
ABSTRACT
Introduction
Targeting RNAs with small molecules offers an alternative to the conventional protein-targeted drug discovery and can potentially address unmet and emerging medical needs. The recent rise of interest in the strategy has already resulted in large amounts of data on disease associated RNAs, as well as on small molecules that bind to such RNAs. Artificial intelligence (AI) approaches, including machine learning and deep learning, present an opportunity to speed up the discovery of RNA-targeted small molecules by improving decision-making efficiency and quality.
Areas covered
The topics described in this review include the recent applications of AI in the identification of RNA targets, RNA structure determination, screening of chemical compound libraries, and hit-to-lead optimization. The impact and limitations of the recent AI applications are discussed, along with an outlook on the possible applications of next-generation AI tools for the discovery of novel RNA-targeted small molecule drugs.
Expert opinion
Key areas for improvement include developing AI tools for understanding RNA dynamics and RNA – small molecule interactions. High-quality and comprehensive data still need to be generated especially on the biological activity of small molecules that target RNAs.
1. Introduction
The concept of targeting RNAs with small molecules was first met with skepticism, but the FDA approval of an RNA splicing modulator, risdiplam [Citation1], has kindled a heightened interest in the field. Another force that has driven interest in the field is the belief that it could greatly expand the reach of small molecule drugs, even to diseases linked to proteins that were deemed ‘undruggable.’ It has been estimated that only a small portion (0.05%) of the human genome has been drugged and that this small portion corresponds to only 10–15% of disease-related proteins [Citation2]. Thus, the messenger RNAs (mRNAs) that encode for the remaining 85–90% of the disease-related proteins could be prime targets for drug discovery. In addition, a large portion of the human genome is transcribed as non-coding RNAs (ncRNAs) [Citation3]. Many of these ncRNAs influence disease [Citation4] and may represent promising targets for small molecule drug discovery.
Despite the growing interest by the scientific community, RNA-targeted small molecule drug discovery is still at the nascent stages. What could potentially catapult the field into the mainstream is the use of artificial intelligence (AI), in particular machine and deep learning (ML and DL) technologies, which have already produced a whole host of applications in protein-targeted drug discovery [Citation5,Citation6]. Encouragingly, a number of pharmaceutical and biotech companies have already embraced AI as a way to speed up the RNA-targeted drug discovery process [Citation7]. Progress in the field can be more rapid if the community would draw insight and learn good practices in the use of AI tools from the protein-centric programs.
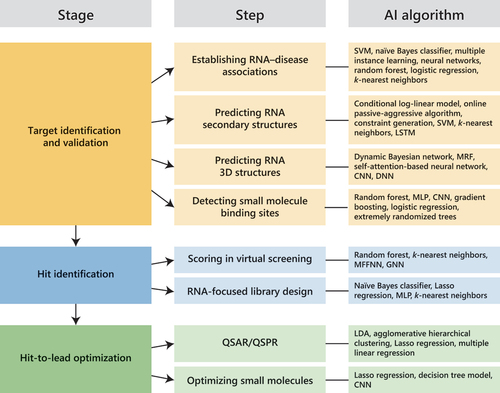
In this review article, we will focus on the recent applications of AI tools in the early stages of RNA-targeted small molecule drug discovery (). First, we will describe the applications of AI in RNA target identification, such as establishing target – disease associations, predicting the structures of potential RNA targets, and detecting small molecule binding sites. Next, we will cover the applications of AI in compound screening and RNA-focused library design. Finally, we will provide examples on the use of AI to understand structure–activity relationships and optimize hit compounds. The references used in this review were retrieved by searching PubMed, Google Scholar, bioRxiv, and arXiv through October 2023.
Figure 1. Artificial intelligence (AI) applications and tools in RNA-targeted small molecule drug discovery. Successful applications of AI tools in the various stages of drug discovery have been reported. CNN, convolutional neural network; DNN, deep neural network; GNN, graph neural network; Lasso, least absolute shrinkage and selection operator; LDA, linear discriminant analysis; LSTM, long short-term memory; MFFNN, multilayer feed-forward neural network; MLP, multilayer perceptron; MRF, Markov random fields; QSAR/QSPR, quantitative structure – activity/property relationship; SVM, support vector machine.
2. Key concepts and terminologies in AI
AI is the emulation of human intelligence by computer systems, which have been designed to think and learn more or less in the same capacity as humans. AI systems execute a specific task or series of tasks that traditionally rely on human intelligence. One can visualize AI, ML, and DL as a set of Russian dolls nested within each other – with DL being a specialized subset of ML, which is a subset of AI.
