
Abstract
Multiple diseases are treated with carbohydrate-based medicinal products worldwide. Direct regioselective acylation of methyl α-D-mannopyranoside (MDMP) derivatives 2-6 afforded from the 6-O-butyryl derivative. This isolated 6-O-derivative was converted to 2,3,4-tri-O-acyl derivatives, and the resulting compounds were analyzed using FTIR, 1H-NMR, 13C-NMR, and elemental analysis. The acylated derivatives showed moderate to good antimicrobial activity. Cytotoxicity assessment indicated that compound 2 had the lowest toxicity. A SAR study demonstrated that lauroyl and myristoyl acyl chains combined with mannopyranose were particularly effective against bacteria. In this context, molecular docking analysis demonstrated crucial interactions involved in assessing the binding affinity of ligands 1-6 for the active sites of Escherichia coli (4XO8) and Aspergillus flavus (1R51). A 100-ns molecular dynamics simulation showed that all the compounds were stable at the active site of protein 1R51. In silico ADMET prediction revealed greater drug similarity for MDMP derivatives. The results of this investigation may help create MDMP derivative-based multidrug-resistant antimicrobial agents.
Highlights
Methyl α-D-mannopyranoside (MDMP) derivatives were designed, and synthesized, and their structures were ascertained via spectral analyses.
MDMPs were assessed in vitro to identify potential antibacterial or antifungal potential antimicrobial candidate(s) against human and plant organisms.
Molecular docking results revealed significant interactions between compounds 1-6 and the active sites of Escherichia coli (4XO8) and Aspergillus flavus (1R51).
A 100 ns molecular dynamic simulation demonstrated that the docked ligand–receptor complex had better dynamic stability, as determined through the RMSD, RMSF, SASA, and Rg profiles.
ADMET prediction revealed an improved drug-likeness profile for all MDMP derivatives.
GRAPHICAL ABSTRACT
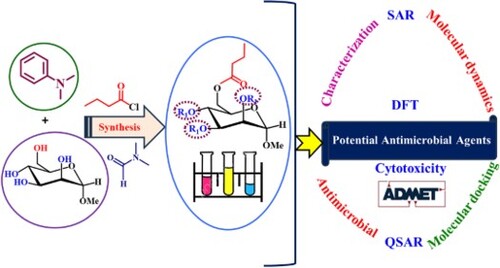
1. Introduction
Carbohydrates are vital compounds found in nature that serve various functions in biological processes. Researchers have long been interested in studying carbohydrates because of their important role in various biological systems. Carbohydrates play a crucial role in viral and bacterial infections, cell growth and proliferation, cell–cell interactions, and immunity [Citation1,Citation2]. They serve as a source of metabolic energy while also playing a role in refining cell–cell connections [Citation3,Citation4] and other vital biological processes [Citation5,Citation6]. Carbohydrates possess a range of beneficial biological effects in fighting infections. These effects include antibacterial, antiviral, antineoplastic, antiprotozoal, and antifungal effects [Citation7]. A literature review revealed the presence of biologically active substances that possess aromatic, heteroaromatic, and acyl substituents [Citation8,Citation9]. The biological activity of the parent molecule is known to be enhanced by various substituents, such as benzene, substituted benzene, nitrogen, sulfur, or halogen [Citation10]. It is widely recognized that when two active nuclei combine, the resulting molecule may exhibit greater promise for biological action [Citation11]. Moreover, the biological activity of carbohydrates can be greatly enhanced through selective acylation and microbiological activity evaluation. Specifically, when two or more heteroaromatic nuclei and acyl chains are combined, the resulting compound exhibits significantly greater biological activity than the distinct nucleus [Citation12,Citation13].
Computational chemistry is a discipline within the field of chemistry that employs computer models to address chemical challenges. The physiochemical characteristics of the compounds were calculated using theoretical chemistry methods combined with efficient computer programmes [Citation14,Citation15]. Chemical descriptors such as the HOMO–LUMO energy gap, ionization potential, electronegativity (χ), electron affinity (A), chemical potential (µ), electrophilicity (ω), hardness (η), softness (σ), DOS plot, and molecular electrostatic potential (MESP) have been calculated to determine chemical characteristics [Citation16–18].
Structure–activity relationships (SARs) and quantitative structure–activity correlations (QSARs) are predictive models that can be utilized to forecast the physicochemical and biological characteristics of particles [Citation19,Citation20]. The biological effect of a novel or unproven molecule can be deduced from its molecular composition or other characteristics of similar compounds that have already been assessed, forming the foundation for any QSAR [Citation21]. Modifications in the composition of a substance might modify the kind and strength of its harmful impact. Therefore, models such as the SAR model have been employed to illustrate, elucidate, and significantly predict relevant events [Citation22]. Computational studies are currently employed to examine the medicinal properties of natural plant compounds in the domains of cancer and COVID-19 research [Citation23,Citation24]. A comprehensive analysis of the molecular pathways involved in cancer was conducted, and prospective targets for pharmaceutical intervention were identified. Additionally, we aimed to examine the viral structure and provide predictions regarding potential treatment strategies for COVID-19 [Citation25]. The QSAR technique offers a quick means of addressing data deficiencies due to the scarcity or absence of experimental data. The computational method has been effectively utilized in various domains, including drug discovery, toxicity assessment, and pharmacy [Citation26,Citation27]. Carbohydrate moieties containing drugs, e.g. Molnupiravir, Nelarabine, Cethromycin and Sodium oligomannate, may be considered potential therapeutics for treating antiviral, anticancer, antibacterial and Alzheimer disease in both previously treated and untreated patients (Figure ) [Citation28].
Figure 1. Marketed drugs that have carbohydrate moieties in their structure.

