
ABSTRACT
Introduction
Hereditary transthyretin (ATTRv) amyloidosis is a progressive, fatal disorder caused by mutations in the transthyretin (TTR) gene leading to deposition of the misfolded protein in amyloid fibrils. The main phenotypes are peripheral neuropathy (PN) and cardiomyopathy (CM).
Areas covered
Gene silencing therapy, by dramatically reducing liver production of TTR, has transformed ATTRv-PN patient care in the last decade. In this drug discovery case history, the authors discuss the treatment history of ATTRv-PN and focus on the latest siRNA therapy: vutrisiran. Vutrisiran is chemically enhanced and N-acetylgalactosamin-conjugated, allowing increased stability and specific liver delivery. HELIOS-A, a phase III, multicenter randomized study, tested vutrisiran in ATTRv-PN and showed significant improvement in neuropathy impairment, disability, quality of life (QoL), gait speed, and nutritional status. Tolerance was acceptable, no safety signals were raised.
Expert opinion
Vutrisiran offers a new treatment option for patients with ATTRv-PN. Vutrisian’s easier delivery and administration route, at a quarterly frequency, as well as the absence of premedication, are major improvements to reduce patients’ disease burden and improve their QoL. Its place in the therapeutic strategy is to be determined, considering affordability.
1. Introduction
Hereditary transthyretin-mediated amyloidosis (ATTRv) is a rare, autosomal dominant, life-threatening systemic disease caused by mutations in the TTR gene [Citation1]. The TTR protein, also known as pre-albumin, is a 55 kDa tetramer with four identical monomers. It is mainly synthesized by the liver (95%), though a small amount is synthesized by the retinal pigment epithelium and choroid plexus of the brain [Citation1]. In ATTRv patients, the mutation leads to destabilization of the protein into misfolded TTR monomers that ultimately aggregate and form amyloid fibrils in tissues, resulting in progressive organ dysfunction.
In a recent survey, it was estimated that 50000 people had ATTRv worldwide distributed heterogeneously. Among 130 variants described so far, the TTRV30M is the most common one in Europe. Overall, one observes a high range of genotype-phenotype variability [Citation2]. Penetrance of the disease as well as age of onset (which varies broadly between 30 and 80 years old) depend on the TTR variant and, for the TTRV30M, on the geographic origin [Citation3]. While ATTRv is a multisystemic disorder with heterogeneous clinical presentations, the phenotype is dominated by peripheral neuropathy and cardiac involvement.
ATTRv-polyneuropathy (PN) is typically characterized by a symmetric, axonal sensori-motor and autonomic neuropathy. In the early onset TTRV30M phenotype, inaugural symptoms are generally sensory manifestations including pains or autonomic findings such as severe gastrointestinal symptoms, orthostatic hypotension, bladder, or erectile dysfunction. As the disease progresses, weakness will occur with gradual gait impairment and ambulatory loss [Citation1]. However, phenotypes vary worldwide and late onset cases often develop more predominant large fiber neuropathies, including early proprioceptive ataxia and motor deficit. ATTRv-PN is a severe disease with a mean survival of 7 to 12 years, depending on the mutation and age of onset [Citation4].
Cardiac impairment is characterized by conduction abnormalities, arrhythmias, and symptoms of heart failure due to a hypertrophic cardiomyopathy with preserved ejection fraction. ATTRv-cardiomyopathy (CM) is associated with an even worse prognosis, death occurring 3 to 5 years after diagnosis [Citation5].
Other organ involvement of ATTRv may include renal disease and, less frequently, ocular and central nervous system (CNS) manifestations due to local production of transthyretin from the retinal and choroid plexus cells.
In the past two decades, outstanding advances have been made in the treatment of ATTRv, particularly in ATTRv-PN, which we will focus on. So far, the successive strategies aimed to limit formation of ATTR fibrils, preventing their deposition and disease progression. At first, liver transplantation aimed to suppress the mutant TTR synthesis. Next, the development of noninvasive oral TTR stabilizers prevented tetrameric dissociation of the protein. More recently, the era of TTR gene silencing therapy represented a major advance with the development of various agents able to dramatically reduce liver production of both mutant and wild type TTR.
Simultaneously, important refinement in the functional clinical and neurophysiological tests enabled close monitor of the course of the disease and detection of disease progression (). Such tools were essential to the achievement of clinical trials.
Table 1. Main phase III clinical trials of gene silencing drugs in ATTRv-PN.
After a brief overview of the main therapeutic milestones of ATTRv-PN, we will focus on the most recently developed small interfering ribonucleic acid (siRNA) therapy, vutrisiran, and its place in treatment strategy of the disease.
2. Overview of the market
2.1. Liver transplantation and TTR stabilizers
In 1990, liver transplantation was the first therapeutic approach performed in ATTRv-PN to stop the mutant TTR synthesis [Citation6]. Although highly invasive, with a morbidity rate up to 25% in the patients most severely affected, the procedure was shown to significantly increase survival in ATTRV30M early-onset patients after 10 years [Citation7]. In contrast, the 10-year survival remained below 50% in ATTRnonV30M and late onset V30M cases, due to a progression of both the CM and the PN [Citation6]. Notably, the progression of the cardiomyopathy was clearly linked to ongoing amyloid deposition of wild type TTR fibrils [Citation8] affecting up to 45% of transplanted patients [Citation6]. Similar findings were observed in ATTRv-PN patients [Citation9].
In a next step, TTR stabilizers were developed to treat ATTRv-PN such as diflunisal or tafamidis. Such molecules act directly at the protein level by binding to the T4-sites of the TTR protein, preventing its dissociation into monomers which ultimately misfold and form amyloid fibrils [Citation10]. The 18-month pivotal phase II-III-controlled trial tested tafamidis 20 mg specifically in stage 1 TTRV30M-PN, meaning patients with no walking disability [Citation11]. This study led to the drug approval in 2011 for ATTRv patients with a stage 1 PN in Europe, Japan, and South America. However, the level of evidence was considered insufficient in other countries such as the U.S.A. and UK.
