
ABSTRACT
Introduction
Brain development is highly dependent on hormonal regulation. Exposure to chemicals disrupting endocrine signaling has been associated with neurodevelopmental impairment. This raises concern about exposure to the suspected thousands of endocrine disruptors, and has resulted in efforts to improve regulation of these chemicals. Yet, the causal links between endocrine disruption and developmental neurotoxicity, which would be required for regulatory action, are still largely missing.
Areas covered
In this review, we illustrate the importance of two endocrine systems, thyroid hormone and retinoic acid pathways, for neurodevelopment. We place special emphasis on TH and RA synthesis, metabolism, and how endocrine disrupting chemicals known or suspected to affect these systems are associated with developmental neurotoxicity.
Expert opinion
While it is clear that neurodevelopment is dependent on proper hormonal functioning, and evidence is increasing for developmental neurotoxicity induced by endocrine disrupting chemicals, this is not grasped by current chemical testing. Thus, there is an urgent need to develop test methods detecting endocrine disruption in the context of neurodevelopment. Key to this development is further mechanistic insights on the involvement of endocrine signaling in neurodevelopment as well as increased support to develop and validate new test methods for the regulatory context.
1. Introduction
Hormones play a fundamental role during fetal brain development influencing key processes such as proliferation, apoptosis, differentiation, migration, and myelination [Citation1–5]. Thus, the developing brain is rendered vulnerable to environmental stimuli that can disturb the hormonal milieu. This raises concerns about health consequences of developmental exposures to a group of chemicals known as endocrine disrupting chemicals (EDCs), which are capable of interacting with various hormonal pathways at different levels. EDCs are exogenous substances or mixtures capable of interacting with the endocrine system and leading to adverse effects in intact organisms, their progeny or (sub) populations [Citation6].
EDCs have been shown to be present in biological fluids of pregnant women [Citation7,Citation8] and there is emerging epidemiological evidence suggesting associations between exposure to EDCs and negative neurodevelopmental outcomes in children. For instance, exposure to EDCs has been associated with disorders indicative of developmental neurotoxicity (DNT) such as autism spectrum disorder (ASD), attention deficit hyperactivity disorder (ADHD), and intellectual disabilities (for a comprehensive review see [Citation9]). Briefly, Bisphenol A (BPA), phthalates, and perfluoroalkyl substances (PFAS), have been associated with ASD [Citation10] and social behavior disturbances which may be significant indicators of ASD [Citation11–17]. Likewise, links between ADHD-related neurobehavioral traits and BPA [Citation18], phthalates [Citation19,Citation20] or organic persistent pollutants [Citation21] have been established. Intellectual disabilities have also been reported to be associated with EDC exposure, whereby intellectual quotient (IQ) was used in many studies as a measure of intellectual capacity. Lower IQ scores have been correlated to some metabolites of phthalates [Citation22], bisphenol F (BPF) [Citation23] and different mixtures of EDCs [Citation24,Citation25] in different settings. BPA has been shown to have detrimental effects on executive functions [Citation26], working memory [Citation12] and language development [Citation27]. Moreover, behavioral disturbances [Citation28,Citation29] and academic achievement [Citation30] have also been shown to be associated with EDC exposure.
Although many epidemiological studies have shown associations between neurodevelopmental outcomes and prenatal exposure to EDCs, at present the epidemiological literature remains inconclusive due to the heterogeneity and inherent limitations of such studies. However, the epidemiological evidence is supported by data obtained from animal studies showing that in utero/perinatal exposure to EDCs can lead to effects on behavior, memory, and motor activity (reviewed in [Citation31–34]).
In Europe, EDCs are currently identified using hazard-based criteria in the context of EU Regulations (EC) No. 1107/2009 Plant Protection Products Regulation (PPPR) and (EU) No. 528/2012 Biocidal Products Regulation (BPR). The requirements for determining whether a substance meets the definition specified in the EDC criteria for PPPR and BPR include evidence that the substance has endocrine activity and that there is a biologically plausible link between the endocrine mode of action and the adverse effect. However, the few available in vivo DNT tests do not address effects induced through endocrine modes of action, which highlights the importance of, scientifically and methodologically, bridging the gap between EDCs and DNT from a regulatory perspective.
In this review, we aim at illustrating the importance of hormonal functioning for neurodevelopment, and thus the need to consider endocrine disruption in the field of DNT. There are a number of endocrine pathways involved in neurodevelopment. For example, sex steroids, the arguably most studied hormones in the context of endocrine disruption, are involved in the development of sexual dimorphism of the brain and mediate neurodevelopmental processes such as neuronal apoptosis, neurite growth, differentiation, and activity of dopaminergic neurons, oligodendrocyte maturation, myelination, synaptogenesis, and synaptic plasticity [Citation35,Citation36]. This review is focused on two other examples, the thyroid hormone and retinoic acid systems, which we use to describe the molecular mechanisms linking hormonal signaling with neurodevelopment. Furthermore, we present evidence clearly linking their disruption by EDCs with DNT outcomes, and provide an overview of current efforts to develop models capable of detecting ED-induced DNT.
2. Thyroid hormone disruption and developmental neurotoxicity
Thyroid hormone (TH) plays a crucial role in prenatal development, and many adverse consequences arising from thyroid diseases during pregnancy are well described in the literature. Fetal hypothyroidism can, in the most extreme cases lead to cretinism, which manifests, among others, with intellectual disability [Citation37]. Also maternal hypothyroidism is associated with lower IQ scores, as well as with ASD and ADHD in different populations [Citation38–40]. Brain morphology has also been demonstrated to be affected by maternal hypothyroidism [Citation2,Citation41]. Correspondingly, the link between maternal hyperthyroidism and cortical gray matter alterations has been established [Citation2,Citation41]. At the cellular level, TH is involved in neuronal and oligodendrocyte differentiation and maturation [Citation42,Citation43].
2.1. TH synthesis, signaling, and metabolism
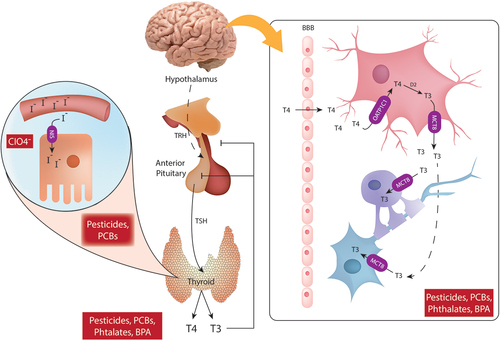
When stimulated by thyroid stimulating hormone (TSH) secreted by the pituitary gland, the thyroid gland produces the thyroid hormones thyroxine (T4), and triiodothyronine (T3) [Citation44] (). Acquired from seafood, dairy, and fortified food items such as salt, iodine is essential for TH synthesis. Iodine is absorbed in the stomach and small intestine and stored in iodine concentrating tissues such as the thyroid gland, the salivary gland, lactating breast, intestine, and stomach [Citation45]. Once in the bloodstream, iodine is transported into storing tissues by the action of the sodium/iodide symporter (NIS), whose transcription and insertion in cellular membranes is regulated by TSH [Citation46,Citation47] (). Congenital iodine transport defects associated with hypothyroidism and goiter have been linked to mutations in SLC45A, which encodes for NIS [Citation48]. Iodine in thyroid follicular cells is transported through the cellular apical membrane toward the follicular lumen by pendrin, a member of the SLC26A family [Citation49,Citation50]. Upon uptake by the thyroid, iodine is oxidized and added to tyrosine residues in thyroglobulin by thyroid peroxidase (TPO), resulting in the formation of monoiodothyronine (MIT), diiodothyronine (DIT), T3, reverse T3 (an isomer of triiodothyronine), and T4; although T3 is mainly produced by T4 deiodination (reviewed in [Citation51]). During gestation, maternal iodine intake and placental transport are the main contributors to the fetal iodine supply [Citation52].