This study aimed to investigate the molecular docking of several MDMP derivatives against Escherichia coli (PDB ID: 4XO8) and Aspergillus flavus (PDB ID: 1R51), as well as to predict QSAR data, to identify novel drugs [Citation29,Citation30]. This study investigated the in vitro antimicrobial effects of several mannopyranoside analogs, 2–6, with different aliphatic and aromatic chains against seven pathogens. To confirm the stability of the docked complexes, molecular dynamics were performed for 100 ns. Furthermore, density functional theory (DFT) was employed to optimize the synthesized analogs along with the prediction of pharmacokinetic and drug likeness properties. Moreover, molecular docking and prediction of the activity spectra for substances (PASS) were conducted. This paper emphasizes the need for future drug design to prioritize the development of new compounds effective against various types of pathogens, including bacteria, viruses, cancer cells, and fungi. By encouraging the development of versatile chemical structures that can target multiple diseases, this research aims to advance global health efforts and improve treatments in various medical areas.
2. Materials and methods
2.1. Chemicals and apparatus
This study utilized high-quality commercial reagents and solvents from Sigma—Aldrich, Fluka, Merck, and Wako Pure Chemical Ind. Ltd. for synthesis, purification, and biological activity. Silica gel GF254 (Kieselgel/Millipore Sigma, Germany) was utilized for TLC. The column chromatography fluids used were silica gel G60 and CHCl3/CH3OH integrates in varying quantities. 1H-NMR spectra were acquired in CDCl3 using a Brucker Avance DPX-400 MHz instrument, with chemical shifts (δ) quantified in ppm. At 200–4000 cm-1, a Fourier transform infrared (FTIR) spectrophotometer (IR Prestige-21, Shimadzu, Japan) was used to study the bands. The melting points (m.p.) were estimated via electrothermal treatment, and all the solvents were purified according to the manufacturer’s instructions.
2.2. Synthesis of methyl α-D-mannopyranoside (MDMP) derivatives
2.2.1. Synthesis of methyl 6-O-butyryl-α-D-mannopyranoside (2)
To prepare the solution, methyl α-D-mannopyranoside (1) (200 mg, 1.03 mmol) in dry dimethylformamide (DMF) (3 mL)/N,N-dimethylaniline (DMA) was cooled to −5°C, and butyryl chloride (0.06 mL, 1.1 molar eq.) was added. The mixture was stirred continuously for 6 h at 0 °C and 12 h at room temperature. TLC (1:10; methanol: chloroform) was used to track how the reaction progressed. All of the starting materials were converted to a single product (Rf = 0.52). The solution was added to ice water while it was being stirred constantly. The mixture was cleaned using chromatography with CH3OH:CHCl3 (1:10) as the eluant, which yielded the butyryl derivative (2) (155 mg) as a crystalline solid with a melting point of 56-58 °C (recrystallization, EtOAc-n-C6H14), which was used in the next step.
Yield %79.0, white, m.p.: 56-58 oC. IR (KBr) ν/cm−1 1705 (C = O), 3405-3510 (br, -OH); 1H-NMR (400 MHz, CDCl3) δH (ppm) 4.86 (1H, m, H-6a), 4.84 (1H, m, H-6b), 4.35 (1H, s, H-1), 4.32 (1H, d, J = 3.0 Hz, H-2), 4.18 (1H, t, J = 9.2 Hz, H-4), 3.92 (1H, dd, J = 3.1 and 9.3 Hz, H-3), 3.61 (1H, m, H-5), 3.02 (3H, s, 1-OCH3), 2.45 {2H, m, CH3CH2CH2CO-}, 1.6 (2H, m, CH3CH2CH2CO-), 0.90 {3H, m, CH3(CH2)2CO-}. 13C-NMR (100 MHz, CDCl3): δ 174.51 {CH3(CH2)2CO-}, 104.10 (C-1), 77.20 (C-2), 77.09 (C-4), 75.28 (C-3), 69.21 (C-5), 62.16 (C-6), 57.11 (1-OCH3), 34.09, 24.53, 13.81 {CH3(CH2)2CO-}; LC-MS [M + 1]+ 265.23; Calcd. For C11H20O7: C, 49.95, H, 7.63; found: C, 49.97, H, 7.64%.
2.2.2. General procedure for the synthesis of methyl 6-O-butyryl-α-D-mannopyranoside derivatives (3-6)
Lauroyl chloride (0.26 ml, 5 molar eq.) was used to cool the derivative (2) (60 mg, 0.227 mmol) in anhydrous DMF (3 mL)/DMA to 0 °C. After 5-6 h of stirring, the solution was kept overnight at ambient temperature. T.L.C. (methanol–chloroform, 1:10 v/v) showed full conversion of the materials to a single product (Rf = 0.5). Regular workup and silica gel column chromatography with methanol–chloroform (1:10) eluent gave title compound 3 as a crystalline solid (127 mg). Recrystallization of EtOAc-n-C6H14 released solid lauroyl derivatives (3).
Similar reactions of compounds 4, 5 and 6 afforded crystalline solids of myristoyl (190 mg), palmitoyl (245 mg), and trityl (265 mg), respectively.
2.2.3. Methyl 6-O-butyryl-2,3,4-tri-O-lauroyl-α-D-mannopyranoside (3)
Yield %67.12, white, m.p.: 46-48 oC. IR (KBr) ν/cm−1 1697 (C = O); 1H-NMR (400 MHz, CDCl3) δH (ppm) 5.48 (1H, s, H-1), 5.12 (1H, d, J = 3.3 Hz, H-2), 4.98 (1H, dd, J = 3.2 and 9.0 Hz, H-3), 4.75 (1H, t, J = 9.2 Hz, H-4), 4.45 (1H, m, H-6a), 4.37 (1H, m, H-6b), 4.01 (1H, m, H-5), 3.56 (3H, s, 1-OCH3), 2.41 {6H, m, 3×CH3(CH2)9CH2CO-}, 2.35 {6H, m, 3×CH3CH2CH2CO-}, 1.65 {6H, m, 3×CH3(CH2)8CH2CH2CO-}, 1.62 (6H, m, 3×CH3CH2CH2CO-), 1.28 {48H, m, 3×CH3(CH2)8CH2CH2CO-}, 0.90 {9H, m, 3×CH3(CH2)10CO-}, 0.88 {9H, m, 3×CH3(CH2)2CO-}. 13C-NMR (100 MHz, CDCl3): δ 174.53 {CH3(CH2)2CO-}, 172.52, 172.46, 172.41 {3×CH3(CH2)10CO-}, 104.05 (C-1), 77.20 (C-2), 77.10 (C-4), 75.21 (C-3), 69.16 (C-5), 62.08 (C-6), 58.01 (1-OCH3), 34.31, 34.12 (×2), 31.64 (×3), 29.52 (×3), 29.42, 29.31 (×2), 29.22 (×3), 29.10, 25.0 (×2), 24.86, 22.62(×3), 22.61, 22.60 (×3), 21.65, 21.63, 20.0 (×2) {3×CH3(CH2)10CO-}, 34.07, 24.58 {CH3(CH2)2CO-}, 13.80 {CH3(CH2)2CO-}; 13.55, 13.53, 13.44 {3×CH3(CH2)10CO-}, LC-MS [M + 1]+ 811.69; Calcd. For C47H86O10: C, 69.57, H, 10.69; found: C, 69.59, H, 10.70%.
2.2.4. Methyl 6-O-butyryl-2,3,4-tri-O-myristoyl-α-D-mannopyranoside (4)
Yield % 78.21, white, m.p.: 49-50 oC. IR (KBr) ν/cm−1 1702 (C = O); 1H-NMR (400 MHz, CDCl3) δH 5.29 (1H, s, H-1), 4.86 (1H, d, J = 3.2 Hz, H-2), 4.82 (1H, dd, J = 3.3 and 9.0 Hz, H-3), 4.75 (1H, t, J = 9.1 Hz, H-4), 4.61 (1H, m, H-6a), 4.53 (1H, m, H-6b), 3.98 (1H, m, H-5), 3.45 (3H, s, 1-OCH3), 2.36 {6H, m, 3×CH3(CH2)11CH2CO-}, 2.31 {6H, m, 3×CH3CH2CH2CO-}, 1.68 {6H, m, 3×CH3(CH2)10CH2CH2CO-}, 1.63 (6H, m, 3×CH3CH2CH2CO-), 1.29 {60H, m, 3×CH3(CH2)10CH2CH2CO-}, 0.92 {9H, m, 3×CH3(CH2)12CO-}, 0.89 {9H, m, 3×CH3(CH2)2CO-}. 13C-NMR (100 MHz, CDCl3): δ 174.48 {CH3(CH2)2CO-}, 172.50, 172.47, 172.41 {3×CH3(CH2)12CO-}, 104.09 (C-1), 77.25 (C-2), 77.14 (C-4), 75.13 (C-3), 69.19 (C-5), 62.03 (C-6), 57.91 (1-OCH3), 34.26, 24.50 {CH3(CH2)2CO-}, 34.20, 34.18, 34.10 (×2), 31.89, 31.88 (×2), 29.57 (×2), 29.48, 29.34, 29.30 (×2), 29.21 (×3), 29.09, 25.0 (×2), 24.93, 24.87, 22.64(×3), 22.60, 22.58(×3), 22.54 (×3), 21.71, 21.64, 20.06 (×2), 20.0 {3×CH3(CH2)12CO-},14.06, 14.03, 14.0 {3×CH3(CH2)12CO-}, 13.81 {CH3(CH2)2CO-}; LC-MS [M + 1]+ 895.78; Calcd. For C53H98O10: C,71.08, H, 11.04; found: C,71.09, H, 11.03%.
2.2.5. Methyl 6-O-butyryl-2,3,4-tri-O-palmitoyl-α-D-mannopyranoside (5)
Yield % 83.32, white, m.p.: 58-60 oC. IR (KBr) ν/cm−1 1709 (C = O); 1H-NMR (400 MHz, CDCl3) δH 5.18 (1H, s, H-1), 5.0 (1H, d, J = 3.1 Hz, H-2), 4.96 (1H, dd, J = 3.3 and 9.0 Hz, H-3), 4.51 (1H, t, J = 9.0 Hz, H-4), 4.05 (1H, m, H-6a), 4.0 (1H, m, H-6b), 3.94 (1H, m, H-5), 3.31 (3H, s, 1-OCH3), 2.38 {6H, m, 3×CH3(CH2)11CH2CO-}, 2.35 {6H, m, 3×CH3CH2CH2CO-}, 1.66 {6H, m, 3×CH3(CH2)10CH2CH2CO-}, 1.62 (6H, m, 3×CH3CH2CH2CO-), 1.28 {60H, m, 3×CH3(CH2)10CH2CH2CO-}, 0.91 {9H, m, 3×CH3(CH2)12CO-}, 0.88 {9H, m, 3×CH3(CH2)2CO-}. 13C-NMR (100 MHz, CDCl3): δ 174.53 {CH3(CH2)2CO-}, 172.27, 172.10, 171.88 {3×CH3(CH2)14CO-}, 103.88 (C-1), 77.21 (C-2), 77.13 (C-4), 75.19 (C-3), 69.06 (C-5), 62.07 (C-6), 57.97 (1-OCH3), 33.94, 24.51 {CH3(CH2)2CO-}, 33.27, 33.25, 33.23, 33.10 (×2), 31.92, 31.90(×2), 31.83 (×3), 29.52 (×2), 29.13, 29.26, 29.21 (×3), 29.09 (×3), 29.04, 25.10 (×2), 24.77, 24.62, 22.60 (×3), 22.57, 22.52 (×3), 22.30 (×2), 21.61(×2), 21.55, 20.01 (×3), 20.0 {3×CH3(CH2)14CO-}, 14.10, 14.09, 14.04 {3×CH3(CH2)14CO-}, 13.81 {CH3(CH2)2CO-}; LC-MS [M + 1]+ 979.88; Calcd. For C59H110O10: C, 72.33, H, 11.33; Found: C, 72.35, H, 11.31%.
2.2.6. Methyl 6-O-butyryl-2,3,4-tri-O-trityl-α-D-mannopyranoside (6)
Yield % 85.07, white, m.p.: 138-140 oC. IR (KBr) ν/cm−1 1699 (C = O); 1H-NMR (400 MHz, CDCl3) δH 7.35 (18H, m, 3×Ar-H), 7.31 (27H, m, 3×Ar-H), 4.98 (1H, s, H-1), 4.58 (1H, d, J = 3.5 Hz, H-2), 4.40 (1H, dd, J = 3.2 and 9.0 Hz, H-3), 4.38 (1H, t, J = 9.1 Hz, H-4), 4.0 (1H, m, H-6a), 3.82 (1H, m, H-6b), 3.76 (1H, m, H-5), 3.49 (3H, s, 1-OCH3), 2.43 {6H, m, 3×CH3CH2CH2CO-}, 1.63 (6H, m, 3×CH3CH2CH2CO-), 0.92 {9H, m, 3×CH3(CH2)2CO-}. 13C-NMR (100 MHz, CDCl3): δ 171.82 {CH3(CH2)2CO-}, 145.69 (×3, C-1), 145.56 (×3, C-1), 144.60 (×3, C-1), 130.64 (×6, C-2), 130.23 (×6, C-2), 129.60 (×6, C-2), 128.96 (×3, C-3), 128.83 (×3, C-3), 128.45 (×3, C-3), 126.55 (×6, C-4), 126.20 (×6, C-4), 126.07 (×6, C-4) {3×(C6H5)3C-}, 77.63 (×3, C-5), 77.05 (×3, C-5), 76.73 (×3, C-5) {3×(C6H5)3C-}, 34.42, 24.55 {CH3(CH2)2CO-}, 13.98 {CH3(CH2)2CO-}, 104.03 (C-1), 77.24 (C-2), 77.11 (C-4), 75.17 (C-3), 69.10 (C-5), 62.06 (C-6), 57.76 (1-OCH3); LC-MS [M + 1]+ 991.49; Calcd. For C68H62O7: C, 82.38, H, 6.31; Found: C, 82.39, H, 6.30.
2.3. In vitro antimicrobial activity test
The bacteria and fungi were provided Department of Microbiology, University of Chittagong. Table shows the antimicrobial evaluation results for the human and plant pathogens.
Table 1. Name of the pathogenic microorganism.
2.4. Disk diffusion antimicrobial susceptibility test
A disc diffusion study [Citation31] tested new acylated MDMP esters against five human pathogenic bacteria in vitro. An experiment employed 4 mm long paper discs and a 90 mm long glass Petri dish. Sterile 5% (w/v) dimethyl sulfoxide (DMSO) was used to concentrate the synthetic compounds and traditional antibiotics. Antibacterial testing was performed on paper discs with 20 mg/mL test chemicals. Swab bacterial suspensions were spread on Mueller–Hinton agar media (MHA), and sterile discs were arranged on plates. Mueller Hinton agar media was prepared by dissolving 38 g of Mueller Hinton agar powder in 1 L of distilled water. The mixture was boiled for 10 min and then transferred to a 500 mL conical flask, which was sealed with a cotton plug. The medium was autoclaved at 121 °C and 15 psi for 15 min. Following autoclaving, the medium was ready for use in the antibacterial test. The test organisms were grown on 24-hour-incubated plates at 37°C. The positive control was BEXIMCO (Bangladesh) Ltd. Azithromycin, and the negative control was DMSO.