Post-marketing studies clarified the effect of tafamidis 20 mg on PN in the long term. A slowed but nonetheless persistent progression of the disease was observed in the original pivotal ATTRV30M cohort [Citation12] as well as in more heterogeneous ATTRnonV30M populations [Citation13,Citation14]. Also, in a different TTRV30M-PN cohort of Portuguese origin (n = 210), expert evaluation based on a battery of neurological and functional tests, estimated that 34% of the patients were stable, 36% slightly progressed and 30% deteriorated, after at least 18 months of treatment [Citation15]. Then, the 30-month placebo-controlled phase III ATTR-ACT study tested the drug in ATTRv-CM patients at 20 and 80 mg (bioequivalent to 61 mg of tafamidis-free acid) [Citation16]. The primary endpoint, a hierarchical combination of all-cause mortality and frequency of cardiovascular-related hospitalizations was met. In 2019–2020, the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) approved tafamidis at 80 mg for ATTR-CM [Citation17]. Overall, the drug is very well tolerated and safe both at 20 and 80 mg doses ().
Table 2. Prescription guidelines and most frequent adverse effects of main commercialized drugs for ATTRv-PN.
Diflunisal, an FDA-approved generic nonsteroidal anti-inflammatory drug (NSAID), was found to be an effective treatment of ATTRv in a randomized, double-blind placebo-controlled study in 2013 [Citation18]. It obtained an orphan designation by the European Medicines Agency (EMA) and is known to be used off-label in patients without access to tafamidis across the world [Citation19].
2.2. Gene silencing
In the last decade, the successful development of silencing messenger RNA (mRNA) therapies in ATTRv-PN represents a major step forward [Citation20]. Both methods, antisens oligonucleotides (ASO) and siRNA, were developed concomitantly. Their mode of action is summarized in .
Figure 1. Mechanisms of action of gene silencing therapies in ATTRv.
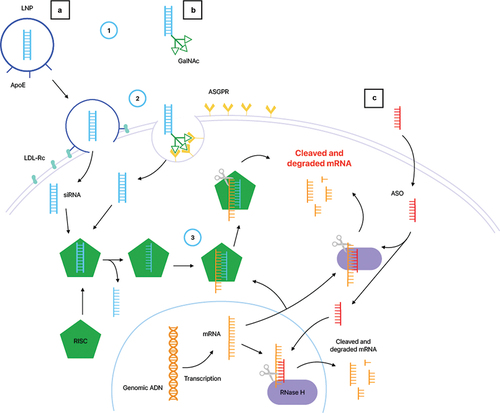
Briefly, ASO are 16–20 bases, single-strand, synthetic oligonucleotides which bind the 3’ untranslated region of human TTR mRNA in order to induce its destruction by a variety of mechanisms. They can enter the cells without the use of transfecting agents. Once in the hepatocyte, this heteroduplex activates endogenous ribonuclease (RNase) H mediated degradation in the cytoplasm and the nucleus () [Citation21].
SiRNAs are 20 to 30 bases, double-stranded RNA molecules binding to the complementary TTR mRNA, which trigger their degradation through the RNA-induced silencing complex (RISC) by binding to Argonaute-2 (AGO2) protein [Citation22]. The siRNA duplex is composed of a sense strand called ‘passenger’ and an antisens strand or ‘guide’. Once in the cell, the antisens strand which is complementary to the mRNA of the targeted gene binds to AGO2 and separates from the passenger strand. SiRNA are large, hydrophilic, and negatively charged molecules that must be delivered into the cytoplasm of hepatocytes while avoiding rapid degradation by circulating nucleases, phagocytes, and renal filtration. Hence, first generation anti-TTR siRNA were formulated in lipidic nanoparticles (LNP) which bind with high affinity to hepatocytes’ ApoE receptors, allowing its specific and safe delivery () [Citation23].
2.3. Inotersen
Inotersen was the first generation TTR-ASO tested in the Neuro-TTR, a 15-month phase III double-blind placebo-controlled study. A total of 173 ATTRv-PN patients were enrolled, with a wide range of disease severity and TTR variants. The study met co-primary end points with treated patients showing a significant reduction from baseline in the modified neuropathy impairment (mNIS +7) and Norfolk Quality Of Life – Diabetic Neuropathy (QOL-DN) scores compared to the placebo group (). Herein, 36% of treated patients had a stable or improved mNIS + 7 versus 19% in the placebo arm. The mean steady state serum TTR reduction was 84% from baseline [Citation24]. An open-label extension (OLE) study over 3 years followed 109 patients from the initial trial and showed a persistent benefit in the inotersen-inotersen group (n = 70) and better neurological outcomes than expected from natural history in the placebo-inotersen group (n = 39) [Citation25]. The safety profile in the Neuro-TTR trial showed mild to moderate adverse effects (AE) in 20% of the treated patients, including headache, nausea, fever, injection site reaction, and thrombocytopenia, with drug withdrawal in 14% of them. Notably, one death in the inotersen group was due to intracranial hemorrhage in a patient with severe thrombocytopenia [Citation24]. Overall, 54% of patients receiving inotersen developed a thrombocytopenia below 1400000/mm2. This side effect is most likely immune-mediated with evidence of antiplatelets antibodies. In addition, glomerulonephritis occurred in three ATTRV30M inotersen treated patients with crescentic glomerulonephritis identified on renal biopsy. Two of them recovered after immunosuppressive treatment. Thus, close platelet and renal function monitoring are mandatory with inotersen treatment (). Similar findings were reported during the OLE study with 22% of patients discontinuing inotersen because of treatment-emergent adverse effects [Citation25].
Inotersen was approved in 2018 for the treatment of ATTRv-PN by the FDA (stage 1) and the EMA (stages 1 and 2) as a weekly subcutaneous (SC) injection of 300 mg (1.5 mL) associated with oral vitamin A daily supplementation (). In France, given the risk of serious adverse effects, inotersen was accepted as a second-line therapy when patients are ineligible to patisiran, its siRNA competitor.
For ATTR-CM, two studies of 30 and 33 patients [Citation26,Citation27] receiving inotersen during 12 and 36 months, respectively, showed a beneficial effect on the cardiopathy, measured by 6 min walking test and echocardiographic parameters such as left ventricle (LV) mass. An ongoing 24-month phase II open-label study of 50 patients is evaluating inotersen for the treatment of ATTR-CM by assessing the change in echocardiographic parameters after 6, 12 and 24 months [Citation28,Citation29].