ML is an approach that employs algorithms for interpreting data, learning from them, and then making predictions about the significance of new and previously unseen data. In other words, rather than just carrying out what the programmer tells it to do, the machine learns how to use the vast amount of data that it has been trained on to perform a task. Instead of coding for expert rules, the programmer builds the algorithm used to train the machine and provides it with data. It is worth noting that the performance of the ML algorithm is largely dependent on the quantity and quality of the training dataset. At present, the amount of high-quality data pertaining to RNA-targeted small molecule drug discovery is still limited, and this poses a challenge for programmers looking to build accurate and reliable ML algorithms. Another challenge is bias that can be caused by the data source (the manner by which the data was collected) and poorly representative training datasets. Thus, it is imperative to set standards to avoid bias in data collection, or if they cannot be avoided due to technical limitations of the method or equipment, then delineate rules for eliminating bias from the training dataset.
The primary difference between ML and DL is how much each learns and how much data each uses. DL models are built on highly complex neural networks that mimic how the neurons in the brain signal one another. With many layers of processing units, DL automates much of the feature extraction piece of the process, eliminating some of the manual human intervention required. It also enables the use of large datasets. Therefore, DL is often used to complete complex tasks and train models using unstructured data. One tradeoff of the high performance of DL models is their lack of interpretability (also known as the ‘black-box’ problem). For example, it is difficult to tell what the models rely on and which features are important for a particular task. This issue of poor interpretability of DL models makes scientists, particularly in the drug discovery field, wary of entrusting to DL algorithms the task of making crucial decisions. In view of this, explainable AI is increasingly being adopted to improve the transparency of decision-making processes, prevent incorrect predictions due to faulty reasoning, and reduce bias [Citation8].
There are two main ML paradigms: supervised and unsupervised learning. In supervised learning, the machine is given a training dataset to teach it how to generate the desired output. The training dataset is labeled, which means that it contains both the correct and incorrect outputs, enabling the machine to learn over time. Hence, supervised learning is used to create models that predict the potential values of additional data. In unsupervised learning, the machine is not provided with a labeled dataset. Thus, unsupervised learning is for exploratory purposes and is used to build models that allow for clustering and/or reducing the dimensionality of the data in ways that are not explicitly specified by the programmer.
A successful ML model should be able to generalize well the training dataset onto the new test dataset. There are various algorithms within each ML paradigm and each of them have their own strengths and weaknesses (). When selecting the algorithm to use it is important to consider: (1) the size, number of features, and linearity of the training dataset, (2) the accuracy and/or interpretability of the output, and (3) speed of training.
Table 1. Advantages and disadvantages of the ML and DL algorithms used in RNA-targeted small molecule drug discovery.
Overfitting is a consequence of the AI model being too complex and learning not just the signal but also the peculiarities in the training dataset. The model is overfitting the training dataset when it performs well on the training dataset but not on the test dataset. Some solutions for solving the issue of overfitting are using a more complete training dataset, artificially increasing the size of the dataset by applying transformations to the existing data (data augmentation), adding penalties as the model complexity increases (regularization), and removing some features from the data. Underfitting, on the other hand, occurs when the model is overly simplified and fails to recognize the patterns and relationships in the training dataset. When faced with the issue of underfitting, one can try increasing the complexity of the model, reducing regularization, and adding some features to the training data. It should be pointed out that it is counterproductive to add more data when faced with the problem of underfitting. Taking a step back to diagnose the problem and think about the right course of action is an essential part of the process of developing a good ML model.
3. Applications of AI in RNA target identification
3.1. Identification of disease-relevant RNAs
It has been said that the leading causes for failure of drugs in the clinic are lack of clinical efficacy and toxicity [Citation9]. Therefore, at the beginning of a drug discovery program, it is crucial to examine whether targeting a particular RNA (or RNA – protein complex) would be effective in treating the disease or preventing its progression by establishing target RNA – disease association. In the case of targeting mRNAs, this step may be omitted if the protein it encodes has an already well-established role in the disease. However, in the case of targeting ncRNAs, one must first demonstrate that modulating the activity of the ncRNA will alter the disease state.