2.5. Determination of the MIC and MBC
The Clinical and Laboratory Standards Institute (CLSI) method was used to determine the MIC in 96-well microtiter plates [Citation32]. Eight millilitres of MHB (Mueller Hinton Broth) was autoclaved in clean screw cap test tubes. A loopful of 48-hour-old bacterial culture was added to each tube and mixed well. The bacterial suspensions were standardized against McFarland standards and inoculated on pour plates for the sensitivity test. Twofold serial dilutions were used in Mueller–Hinton agar (MHA) at pH 7.4. Standardized microorganism suspensions were deposited in 0.5-mL tubes. After a 24-hour incubation, 10 µl of a 0.5% (w/v) 2,3,5-triphenyltetrazolium chloride indicator (Millipore Sigma) was added to each well to detect microbial pathogen development. The final concentration at which no microbiological growth occurred was the MIC. The MHA plates were inoculated with the appropriate concentrations of bacteria to measure the minimum bactericidal concentration (MBC), which prevents microbial growth. The analyses were performed in duplicate. In one row, well 1 was the negative control, and well 8 was the azithromycin-added positive control.
2.6. Screening of mycelial growth
To assess the mycelial growth of the synthesized MDMP derivatives against two fungi (Table ), a “food poisoning” approach was used [Citation33]. Standard potato dextrose agar (PDA) medium was used in the present study and was prepared by boiling 200 g of sliced potato plants in 500 mL of distilled water. After the extract was decanted, 20 g of dextrose and 15 g of agar powder were added and mixed thoroughly. The medium was then autoclaved for 50 min at 121 °C and 15 psi. Test tube slants of PDA medium were inoculated with small portions of mycelia from the collected pathogens and incubated at room temperature for maintenance. Each sanitized Petri dish received 20 mL of sterilized melted potato dextrose agar (PDA) medium (45°C) (90 mm). The 5 mm long mycelial blocks were placed in Petri dish centres when the medium solidified. The equation below was used to measure mycelial growth inhibition after 48 h at 37°C.
2.7. Evaluation of cytotoxic activity by the BSLA assay
The toxicity of the MDMP derivatives was assessed using the brine shrimp lethality test (BSLA) [Citation34]. The MDMP derivatives were dissolved in DMSO and prepared at concentrations of 20, 40, 80, and 160 µL in a total volume of 5 mL in each vial. Vials A, B, C, and D contained samples with concentrations of 4, 8, 16, and 32 µL, respectively. Every vial contained a total of 10 brine shrimp nauplii, and three tests were performed for each concentration. A control experiment was conducted using a vial containing 10 nauplii suspended in 5 mL of seawater. Following the incubation period, we counted the viable cells in each vial using a magnifying lens. The concentration was used to compute the mortality rate of nauplii based on the data. No deceased controls were included.
2.8. Structure–activity relationship (SAR)
Structure—activity relationship (SAR) analysis was used to estimate whether a pharmaceutical target would kill organisms based on its molecular structure. Researchers who make remedies use SAR analysis to find or make new compounds with useful properties. Methods for studying structure—activity relationships involve changing a molecule's structure and determining how it affects its biological activity. This includes making similar molecules, testing them, and using computer modelling to understand the connection between structure and function. The SAR study used the ideas of Hunt [Citation35] and Kim [Citation36] for evaluating membrane penetration.
2.9. Calculation of the chemical reactivity descriptor
The optimized cocrystal structures, energies, and geometries were computed using density functional theory (DFT) [Citation37], Gaussian 09 [Citation38], and B3LYP hybrid exchange correlation (local, nonlocal) with the Lee–Yang–Parr (B3LYP) correlation functional [Citation39] on the 6-311G(d,p) basis set [Citation40]. Multiple suitable calculation formulas were employed to determine chemical reactivity and descriptor values. For the energy gap, Δε was calculated as εLUMO – εHOMO. Similarly, ionization potential, I = −εHOMO; electron affinity, A = −εLUMO; and electronegativity, χ = (I + A)/2. Moreover, chemical potential, µ from −(I + A)/2; hardness, η = (I-A)/2; and electrophilicity, ω = µ2/2η. Finally, softness, σ, was calculated as 1/η. We used GaussSum 3.0 to obtain a DOS plot. To visualize the MEPs, the online WebMO demo server (https://www.webmo.net) was used for all the compounds.
2.10. Molecular docking exploration
Molecular docking was conducted to study the interaction between potential drugs and the active site of the target protein and determine the crucial structural requirements of a model based on binding affinity [Citation41]. The 3D crystal structures of the target proteins Escherichia coli (PDB ID: 4XOB) [Citation42] and Aspergillus flavus (PDB ID: 1R51) [Citation43] were obtained from the Protein Data Bank (PDB) database (https://www.rcsb.org/). Discovery Studio software was used to remove water molecules, ligands, and nonprotein components. PyRx software was used to analyze ligand—protein interactions, while AutoDock Vina [Citation44] was used to create a 3D grid and measure the energies of ligand—protein interactions. The centre grid box sizes for 4XO8 were (−20.461, −10.721, and −4.502), and for 1R51, they were set to (26.532, 31.527, and 36.713) to localize the ligand in the complex. After completion of the docking process, the binding interactions of the docked ligand conformations were analyzed using 2D and 3D visualizations in Discovery Studio.
2.11. Docking prediction validation
Molecular docking was validated using a redocking strategy applied to the crystallized ligands of specific target proteins. This step was crucial for assessing the accuracy of the grid maps associated with each protein. Initially, the native ligand was detached from its receptor using BIOVIA Discovery Studio Visualizer 2020 software. After this separation, the native ligand underwent a redocking procedure with its corresponding receptor. The redocked ligand was then aligned in its most energetically favourable position with the native ligand, enabling the calculation of the root mean square distance (RMSD) between these precisely aligned configurations. A docking operation was considered successful when the RMSD was less than 2 Å, confirming the accuracy and efficiency of the molecular docking technique [Citation45,Citation46].
2.12. Molecular dynamic simulation
In the present study, molecular dynamics simulations were carried out using the NAMD programme and the CHARMM36 force field [Citation47,Citation48]. The simulation system was solvated in a 10 Å cubic box of TIP3P water and neutralized with a 0.154 M NaCl solution of Na+ and Cl- ions using the Monte Carlo method [Citation49]. The energy of the system was first minimized for 10,000 steps using the steepest descent method, followed by an equilibration process for 100 ns at 310 K in a standard number of particles, volume, and temperature (NVT) ensemble. Finally, the system underwent an unrestrained 100 ns-production molecular dynamics simulation in a constant number of atoms, constant pressure, and constant temperature (NPT) ensemble. The stability of the systems was evaluated by analyzing the molecular dynamics trajectory using visual molecular dynamics (VMD) [Citation50,Citation51] to compute the root mean square deviation (RMSD), root mean square fluctuation (RMSF), protein solvent accessible surface area (SASA), and radius of gyration (Rg).
2.13. In silico pharmacokinetics ADMET
The evolution of computer technology has had a significant impact on the field of drug discovery, enabling new drug candidates to be developed more efficiently and accurately. In silico studies have become valuable approaches for better understanding the absorption, distribution, metabolism, excretion and toxicity (ADMET) of compounds. This method relies on pharmacokinetic parameters and drug similarity to perform initial assessments during the drug discovery process. Using the online tool pkCSM [Citation52], we were able to assess the potential of a compound for absorption in the human gut, distribution in the body, metabolic transformation, elimination routes and toxicity levels. Therefore, computer technology plays an essential role in the evaluation of ADMET pharmacokinetic parameters in pharmaceutical research.
2.14. Statistical analysis
For each measurement, the test results are given as the mean ± standard error of three distinct trials. Student’s t tests with two-tailed results were used to evaluate the statistical data. Only ρ values less than 0.05 were considered to indicate statistical significance.
3. Results and discussion
3.1. Chemistry
In this study, the antibacterial activities of various mannopyranoside derivatives were tested against human and plant pathogenic bacteria and fungi. Figure shows the study process's essential components, techniques, and sequential procedures in a flow diagram. For testing, we needed a wide range of substituted mannopyranosides in a single-molecule framework. Numerous acylated monosaccharide compounds show biological effectiveness [Citation53]. We selectively butyrylated methyl α-D-mannopyranoside (1) using direct acylation (Scheme 1 and Figure ).
Figure 2. Flow diagram of the current study.