Figure 1. Schematic representation of thyroid hormone disruption by endocrine disruptors. Thyrotropin-releasing hormone (TRH) is produced by the hypothalamus and sensed by the anterior pituitary gland where the thyroid-stimulating hormone (TSH) is produced and released into the general circulation. TSH promotes the secretion of thyroxine (T4) and triiodothyronine (T3) from the thyroid gland. Thyroid follicular cells uptake iodine (I-) from the circulation by sodium/iodide symporter (NIS), iodine will be later used for the production of thyroid hormones. NIS is blocked by CIO4-. A negative feedback loop for T4 and T3 is mediated by the sensing of circulation T4 and T3 levels by the anterior pituitary and the hypothalamus. Pesticides, Polychlorinated biphenyls (PCBs), phthalates and bisphenol A (BPA) can interfere with thyroid hormone signaling and affect circulating levels of T4 and T3. In the central nervous system, T4 crosses the brain-blood barrier (BBB) and is taken up by astrocytes via OATP1C1. Once inside the astrocytes, T4 is converted to T3 by the action of D2. T3 exits the astrocytes via MCT8 and enters oligodendrocytes and neurons via the same MCT8 transporter.
T3 exerts most of its functions by binding to the Thyroid hormone receptor (THR), which can then dimerize and form homodimers or heterodimers with retinoic X receptors (RXRs). The dimers then bind to thyroid hormone response elements (TRE) in the DNA and initiate gene transcription of target genes [Citation4]. On the other hand, low intracellular T3 levels induce the recruitment of nuclear corepressors leading to repression of target genes. THRs are divided into two subtypes, encoded by two genes, named thyroid hormone receptor alpha (THRα) and thyroid hormone receptor beta (THRβ) with the main differences at the level of amino acids present in their DNA binding domain (reviewed in [Citation53]). In the developing rat brain, 60% of THRs are THRα, while THRβ appears in the brain at late developmental stages and its expression is more limited to specific areas (e.g. hypothalamus, pituitary, retina, and cochlea) [Citation54]. It is, however, important to note that limitations regarding the specificity of antibodies for THRα and THRβ have been reported [Citation55] posing a problem for the identification of these receptors in the developing brain. In humans, germ-line mutations in the THRα lead to a condition similar to congenital hypothyroidism [Citation56]. On the other hand, germ-line mutations in THRβ produce a condition characterized by resistance to TH with elevated T3 levels and varied tissue sensitivity to the hormone. In this case, manifestations include a myriad of symptoms typical of both hypo- and hyperthyroidism (reviewed in [Citation57]). TH can also modulate important cellular responses via non-canonical pathways such as the PKA pathway, modulation of the expression of epidermal growth factor (EGF) and fibroblast growth factor (FGF), and the interaction between T4 and integrin αvβ3 [Citation58]. Likewise, T3 has been reported to trigger generation of Ca2+, NO, inositol triphosphate and cAMP within minutes after its administration, suggesting that these effects are not mediated by transcriptional changes [Citation59].
TH production is regulated by negative feedback in the hypothalamic-pituitary-thyroid (HPT) axis (). Circulating TH levels are sensed in the hypothalamus and pituitary gland, which in turn decrease the production of TSH by the anterior pituitary and/or of thyrotropin-releasing hormone (TRH), produced in the hypothalamus [Citation4]. Due to its hydrophobicity, TH binds to plasmatic proteins in the circulation (thyroxine-binding protein, transthyretin, and albumin), which facilitate hormonal transit. At the cellular level, T4 and T3 are taken up by the cells via monocarboxylate transporters (MCT), which prefer T3, l-amino acid transporters (LAT) which transports both T3 and T4, and organic anion transporters polypeptides (OATP) which transport mainly T4 and reverse T3 [Citation60]. In the brain, MCT8 and MCT10 are the main transporters, and a mutation in MCT8 is linked to the Allan-Herndon-Dudley syndrome (AHDS), characterized by intellectual impairment and a myriad of neurological symptoms [Citation61]. In the fetus, the thyroid gland develops from weeks 5 to 6 of gestation, but fetal TH synthesis starts later on (weeks 14–16 of gestation) [Citation62,Citation63]. Fetal hypothalamic–pituitary–thyroid axis (HPT) starts to function at the beginning of the third trimester [Citation63] and TH reaches adult levels after birth. Thus, fetal TH levels in early gestation are dependent on maternal transfer regulated by placental transporters, deiodinases D2 and D3, and placental synthesis and secretion of transthyretin and albumin [Citation64,Citation65].
2.2. TH signaling and neurodevelopment
In the developing brain, TH is involved in cell proliferation, neurogenesis myelination and proliferation of oligodendrocyte precursors, glia-neuron communication, and the formation of Bergmann’s glia [Citation4]. T4 is actively transported across the brain-blood barrier (BBB), whereas BBB is mostly impermeable to T3 (reviewed in [Citation66]). T4 entering the brain after moving across the endothelial cells, is taken up by astrocytes by an OATP transporter (Oatp1c1), where it is deiodinated by D2 producing T3, which is then able to exit the astrocytes via MCT8 and be taken up by neurons and oligodendrocytes via MCT8 [Citation67]) (). Once inside the cell, 5’-deiodinases will remove iodide from TH depending on the current cellular needs. Three types of deiodinase enzymes have been described, D1 and D2 are responsible for the conversion of T4 to T3, D1 and D3 convert T4 to reverse T3, and D1, D2 and D3 convert T3 and reverse T3 to T2. The main deiodinases found in the brain are D2 and D3, in fact most of the T3 in the brain comes from D2 expressed in astrocytes, tanycytes and oligodendrocyte progenitor cells [Citation68]. The expression of TH transporters MCT8, MCT10, OATP1A2, OATP4A1, LAT1 and LAT2 have been reported in placenta [Citation69,Citation70]. At the cellular level, TH regulates neural stem cell fate, oligodendrocyte differentiation, and differentiation of dopaminergic neurons. Oligodendrocyte differentiation is dependent on T3 in part due to T3 mediated secretion of neurotrophins by other cell types (reviewed in [Citation55,Citation66]).
2.3. Chemical exposures causing disruption in the thyroid hormone signaling pathway leading to DNT
EDCs can interfere with TH signaling by interfering with TH synthesis and circulating levels, blocking TPO enzymatic activity or iodine transport, or disrupting nuclear receptor signaling [Citation71] (). For instance, perchlorate (ClO4−) compounds interfere with NIS, inhibiting iodine transport due to the increased affinity ClO4− has for NIS in comparison with iodide [Citation72]. ClO4− has been used in detonators, pyrotechnics, explosives, airbags, rocket fuels, PVC production, fertilizers, and it is also produced by degradation of hypochlorite (reviewed in [Citation73,Citation74]). Taylor P.N. et al investigated the correlation between urinary ClO4− in the first trimester of pregnancy in mothers with hypothyroidism and supplementation with levothyroxine, finding increased odds for lower IQs in their children at 3 years of age [Citation75]. This association is supported by experimental data where gestational exposure to ClO4− decreased T4 levels and reduced hippocampal synaptic transmission in rats [Citation76].
Another group of chemicals affecting TH homeostasis are pesticides from the organochlorine, organophosphate, carbamate, pyrethroid and neonicotinoid families. A recent review by Leemans et al reported that organochlorine pesticides such as DDT and hexachlorobenzene have been associated with decreased total and free T3, decreased free T4 and increased TSH in multiple mother and child cohorts [Citation77]. Likewise, decreased levels of T4 and altered intellectual development have been associated with exposure to organophosphates [Citation77]. These associations are supported by experimental in vivo data where pesticides have been found to affect TH levels, decrease circulating transthyretin, affect the hepatic metabolism of T4, induce histological changes in the thyroid and reduce brain weight. For a comprehensive review the reader is referred to [Citation77].
Polychlorinated biphenyls (PCBs) are persistent organic pollutants which were used for varied applications and were present in transformers, electrical capacitors, hydraulic fluids and paints. Although they are no longer made, PCBs can still be found as part of products produced before the ban [Citation78]. PCBs act on THR as agonists or antagonists and can affect TH levels in humans and experimentally in vivo [Citation78,Citation79]. In epidemiological studies, PCBs have been associated with increased risk of hyperactivity and attention deficit among others (reviewed in [Citation80]). Exposure to a PCB mixture induced changes in serum TH concentration in dams together with altered cell cycle exit of neuronal progenitors and delayed radial glia migration in the cortex of the developmentally exposed pups [Citation79].
