overview and treatment options. P T. 39(9):638–645.
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J. 2019. g: profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47(W1): w191–W198. doi: 10.1093/nar/gkz369.
- Sayers EW, Beck J, Bolton EE, Bourexis D, Brister JR, Canese K, Comeau DC, Funk K, Kim S, Klimke W, et al. 2021. Database resources of the national center for biotechnology information. Nucleic Acids Res. 49(D1):D10–D17. doi: 10.1093/nar/gkaa892.
- Schwartz TL, Sachdeva S, Stahl SM. 2012. Glutamate neurocircuitry: theoretical underpinnings in schizophrenia. Front Pharmacol. 3:195. doi: 10.3389/fphar.2012.00195.
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11):2498–2504. doi: 10.1101/gr.1239303.
- Tamminga CA, Holcomb HH. 2005. Phenotype of schizophrenia: a review and formulation. Mol Psychiatry. 10(1):27–39. doi: 10.1038/sj.mp.4001563.
- Trubetskoy V, Pardiñas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, Bryois J, Chen C-Y, Dennison CA, Hall LS, et al. 2022. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. 604(7906):502–508. doi: 10.1038/s41586-022-04434-5.
- UniProt C. 2021. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49(D1):D480–D489.
- van Iersel MP, Pico AR, Kelder T, Gao J, Ho I, Hanspers K, Conklin BR, Evelo CT. 2010. The BridgeDb framework: standardized access to gene, protein and metabolite identifier mapping services. BMC Bioinformatics. 11(1):5. doi: 10.1186/1471-2105-11-5.
- Waagmeester A, Stupp G, Burgstaller-Muehlbacher S, Good BM, Griffith M, Griffith OL, Hanspers K, Hermjakob H, Hudson TS, Hybiske K, et al. 2020. Science forum: wikidata as a knowledge graph for the life sciences. Elife. 9:e52614. doi: 10.7554/eLife.52614.
- Woo JJ, Pouget JG, Zai CC, Kennedy JL. 2020. The complement system in schizophrenia: where are we now and what’s next? Mol Psychiatry. 25(1):114–130. doi: 10.1038/s41380-019-0479-0.