Figure 3. Acylating agents used for the synthesis of MDMP derivatives 2-6.

Scheme 1. Synthetic pathway of MDMP derivatives 2-6.

3.2. Characterization
In Scheme 1, butyryl chloride was applied to methyl α-D-mannopyranoside (1) at −5 °C in dry DMF/Et3N. Recrystallization afforded compound 2 as a crystalline solid at 79.0%, m.p. 56-58 °C. There were -CO and -OH stretching absorption bands at 1705cm-1 and 3405-3510 cm−1 in the FTIR spectra (Figure ). The compound's 1H-NMR spectra (Figure ) support butyryl group binding with two proton multiplets at δ 2.45 and δ 1.60 and three at δ 0.90. A downfield shift of the C-6 protons occurred at δ 5.21 and 5.07 from the precursor (1) values. Proton resonances showed that butyryl group attachment was less inhibited and more reactive at position 6. In compound 2, the 13C-NMR spectrum showed butyryl groups at δ 174.51, 34.09, 24.53, and 13.81. At m/z [M + 1]+, 265.23 corresponds to the C11H20O7 peak. FTIR, 1H-NMR, 13C-NMR and mass spectra confirmed the structure of methyl 6-O-butyryl-α-D-mannopyranoside (2) (Figures and ).
Figure 4. The FTIR spectra of the compounds 2-6.

Figure 5. The 1H-NMR spectrum of the compound 2.

Figure 6. The 13C-NMR spectrum of the compound 2.

When the butyryl derivative (2) was allowed to react with an equal amount of fatty acid halides, at freezing temperature, the tri-O-lauroyl derivative (3), myristoyl derivative (4), and tri-O-palmitoyl derivative (5) were produced as crystalline solids. The FTIR spectrum of the lauroyl derivative (3) (Figure ) showed a carbonyl stretching absorption band at 1697 cm−1. The 1H-NMR spectrum (Figure S1) of compound 3 shows three lauroyl groups: two six-proton multiplets at δ 2.35 and 1.65, a forty-eight-proton multiplet at δ 1.28, and a nine-proton multiplet at δ 0.90. Therefore, C-2, C-3, and C-4 were relocated downfield to the triol (2) level, indicating the presence of three lauroyl groups in the molecule. The structures of the myristoyl (4) and palmitoyl (5) derivatives were validated by FTIR, 1H-NMR, 13C-NMR (Figure S2), mass spectrometry, and elemental data. The spectra were consistent with the structures of lauroate (3), myristoate (4), and palmitoate (5) (Figures S3-S6).
Compound 2 was stabilized by converting it to trityl (6). Following a freezing reaction of compound 2 with trityl chloride, conventional aqueous work-up and chromatography produced 85.07% of compound (6) as a crystalline solid (m.p. 138-140 °C). FTIR (Figure ) indicated a peak at 1699 cm−1 (-CO stretching) for this compound. In the molecule's 1H-NMR spectrum, two peaks are observed: an eighteen-proton multiple at δ 7.35 and a twenty-seven-proton multiple at δ 7.31 due to the three downfield shifts of C-2, C-3, and C-4 protons to δ 4.58, δ 4.40, and δ 4.38, respectively, compared to the precursor molecule (2). Based on the protons’ predicted resonance sites (Figures S7 and S8), methyl 6-O-butyryl-2,3,4-tri-O-trityl-α-D-mannopyranoside (6) was selected.
3.3. Antibacterial potential
The screening showed that most of the produced compounds were efficacious against the microorganisms (Table S1) (Figures and ). Compound 3 had the greatest inhibitory effect on B. subtilis (17 ± 0.25 mm) and B. cereus (15 ± 0.50 mm), while compounds 2, 3 and 4 had much greater inhibitory effects. However, gram-positive organisms did not react with compound 5.
Figure 7. MDMP derivatives inhibit gram-positive bacteria.

Figure 8. MDMP derivatives inhibit gram-negative bacteria.

The disc diffusion test showed that the produced compounds had outstanding effects on the three gram-negative bacteria. Compound 4 inhibited Salmonella typhi the most (18 ± 0.0 mm). Compounds 2, 3 and 4 inhibited E. coli and S. typhi well, whereas compound 5 did not. According to previous research [Citation54], compound 6 suppressed all gram-negative pathogens less than azithromycin did.
In general, the selectively acylated MPDP compounds generated by the application of various acylating agents have antibacterial effects on the same order of bacteria as gram-positive bacteria: gram-negative bacteria are ordered as follows: 4 > 3 > 2 > 6. For the five test organisms, chemical 3 was the most active.
3.4. Assessment of MIC and MBC activity
The MIC and MBC of the most effective compound, methyl α-D-mannopyranoside, were measured to evaluate its antibacterial activities against pathogenic bacteria (Figure ). MDMP derivatives 3 and 6 showed the strongest antibacterial activity against the research microorganisms, with MIC values ranging from 0.25 to 2.0 μg/mL. The test compounds had positive results against all the tested pathogens, with the maximum activity against E. coli occurring at 0.25 μg/mL. Both analogs had an MBC of 8.00 μg/mL against E. coli, B. cereus, and S. typhi (Figure ). The compounds had the highest MBC values (16.00 μg/mL) against B. subtilis and P. aeruginosa. The MDMP compounds that eradicated other species had MBCs of 8.00-16.00 μg/mL. In their study, Tamokou et al. [Citation55] discovered that Compound 3 exhibited the highest activity, with a minimum inhibitory concentration (MIC) ranging from 6.25 to 100 µg/mL. Additionally, Staphylococcus aureus, Enterococcus faecalis, Candida tropicalis, and Cryptococcus neoformans showed the greatest sensitivity to all the compounds tested.
Figure 9. MIC values of 3 and 4 derivatives against the tested species.

Figure 10. MBC values of 3 and 4 derivatives against the tested species.

3.5. Antifungal susceptibility
Table S2 and Figure S9 show that most of the MPDP derivatives inhibited A. niger and A. flavus mycelial growth well. Among the tested derivatives, compound 2 inhibited A. niger (77 ± 0.10%) and A. flavus (80 ± 0.05%). Compounds 3 and 6 showed significant resistance to A. niger (75 ± 0.24% and 68 ± 0.07%, respectively) and A. flavus (76 ± 0.54% and 65 ± 0.20%, respectively) in mycelial growth tests. These compounds were more potent than nystatin were; compounds 2, 3, and 6 inhibited A. niger and A. flavus. These pathogens were unaffected by compounds 4 and 5. The antifungal activities of the synthesized MDMP compounds are shown in Figure .
Figure 11. The antifungal activity of the synthesized MDMP derivative.

3.6. Determination of the cytotoxic activity of the MDMP derivatives
The cytotoxic effects of the developed MDMP derivatives (2-6) were assessed using brine shrimp lethality assays [Citation56]. The proportions of shrimp mortality at 24 and 48 h are shown in Figure . Long alkyl chains are thought to increase hydrophobicity and cytotoxicity [Citation35]. The findings showed that derivative 2 [methyl 6-O-butyryl-α-D-mannopyranoside] had the lowest toxicity, with a mortality rate of 27.21%. Derivatives 3 and 4 showed the highest acute toxicity, with fatality rates of 42.04% and 40.68%, respectively. The remaining MDMP compounds, 5 and 6, were less lethal to brine shrimp, indicating that the phenyl and palmitoyl groups were less cytotoxic [Citation57]. The cytotoxicity of alkyl chain derivatives increases proportionally with concentration.
Figure 12. Cytotoxicity of MDMP derivatives 2-6. The investigated compound concentrations were A, B, C, and D.