2.4. Patisiran
Patisiran was the first siRNA developed in ATTRv-PN. In phase I and II clinical studies, a maximum reduction in serum TTR levels of 87.6% was obtained for intravenous (IV) administration of 0.3 mg/kg dose every 3 weeks [Citation30,Citation31]. The phase III APOLLO trial, an 18-month, 2:1 randomized, double-blind placebo-controlled study, tested the efficacy and safety of patisiran (). A total of 225 patients, median age of 62 years old, with a wide range of disease severity and TTR variants, were enrolled. The administration of patisiran resulted in a sustained reduction of 81% in serum TTR levels [Citation32]. The study met its primary outcome, with an improvement from baseline in the mNIS + 7. Among patients treated 56% had stable or improved mNIS + 7 at month 18 versus 4% in the placebo arm. All secondary endpoints were met, including the Norfolk QOL-DN, gait speed (10-m walking test), modified body mass index (BMI) and COMPASS 31 which evaluates autonomic neuropathy () [Citation32]. The phase II and APOLLO OLE 24-month studies showed a maintained efficacy and sustained serum TTR reduction in patients receiving patisiran, including data from up to 48 months of treatment [Citation33,Citation34].
Patisiran is administered with a premedication which includes dexamethasone, paracetamol, H1 and H2 blockers, to limit the risk of infusion-related reaction (IRR) [Citation32] as well as vitamin A supplementation (2500 IU per day). Total duration of the infusion is 140 min: 60 min of premedication followed by 80 min of patisiran. The drug demonstrated an acceptable safety profile [Citation32]. Common AE in the patisiran group were peripheral edema and IRR, including flushing, nausea and back or abdominal pain (). In the 24-month OLE studies, there were no serious IRRs or discontinuation due to IRR [Citation34]. Several post-marketing studies further confirmed its efficacy on the PN and safety profile since 2018 [Citation35].
Patisiran has been approved in more than 30 countries for ATTRv-PN stage 1 and 2, including the US, EU, Canada, UK, Brazil, and Japan.
Recently, the main results of the APOLLO-B trial, a 12-month placebo-controlled trial assessing patisiran for the treatment of ATTR-CM, have been released. Though the study met its primary endpoint, a significant reduction in the decline of 6-min-walk-test distance under patisiran, the composite endpoint which included cardiovascular events did not differ significantly between the groups at 12 months, illustrating a lack of effect on clinical ground [Citation36,Citation37]. Secondary endpoints as functional cardiac symptoms, echographic data, and cardiac biomarkers (NT-proBNP and troponin I) were met. Given these discrepancies, the FDA denied approval of patisiran for ATTRv-CM.
3. Vutrisiran
3.1. Discovery strategy and mode of action
Vutrisiran is the next generation siRNA developed for the treatment of ATTRv-PN using the same approach as patisiran () [Citation38]. However, the phosphodiester backbone used in patisiran has been replaced by a phosphorothioate (PS) [Citation39,Citation40], leading to increased stability and tolerability [Citation22]. Also, vutrisiran is conjugated with a triantennary N-acetylgalactosamine (GalNAc) ligand. GalNAc is a specific nanocarrier binding with high affinity to asialoglycoprotein receptors (ASGPR), abundantly expressed on the surface of hepatocytes [Citation22,Citation40]. The covalent conjugation of the siRNA with GalNac enhances hepatocyte delivery and uptake and stabilizes the molecule during its systemic circulation. These characteristics offer an increased potency, higher metabolic stability, and enhanced delivery, allowing a single subcutaneous administration of smaller doses with a longer dosing interval: once every 3 months. Additionally, GalNAc-siRNA are easier to synthesize and refine [Citation23] and can be administered subcutaneously with no need for anti-inflammatory premedication [Citation22]. Recently, other GalNAc-siRNA have been developed, such as givosiran for acute hepatic porphyria, nedosiran for primary hyperoxaluria, and inclisiran for heterozygous familial hypercholesterolemia.
3.1.1. Clinical development
In a randomized phase I trial, 60 healthy volunteers received vutrisiran administered as a single injection SC with escalation dose from 5 to 300 mg compared to a placebo group (n = 20) [Citation38]. A dose dependent TTR knockdown was observed and a single dose of vutrisiran 25 mg led to a maximum reduction in serum TTR levels of 80%, sustained for 90 days. Adverse effects reported in the vutrisiran group were mild and none led to withdrawal from the study [Citation38]. The dose of 25 mg quarterly was retained for both phase-III trials, HELIOS-A and HELIOS-B, launched to test vutrisiran in ATTRv-PN and CM, respectively.
The HELIOS-A was a multi-center, open-label, randomized 18-month study, evaluating vutrisiran in ATTRv-PN patients. The trial, conducted at 57 sites in 22 countries, enrolled patients with ATTRv-PN randomized 3:1 for vutrisiran 25 mg SC quarterly or patisiran 0.3 mg/kg IV every three weeks with premedication before each infusion. All patients received a vitamin A supplementation of 2500 IU daily [Citation41]. Patients with prior liver transplantation as well as patients who had received previous gene-silencing therapy were not eligible, whereas previous use of TTR stabilizers was permitted. The primary endpoint was the change in neuropathy from baseline measured by the mNIS + 7 compared with the external placebo group from the APOLLO trial (n = 77), at month 9 () [Citation32,Citation41]. Between February 2019 and March 2020, 164 patients were randomized and treated (vutrisiran, n = 122; patisiran, n = 42). Respectively, 117 (95.9%) and 38 (90.5%) patients completed the 18-month treatment period. The patient population presented 26 TTR variants (45% TTRV30M) and a wide range of disease severity, with a median age (IQR) of 60 years old (20), reflecting the global spectrum of the disease. The median time since ATTRv amyloid diagnosis was 2.2 years. Most patients (61.5%) had previously received TTR stabilizers. Baseline characteristics were comparable across HELIOS-A and APOLLO placebo groups, except for the proportion of patients with mPND I-II which was greater in the vutrisiran group (77%) than in the external placebo group (55.9%) [Citation41].