Also chemicals used in plastic production, phthalates and bisphenols, are suspected to interfere with TH signaling. Phthalates have been shown to affect TH signaling in different ways, including altering circulating TH levels, increasing thyroglobulin expression, upregulating D1 and D2, decreasing transthyretin levels and decreasing the expression of THRα and THRβ (reviewed in [Citation81]). A recent study by Derakhshan et al, showed that exposure to phthalates was associated with lower circulating levels of TH including free T4 and free T3 in pregnant women [Citation8]. Furthermore, prenatal exposure to phthalates has been associated with disturbances in child motor development and lower scores in mental and psychomotor indices assessed with the Bayley scales [Citation82,Citation83]. In a recent study, Bornehag et al found associations between first-trimester maternal urinary levels of dibutyl phthalate (DBP) and butyl benzyl phthalate (BBzP), and language delay in children 2.5 to 3 years old [Citation84]. Finally, BPA, used, e.g. as starting material for most epoxy resins, and its analogues BPF and BPS can bind to THR and act as either agonist or antagonist. BPA has been associated with altered serum TH levels in pregnant women [Citation85,Citation86] as well as with neurodevelopmental impacts in epidemiological and experimental data (reviewed in [Citation87]). These studies show, on occasion, contrasting results, in part explained by differences in species, sex, administered doses and tests used for evaluation of neurodevelopment.
In conclusion, there is evidence from an increasing number of studies for TH disruption by industrial chemicals on one hand, and associations between exposure to these chemicals and neurodevelopmental impacts on the other hand. Yet, studies clearly linking DNT and TH disruption in the same population or experimental setting, are very rare.
3. Involvement of retinoids in brain development
Retinoids are essential for both embryonic and adult growth. Retinoic acid (RA), a metabolite of vitamin A, is involved in the development of the heart, limbs, nervous system, and through the regulation of Homeobox (HOX) genes, RA directs anterior-posterior patterning in the embryo [Citation88]. At the cellular level, RA has been widely demonstrated to induce differentiation of neurons and glia, and to increase the number and the length of neurites [Citation89].
3.1. RA synthesis, signaling, and metabolism
Retinoids are acquired from the diet and are precursors for at least two important metabolites, all-trans-retinoic acid, and 9-cis-retinoic acid. In order to become biologically active, retinol has to be absorbed in the gut, metabolized and transported to various tissues. The transport of retinoids into the cells is facilitated by retinoid binding proteins (RBP) that bind to the STRA6 receptor (stimulated by retinoic acid 6), enabling the hydrophobic retinoids to enter the cells [Citation90] (). The catabolism of retinol to retinoic acid consists in a two-step oxidation. Initially, retinol is converted to retinaldehyde by two enzyme families, retinol dehydrogenases (Rhd10) and alcohol dehydrogenases (ADHs) [Citation91]. Retinaldehyde can be converted back to retinol by dehydrogenase/reductase enzymes (Dhr), regulating the amount of substrate available for retinoic acid synthesis [Citation92]. The second and final step in this synthesis is the oxidation of retinaldehyde to retinoic acid facilitated by retinaldehyde dehydrogenases (Raldh1–3) [Citation91]. Retinoic acid is metabolized by oxidation to the less bioactive metabolite 4-oxo-retinoic acid by CYP26A-C1 enzymes that belong to the cytochrome P450 hydroxylase family [Citation93]. In addition, retinoic acid enhances its own degradation by activating the RAR and RXR receptors, leading to downregulation of Rdh10 and Raldh2 and upregulation of CYP26 and CRABP2 [Citation94–96].
Figure 2. Schematic representation of Retinoic acid (RA) signaling disruption mediated by xenobiotics. Retinoid binding protein (RBP) facilitate the cellular uptake of retinoids. Once inside the cell, the conversion of retinol to RA is mediated by alcohol dehydrogenases (ADH) and retinaldehyde dehydrogenases (RALDH). Cyp26 metabolized RA into the less bioactive 4-oxo-retinoic acid. Retinoic acid binding proteins (CRABPs) transport RA into the nucleus where RA binds to corresponding response elements. Acrylamide has been shown to downregulate the expression of Crabp2 and Rpb7, likewise VPA downregulates Cyp26a1 and affects Aldh1a2 altering RA signaling.
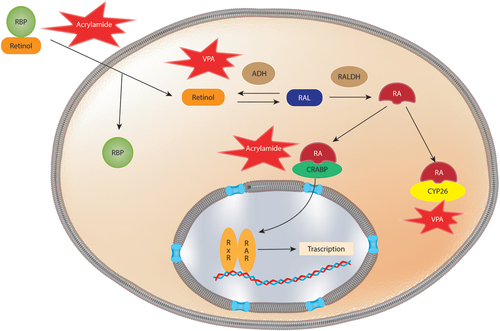
Once retinoic acid is synthesized, it is transferred to the nucleus by cellular retinoic acid binding proteins (CRABPs), where it acts as a transcriptional activating ligand by binding to the nuclear retinoic acid receptors (RARs) and RXRs (). RARs recognize both all-trans-retinoic acid and 9-cis-retinoic acid, while RXRs only recognize 9-cis-retinoic acid. Upon binding, the receptors form a heterodimer complex, activating gene transcription by binding to retinoic acid response elements (RARE) in the genome [Citation97]. RXR forms heterodimers with other hormonal nuclear receptors such as the thyroid hormone receptors (TRs), vitamin D receptors, peroxisome proliferator-activated receptors (PPARs) and liver X receptors (LXR) [Citation98,Citation99].
3.2. RA signaling and neurodevelopment
RARs and RXRs have been found to be expressed from early developmental stages in zebrafish and xenopus (Reviewed by [Citation100]). In mammals, a study in mouse totipotent-like cells resembling the blastomere of 2-cell embryos showed expression of Rars and Rxr and activity in response to RA, implying activation of the RA pathway at this developmental stage [Citation3]. During gastrulation, RA is restricted to the posterior region by the presence of CYP26A1 and CYP26C1 in the anterior region. Once the neural plate is formed, RALDH2 is transiently expressed in the rostral forebrain leading to activation of RA in this region and regulation of cellular signals such as Wnts, fibroblast growth factor 8 (FGF8), and sonic hedgehog (SHH), which are needed for the development of telencephalic, diencephalic and optic vesicles [Citation100–102]. As the embryo continues developing, RA-dependent signaling is present in the primitive streak, primitive node and in the mesoderm, while at the same time CYP26A1 and CYP26C1 maintain anterior embryonic regions free from RA [Citation103–108]. At this stage the expression of RA and RXR receptors centers on the neural ectoderm and a differential expression of RAR subtypes is observed with a strong downregulation of Rarg in the neural plate and mesoderm. At this point, the developing brain subdivides into three regions: the forebrain (which will become the cerebral cortex, basal ganglia and hippocampus), the midbrain, and the hindbrain (which will become the cerebellum, pons and medulla). RA concentrates in the hindbrain, where it is essential for the organization of segmented regions called rhombomeres. As embryonic development progresses, RA becomes indispensable for the development of anterior structures and forebrain growth [Citation100,Citation109], with both RALDH2 and RALDH3 as the main RA producing enzymes in the frontonasal region. During late brain development, Rara is expressed almost exclusively in regions derived from the hindbrain (medulla oblongata, pons and cerebellum), the corpus striatum and corpus pallidum, whereas Rarb expression is found in the medulla oblongata, pons, cerebellum, choroid plexuses, and the developing meninges [Citation110,Citation111].
The balance of bioavailable retinoic acid is primarily driven by Rhd, Raldh and CYP26 enzymes, by regulating the synthesis and degradation of retinoic acid. The expression of these enzymes is tissue-dependent and highly regulated during neuronal development. Loss of Rdh10 activity has been shown to cause a deficiency of bioavailable retinoic acid and leads to craniofacial and limb developmental defects in mice [Citation112,Citation113]. Similarly, loss of Raldh2 and Raldh3 function in mice leads to cardiac and forelimb development deficiency [Citation114,Citation115]. Disruption of the CYP26A1 and CYP26B1 enzymes in mice, leading to an increase of retinoic acid in the cell, causes abnormalities in the hindbrain patterning, proximal limb patterning, resembling the teratogenic effects of RA [Citation116,Citation117].