3.7. Investigation of SAR
The emergence of multidrug-resistant microorganisms and unexpected diseases has accelerated the progress of antimicrobial medication research. The majority of antimicrobial agents are heterocyclic molecules that control the metabolism of all living cells [Citation58]. Condensed ring systems have attracted interest due to their varied physiological activities and effectiveness as exclusive therapeutic frameworks. Carbohydrate analogs can selectively interact with enzymes that play a role in the manufacture of bacterial peptidoglycan and fungal chitin and in protein synthesis. This is because nucleosides are crucial for several metabolic processes within cells. The antibacterial properties of the glucopyranoside derivatives were elucidated by their SARs, as shown in Figure . The association between the structure and activity of glycopyranoside analogs, known as the structural-activity relationship (SAR), can be deduced from their antibacterial properties, as presented in Tables S1 and S2. The glucopyranoside compound was ineffective at eliminating dangerous germs, resulting in significant modifications to its antibacterial properties. An observable decrease in influence can be noted in this sequence. The order of effectiveness against gram-positive bacteria was lauroyl > myristoyl > trityl, while that against gram-negative bacteria was myristoyl.
Figure 13. Structure—activity relationship study of the synthesized MDMP derivatives.

In contrast to gram-positive bacteria, gram-negative bacteria require higher concentrations (MIC values of 256 µg/L) of each compound to inhibit growth. This tendency may be linked to the unique cell wall structures of gram-positive and gram-negative bacteria. The membrane of gram-negative bacterial peptidoglycan suppresses lipopolysaccharide (LPS) transfer, which may explain this difference. Selective permeability [Citation59] prevents chemical penetration quickly. Gram-negative bacteria disintegrate ambient substances with periplasmic hydrolytic enzymes. Thicker, hydrophilic, porous gram-positive bacteria have no outer membrane, increasing permeability. Gram-positive bacteria should be more vulnerable to the derivatives produced than gram-negative organisms are. The presence of myristoyl and lauroyl groups increased the hydrophobicity of the MDMP derivatives. Permeability and compound hydrophobicity affect toxicity, membrane integrity, and bioactivity. Hunt [Citation35] and Judge [Citation60] claim that hydrophobic associations between alcohol alkyl chains and lipid membranes regulate aliphatic alcohol potency and solubility. When MDMP acyl chains in lipid-like bacterial membranes react hydrophobically due to a decrease in membrane permeability, hydrophobic bacteria are eliminated.
3.8. Chemical descriptors
In-depth analysis of molecular orbitals using density functional theory (DFT) reveals crucial details about the electronic configuration of the compounds studied [Citation61,Citation62]. The energies of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) molecular orbitals provide a direct insight into molecular stability and reactivity, characterized by the energy gap (E(gap)). A high E(gap) is synonymous with greater stability and less reactivity, reflecting a propensity for molecular softness [Citation63]. Conversely, a low E(gap) indicates a tendency toward high polarity and reactivity, negatively influencing molecular efficiency in biological interactions. In our study, the chemical descriptors as defined in section 2.9 - chemical potential (μ), hardness (η), softness (σ), and electrophilicity coefficient (ω) - serve as the basis for assessing the predictive biological activity of compounds [Citation64]. Compound 4, with the most significant HOMO–LUMO gap of 10.962 eV, exemplifies the impact of these parameters on molecular stability and, by extension, on their bioactive potential. The density of states (DOS) profiles relative to the HOMO–LUMO gaps for compounds 4 and 6, shown in Figure , together with the values of the frontier orbitals (HOMO, LUMO) reported in Table , refine our understanding of the links between the electronic structure and biological functionality.
Figure 14. DOS plot representing the HOMO-LUMO gap of compounds 4 and 6.

Table 2. Chemical descriptor data.
3.9. Frontier molecular orbitals (HOMO and LUMO)
Figure shows the frontier orbital representations of the HOMO and LUMO, which are marked by unique colours. The deep green colour represents the positive node in the HOMO, whereas the radish colour represents the negative node. In the LUMO, yellow symbolizes the negative orbital portion, and the lightest maroon represents the positive portion. Specific-colored images (Figure ) can be used to find and access all extra compounds. According to these observations, the HOMO may lie near the benzene ring terminal, explaining the aromatic ring resonance. Oxygen atoms signify the LUMO, while benzene rings and alkyl groups suggest the HOMO.
Figure 15. HOMO-LUMO images of the compounds 1-6.

3.10. Molecular docking analysis
The molecular docking approach is used to identify binding geometries and interactions between drugs and the active site of a protein. Therefore, we carried out molecular docking studies of synthetic ligands against the proteins of Escherichia coli (4XO8) and Aspergillus flavus (1R51) to determine the binding mode and nonbinding interactions of the ligands. Table presents the results of our calculations for the binding affinities between the ligands and the bacterial and fungal proteins.
Table 3. Molecular docking scores (binding affinities) of ligands for E. coli (4XO8) and A. flavus (1R51).
The binding affinities of all the derivatives of the protein 4XO8 receptor ranged from −3.8 to −5.7 kcal/mol. Among these ligands, ligand 2 has the strongest binding affinity (−5.7 kcal/mol), surpassing that of the drug azithromycin (−5 kcal/mol). For the protein 1R51 receptor, the binding affinity ranged from −5.1 to −5.9 kcal/mol. Ligand 6 had the highest binding affinity (−5.9 kcal/mol) of the 1R51 receptor derivatives, although it was still lower than that of the drug nystatin (−7.7 kcal/mol). This observation correlates with the in vitro activities observed. All the ligands exhibited binding affinities greater than −5 kcal/mol and demonstrated convergent complexation energies. To confirm the geometry of ligands within the active site of receptors, it is crucial to assess the affinity of the binding site by comparing it with the optimal placement of ligands in the active site of each receptor. Understanding the interaction between bioactive molecules and their protein targets has led to the study of intermolecular interaction modes and the prediction of biological activity, focusing specifically on two bacterial proteins, 4XO8 and 1R51. This analysis revealed the key sites in 4XO8 that form the active site pocket, namely, Phe1, Asp47, Asp54, Gln133, Asn135, and Asp140. For 1R51, the crucial sites identified were Lys10, Thr57, Arg176, Gln228, Asn254, and His256, highlighting the importance of these sites in ligand—protein interactions and their potential role in predicting biological activity (Table ).
Table 4. Hydrogen bonding interactions of ligands against the bacterial protein Escherichia coli (4XO8) and the fungal protein Aspergillus flavus (1R51).
Figure 16. 2D visualization of the interaction types between the ligands and Escherichia coli (4XO8).


Figure 17. 2D visualization of the interaction types between the best ligands and Aspergillus flavus (1R51).


Table summarizes the distances, angles, and residues involved in the hydrogen bonds between the target proteins and the ligands. Figures and provide 2D visualizations of the hydrogen bonding interactions between the bioactive molecules and the critical amino acids of the 4XO8 and 1R51 bacterial proteins, respectively. Figure demonstrates that all the ligands form multiple hydrogen bonds with the Phe1, Asp47, Asp54, Gln133, Asn135, and Asp140 residues within the active site of the 4XO8 protein. This highlights the ability of the ligands to effectively occupy the binding pocket of this protein. Our findings also revealed a strong correlation between the best interactions formed with the protein 4XO8 and the high docking scores obtained for these compounds. Specifically, ligands 1 and 2, which had the highest docking scores, formed hydrogen bonds with nearly all the active site residues. The other ligands exhibit hydrogen bonding interactions with crucial residues, such as Phe1, Asn135, and Gln133. Additionally, Figure indicates that all the ligands, except for ligand 1, form several hydrogen bonds with protein 1R51 (as shown in Table ). The most favourable hydrogen bonding positions within the active site of protein 1R51 were found to be residues Arg176, Gln228, Asn254, and His256. While ligands 3 and 5, which have low binding affinities, form multiple hydrogen bonds with crucial residues, ligand 6, which has the highest binding affinity, forms only a single hydrogen bond with residue Gln228. However, a high docking score does not necessarily reflect the position or strength of the interactions with the active site of protein 1R51. These results highlight the similarity between the binding sites of the synthesized α-D-mannopyranoside and the selected bacterial proteins.
3.11. Reliability of docking predictions
To assess the effectiveness of the docking algorithms in predicting the conformations of protein-bound ligands, a series of redockings were performed on the crystallized ligands of the target proteins E. coli (4XOB) and A. flavus (1R51), with the aim of verifying the accuracy of the predicted poses. As shown in Figure , the results demonstrate a precise superposition of the docked conformations with respect to the structures of the native crystallized ligands, displaying RMSD values of 0.449 Å for E. coli and 0.409 Å for A. flavus. These results, significantly below the 2 Å threshold, not only confirm the precision and reliability of our molecular docking protocol but also attest to the accuracy of the grid maps generated for each target protein. Consequently, these observations validate our docking method as a robust and reliable technique for predicting ligand—protein configurations, playing a crucial role in the elucidation of molecular interaction mechanisms.
Figure 18. (a) Redocking pose of E. coli with its native ligand. (b) Redocking pose of A. flavus with its native ligand, showing the original (blue) and docked (green) configurations for both.