After 9 months, vutrisiran resulted in significant improvement in mNIS + 7 (mean change from baseline −2,24 versus +14,76 in the placebo group), meeting the primary endpoint. Notably, 50.4% of patients in the vutrisiran group showed improvement, versus 18.2% in the external placebo group. This proportion was 48.3% versus 3.9%, respectively, at month 18. A significant improvement with vutrisiran compared to external placebo was observed for all secondary endpoints: quality of life (QoL) measured by the Norfolk QOL-DN questionnaire and gait speed by the 10-m walk test at months 9 and 18, neuropathy impairment and disability assessed by mNIS + 7 and R-ODS and nutritional status by modified BMI at month 18 [Citation41]. The benefits of vutrisiran versus placebo on mNIS + 7 and Norfolk QOL-DN were significant across all prespecified subgroups, including age (<65 or ≥65 years), sex, baseline NIS (<50 or ≥50), TTR variant (V30M or nonV30M), previous use of TTR stabilizer, mPND stage (I or II-III), at months 9 and 18. In exploratory analysis, vutrisiran showed a benefit in the subcomponents of mNIS + 7, in each of the 5 domains of Norfolk-QOL-DN, and in other measures of QoL (EQ-VAS) and functional status (KPS). Interestingly, a lower neuropathy disability score at baseline was associated with a better Norfolk-QOL-DN score after 18 months of treatment [Citation42]. Serum TTR reduction level under vutrisiran occurred in less than 3 weeks and was sustained over 18 months. Steady-state mean peak and trough serum TTR reductions from baseline were 87.6% and 81%, respectively, in the vutrisiran group and the within-study patisiran group. Additionally, vutrisiran was associated with less peak-to-trough fluctuation (5.4%) than patisiran (10.1%), reflecting a reduced variability in TTR level across the 18-month study.
Vutrisiran was well tolerated, with the majority of AE reported being mild to moderate and consistent with ATTRv amyloidosis. Two deaths (1.6%) occurred in the vutrisiran group and three in the patisiran (7.1%), none of which were considered drug related. One death in each group was due to COVID-19. Two patients experienced serious adverse effect considered related to vutrisiran (one dyslipidemia and one urinary tract infection), but this did not lead to discontinuation of the treatment. Injection site reactions occurred in 4.1% of the vutrisiran treated patients versus 23.8% of the patisiran group. There were no safety signals regarding liver function tests, hematology, or renal function. Four patients treated by vutrisiran developed anti-drug antibodies, titers were low and transient, with no evidence of clinical effect or pharmacodynamic modification detected.
In the HELIOS-A trial, cardiac outcomes analysis which were performed in a predefined subgroup including 40 patients (25% TTRV30) showed a decrease of NT proBNP levels and a trend toward improvement in echocardiographic parameters at month 18 [Citation43]. The specific effects of the drug on cardiomyopathy are currently investigated in HELIOS-B, a 36-month randomized, double-blind, placebo-controlled study of vutrisiran in 655 patients with ATTRv and ATTRwt-CM [Citation44]. The primary outcome is a composite score of all-cause mortality, cardiovascular hospitalizations, and urgent hospital visits related to heart failure after 30 to 36 months.
3.1.2. Vutrisiran in clinical practice
In 2022, vutrisiran has been approved in the U.S.A. for the treatment of ATTRv-PN in adults and in the EU for the treatment of ATTRv in adults with stage 1 or 2 PN [Citation45]. The treatment should be administered at a dosage of 25 mg SC every 3 months by a health-care professional. The product is ready to use in a 1 mL prefilled syringe for SC administration only, without premedication (. Contraindications are severe hypersensitivity to active substance or to any of the excipients listed [Citation46]. As with patisiran, daily vitamin A supplementation of 2500 IU is necessary to compensate for the reduction of serum vitamin A level related to TTR decrease. In this setting, baseline ophthalmologic evaluation and search of vitamin A deficiency symptoms, such as night blindness, dry eyes, or eye inflammation, is recommended with an annual follow-up.
Also, the use of vutrisiran is detailed in specific populations. In the absence of data on the effect of vutrisiran during pregnancy, contraception should be used in women of childbearing age and pregnancy should be excluded before initiation of treatment [Citation46]. Based on its pharmacokinetic properties, there is no impact of moderate hepatic or renal impairment (eGFR > 30 to <90 ml/mn) on the use of the product. Vutrisiran should be used in patients with moderate or severe hepatic impairment or with severe renal insufficiency if the anticipated clinical benefit outweighs the potential risks. Likewise, use in elderly patients >65 years old does not required dose adjustments, given the absence of difference in pharmacokinetic of TTR reduction in this age group [Citation41,Citation46]. Although patisiran can be administered in post-liver transplant patients based on a small open-label study, the use of vutrisiran in this population of patients has not yet been tested. There were no clinical drug–drug interaction studies but in vitro vutrisiran does not interact with cytochrome P450 enzyme and should not affect drug transporters. Therefore, no significant interaction with other drugs is expected.
The specific place of Vutrisiran in the ATTRv-PN treatment strategy is still subject to controversy. A cost comparaison analysis led by the National Institute for Health and Care Excellence (NICE) between vutrisiran and patisiran was in favor of vutrisiran, making it a first-choice therapy in the United Kingdom [Citation47]. In contrast, because vutrisiran was compared to an external placebo group and was only shown non-inferior to patisiran on a biological secondary endpoint, the ‘Haute Autorité de Santé’ (HAS) in France recommends the use of vutrisiran as a second-choice therapy after patisiran, when patients are eligible for both [Citation48]. Real world data will be crucial to evaluate the overall benefit of vutrisiran on patients’ lives [Citation49].