Although RA is essential for neurogenesis in both embryo and adult, this morphogen was demonstrated to be teratogenic. Thus, both a deficiency and an excess of RA can lead to negative effects on the neurodevelopmental process [Citation88,Citation118]. For example, the removal of vitamin A from maternal diet in animals caused problems such as lack of eyeballs in pig offspring, hydrocephalus, spina bifida and microphthalmia [Citation89,Citation119–122]. Furthermore, the lack of this vitamin during in utero development led to abnormalities at different levels in quails, rats, chick and mouse embryos [Citation1]: in the development of the caudal hindbrain [Citation2], in the formation of the spinal cord [Citation3], and by causing the death of the neural crest cells [Citation89,Citation123–127]. In addition, Bao et al. (2012) reported an increase in the risk of developing schizophrenia or other schizophrenia spectrum disorders after retinol deficiency during the second trimester of pregnancy [Citation118,Citation128–131].
On the contrary, excess of RA and vitamin A have been demonstrated to be teratogenic [Citation89,Citation132–135] producing defects at the level of the eye, the ear, the spinal cord and the hindbrain [Citation89,Citation123,Citation136–140]. 13-cis RA causes on the one hand hydrocephaly and malformations in all the hindbrain structures especially in the precerebellar nuclei [Citation89] leading to motor and sensory developmental delays and to severe mental retardation [Citation89,Citation141,Citation142]. On the other hand, it can also lead to microcephaly or a decrease in size of specific forebrain regions, together with occasional cortical heterotopias [Citation89]. It is also noteworthy that children embryonically exposed to 13-cis RA and born without major malformations often exhibit cognitive impairments, such as reduced mental ability [Citation89,Citation141–143]. In addition, human males seem to be more vulnerable to cognitive impairments than females, although the reason for this difference has not been elucidated yet [Citation142].
3.3. Chemical exposures correlated with disruption in the retinoic signaling pathway leading to DNT
The inhibition of RA signaling has been proposed as one of the mechanisms underlying the changes evidenced in the fetal alcohol spectrum disorder (FASD). This link has been made based on the role of ADH and CYP450 enzymes in both RA and alcohol metabolism [Citation144]. FASD is characterized by anatomical and neurodevelopmental abnormalities and, in the most severe cases, microcephaly and growth restriction [Citation144]. Animal data supports these claims as some teratogenic effects induced by ethanol have been rescued by supplementation with retinoic acid [Citation145–147].
To the best of our knowledge, the effect of anthropogenic chemicals on RA signaling in correlation to DNT has not been addressed epidemiologically or in vivo experiments. However, in vitro, chemicals known to produce DNT have been reported to alter RA homeostasis (). For example, exposure to valproic acid (VPA), an anticonvulsant commonly used for epilepsy treatment [Citation148], has shown to cause neural tube malformation and significantly downregulate Cyp26a1 gene expression in mouse embryos and pluripotent mouse embryonal carcinoma cells (P19), resulting in altered retinoic acid signaling [Citation149–152]. Connection between VPA exposure and RA signaling has also been found in an in vitro mouse gastrulation model of P19C5 stem cell derived embryoid bodies. In this model, several genes involved in retinoic acid metabolism were altered (Aldh1a2 and Cyp26a1) and axial patterning and elongation were inhibited, moreover these effects were partially rescued by the use of a RA antagonist [Citation153]. Acrylamide, is an organic compound used to produce polyacrylamide widely used in petroleum applications, in water and wastewater treatment, as a soil conditioner [Citation154]. Acrylamide has also been used in cosmetics and textiles and it is formed when starch rich foods are cooked at high temperatures (reviewed in [Citation155]). Maternal exposure to acrylamide has been associated with reduced fetal growth and head circumference, whereas in adults, it has been associated with hearing loss and mild cognitive decline [Citation155]. Since other DNT effects have not been studied, acrylamide is a suspected DNT. In vitro, acrylamide has shown to alter neuronal differentiation and downregulate the expression of genes involved in RA signaling (e.g. Crabp2 and Rpb7) in differentiated SH-SY5Y cells [Citation156].
Altogether a clear link between chemically induced RA disruption and DNT has not been established. Although there is vast knowledge about the fundamental role of RA signaling during neurodevelopment, there is a lack of studies addressing RA disruption in relation to DNT.
4. Expert opinion
The two pathways reviewed here illustrate the dependence of neurodevelopment on proper hormonal functioning, and thus the link between endocrine disruption and developmental neurotoxicity. However, this link remains largely unaddressed in a correlative, let alone causative manner. Yet, a causative link between an endocrine mode of action and a DNT outcome would be the requirement to regulate EDCs according to the criteria currently used by the EU. Thus, DNT induced by ED is not appropriately covered in the current regulations on chemical testing. In fact, at present, the Organization for Economic Co-Operation and Development (OECD) does not have approved test guidelines (TGs) which can properly address ED induced DNT. The two OECD approved TGs addressing DNT in vivo, TG426 and TG443, are not required to be performed unless a compound shows evidence of neurotoxicity in repeated dose studies, or evidence of thyroid disrupting activity. The endpoints measured in TG426 and TG443 consist in gross anatomical evaluation of brain tissue, histopathological examination, and a very limited number neurobehavioral tests, with only optional evaluations of social and cognitive impairments, which are often seen with exposures to EDCs. Therefore, ED-induced manifestations of developmental neurotoxicity are likely missed by the chemical assessment required by current regulations.
Furthermore, the in vivo rodent chemical testing has significant cost and time requirements, as well as translational issues, an approach which is unsustainable and unsuitable for the evaluation of thousands of chemicals for DNT potential [Citation157,Citation158]. As a result, there has been a great interest in developing New Approach Methods (NAMs) for DNT, which represent non-animal based approaches that can be used to provide information in the context of chemical hazard and risk assessment [Citation159,Citation160]. For DNT, efforts have been especially dedicated to the development of in vitro models which recapitulate key events during neurodevelopment, such as proliferation, migration, neural differentiation, neurite outgrowth, and neural network formation and function [Citation157,Citation161]. At present, these models are in different stages of development and still require international validation to be used for regulatory decision-making [Citation161]. Ideally, the future toxicological testing strategy will shift toward Integrated Approaches to Testing and Assessment (IATA), implying integration of data from in vitro testing batteries with other streams of evidence to guide risk management decisions and the prioritization of chemicals for in vivo DNT testing [Citation161].
To aid in the development of IATA for ED-induced DNT, the Adverse Outcome Pathway (AOP) concept has been introduced as a promising tool, as it represents an objective systematic approach and a practical framework for the organization and understanding of toxicological knowledge [Citation162]. AOPs are constructed based on a sequence of causally linked and measurable Key Events (KEs), which connect a molecular initiating event (MIE) to an adverse outcome (AO) through different levels of biological organization [Citation162]. Although AOPs contain an immense potential to integrate basic research with regulatory needs, AOPs for ED-induced DNT are difficult to develop at this time due to the lack of mechanistic knowledge related to endocrine mediated processes driving neurodevelopment, the importance of timing of such processes, and crosstalk between endocrine and other physiological systems. It is also worth mentioning that in the two cases exposed in this review (TH and RA) there is a good amount of knowledge available on their role in neurodevelopment; however, this is not the case for other hormonal pathways.
Building on these efforts, the EU, through the European Cluster to Improve Identification of Endocrine Disruptors (EURION: https://eurion-cluster.eu/), is currently supporting the development and validation of NAMs for the assessment of the effects of EDCs on neurodevelopment (ENDpoiNTs project) and also on TH disruption (ATHENA: http://athenaedctestmethods.net/; ERGO: https://ergo-project.eu/; and SCREENED: https://www.screened-project.eu/ projects) [Citation163–166]. Other efforts at further studying TH disruption outside the EURION cluster include the EC thyroid feasibility study which finished in 2019 with a report [Citation167] and the Cefic-Lri – Long-Range Research Initiative. Likewise, efforts leading to the development of AOPs involving the endocrine system are currently taking place within and among these projects. Ultimately, it is expected that these efforts will help clarify the link between ED and DNT leading to the development of in vitro and in silico testing batteries which will in turn lead to a better evaluation of EDC exposure, moving the toxicological field forward and preventing adverse effects.