3.12. Molecular dynamic simulation
After evaluating the molecular docking results of the synthesized compounds, we performed MD simulations to assess the dynamic behaviour and stability of the target protein by analyzing the RMSD, RMSF, SASA, and Rg parameters. The results presented in Figures present the curves of these parameters for the complexes formed between the bacterial protein 4XO8, the fungal protein 1R51, and all the ligands.
Figure 19. a) The RMSD values of Escherichia coli with its ligands. b) The RMSD values of Aspergillus flavus with its ligands.

Figure 20. a) The RMSF values of Escherichia coli with its ligands. b) The RMSF values of with Aspergillus flavus its ligands.

Figure 21. a) The Radius of gyration (Rg) values of Escherichia coli with its ligands. b) The Radius of gyration (Rg) values of Aspergillus flavus with its ligands.

Figure 22. a) SASA values of Escherichia coli with its ligands. b) SASA values of Aspergillus flavus with its ligands.

For the protein 4XO8 (Figure a), analysis of the RMSD plots indicated that all the systems exhibited a rapid increase in RMSD values from 0.6 to 1 Å within a 10 ns period. Afterwards, all the systems fluctuated within a similar distance range of 1 to 1.5 Å, suggesting that all the systems reached a steady state and equilibrium. The RMSD values of ligands 1-6 complexed with protein 4XO8 were 1.269 , 1.368 , 1.290 , 1.511 , 1.279 , and 1.298 Å, respectively, while the RMSD value of azithromycin complexed with protein 4XO8 was 1.330 Å, serving as the reference ligand in this study. According to a previous study [Citation65–67], an RMSD value less than 3 Å is an indicator of the conformational stability of protein–ligand complexes. All the compounds had values less than 2 Å, indicating that all the compounds were stable at the active sites of the protein and that ligand 1 displayed the best stability. The analysis of RMSD plots for protein 1R51 (Figure b) revealed that all the systems displayed a quick increase in RMSD values from 0.7 to 1.8 Å within a 40 ns time frame. Afterwards, all the systems fluctuated within a similar range of 1.3 to 2.8 Å, indicating that they reached a stable state and equilibrium. The RMSD values for ligands 1-6 complexed with protein 1R51 were 2.083 , 2.247 , 1.967 , 2.874 , 2.256 and 2.151 Å, respectively. Moreover, the RMSD for the interaction of the nystatin complex with protein 1R51 was 2.438 Å, which served as the reference ligand in the study. As all RMSD values were less than 3 Å, it can be concluded that all the compounds are stable in the active site of protein 1R51, with ligand 3 being the most stable, having an RMSD value of less than 2 Å. This in silico result is consistent with the in vitro experiments, where ligand 3 was found to be the only active compound against Aspergillus flavus bacteria. In conclusion, the complexation of the predicted ligand with the bacterial proteins 4XO8 and 1R51 indicated increased the conformational stability of the complex.
The evaluation of RMSF tracks the impact of ligand binding on the protein's flexibility over the course of a 100 ns molecular dynamics simulation. This information is critical in determining the stability, rigidity, and compactness of the receptor. Flexible residues have high RMSF values, while stable residues have low RMSF values. In protein 4XO8 (Figure a), the majority of residues exhibited similar RMSF values, with greater fluctuations appearing in different regions, such as ASN7 (2.076 Å), ILE13 (2.07 Å), GLY65 (2.028 Å), SER114 (1.706 Å), and THR158 (1.535 Å), which are not crucial because they are located in the inactive regions of protein 4XO8. However, key active site residues such as Phe1, Gln133, and Asn135 exhibited smaller fluctuations, with RMSF values less than 0.6 Å, indicating that the established hydrogen bonds stabilize the ligands with the 4XO8 protein. Analysis of the RMSF values of the protein 1R51 (Figure b) revealed that residues with larger fluctuations, such as GLU22 (4.953 Å), TYR46 (2.874 Å), ARG122 (3.119 Å), GLU166 (4.363 Å), LYS266 (4.387 Å), and SER296 (4.290 Å), are located in inactive regions of the protein and are not involved in the active site. Conversely, crucial active site residues, such as Arg176, Gln228, Asn254, and His256, exhibited smaller fluctuations with RMSF values less than 1.2 Å, suggesting that the established hydrogen bonds stabilize the ligand complexations with the protein 1R51. These results demonstrated that all the complexes formed between the ligand and both proteins exhibited increased conformational stability due to hydrogen bonding.
The radius of gyration (Rg) is a measure of the compactness of protein–ligand complexes. The smaller the Rg value is, the more compact the structure. The results shown in Figure indicate that the structures of the complexes with the proteins 4XO8 and 1R51 remained relatively stable throughout the simulations. The limited variations in Rg values (Figure a and b) suggest that no significant conformational changes occurred in any of the systems, which indicates that all the complexes were compact and stable. Overall, the docking data suggest that the predicted ligand complexes with bacterial and fungal proteins exhibit high levels of conformational stability [Citation68,Citation69].
SASA provides insight into how much of a protein–ligand complex is exposed to solvent molecules. A lower SASA indicates a more compact protein with less exposure to solvents. The SASA values for the protein 4XO8 (Figure a) were similar across all the complexes, with only a minor increase occurring over the 30 ns period of simulation. This stability suggested that all the complexes were compact. The SASA values for protein 1R51 (Figure b) also remained stable for all the complexes throughout the simulations, indicating no changes in the complex structure or a high level of compactness. The SASA results suggest that both proteins form stable complexes with the respective ligands [Citation70,Citation71].
3.13. ADMET prediction
Naturally occurring compounds against bacterial proteins were tested using ADMET pharmacokinetic criteria [Citation72]. Table shows the prediction results from pkCSM, an online tool.
Table 5. In silico ADMET prediction of the potential inhibitors.
In general, low absorption (less than 30%) is considered a sign of poor absorption. However, the results indicated that all the compounds demonstrated good absorption in the human intestine. The volume of distribution (VDss) can be considered low when logVDss < −0.15 and high when logVDss > 0.45. Standard permeability values for the central nervous system (CNS) and blood—brain barrier (BBB) were between > −2 and < −3 for LogPS and between >0.3 and < −1 for LogBB. For a specific compound, LogBB < −1 indicates poor distribution to the brain, whereas LogBB >0.3 suggests the potential to cross the BBB. A LogPS > −2 is believed to penetrate the CNS, while a LogPS < −3 is considered to indicate difficulty entering the CNS. Hence, ligand 6 has a better barrier-crossing ability.
Drug metabolism refers to the biological transformation of pharmaceutical substances within the body. Drugs undergo multiple enzymatic reactions, resulting in different metabolites with varying pharmacological, pharmacokinetic, and physicochemical properties. Understanding drug metabolism and potential drug interactions is crucial. Cytochrome P450 inhibition, specifically by CYP1A2, 2C19, 2C9, 2D6, and 3A4, plays a significant role in drug metabolism and can result in drug interactions. These cytochrome P450 enzymes can also be targeted as therapeutic targets. Thus, we demonstrated the metabolism of each ligand, as presented in Table . Among these cytochromes, CYP3A4 was the most significant inhibitor [Citation66,Citation67]. Most of the compounds we designed were substrates or inhibitors of CYP3A4, with the exception of ligand 1. Moreover, a lower total clearance value suggests that the drug stays in the body for an extended period, which may be beneficial for certain drugs. Finally, the results of this study confirmed that all the compounds are nontoxic, which is an essential step in the drug development process. The outcomes of the in silico ADMET prediction are highly encouraging. The synthesized molecules possess desirable kinetic characteristics, meet the necessary drug-likeness standards, and exhibit valuable biological activity.
An obstacle in the acylation procedure is the difficulty in obtaining a single product due to the presence of numerous hydroxyl groups, which ultimately affects the total yield. Furthermore, the challenge of sustaining low temperatures throughout the reaction was considerable. Additionally, the use of reagents and chemicals results in significant expenses, which further adds to the limitations of the study. In the future, it is important for research in drug design to focus on synthesizing new chemicals that are very effective against a broad range of pathogens, including bacteria, viruses, cancer cells, and fungi.