4. Compound competitors: molecules under development
4.1. Eplontersen
Eplontersen is a new generation ASO with a GalNac conjugated delivery system. The phase I clinical trial showed eplontersen to be 30 to 50-fold more potent than inotersen in reducing TTR expression allowing lower dosages less frequently [Citation50]. Results of Neuro-TTRansform, an open-label phase III multicentre trial designed to assess the efficacy and safety of eplontersen in ATTRv-PN, have been reported recently [Citation51]. A random assignation 6:1 was performed for eplontersen (45 mg every 4 weeks, n = 144) or inotersen (300 mg weekly, n = 24). The eplontersen group had a mean age of 53 years old, comprised of 69% of males and 59% of TTRV30M. The placebo group from Neuro-TTR served as an external placebo group. The study showed significant effect of eplontersen in the co-primary endpoints including change from baseline in serum TTR level, in mNIS + 7 and Norfolk QOL-DN score, at 66 weeks (). Among study completers, 53% of patients receiving eplontersen had an improved mNIS + 7 versus 19% in the placebo arm. It is important to note that slightly different versions of the mNIS + 7 were used in APOLLO and HELIOS-A from that used in Neuro-TTR and Neuro-TTRansform [Citation52]. All secondary endpoints, evaluating neuropathy impairment, disability, and nutritional status (), were in favor of eplontersen. Its safety profile was acceptable with no serious AEs related to study drug. Three deaths occurred in the eplontersen group but none were considered drug related. In each group, thrombocytopenia occurred in 2% of the patients. In the eplontersen group, it was always mild and recovered spontaneously without dosage change or interruption. No glomerulonephritis occurred in the eplontersen group. These results led to FDA approval of eplontersen for ATTRv-PN in December 2023.
CardioTTRansform is an ongoing phase III double-blind randomized placebo-controlled study assessing the efficacy and safety of eplontersen in both hereditary and wild-type ATTR-CM, in approximately 1500 patients. Its primary endpoint is a composite of cardiovascular mortality and recurrent cardiovascular events occurring within 140 weeks of follow-up [Citation53].
4.2. NTLA-2001
The CRISPR-Cas9 system is a recent breakthrough technology that permits targeted genome editing in vivo [Citation54]. ATTRv is an ideal candidate for this technique, as a monogenic disease with circulating TTR specifically produced by the liver. Moreover, TTR knockdown is now proven to be clinically beneficial in various mRNA-aimed treatments. NTLA-2001 is composed of an LNP delivery system carrying a single guide RNA that targets TTR and a human-codon-optimized mRNA sequence of Cas9 protein. Preliminary results show effective TTR gene silencing in hepatocytes of animal models as well as in six patients with ATTRv-PN that lasted 12 months after a single injection [Citation54]. A phase I, open-label, single-dose of NTLA-2001 study in patients with ATTRv-PN and ATTR-CM is ongoing [Citation55].
5. Expert opinion
Vutrisiran, the second generation TTR-siRNA agent, is a step forward in the treatment of ATTRv-PN by silencing TTR mRNA in a sustainable way.
It uses N-acetylgalactosamine conjugation to specifically target the liver, and enhanced stabilization chemistry to increase its delivery. These characteristics allow subcutaneous administration of Vutrisiran 25 mg quaterly to adequately silence TTR mRNA.
The efficacy of Vutrisiran was demonstrated in a phase III open-label trial in ATTRv-PN patients randomized 3:1 for vutrisiran or patisiran. At month 9, vutrisiran resulted in a significant difference of mNIS + 7 versus the external placebo group from the APOLLO study, reflecting an improvement or stabilization of the PN. This was confirmed after 18 months along with the secondary endpoints including Norfolk QOL-DN, gait speed, nutritional status, and different functional scores of the PN. Also, a rapid and sustained reduction of serum transthyretin levels of 83% was obtained with vutrisiran across 18 months, similar to patisiran. By using an indirect comparison with an external placebo group, the data supports a therapeutic non-inferiority of vutrisiran compared to patisiran. Patterns of stabilization or improvement across endpoints which were observed in APOLLO (patisiran) were also observed in HELIOS-A (vutrisiran and patisiran). However, it must be acknowledged that this indirect approach limits the data interpretation. More recently, these results were corroborated in a post-hoc analysis of the HELIOS-A study. Herein, a direct comparison of the two groups enrolled in HELIOS-A demonstrated that vutrisiran showed comparable efficacy to patisiran for treating ATTRv-PN at months 9 and 18 [Citation56].
In terms of safety, the tolerability of vutrisiran appears generally good. Most treatment AE were mild or moderate, consistent with ATTRv amyloidosis. The injection site reactions were significantly less frequent compared to patisiran’s IRR and premedication is not required. Vutrisiran allows an easier monitoring of patients with no need for platelet count and renal function surveillance unlike inotersen.
Vutrisiran has been approved in the U.S.A. for the treatment of ATTRv-PN in adults and in the EU for the treatment of ATTRv in adults with stage 1 or 2 PN. Altogether, the easier subcutaneous route, the longer administration interval along with an efficacy comparable to patisiran and a good safety profile point out vutrisiran as an effective noninvasive agent in the therapeutic landscape of ATTRv-PN going foward. As shown in the HELIOS-A study, vutrisiran has a dramatic effect on the patients’ QoL with approximately half of vutrisiran-treated patients showing improvement in QoL relative to baseline. In addition, its efficacy appears higher if administered at an earlier stage of ATTRv-PN, in ambulatory patients [Citation42]. Eventually, the treatment is expected to have a positive impact on patient care pathways, although no data is yet available to substantiate this.
Number of points need to be unraveled about vutrisiran. First, its cardiac effect remains to be investigated. Recently, the results of APOLLO B study were released, showing that patisiran preserved cardiac functional capacity over a 12-month period. However, there was no significant effect observed on cardiovascular events, leading the FDA to decline the approval of the drug [Citation36]. The ongoing HELIOS-B trial designed to evaluate the efficacy and safety of vutrisiran in 665 ATTR-CM patients (variant and wild type) during 36 months should soon provide insights on the question [Citation44].
Additionally, patients’ follow-up will provide information on the effect of vutrisiran in ATTRv-PN on the long term. Given the results observed with patisiran, available on the market since 2018, one would expect a sustained efficacy across time [Citation34]. Overall, questions remain regarding minimum threshold of serum TTR knockdown necessary to correlate with a clinical response. In this context, the development of specific biomarkers predictive of TTR amyloid deposition are essential to further research. Recently, the neurofilament light chain (NfL) has appeared as a potential marker of active axonal lesions and might be useful to monitor ATTRv-PN [Citation34].