Article highlights
The importance of hormonal signaling, e.g. thyroid hormone and retinoic acid, during brain development is well established
Epidemiological and experimental data support association between exposure to man-made chemicals and thyroid hormone disruption on one hand, and neurodevelopmental impairments on the other hand
Disruption of retinoic acid signaling has been studied in relation to fetal alcohol spectrum disorder, however it is not commonly addressed in the context of man-made chemicals
Causative links between endocrine mode of action and impaired neurodevelopment are not established, not even for hormonal pathways that are clearly involved in neurodevelopment
As such causative links for regulation of chemicals with endocrine disrupting properties are required, further knowledge on the involvement of endocrine signaling in neurodevelopment is needed
Such insights facilitate development of new test methods for the regulatory context
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
The authors would like to thank Manuel Merinero de los Santos, for his assistance with the design of the figures.
Additional information
Funding
References
- Schug TT, Blawas AM, Gray K, et al. Elucidating the links between endocrine disruptors and neurodevelopment. Endocrinology. 2015;156(6):1941–1951.
- Jansen TA, Korevaar TIM, Mulder TA, et al. Maternal thyroid function during pregnancy and child brain morphology: a time window-specific analysis of a prospective cohort. Lancet Diabetes Endocrinol. 2019;7(8):629–637.
- Iturbide A, Ruiz Tejeda Segura ML, Noll C, et al. Retinoic acid signaling is critical during the totipotency window in early mammalian development. Nat Struct Mol Biol. 2021;28(6):521–532.
- Giannocco G, Kizys MML, Maciel RM, et al. Thyroid hormone, gene expression, and central nervous system: where we are. Semin Cell Dev Biol. 2021;114:47–56.
- Chatonnet F, Guyot R, Benoît G, et al. Genome-wide analysis of thyroid hormone receptors shared and specific functions in neural cells. Proc Natl Acad Sci U S A. 2013;110(8):E766–75.
- International Programme on Chemical Safety, World Health Organization.Global assessment on the state of the science of endocrine disruptors; 2002. Retrieved 2022 03 03. https://apps.who.int/iris/handle/10665/67357
- Woodruff TJ, Zota AR, Schwartz JM. Environmental chemicals in pregnant women in the United States: NHANES 2003–2004. Environ Health Perspect. 2011;119(119.6):878–885.
- Derakhshan A, Shu H, Broeren MAC, et al. Association of phthalate exposure with thyroid function during pregnancy. Environ Int. 2021;157:106795.
- Rivollier F, Krebs MO, Kebir O. Perinatal exposure to environmental endocrine disruptors in the emergence of neurodevelopmental psychiatric diseases: a systematic review. Int J Environ Res Public Health. 2019;16(8):1318.
- Stein TP, Schluter MD, Steer RA, et al. Bisphenol a exposure in children with autism spectrum disorders. Autism Res. 2015;8(3):272–283.
- Lim YH, Bae S, Kim BN, et al. Prenatal and postnatal bisphenol A exposure and social impairment in 4-year-old children. Environ Health. 2017;16(1):79.
- Braun JM, Muckle G, Arbuckle T, et al. Associations of prenatal urinary bisphenol a concentrations with child behaviors and cognitive abilities. Environ Health Perspect. 2017;125(6):067008.
- Hansen JB, Bilenberg N, Timmermann CAG, et al. Prenatal exposure to bisphenol A and autistic- and ADHD-related symptoms in children aged 2 and5 years from the odense child cohort. Environ Health. 2021;20(1):24.
- Oulhote Y, Lanphear B, Braun JM, et al. Gestational exposures to phthalates and folic acid, and autistic traits in canadian children. Environ Health Perspect. 2020;128(2):27004.
- Alampi JD, Lanphear BP, Braun JM, et al. Association between gestational exposure to toxicants and autistic behaviors using bayesian quantile regression. Am J Epidemiol. 2021;190(9):1803–1813.
- Shin HM, Bennett DH, Calafat AM, et al. Modeled prenatal exposure to per- and polyfluoroalkyl substances in association with child autism spectrum disorder: a case-control study. Environ Res. 2020;186:109514.
- Oh J, Bennett DH, Calafat AM, et al. Prenatal exposure to per- and polyfluoroalkyl substances in association with autism spectrum disorder in the MARBLES study. Environ Int. 2021;147:106328.
- Arbuckle TE, Davis K, Boylan K, et al. Bisphenol A, phthalates and lead and learning and behavioral problems in Canadian children 6–11 years of age: CHMS 2007–2009. Neurotoxicology. 2016;54:89–98.
- England-Mason G, Martin JW, MacDonald A, et al. Similar names, different results: consistency of the associations between prenatal exposure to phthalates and parent-ratings of behavior problems in preschool children. Environ Int. 2020;142:105892.
- Ku HY, Tsai TL, Wang PL, et al. Prenatal and childhood phthalate exposure and attention deficit hyperactivity disorder traits in child temperament: a 12-year follow-up birth cohort study. Sci Total Environ. 2020;699:134053.
- Lenters V, Iszatt N, Forns J, et al. Early-life exposure to persistent organic pollutants (OCPs, PBDEs, PCBs, PFASs) and attention-deficit/hyperactivity disorder: a multi-pollutant analysis of a Norwegian birth cohort. Environ Int. 2019;125:33–42.
- Jankowska A, Polańska K, Koch HM, et al. Phthalate exposure and neurodevelopmental outcomes in early school age children from Poland. Environ Res. 2019;179(Pt B):108829.
- Bornehag CG, Engdahl E, Unenge Hallerbäck M, et al. Prenatal exposure to bisphenols and cognitive function in children at 7 years of age in the Swedish SELMA study. Environ Int. 2021;150:106433.
- Tanner EM, Hallerbäck MU, Wikström S, et al. Early prenatal exposure to suspected endocrine disruptor mixtures is associated with lower IQ at age seven. Environ Int. 2020;134:105185.
- Guo J, Wu C, Zhang J, et al. Prenatal exposure to mixture of heavy metals, pesticides and phenols and IQ in children at 7 years of age: the SMBCS study. Environ Int. 2020;139:105692.
- England-Mason G, Liu J, Martin JW, et al. Postnatal BPA is associated with increasing executive function difficulties in preschool children. Pediatr Res. 2021;89(3):686–693.
- Pan R, Wang C, Shi R, et al. Prenatal Bisphenol A exposure and early childhood neurodevelopment in Shandong, China. Int J Hyg Environ Health. 2019;222(5):896–902.
- Minatoya M, Araki A, Nakajima S, et al. Cord blood BPA level and child neurodevelopment and behavioral problems: the hokkaido study on environment and children’s health. Sci Total Environ. 2017;607–608:351–356.
- Li N, Papandonatos GD, Calafat AM, et al. Gestational and childhood exposure to phthalates and child behavior. Environ Int. 2020;144:106036.
- Jackson-Browne MS, Papandonatos GD, Chen A, et al. Gestational and childhood urinary triclosan concentrations and academic achievement among 8-year-old children. Neurotoxicology. 2020;78:170–176.
- Dingemans MM, van den Berg M, Westerink RH. Neurotoxicity of brominated flame retardants: (in)direct effects of parent and hydroxylated polybrominated diphenyl ethers on the (developing) nervous system. Environ Health Perspect. 2011;119(7):900–907.
- Quinnies KM, Harris EP, Snyder RW, et al. Direct and transgenerational effects of low doses of perinatal di-(2-ethylhexyl) phthalate (DEHP) on social behaviors in mice. PLoS One. 2017;12(2):e0171977.
- Xu XH, Zhang J, Wang YM, et al. Perinatal exposure to bisphenol-A impairs learning-memory by concomitant down-regulation of N-methyl-D-aspartate receptors of hippocampus in male offspring mice. Horm Behav. 2010;58(2):326–333.
- André SM, Markowski VP. Learning deficits expressed as delayed extinction of a conditioned running response following perinatal exposure to vinclozolin. Neurotoxicol Teratol. 2006;28(4):482–488.
- Nalvarte I, Varshney M, Inzunza J, et al. Estrogen receptor beta and neural development. Vitam Horm. 2021;116:313–326.
- Küppers E, Ivanova T, Karolczak M, et al. Classical and nonclassical estrogen action in the developing midbrain. Horm Behav. 2001;40(2):196–202.