4. Conclusions and future perspective
In the present study, we synthesized MDMP derivatives via spectral analysis, physicochemical analysis, antimicrobial activity, cytotoxicity evaluation, molecular docking, molecular dynamics, and SAR, QSAR, and ADMET studies. The results of the biological tests revealed that both compounds 3 and 4 had potent inhibitory effects on certain bacterial and fungal organisms. The different reactivity parameters indicated that these compounds have moderate reactivities. The biological activity of MDMP can be greatly improved by the addition of aliphatic and aromatic groups. Molecular docking studies reveal ligand—protein interactions and validate the efficacy of medicines against bacteria and fungi. The results of the MD simulation further affirm the stability of the ligand—protein complex under physiological conditions. Binding affinity analysis revealed that ligands 2, 3, 4, and 6 had the highest binding affinities for microbial proteins. The in silico pharmacokinetic predictions revealed optimized pharmacokinetic profiles for the compounds. This work enhances the understanding of the chemical, thermal, physicochemical, biological, and bioactivity characteristics of MDMP derivatives. This study lays the foundation for synthesizing novel antimicrobial agents that fight pathogenic microbes by revealing structural modifications that enhance their effectiveness. Additionally, this work offers significant data that might be utilized in the field of antibacterial drug discovery research. Understanding the structure—activity correlations and pharmacokinetic features of MDMP derivatives allows researchers to develop and improve antimicrobial medicines with increased efficiency and effectiveness. These findings help to characterize MDMP derivatives and aid in the rational development of antimicrobial drugs with enhanced effectiveness, reduced toxicity, and improved pharmacokinetics. These discoveries have the potential to stimulate the advancement of novel therapeutics for infectious diseases, addressing a pressing global healthcare demand. ADMET and QSAR showed good drug likeness and biological activity. This research may reveal the chemical, thermal, physicochemical, biological, and bioactivity features of MDMP derivatives. Thus, focusing on these data and compounds will help construct antimicrobial agents for use against pathogenic microbes.
Availability of data and materials
All relevant data are within the manuscript and available from the corresponding author upon request.
Consent to publish
The authors agree to publish the article under the Creative Commons Attribution License.
Supplemental Material
Download MS Word (5.1 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Naika HR, Lingaraju K, Chandramohan V, et al. Evaluation of phytoconstituents and molecular docking against NS3 protease of hepatitis C virus. J Pharm Sci Pharmacol. 2016;2:96–103. doi:10.1166/jpsp.2015.1057
- Choo QL, Kuo G, Weiner AJ, et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi:10.1126/science.2523562
- Bisceglie AD. Hepatitis C–related hepatocellular carcinoma in the United States: influence of ethnic status. Am J Gastroenterol. 2003;98:2060–2063. doi:10.1111/j.1572-0241.2003.t01-1-07641.x
- Pately PD, Pately MR, Kaushik-Basu N, et al. 3D QSAR and molecular docking studies of benzimidazole derivatives as hepatitis C virus NS5B polymerase inhibitors. J Chem Inf Model. 2008;48:42–55. doi:10.1021/ci700266z
- Sears P, Wong CH. Intervention of carbohydrate recognition by proteins and nucleic acids. Proc Natl Acad Sci. USA. 1996;93:12086–12093. doi:10.1073/pnas.93.22.12086
- Seeberger PH, Werz DB. Synthesis and medical applications of oligosaccharides. Nature. 2007;446:1046–1051. doi:10.1038/nature05819
- Chen S, Fukuda M. Cell type-specific roles of carbohydrates in tumor metastasis. Meth Enzymol. 2006;416:371–380. doi:10.1016/S0076-6879(06)16024-3
- Kawsar SMA, Hosen MA, El Bakri Y, et al. In silico approach for potential antimicrobial agents through antiviral, molecular docking, molecular dynamics, pharmacokinetic and bioactivity predictions of galactopyranoside derivatives. Arab J Basic Appl Sci. 2022;29:99–112. doi:10.1080/25765299.2022.2068275
- Qais FA, Khan MS, Ahmad I, et al. Plumbagin inhibits quorum sensing-regulated virulence and biofilms of gram-negative bacteria: in vitro and in silico investigations. Biofouling. 2021;37:724–739. doi:10.1080/08927014.2021.1955250
- Kabir AKMS, Kawsar SMA, Bhuiyan MMR, et al. Antimicrobial screening studies of some derivatives of methyl α-D-glucopyranoside. Pakistan J Sci Ind Res. 2009;52:138–142.
- Kawsar SMA, Hamida AA, Sheikh AU, et al. Chemically modified uridine molecules incorporating acyl residues to enhance antibacterial and cytotoxic activities. Int J Org Chem. 2015;5:232–245. doi:10.4236/ijoc.2015.54023
- Hosen MA, Alam A, Islam M, et al. Geometrical optimization, PASS prediction, molecular docking, and in silico ADMET studies of thymidine derivatives against FimH adhesin of escherichia coli. Bulg Chem Commun. 2021;53:327–342.
- Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3:97–130. doi:10.1093/glycob/3.2.97
- Chtita S, Larif M, Ghamali M, et al. Quantitative structure–activity relationship studies of dibenzo [a,d] cycloalkenimine derivatives for non-competitive antagonists of N-methyl-D-aspartate based on density functional theory with electronic and topological descriptors. J Taibah Univ Sci. 2015;9:143–154. doi:10.1016/j.jtusci.2014.10.006
- Ghamali M, Chtita S, Aouidate A, et al. Combining DFT and QSAR computation to predict the interaction of flavonoids with the GABA (A) receptor using electronic and topological descriptors. J Taibah Univ Sci. 2017;11:422–433. doi:10.1016/j.jtusci.2016.06.005
- Tabti K, Sbai A, Maghat H, et al. Computational assessment of the reactivity and pharmaceutical potential of novel triazole derivatives: an approach combining DFT calculations, molecular dynamics simulations, and molecular docking. Arabian J Chem. 2024;17:105376, doi:10.1016/j.arabjc.2023.105376
- Nour H, Daoui O, Abchir O, et al. Combined computational approaches for developing new anti-Alzheimer drug candidates: 3D-QSAR, molecular docking and molecular dynamics studies of liquiritigenin derivatives. Heliyon. 2022;8:e11991, doi:10.1016/j.heliyon.2022.e11991
- Abdessadak O, Alaqarbeh M, Zaki H, et al. Computational approaches to discover a kaempferol derivative extracted from senna alexandrina as escherichia coli enzyme (MurF) inhibitor by molecular docking, molecular dynamics simulation, and ADME-Tox. Struct Chem. 2023;34:1173–1187. doi:10.1007/s11224-022-02068-x
- Chtita S, Hmamouchi R, Larif M, et al. QSPR studies of 9-aniliioacridine derivatives for their DNA drug binding properties based on density functional theory using statistical methods: model, validation and influencing factors. J Taibah Univ Sci. 2016;10:868–876. doi:10.1016/j.jtusci.2015.04.007
- Belhassan A, Chtita S, Lakhlifi T, et al. QSPR study of the retention/release property of odorant molecules in pectin gels using statistical methods. J Taibah Univ Sci. 2017;11:1030–1046. doi:10.1016/j.jtusci.2017.05.004
- Nour H, Abchir O, Belaidi S, et al. 2D-QSAR and molecular docking studies of carbamate derivatives to discover novel potent anti-butyrylcholinesterase agents for Alzheimer’s disease treatment. Bull Korean Chem Soc. 2022;43:277–292. doi:10.1002/bkcs.12449
- Dearden JC. The history and development of quantitative structure-activity relationships (QSARs). Int J Quant Struct Relation. 2017;2:36–46. doi:10.4018/IJQSPR.2016010101
- Chtita S, Belhassan A, Bakhouch M, et al. QSAR study of unsymmetrical aromatic disulfides as potent avian SARS-CoV main protease inhibitors using quantum chemical descriptors and statistical methods. Chemometr Intell Lab Sys. 2021;210:104266, doi:10.1016/j.chemolab.2021.104266
- Nour H, Hashmi MA, Belaidi S, et al. Design of acetylcholinesterase inhibitors as promising anti-Alzheimer’s agents based on QSAR, molecular docking, and molecular dynamics studies of liquiritigenin derivatives. ChemistrySelect. 2023;8:e202301466, doi:10.1002/slct.202301466
- Abchir O, Daoui O, Nour H, et al. Exploration of cannabis constituents as potential candidates against diabetes mellitus disease using molecular docking, dynamics simulations and ADMET investigations. Sci Afr. 2023;21:e01745, doi:10.1016/j.sciaf.2023.e01745
- Hassan SA, Aziz DM, Abdullah MN, et al. In vitro and in vivo evaluation of the antimicrobial, antioxidant, cytotoxic, hemolytic activities and in silicoPOM/DFT/DNA-binding and pharmacokinetic analyses of new sulfonamide bearing thiazolidin-4-ones. J Biomol Struct Dyn. 2023: 1–17. doi:10.1080/07391102.2023.2226713