The place of vutrisiran in the therapeutic strategy of ATTRv-PN is still to be determined. Its close competitor, the new generation TTR-ASO silencer eplontersen, was tested in Neuro-TTRansform at 45 mg SC monthly. The study used a similar design to HELIOS-A and favorable results have recently been reported with a significant lowering of TTR serum concentration, less neuropathy impairment, and better QoL [Citation51]. Approval from FDA has now been obtained for ATTRv-PN. An analysis comparing eplontersen and vutrisiran would be essential for patients, clinicians, and payers. As of yet, we must rely on indirect comparisons and take into account variations between the pivotal studies such as differences in the baseline patients’ characteristics, in the mNIS + 7 versions used and in the duration of studies [Citation57,Citation58]. Here, post-marketing studies will be important to better specify the drug effect in the different scenarios.
Another important concern in the development of siRNAs is extra-hepatic sources of TTR such as the retinal pigment epithelium causing independent amyloid deposition in the eye and disabling ocular manifestations. So far, all gene silencing therapies aimed at the liver production of TTR and none were shown to reach the eye or the CNS compartment. Recently, intravitreal TTR siRNA conjugate injection has been successfully tested in rabbit eyes, introducing a possible treatment of ocular ATTR amyloidosis [Citation59].
Overall, Vutrisiran is a milestone in the development of next generation RNAi therapeutic silencing of the TTR gene. Additional TTR-RNAi agents are already in the pipeline, such as the ALN-TTRsc04 which, by using Alnylam’s IKARIA technology, provides the potential of over 90% gene knockdown with a single annual dose. A phase I study is ongoing [Citation60].
In an ultimate goal to silence the TTR gene and suppress the main production of TTR durably, the CRISPR/Cas9 in vivo genome editing with specific hepatocytes delivery via LNP might be another important development in the field. In this setting, data of the ongoing dose expansion phase I trial are eagerly awaited [Citation29,Citation55]. Moreover, in a phase I trial, administration of a recombinant human antibody NI006 for ATTR cardiomyopathy was deemed to be safe and associated with reduction in imaging-based surrogate markers of cardiac amyloid load. Although very preliminary, these data could provide insight into therapies that deplete ATTR amyloid deposits [Citation61]. In the future, synergy between gene silencing and other treatments might be conceivable. Advances in the treatments of ATTR-PN have been remarkable over the past decades and developments continue at a rapid pace. Going forward, an important challenge to manage this devastating disease is to ensure drug accessibility and affordability [Citation47,Citation62].
Article highlights
Progress in ATTRv-PN treatments has transformed the course of this life-threatening disease in the past 20 years.
Liver transplantation (LT) then TTR stabilizers were the first available treatments, however, complications following LT and inconsistent efficacy with tafamidis are strong limitations to their use.
2018 was a pivotal year with the development of two silencing mRNA therapies for ATTRv-PN: patisiran and inotersen.
In 2022, vutrisiran, a new chemically enhanced, GalNAc-conjugated siRNA, was approved for the treatment of ATTRv-PN stage 1 and 2 in the U.S.A. and the EU as a subcutaneous injection every 3 months.
Vutrisiran was shown to improve neuropathy impairment and quality of life.
Its place in treatment strategy is to be determined considering its quarterly SC administration, the absence of premedication and affordability.
Abbreviation
| AE | = | adverse effect |
| AGO2 | = | argonaute-2 |
| AGPR | = | asialoglycoprotein receptor |
| ASO | = | antisens oligonucleotide |
| ATTRv | = | hereditary transthyretin-mediated amyloidosis |
| BMI | = | body mass index |
| CM | = | cardiomyopathy |
| CNS | = | central nervous system |
| DN | = | diabetic neuropathy |
| EMA | = | European Medicines Agency |
| FDA | = | Food and Drug Administration |
| GalNAc | = | N-acetylgalactosamin |
| IRR | = | infusion related reaction |
| IU | = | international unit |
| IV | = | intravenous |
| kDa | = | kilo Dalton |
| LNP | = | lipidic nanoparticles |
| LV | = | left ventricle |
| mNIS | = | modified neuropathy impairment score |
| mRNA | = | messenger RNA |
| OLE | = | open-label extension |
| PN | = | polyneuropathy |
| PS | = | phosphorothioate |
| Norfolk QOL-DN | = | Norfolk Quality Of Life – Diabetic Neuropathy |
| QoL | = | quality of life |
| RISC | = | RNA-induced silencing complex |
| R-ODS | = | rasch-built overall disability scale |
| RNA | = | ribonucleic acid |
| RNase | = | ribonuclease |
| SC | = | subcutaneous |
| siRNA | = | small interfering RNA |
| TTR | = | transthyretin |
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
One referee is the inventor of, and receives royalty payments for, a competitor drug that is in competition with vutrisiran while another referee has received a travel grant to attend scientific congresses from Alnylam (the originator company of vutrisiran). Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Carroll A, Dyck PJ, de Carvalho M, et al. Novel approaches to diagnosis and management of hereditary transthyretin amyloidosis. J Neurol Neurosurg Psychiatry. 2022;93(6):668–678. doi: 10.1136/jnnp-2021-327909
- Plante-Bordeneuve V. Transthyretin familial amyloid polyneuropathy: an update. J Neurol. 2018;265(4):976–983. doi: 10.1007/s00415-017-8708-4
- Planté-Bordeneuve V, Gorram F, Olsson M, et al. A multicentric study of the disease risks and first manifestations in hereditary transthyretin amyloidosis (ATTRv): insights for an earlier diagnosis. Amyloid. 2023;30(3):313–320. doi: 10.1080/13506129.2023.2178891
- Mariani L-L, Lozeron P, Théaudin M, et al. Genotype-phenotype correlation and course of transthyretin familial amyloid polyneuropathies in France. Ann Neurol. 2015;78:901–916. doi: 10.1002/ana.24519
- Porcari A, Fontana M, Gillmore JD. Transthyretin cardiac amyloidosis. Cardiovasc Res. 2022;118:3517–3535. doi: 10.1093/cvr/cvac119
- Ericzon B-G, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99(9):1847–1854. doi: 10.1097/TP.0000000000000574
- Yamashita T, Ando Y, Okamoto S, et al. Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology. 2012;78:637–643. doi: 10.1212/WNL.0b013e318248df18
- Olofsson B-O, Backman C, Karp K, et al. Progression of cardiomyopathy after liver transplantation in patients with familial amyloidotic polyneuropathy, Portuguese type. Transplantation. 2002;73(5):745–751. doi: 10.1097/00007890-200203150-00015
- Liepnieks JJ, Zhang LQ, Benson MD. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology. 2010;75(4):324–327. doi: 10.1212/WNL.0b013e3181ea15d4
- Bulawa CE, Connelly S, DeVit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci. 2012;109(24):9629–9634. doi: 10.1073/pnas.1121005109
- Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy. Neurology. 2012;79:785–792. doi: 10.1212/WNL.0b013e3182661eb1
- Coelho T, Maia LF, da Silva AM, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260:2802–2814. doi: 10.1007/s00415-013-7051-7
- Planté-Bordeneuve V, Gorram F, Salhi H, et al. Long-term treatment of transthyretin familial amyloid polyneuropathy with tafamidis: a clinical and neurophysiological study. J Neurol. 2017;264(2):268–276. doi: 10.1007/s00415-016-8337-3
- Cortese A, Vita G, Luigetti M, et al. Monitoring effectiveness and safety of tafamidis in transthyretin amyloidosis in Italy: a longitudinal multicenter study in a non-endemic area. J Neurol. 2016;263(5):916–924. doi: 10.1007/s00415-016-8064-9
- Monteiro C, Mesgazardeh JS, Anselmo J, et al. Predictive model of response to tafamidis in hereditary ATTR polyneuropathy. JCI Insight. 2019;4:e126526. doi: 10.1172/jci.insight.126526.