- Rovet JF. The role of thyroid hormones for brain development and cognitive function. Endocr Dev. 2014;26:26–43.
- Haddow JE, Palomaki GE, Allan WC, et al. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med. 1999;341(8):549–555.
- Andersen SL, Andersen S, Vestergaard P, et al. Maternal thyroid function in early pregnancy and child neurodevelopmental disorders: a danish nationwide case-cohort study. Thyroid. 2018;28(4):537–546.
- Getahun D, Jacobsen SJ, Fassett MJ, et al. Association between maternal hypothyroidism and autism spectrum disorders in children. Pediatr Res. 2018;83(3):580–588.
- Korevaar TI, Muetzel R, Medici M, et al. Association of maternal thyroid function during early pregnancy with offspring IQ and brain morphology in childhood: a population-based prospective cohort study. Lancet Diabetes Endocrinol. 2016;4(1):35–43.
- Remaud S, Ortiz FC, and Perret-Jeanneret M, et al. Transient hypothyroidism favors oligodendrocyte generation providing functional remyelination in the adult mouse brain. Elife. 2017;6:e29996.
- López-Juárez A, Remaud S, Hassani Z, et al. Thyroid hormone signaling acts as a neurogenic switch by repressing Sox2 in the adult neural stem cell niche. Cell Stem Cell. 2012;10(5):531–543.
- Brent GA. Mechanisms of thyroid hormone action. J Clin Invest. 2012;122(9):3035–3043.
- Dai G, Levy O, Carrasco N. Cloning and characterization of the thyroid iodide transporter. Nature. 1996;379(6564):458–460.
- Riedel C, Levy O, Carrasco N. Post-transcriptional regulation of the sodium/iodide symporter by thyrotropin. J Biol Chem. 2001;276(24):21458–21463.
- Kogai T, Curcio F, Hyman S, et al. Induction of follicle formation in long-term cultured normal human thyroid cells treated with thyrotropin stimulates iodide uptake but not sodium/iodide symporter messenger RNA and protein expression. J Endocrinol. 2000;167(1):125–135.
- Wolff J. Congenital goiter with defective iodide transport. Endocr Rev. 1983;4(3):240–254.
- Yoshida A, Taniguchi S, Hisatome I, et al. Pendrin is an iodide-specific apical porter responsible for iodide efflux from thyroid cells. J Clin Endocrinol Metab. 2002;87(7):3356–3361.
- Gillam MP, Sidhaye AR, Lee EJ, et al. Functional characterization of pendrin in a polarized cell system. Evidence for pendrin-mediated apical iodide efflux. J Biol Chem. 2004;279(13):13004–13010.
- Ohara A, Yamada F, Fukuda T, et al. Specific alteration of gene expression profile in rats by treatment with thyroid toxicants that inhibit thyroid hormone synthesis. J Appl Toxicol. 2018;38(12):1529–1537.
- Glinoer D. The regulation of thyroid function in pregnancy: pathways of endocrine adaptation from physiology to pathology. Endocr Rev. 1997;18(3):404–433.
- Vella KR, Hollenberg AN. The actions of thyroid hormone signaling in the nucleus. Mol Cell Endocrinol. 2017;458:127–135.
- Jones I, Srinivas M, Ng L, et al. The thyroid hormone receptor beta gene: structure and functions in the brain and sensory systems. Thyroid. 2003;13(11):1057–1068.
- Flamant F, Gauthier K, Richard S. genetic investigation of thyroid hormone receptor function in the developing and adult brain. Curr Top Dev Biol. 2017;125:303–335.
- Bochukova E, Schoenmakers N, Agostini M, et al. A mutation in the thyroid hormone receptor alpha gene. N Engl J Med. 2012;366(3):243–249.
- Dumitrescu AM, Refetoff S. The syndromes of reduced sensitivity to thyroid hormone. Biochim Biophys Acta. 2013;1830(7):3987–4003.
- Davis PJ, Goglia F, Leonard JL. Nongenomic actions of thyroid hormone. Nat Rev Endocrinol. 2016;12(2):111–121.
- Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31(2):139–170.
- Bernal J. The significance of thyroid hormone transporters in the brain. Endocrinology. 2005;146(4):1698–1700.
- Fischer J, Kleinau G, Müller A, et al. Modulation of monocarboxylate transporter 8 oligomerization by specific pathogenic mutations. J Mol Endocrinol. 2015;54(1):39–50.
- Fernández LP, López-Márquez A, Santisteban P. Thyroid transcription factors in development, differentiation and disease. Nat Rev Endocrinol. 2015;11(1):29–42.
- Patel J, Landers K, Li H, et al. Delivery of maternal thyroid hormones to the fetus. Trends Endocrinol Metab. 2011;22(5):164–170.
- McKinnon B, Li H, Richard K, et al. Synthesis of thyroid hormone binding proteins transthyretin and albumin by human trophoblast. J Clin Endocrinol Metab. 2005;90(12):6714–6720.
- Koopdonk-Kool JM, de Vijlder JJ, Veenboer GJ, et al. Type II and type III deiodinase activity in human placenta as a function of gestational age. J Clin Endocrinol Metab. 1996;81(6):2154–2158.
- Schiera G, Di Liegro CM, Di Liegro I. Involvement of thyroid hormones in brain development and cancer. Cancers (Basel). 2021;13(11). DOI:https://doi.org/10.3390/cancers13112693
- Morte B, Bernal J. Thyroid hormone action: astrocyte-neuron communication. Front Endocrinol (Lausanne). 2014;5:82.
- Fliers E, Unmehopa UA, Alkemade A. Functional neuroanatomy of thyroid hormone feedback in the human hypothalamus and pituitary gland. Mol Cell Endocrinol. 2006;251(1–2):1–8.
- Chan SY, Franklyn JA, Pemberton HN, et al. Monocarboxylate transporter 8 expression in the human placenta: the effects of severe intrauterine growth restriction. J Endocrinol. 2006;189(3):465–471.
- Sato K, Sugawara J, Sato T, et al. Expression of organic anion transporting polypeptide E (OATP-E) in human placenta. Placenta. 2003;24(2–3):144–148.
- Decherf S, Seugnet I, Fini JB, et al. Disruption of thyroid hormone-dependent hypothalamic set-points by environmental contaminants. Mol Cell Endocrinol. 2010;323(2):172–182.
- Tonacchera M, Pinchera A, Dimida A, et al. Relative potencies and additivity of perchlorate, thiocyanate, nitrate, and iodide on the inhibition of radioactive iodide uptake by the human sodium iodide symporter. Thyroid. 2004;14(12):1012–1019.
- Lisco G, De Tullio A, Giagulli VA, et al. Interference on iodine uptake and human thyroid function by perchlorate-contaminated water and food. Nutrients. 2020;12(6):1669.
- Niziński P, Błażewicz A, Kończyk J, et al. Perchlorate - properties, toxicity and human health effects: an updated review. Rev Environ Health. 2020;36(2):199–222.
- Taylor PN, Okosieme OE, Murphy R, et al. Maternal perchlorate levels in women with borderline thyroid function during pregnancy and the cognitive development of their offspring: data from the controlled antenatal thyroid study. J Clin Endocrinol Metab. 2014;99(11):4291–4298.
- Gilbert ME, Sui L. Developmental exposure to perchlorate alters synaptic transmission in hippocampus of the adult rat. Environ Health Perspect. 2008;116(6):752–760.
- Leemans M, Couderq S, Demeneix B, et al. Pesticides with potential thyroid hormone-disrupting effects: a review of recent data. Front Endocrinol (Lausanne). 2019;10:743.
- Ghassabian A, Trasande L. Disruption in thyroid signaling pathway: a mechanism for the effect of endocrine-disrupting chemicals on child neurodevelopment. Front Endocrinol (Lausanne). 2018;9:204.
- Naveau E, Pinson A, Gérard A, et al. Alteration of rat fetal cerebral cortex development after prenatal exposure to polychlorinated biphenyls. PLoS One. 2014;9(3):e91903.
- Ramírez V, Gálvez-Ontiveros Y, González-Domenech PJ, et al. Role of endocrine disrupting chemicals in children’s neurodevelopment. Environ Res. 2022;203:111890.