- Bhat AR, Dongre RS, Almalki FA, et al. Synthesis, Biological activity and POM/DFT/docking analyses of annulated pyrano[2,3-d]pyrimidine derivatives: identification of antibacterial and antitumor pharmacophore sites. Bioorg Chem. 2021;106:104480, doi:10.1016/j.bioorg.2020.104480
- Nour H, Abdou A, Belaidi S, et al. Discovery of promising cholinesterase inhibitors for Alzheimer’s disease treatment through DFT, docking, and molecular dynamics studies of eugenol derivatives. J Chinese Chem Soc. 2022;69:1534–1551. doi:10.1002/jccs.202200195
- Rana KM, Maowa J, Alam A, et al. In silico DFT study, molecular docking, and ADMET predictions of cytidine analogs with antimicrobial and anticancer properties. In Silico Pharmacol. 2021;9:42, doi:10.1007/s40203-021-00102-0
- Ahmed S, Bhat AR, Rahiman AK, et al. Green synthesis, antibacterial and antifungal evaluation of new thiazolidine-2,4-dione derivatives: molecular dynamic simulation, POM study and identification of antitumor pharmacophore sites. J Biomol Struct Dyn. 2023: 1–17. doi:10.1080/07391102.2023.2258404
- Bauer AW, Kirby WMM, Sherris JC. Antibiotic susceptibility testing by a standardized single disc method. Am J Clin Pathol. 1966;45:439–476. doi:10.1093/ajcp/45.4_ts.493
- Clinical Laboratory Standards Institute (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard-Ninth Edition. Wayne (PA): Clinical and Laboratory Standards Institute. CLSI Documents M07-A9; 2012.
- Grover RK, Moore JD. In-vitro efficacy of certain essential oils and plant extracts against three major pathogens of jatropha curcas L. Phytopathology. 1962;52:876–879.
- Kawsar SMA, Huq E, Nahar N. Cytotoxicity assessment of the aerial parts of macrotyloma uniflorum linn. Int J Pharmacol. 2008;4:297–300. doi:10.3923/ijp.2008.297.300
- Hunt WA. The effects of aliphatic alcohols on the biophysical and biochemical correlates of membrane function. Adv Exp Med Biol. 1975;56:195–210. doi:10.1007/978-1-4684-7529-6_9
- Kim YM, Farrah S, Baney RH. Structure–antimicrobial activity relationship for silanols, a new class of disinfectants, compared with alcohols and phenols. Int J Antimicrob Agents. 2007;29:217–222. doi:10.1016/j.ijantimicag.2006.08.036
- Mourik T, Bühl M, Marie-Pierre G. Density functional theory across chemistry, physics and biology. Philos Trans A Math Phys Eng Sci. 2011;372:20120488, doi:10.1098/rsta.2012.0488
- Gaussian RA, Frisch MJ, Trucks GW, et al. Gaussian, Inc, Wallingford CT. 2009.
- Lee C, Yang W, Parr RG. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37:785–789. doi:10.1103/PhysRevB.37.785
- Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A. 1988;38:3098–3100. doi:10.1103/PhysRevA.38.3098
- Hadni H, Elhallaoui M. 2D and 3D-QSAR, molecular docking and ADMET properties in silico studies of azaaurones as antimalarial agents. New J Chem. 2020;44:6553–6565. doi:10.1039/C9NJ05767F
- Yakovenko O, Sharma S, Forero M. Fimh forms catch bonds that are enhanced by mechanical force due to allosteric regulation. J Biol Chem. 2008;283:11596, doi:10.1074/jbc.M707815200
- Abchir O, Yamari I, Nour H, et al. Structure-based virtual screening, ADMET analysis, and molecular dynamics simulation of Moroccan natural compounds as candidates α-amylase inhibitors. ChemistrySelect. 2023;8:e202301092, doi:10.1002/slct.202301092
- Trott O, Olson AJ. Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi:10.1002/jcc.21334
- Onodera K, Satou K, Hirota H. Evaluations of molecular docking programs for virtual screening. J Chem Inf Model. 2007;47:1609–1618. doi:10.1021/ci7000378
- Hadni H, Elhallaoui M. 3D-QSAR, docking and ADMET properties of aurone analogues as antimalarial agents. Heliyon. 2020;6:e03580, doi:10.1016/j.heliyon.2020.e03580
- Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi:10.1002/jcc.20289
- Jo S, Kim T, Iyer VG, et al. Charmm-Gui: a web-based graphical user interface for Charmm. J Comput Chem. 2008;29:1859–1865. doi:10.1002/jcc.20945
- Im W, Seefeld S, Roux B. A grand canonical monte carlo–Brownian dynamics algorithm for simulating ion channels. Biophys J. 2000;79:788–801. doi:10.1016/S0006-3495(00)76336-3
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi:10.1016/0263-7855(96)00018-5
- En-nahli F, Baammi S, Hajji H, et al. High-throughput virtual screening approach of natural compounds as target inhibitors of plasmepsin-II. J Biomol Struct Dyn. 2022: 1–11. doi:10.1080/07391102.2022.2152871
- Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem.
- Farhana Y, Amin MR, Hosen A, et al. Bromobenzoylation of methyl α-D-mannopyranoside: synthesis and spectral characterization. J Sib Fed Univ Chem. 2021;14:171–183. doi:10.17516/1998-2836-0226
- Islam S, Hosen MA, Ahmad S, et al. Synthesis, antimicrobial, anticancer activities, PASS prediction, molecular docking, molecular dynamics and pharmacokinetic studies of designed methyl α-D-glucopyranoside esters. J Mol Struct. 2022;1260:132761, doi:10.1016/j.molstruc.2022.132761
- Tamokou JDD, Kuiate JR, Tene M, et al. The antimicrobial activities of extract and compounds isolated from brillantaisia lamium. Iran J Med Sci. 2011;36:24–31.
- Maowa J, Alam A, Rana KM, et al. Synthesis, characterization, synergistic antimicrobial properties and molecular docking of sugar modified uridine derivatives. Ovidius Univ Ann Chem. 2021;32:6–21. doi:10.2478/auoc-2021-0002
- Matsumoto R, Fujii Y, Kawsar SMA, et al. Cytotoxicity and glycan-binding properties of an 18 kDa lectin isolated from the marine sponge halichondria okada. Toxins (Basel). 2012;4:323–338.
- Sribalan R, Banuppriya G, Kirubavathi M. Synthesis, biological evaluation and in silico studies of tetrazole-heterocycle hybrids. J Mol Struct. 2019;1175:577–586.
- Li WR, Xie XB, Shi QS, et al. Antibacterial activity and mechanism of silver nanoparticles on escherichia coli. Appl Microbiol Biotechnol. 2010;85:1115–1122. doi:10.1007/s00253-009-2159-5
- Judge V, Narasimhan B, Ahuja M, et al. Synthesis, antimycobacterial, antiviral, antimicrobial activity and QSAR studies of N(2)-acyl isonicotinic acid hydrazide derivatives. Med Chem. 2013;9:53–76. doi:10.2174/157340613804488404
- Marinescu M, Emandi A, Marton G, et al. Structural studies and optical nonlinear response of some pyrazole-5-ones. Nanosci Nanotechnol Lett. 2015;7:846–854. doi:10.1166/nnl.2015.2032
- Parr RG, Donnelly RA, Levy M, et al. Electronegativity: the density functional viewpoint. J Chem Phys. 1978;68:3801–3807. doi:10.1063/1.436185
- Aihara J. Reduced HOMO−LUMO gap as an index of kinetic stability for polycyclic aromatic hydrocarbons. J Phys Chem A. 1999;103:7487–7495. doi:10.1021/jp990092i
- Khedraoui M, Nour H, Yamari I, et al. Design of a new potent Alzheimer’s disease inhibitor based on QSAR, molecular docking and molecular dynamics investigations. Chem Phys Impact. 2023;7:100361, doi:10.1016/j.chphi.2023.100361
- Hadni H, Fitri A, Benjelloun AT, et al. Identification of terpenoids as potential inhibitors of SARS-CoV-2 (main protease) and spike (RBD) via computer-aided drug design. J Biomol Struct Dyn. 2023: 1–14. doi:10.1080/07391102.2023.2245051
- Hasan AH, Shakya S, Hussain FHS, et al. Design, synthesis, anti-ecetylcholinesterase evaluation and molecular modelling studies of novel coumarin-chalcone hybrids. J Biomol Struct Dyn. 2023;41:11450–11462. doi:10.1080/07391102.2022.2162583
- Hussen NH, Hasan AH, Jamalis J, et al. Potential inhibitory activity of phytoconstituents against black fungus: in silico ADMET, molecular docking and MD simulation studies. Comput Toxicol. 2022;100247; doi:10.1016/j.comtox.2022.100247
- Kawsar SMA, Kumer A, Munia NS, et al. Chemical descriptors, PASS, molecular docking, molecular dynamics and ADMET predictions of glucopyranoside derivatives as inhibitors to bacteria and fungi growth. Org Commun. 2022;15:184–203. doi:10.25135/acg.oc.122.2203.2397
- Amin MR, Yasmin F, Dey S, et al. Methyl β-D-galactopyranoside esters as potential inhibitors for SARS-CoV-2 protease enzyme: synthesis, antimicrobial, PASS, molecular docking, molecular dynamics simulations and quantum computations. Glycoconjugate J. 2021;39:261–290. doi:10.1007/s10719-021-10039-3
- Hasan AH, Yusof FSM, Kamarudin NA, et al. Synthesis, anti-ecetylcholinesterase evaluation, molecular docking and molecular dynamics simulation of novel psoralen derivatives. Curr Org Synth. 2023; doi:10.2174/1570179420666230328121554
- Devasia J, Chinnam S, Khatana K, et al. Synthesis, DFT and in silico anti-COVID evaluation of novel tetrazole analogues. Polycyclic Arom Comp. 2023;43:1941–1956. doi:10.1080/10406638.2022.2036778
- Yamari I, Mouhib A, Es-Sounni B, et al. Oxidative functionalization of triterpenes isolated from euphorbia resinifera latex: semisynthesis, ADME-Tox, molecular docking, and molecular dynamics simulations. Chem Phys Impact. 2023;7:100372, doi:10.1016/j.chphi.2023.100372