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi: 10.1056/NEJMoa1805689
- Damy T, Garcia-Pavia P, Hanna M, et al. Efficacy and safety of tafamidis doses in the tafamidis in transthyretin cardiomyopathy clinical trial (ATTR-ACT) and long-term extension study. Eur J Heart Fail. 2021;23(2):277–285. doi: 10.1002/ejhf.2027
- Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310:2658–2667. doi: 10.1001/jama.2013.283815
- Lohrmann G, Pipilas A, Mussinelli R, et al. Stabilization of cardiac function with diflunisal in transthyretin (ATTR) cardiac amyloidosis. J Card Fail. 2020;26(9):753–759. doi: 10.1016/j.cardfail.2019.11.024
- Brannagan TH, Berk JL, Gillmore JD, et al. Liver‐directed drugs for transthyretin-mediated amyloidosis. J Peripher Nerv Syst. 2022;27:228–237. doi: 10.1111/jns.12519
- Bennett CF. Therapeutic antisense oligonucleotides are coming of age. Annu Rev Med. 2019;70(1):307–321. doi: 10.1146/annurev-med-041217-010829
- Zhang MM, Bahal R, Rasmussen TP, et al. The growth of siRNA-based therapeutics: updated clinical studies. Biochem Pharmacol. 2021;189:114432. doi: 10.1016/j.bcp.2021.114432.
- Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. 2017;35(3):222–229. doi: 10.1038/nbt.3802
- Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31. doi: 10.1056/NEJMoa1716793
- Brannagan TH, Coelho T, Wang AK, et al. Long-term efficacy and safety of inotersen for hereditary transthyretin amyloidosis: NEURO-TTR open-label extension 3-year update. J Neurol. 2022;269(12):6416–6427. doi: 10.1007/s00415-022-11276-8
- Dasgupta NR, Rissing SM, Smith J, et al. Inotersen therapy of transthyretin amyloid cardiomyopathy. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. 2020;27:52–58. doi: 10.1080/13506129.2019.1685487
- Benson MD, Dasgupta NR, Rissing SM, et al. Safety and efficacy of a TTR specific antisense oligonucleotide in patients with transthyretin amyloid cardiomyopathy. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. 2017;24:219–225. doi: 10.1080/13506129.2017.1374946
- MD RHF. 24 month open label study of the tolerability and efficacy of an antisense oligonucleotide (inotersen) in patients with transthyretin (TTR) amyloid cardiomyopathy. Available from: https://clinicaltrials.gov/ct2/show/NCT03702829.
- Ioannou A, Fontana M, Gillmore JD. RNA targeting and gene editing strategies for transthyretin amyloidosis. BioDrugs Clin Immunother Biopharm Gene Ther. 2023;37:127–142. doi: 10.1007/s40259-023-00577-7
- Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–829. doi: 10.1056/NEJMoa1208760
- Zhang X, Goel V, Robbie GJ. Pharmacokinetics of patisiran, the first approved RNA interference therapy in patients with hereditary transthyretin-mediated amyloidosis. J Clin Pharmacol. 2020;60(5):573–585. doi: 10.1002/jcph.1553
- Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. doi: 10.1056/NEJMoa1716153
- Adams D, Polydefkis M, González-Duarte A, et al. Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol. 2021;20(1):49–59. doi: 10.1016/S1474-4422(20)30368-9
- Ticau S, Aldinc E, Polydefkis M, et al. Treatment response and neurofilament light chain levels with long-term patisiran in hereditary transthyretin-mediated amyloidosis with polyneuropathy: 24-month results of an open-label extension study. Amyloid. 2023;1–11. doi: 10.1080/13506129.2023.2232520
- Di Stefano V, Fava A, Gentile L, et al. Italian real-life experience of patients with hereditary transthyretin amyloidosis treated with Patisiran. Pharmacogenomics Pers Med. 2022;15:499–514. doi: 10.2147/PGPM.S359851
- Maurer MS, Kale P, Fontana M, et al. Patisiran treatment in patients with transthyretin cardiac amyloidosis. N Engl J Med. 2023;389(17):1553–1565. doi: 10.1056/NEJMoa2300757
- Merlini G. A step forward in solving amyloidosis. N Engl J Med. 2023;389(17):1615–1617. doi: 10.1056/NEJMe2309308
- Habtemariam BA, Karsten V, Attarwala H, et al. Single‐dose pharmacokinetics and pharmacodynamics of transthyretin targeting N‐acetylgalactosamine–small interfering ribonucleic acid conjugate, vutrisiran, in healthy subjects. Clin Pharmacol Ther. 2021;109(2):372–382. doi: 10.1002/cpt.1974
- Berk C, Civenni G, Wang Y, et al. Pharmacodynamic and pharmacokinetic properties of full phosphorothioate small interfering RNAs for gene silencing in vivo. Nucleic Acid Ther. 2021;31:237–244. doi: 10.1089/nat.2020.0852
- An G. Pharmacokinetics and pharmacodynamics of GalNAc-conjugated siRNAs. J Clin Pharmacol. 2023;64(1):45–57. doi: 10.1002/jcph.2337
- Adams D, Tournev IL, Taylor MS, et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid. 2022;30(1):18–26. doi: 10.1080/13506129.2022.2091985