- Zhang X, Qi W, Xu Q, et al. Di(2-ethylhexyl) phthalate (DEHP) and thyroid: biological mechanisms of interference and possible clinical implications. Environ Sci Pollut Res Int. 2022;29(2):1634–1644.
- Polanska K, Ligocka D, Sobala W, et al. Phthalate exposure and child development: the polish mother and child cohort study. Early Hum Dev. 2014;90(9):477–485.
- Kim Y, Ha EH, Kim EJ, et al. Prenatal exposure to phthalates and infant development at 6 months: prospective mothers and children’s environmental health (MOCEH) study. Environ Health Perspect. 2011;119(10):1495–1500.
- Bornehag CG, Lindh C, Reichenberg A, et al. Association of prenatal phthalate exposure with language development in early childhood. JAMA Pediatr. 2018;172(12):1169–1176.
- Aung MT, Johns LE, Ferguson KK, et al. Thyroid hormone parameters during pregnancy in relation to urinary bisphenol A concentrations: a repeated measures study. Environ Int. 2017;104:33–40.
- Romano ME, Webster GM, Vuong AM, et al. Gestational urinary bisphenol A and maternal and newborn thyroid hormone concentrations: the home study. Environ Res. 2015;138:453–460.
- Palanza P, Paterlini S, Brambilla MM, et al. Sex-biased impact of endocrine disrupting chemicals on behavioral development and vulnerability to disease: of mice and children. Neurosci Biobehav Rev. 2021;121:29–46.
- Niederreither K, Dollé P. Retinoic acid in development: towards an integrated view. Nat Rev Genet. 2008;9(7):541–553.
- McCaffery PJ, Adams J, Maden M, et al. Too much of a good thing: retinoic acid as an endogenous regulator of neural differentiation and exogenous teratogen. Eur J Neurosci. 2003;18(3):457–472.
- Kawaguchi R, Yu J, Honda J, et al. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. 2007;315(5813):820–825.
- Kedishvili NY. retinoic acid synthesis and degradation. Subcell Biochem. 2016;81:127–161.
- Haeseleer F, Huang J, Lebioda L, et al. Molecular characterization of a novel short-chain dehydrogenase/reductase that reduces all-trans-retinal. J Biol Chem. 1998;273(34):21790–21799.
- Fujii H,Sato T., Kaneko S., et al. Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos. The EMBO Journal. 1997;16(14):4163–4173.
- Strate I, Min TH, Iliev D, et al. Retinol dehydrogenase 10 is a feedback regulator of retinoic acid signalling during axis formation and patterning of the central nervous system. Development. 2009;136(3):461–472.
- Niederreither K, McCaffery P, Dräger UC, et al. Restricted expression and retinoic acid-induced downregulation of the retinaldehyde dehydrogenase type 2 (RALDH-2) gene during mouse development. Mech Dev. 1997;62(1):67–78.
- White RJ, Nie Q, Lander AD, et al. Complex regulation of cyp26a1 creates a robust retinoic acid gradient in the zebrafish embryo. PLoS Biol. 2007;5(11):e304.
- Mangelsdorf DJ. Vitamin A receptors. Nutr Rev. 1994;52(2 Pt 2):S32–44.
- Umesono K, Evans RM. Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell. 1989;57(7):1139–1146.
- Janowski BA, Grogan MJ, Jones SA, et al. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc Natl Acad Sci U S A. 1999;96(1):266–271.
- Dollé P. Developmental expression of retinoic acid receptors (RARs). Nucl Recept Signal. 2009;7:e006.
- Diez Del Corral R, Olivera-Martinez I, Goriely A, et al. Opposing FGF and retinoid pathways control ventral neural pattern, neuronal differentiation, and segmentation during body axis extension. Neuron. 2003;40(1):65–79.
- Diez Del Corral R, Storey KG. Opposing FGF and retinoid pathways: a signalling switch that controls differentiation and patterning onset in the extending vertebrate body axis. Bioessays. 2004;26(8):857–869.
- Niederreither K, Subbarayan V, Dollé P, et al. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet. 1999;21(4):444–448.
- Ribes V, Le Roux I, Rhinn M, et al. Early mouse caudal development relies on crosstalk between retinoic acid, Shh and Fgf signalling pathways. Development. 2009;136(4):665–676.
- Shiotsugu J, Katsuyama Y, Arima K, et al. Multiple points of interaction between retinoic acid and FGF signaling during embryonic axis formation. Development. 2004;131(11):2653–2667.
- Hernandez RE, Putzke AP, Myers JP, et al. Cyp26 enzymes generate the retinoic acid response pattern necessary for hindbrain development. Development. 2007;134(1):177–187.
- Ribes V, Fraulob V, Petkovich M, et al. The oxidizing enzyme CYP26a1 tightly regulates the availability of retinoic acid in the gastrulating mouse embryo to ensure proper head development and vasculogenesis. Dev Dyn. 2007;236(3):644–653.
- Uehara M, Yashiro K, Mamiya S, et al. CYP26A1 and CYP26C1 cooperatively regulate anterior-posterior patterning of the developing brain and the production of migratory cranial neural crest cells in the mouse. Dev Biol. 2007;302(2):399–411.
- Williams AL, Bohnsack BL. What’s retinoic acid got to do with it? Retinoic acid regulation of the neural crest in craniofacial and ocular development. Genesis. 2019;57(7–8):e23308.
- Mollard R, Viville S, Ward SJ, et al. Tissue-specific expression of retinoic acid receptor isoform transcripts in the mouse embryo. Mech Dev. 2000;94(1–2):223–232.
- Ruberte E, Friederich V, Chambon P, et al. Retinoic acid receptors and cellular retinoid binding proteins. III. Their differential transcript distribution during mouse nervous system development. Development. 1993;118(1):267–282.
- Kurosaka H, Wang Q, Sandell L, et al. Rdh10 loss-of-function and perturbed retinoid signaling underlies the etiology of choanal atresia. Hum Mol Genet. 2017;26(7):1268–1279.
- Sandell LL, Sanderson BW, Moiseyev G, et al. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 2007;21(9):1113–1124.
- Niederreither K, Vermot J, Schuhbaur B, et al. Embryonic retinoic acid synthesis is required for forelimb growth and anteroposterior patterning in the mouse. Development. 2002;129(15):3563–3574.
- Dupé V, Matt N, Garnier JM, et al. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc Natl Acad Sci U S A. 2003;100(24):14036–14041.
- Abu-Abed S, Dollé P, Metzger D, et al. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 2001;15(2):226–240.
- Yashiro K, Zhao X, Uehara M, et al. Regulation of retinoic acid distribution is required for proximodistal patterning and outgrowth of the developing mouse limb. Dev Cell. 2004;6(3):411–422.
- Gagné AM, Hébert M, Maziade M. Revisiting visual dysfunctions in schizophrenia from the retina to the cortical cells: a manifestation of defective neurodevelopment. Prog Neuropsychopharmacol Biol Psychiatry. 2015;62:29–34.
- Mw HEB, McCollum EV, McCollum EV. Further studies on the nutritive deficiencies of wheat and grain mixtures and the pathological conditions produced in swine by their use. J Biol Chem. 1916;25(2):239–259.
- Hughes HJS, Lienhardt HF, Aubel CE. Nerve degeneration resulting from avitaminosis A. J Nutr. 1929;2(2):183–186.
- Corcoran J, So PL, Barber RD, et al. Retinoic acid receptor beta2 and neurite outgrowth in the adult mouse spinal cord in vitro. J Cell Sci. 2002;115(Pt 19):3779–3786.
- Hale F. Pigs Born without Eye Balls. In: Persaud T. V. N., editors. Problems of Birth Defects. Dordrecht: Springer; 1993. p. 166–167.
- Maden M, Gale E, Kostetskii I, et al. Vitamin A-deficient quail embryos have half a hindbrain and other neural defects. Curr Biol. 1996;6(4):417–426.
- White JC, Highland M, Kaiser M, et al. Vitamin A deficiency results in the dose-dependent acquisition of anterior character and shortening of the caudal hindbrain of the rat embryo. Dev Biol. 2000;220(2):263–284.
- Dupé V, Lumsden A. Hindbrain patterning involves graded responses to retinoic acid signalling. Development. 2001;128(12):2199–2208.
- Wendling O, Ghyselinck NB, Chambon P, et al. Roles of retinoic acid receptors in early embryonic morphogenesis and hindbrain patterning. Development. 2001;128(11):2031–2038.
- Niederreither K, Vermot J, Schuhbaur B, et al. Retinoic acid synthesis and hindbrain patterning in the mouse embryo. Development. 2000;127(1):75–85.
- Bao Y, Ibram G, Blaner WS, et al. Low maternal retinol as a risk factor for schizophrenia in adult offspring. Schizophr Res. 2012;137(1–3):159–165.
- Rohde CM, Manatt M, Clagett-Dame M, et al. Vitamin A antagonizes the action of vitamin D in rats. J Nutr. 1999;129(12):2246–2250.
- McGrath JJ, Eyles DW, Pedersen CB, et al. Neonatal vitamin D status and risk of schizophrenia: a population-based case-control study. Arch Gen Psychiatry. 2010;67(9):889–894.
- Valipour G, Saneei P, Esmaillzadeh A. Serum vitamin D levels in relation to schizophrenia: a systematic review and meta-analysis of observational studies. J Clin Endocrinol Metab. 2014;99(10):3863–3872.
- Cohlan SQ. Excessive intake of vitamin A as a cause of congenital anomalies in the rat. Science. 1953;117(3046):535–536.
- Kochhar DM. Teratogenic activity of retinoic acid. Acta Pathol Microbiol Scand. 1967;70(3):398–404.
- Giguere V, Ong ES, Segui P, et al. Identification of a receptor for the morphogen retinoic acid. Nature. 1987;330(6149):624–629.
- Petkovich M, Brand NJ, Krust A, et al. A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature. 1987;330(6147):444–450.
- Shenefelt RE. Morphogenesis of malformations in hamsters caused by retinoic acid: relation to dose and stage at treatment. Teratology. 1972;5(1):103–118.
- Lohnes D, Mark M, Mendelsohn C, et al. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development. 1994;120(10):2723–2748.
- Mallo M. Retinoic acid disturbs mouse middle ear development in a stage-dependent fashion. Dev Biol. 1997;184(1):175–186.
- Hendrickx AG, Tzimas G, Korte R, et al. Retinoid teratogenicity in the macaque: verification of dosing regimen. J Med Primatol. 1998;27(6):310–318.
- Sockanathan S, Jessell TM. Motor neuron-derived retinoid signaling specifies the subtype identity of spinal motor neurons. Cell. 1998;94(4):503–514.
- Adams J, Lammer E.J. Human isotretinoin exposure: the teratogenesis of a syndrome of cognitive deficits Neurotoxicology and Teratology. 1995;3(17):386.
- Adams JBS, Gavin JA, Janulewicz PA, et al. Neuropsychological characteristics of children embryonically exposed to isotretinoin (Accutane®): outcome at age 10. Neurotoxicol Teratol. 2001;23:296.
- Adams Jane, Holson, R. Robert. The Neurobehavioral Teratology of Vitamin A Analogs. In Slikker Jr, William, Paule Merle G, Wang, Cheng, editors. Handbook of Developmental Neurotoxicology. 1998;631–642.
- Petrelli B, Bendelac L, Hicks GG, et al. Insights into retinoic acid deficiency and the induction of craniofacial malformations and microcephaly in fetal alcohol spectrum disorder. Genesis. 2019;57(1):e23278.
- Satiroglu-Tufan NL, Tufan AC. Amelioration of ethanol-induced growth retardation by all-trans-retinoic acid and alpha-tocopherol in shell-less culture of the chick embryo. Reprod Toxicol. 2004;18(3):407–412.
- Twal WO, Zile MH. Retinoic acid reverses ethanol-induced cardiovascular abnormalities in quail embryos. Alcohol Clin Exp Res. 1997;21(6):1137–1143.
- Marrs JA, Clendenon SG, Ratcliffe DR, et al. Zebrafish fetal alcohol syndrome model: effects of ethanol are rescued by retinoic acid supplement. Alcohol. 2010;44(7–8):707–715.
- Romoli M, Mazzocchetti P, D’Alonzo R, et al. Valproic Acid and Epilepsy: from Molecular Mechanisms to Clinical Evidences. Curr Neuropharmacol. 2019;17(10):926–946.
- Nau H, Hauck RS, Ehlers K. Valproic acid-induced neural tube defects in mouse and human: aspects of chirality, alternative drug development, pharmacokinetics and possible mechanisms. Toxicol Pharmacol. 1991;69(5):310–321.
- Kultima K, Jergil M, Salter H, et al. Early transcriptional responses in mouse embryos as a basis for selection of molecular markers predictive of valproic acid teratogenicity. Reprod Toxicol. 2010;30(3):457–468.
- Jergil M, Kultima K, Gustafson AL, et al. Valproic acid-induced deregulation in vitro of genes associated in vivo with neural tube defects. Toxicol Sci. 2009;108(1):132–148.
- Piersma AH, Hessel EV, Staal YC. Retinoic acid in developmental toxicology: teratogen, morphogen and biomarker. Reprod Toxicol. 2017;72:53–61.
- Li AS, Marikawa Y. Adverse effect of valproic acid on an in vitro gastrulation model entails activation of retinoic acid signaling. Reprod Toxicol. 2016;66:68–83.
- Xiong B, Loss RD, Shields D, et al. Polyacrylamide degradation and its implications in environmental systems. Npj Clean Water. 2018;1(1):17.
- Lindeman B, Johansson Y, Andreassen M, et al. Does the food processing contaminant acrylamide cause developmental neurotoxicity? A review and identification of knowledge gaps. Reprod Toxicol. 2021;101:93–114.
- Attoff K, Johansson Y, Cediel-Ulloa A, et al. Acrylamide alters CREB and retinoic acid signalling pathways during differentiation of the human neuroblastoma SH-SY5Y cell line. Sci Rep. 2020;10(1):16714.
- Fritsche E,Crofton K.M, Hernandez A.F, et al. OECD/EFSA workshop on developmental neurotoxicity (DNT): the use of non-animal test methods for regulatory purposes. Altex. 2017;34(2):311–315.
- Sachana M, Bal-Price A, Crofton KM, et al. International regulatory and scientific effort for improved developmental neurotoxicity testing. Toxicol Sci. 2019;167(1):45–57.
- Fritsche E, Grandjean P, Crofton KM, et al. Consensus statement on the need for innovation, transition and implementation of developmental neurotoxicity (DNT) testing for regulatory purposes. Toxicol Appl Pharmacol. 2018;354:3–6.
- Krebs A, van Vugt-lussenburg BMA, Waldmann T, et al. The EU-ToxRisk method documentation, data processing and chemical testing pipeline for the regulatory use of new approach methods. Arch Toxicol. 2020;94(7):2435–2461.
- Bal-Price A,Hogberg H.T, Crofton K.M, et al. Recommendation on test readiness criteria for new approach methods in toxicology: exemplified for developmental neurotoxicity. Altex. 2018;35(3):306–352.
- OECD. Guidance Document for the Use of Adverse Outcome Pathways in Developing Integrated Approaches to Testing and Assessment (IATA) 2017.
- Lupu D, Andersson P, Bornehag CG, et al. The endpoints project: novel testing strategies for endocrine disruptors linked to developmental neurotoxicity. Int J Mol Sci. 2020;21(11):3978.
- Kortenkamp A, Axelstad M, Baig AH, et al. Removing critical gaps in chemical test methods by developing new assays for the identification of thyroid hormone system-disrupting chemicals-the athena project. Int J Mol Sci. 2020;21(9):3123.
- Holbech H, Matthiessen P, Hansen M, et al. Ergo: breaking down the wall between human health and environmental testing of endocrine disrupters. Int J Mol Sci. 2020;21(8):2954.
- Moroni L, Barbaro F, Caiment F, et al. Screened: a multistage model of thyroid gland function for screening endocrine-disrupting chemicals in a biologically sex-specific manner. Int J Mol Sci. 2020;21(10):3648.
- Commission E, Environment D-Gf, Ramhøj L, Martin O, Axelstad M, et al. Development of a study protocol for thyroid disruptor testing in the mammalian system: deliverable 16: final feasibility study report: Publications Office; 2019.