- Obici L, Ajroud-Driss S, Lin K-P, et al. Impact of vutrisiran on quality of life and physical function in patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy. Neurol Ther. 2023;12(5):1759–1775. doi: 10.1007/s40120-023-00522-4
- Garcia-Pavia P, Gillmore JD, Kale P, et al. HELIOS-A: 18-month exploratory cardiac results from the phase 3 study of vutrisiran in patients with hereditary transthyretin-mediated amyloidosis. ESC Heart Failure. 2022;15(1):31–32. doi: 10.1016/j.acvdsp.2022.10.056
- Alnylam Pharmaceuticals. HELIOS-B: a phase 3, randomized, double-blind, placebo-controlled, multicenter study to evaluate the efficacy and safety of vutrisiran in patients with transthyretin amyloidosis with cardiomyopathy (ATTR amyloidosis with cardiomyopathy). Available from: https://clinicaltrials.gov/ct2/show/NCT04153149
- Keam SJ. Vutrisiran: First approval. Drugs. 2022;82:1419–1425. doi: 10.1007/s40265-022-01765-5
- EMA. Amvuttra. European Medicines Agency. 2022. Available from. https://www.ema.europa.eu/en/medicines/human/EPAR/amvuttra
- National Institute for Health and Care Excellence (NICE). Evidence | vutrisiran for treating hereditary transthyretin-related amyloidosis | guidance. NICE; 2023. Available from: https://www.nice.org.uk/guidance/ta868/evidence
- de Santé HA. AMVUTTRA (vutrisiran) - first evaluation. Haute Aut. Santé. Available from: https://www.has-sante.fr/jcms/p_3402366/fr/amvuttra-vutrisiran-amylose-hereditaire-a-transthyretine.
- Nie T, Heo Y-A, Shirley M. Vutrisiran: a review in polyneuropathy of hereditary transthyretin-mediated amyloidosis. Drugs. 2023;83:1425–1432. doi: 10.1007/s40265-023-01943-z
- Diep JK, Yu RZ, Viney NJ, et al. Population pharmacokinetic/pharmacodynamic modelling of eplontersen, an antisense oligonucleotide in development for transthyretin amyloidosis. Br J Clin Pharmacol. 2022;88(12):5389–5398. doi: 10.1111/bcp.15468
- Coelho T, Marques W, Dasgupta NR, et al. Eplontersen for hereditary transthyretin amyloidosis with polyneuropathy. JAMA. 2023;330(15):1448. doi: 10.1001/jama.2023.18688
- Dyck PJB, González-Duarte A, Obici L, et al. Development of measures of polyneuropathy impairment in hATTR amyloidosis: from NIS to mNIS + 7. J Neurol Sci. 2019;405:116424. doi: 10.1016/j.jns.2019.116424
- Ionis Pharmaceuticals, Inc. A phase 3 global, double-blind, randomized, placebo-controlled study to evaluate the efficacy and safety of ION-682884 in patients with transthyretin-mediated amyloid cardiomyopathy (ATTR CM). Available from: https://clinicaltrials.gov/ct2/show/NCT04136171
- Gillmore JD, Gane E, Taubel J, et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. 2021;385(6):493–502. doi: 10.1056/NEJMoa2107454
- Intellia Therapeutics. Phase 1 two-part (open-label, single ascending dose (part 1) and open-label, single dose expansion (part 2)) study to evaluate safety, tolerability, pharmacokinetics, and pharmacodynamics of NTLA-2001 in patients with hereditary transthyretin amyloidosis with polyneuropathy (ATTRv-PN) and patients with transthyretin amyloidosis-related cardiomyopathy (ATTR-CM) Available from: https://clinicaltrials.gov/ct2/show/NCT04601051
- Polydefkis M, Birklein F, Sekijima Y, et al. Comparison of efficacy outcomes with vutrisiran vs. Patisiran in hATTR amyloidosis with polyneuropathy: post-hoc analysis of the HELIOS-A study (S14.003). Lippincott Williams & Wilkins; 2023. Available from: http://www.neurology.org/lookup/doi/10.1212/WNL.0000000000202920
- González-Duarte A. Novel treatments for hereditary ATTRv amyloidosis. JAMA. 2023;330(15):1433. doi: 10.1001/jama.2023.15087
- Merkel M, Danese D, Chen C, et al. Indirect treatment comparison (ITC) of the efficacy of vutrisiran and tafamidis for hereditary transthyretin-mediated amyloidosis with polyneuropathy. Expert Opin Pharmacother. 2023;24(10):1205–1214. doi: 10.1080/14656566.2023.2215925
- Watanabe T, Takihara Y, Jono H, et al. Silencing of ocular transthyretin, a gene responsible for hereditary transthyretin amyloidosis, by intravitreal injection of an siRNA conjugate into rabbit eyes. Biochem Biophys Res Commun. 2024;694:149397. doi: 10.1016/j.bbrc.2023.149397
- Alnylam Pharmaceuticals. A phase 1, randomized, double-blind, placebo controlled, single-ascending dose study to evaluate the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of subcutaneously administered ALN-TTRSC04 in healthy subjects. Available from: https://clinicaltrials.gov/study/NCT05661916
- Garcia-Pavia P, Aus Dem Siepen F, Donal E, et al. Phase 1 trial of antibody NI006 for depletion of cardiac transthyretin amyloid. N Engl J Med. 2023;389(3):239–250. doi: 10.1056/NEJMoa2303765
- Wardhere A, Bampatsias D, Fine N, et al. Heterogeneous worldwide access and pricing of Tafamidis. Amyloid. 2023;1–3. doi: 10.1080/13506129.2023.2263620