Target identification can be approached by experimental, multi-omic, and computational methods. AI, which falls under the third approach, can be utilized to mine the vast literature obtained from experiments and identify ncRNA – disease associations that may not immediately be apparent. For example, Bagewadi et al. [Citation10] extracted mentions of microRNAs (miRNAs) – a group of short (17–22 nt long) single-stranded ncRNAs – and other entities, such as genes, proteins, and diseases from MEDLINE. Then, they used different relation extraction approaches including ML-based approaches, namely linear support vector machine (SVM) and naïve Bayes classifiers, to extract miRNA – gene relations. After extracting the relations, they focused on relations concerning Alzheimer’s disease and compared their results with the data from two repositories, miR2Disease [Citation11] and miRSel [Citation12]. The comparison revealed that their approach can extract miRNAs related to Alzheimer’s disease that were not in miR2Disease and miRSel, suggesting its utility in updating miRNA – disease associations listed in data repositories. Lamurias et al. [Citation13] developed a method that they called Identifying Biomedical Relations (IBRel) to identify miRNA – gene relations based on distantly supervised multiple instance learning. IBRel was trained on a corpus of 4,000 documents related to miRNAs and evaluated using co-occurrence, supervised learning, and multiple instance learning. After evaluating IBRel, they used it to extract relations from 51 abstracts about cystic fibrosis and found that 21 of the 27 relations IBRel produced were correct. Joppich et al. [Citation14] built an approach that they named atheMir to identify miRNA – gene associations in atherosclerosis. They first obtained the miRNA – gene relations by context mining of 28 million abstracts in PubMed. Then, they applied a neural network model, which allowed the evaluation of miRNA – gene relations based on the semantic structure of a sentence. In total, atheMir identified 643 miRNA – gene relations relevant to atherosclerosis, providing further insights into the pathological mechanisms of atherosclerosis.
Aside from mining text from literature, AI can also be used to analyze large amounts of experimental data on ncRNA – disease associations stored in data repositories (). Zhao et al. [Citation26] used genome, regulome, and transcriptome data and developed a naïve Bayes classifier to identify long non-coding RNAs (lncRNAs) with potential cancer relevance. The 707 potentially cancer-related lncRNAs identified by their study exhibited significant differential expression and DNA methylation profiles across various cancer types. Zhang et al. [Citation27] likewise developed a cancer-related lncRNA classifier that they called CRlncRC by integrating genomic, expression, epigenetic, and network features with five ML techniques (naïve Bayes, random forest, SVM, logistic regression, and k-nearest neighbors). In all, 121 cancer-related lncRNAs were identified by CRlncRC. In addition, six in the top 10 cancer-related lncRNAs in their prediction results were already reported to be associated with cancer, demonstrating the applicability of their method. Khalid et al. [Citation28] implemented SVM and random forest classifiers on data for lncRNAs related to cancer and other diseases in the Lnc2Cancer [Citation29] and LncRNADisease [Citation30] data repositories. To differentiate between disease-related and non-disease-related lncRNAs, they extracted sequential (e.g. sequence conservation, GC content, mutation count) and structure-based features (e.g. minimum free energy, number of base pairs) and combined them to build the ML models. Overall, 362 lncRNAs were identified as disease-related, 102 of which were also found in another ncRNA – disease repository, MNDR [Citation31]. As shown in the examples provided above, AI can be used to generate hypotheses on the role of ncRNAs in disease that are important for target identification and prioritization. However, further validation is necessary prior to starting the drug discovery process to establish the therapeutic potential of the ncRNA target.
Table 2. Data repositories containing ncRNA – disease associations*.
3.2. RNA secondary structure prediction
It is now widely accepted that, within RNA molecules, there are structural elements or what we often refer to as ‘motifs’ that allow the RNA to perform a variety of functions. The idea of drugging the RNA is based on the premise that small molecules can modulate function, thereby altering the outcome of a disease process, by binding to crevices or pockets on the RNA structural motif. Therefore, aside from establishing the target RNA – disease association, it is crucial to identify promising motifs within RNA targets that are both druggable and functional.
Functional RNA motifs can be identified computationally or experimentally. The most widely used traditional computational method, free energy minimization, can predict RNA secondary structure from sequence [Citation32]. The free energy minimization method is based on thermodynamic energy parameters derived from wet experiments on model systems (thermodynamic data) [Citation33–37]. As an alternative, ML methods have been used to extract the energy parameters from a large dataset of pairs of RNA sequences and their secondary structures (structural data) (). For example, Do et al. [Citation39] employed a flexible probabilistic ML model, conditional log-linear model, to estimate and optimize parameters obtained from structural data. Compared to the free energy minimization and other probabilistic methods that they analyzed, CONTRAfold exhibited higher prediction accuracy, but its low computational efficiency prevents its application to larger datasets [Citation55]. Zakov et al. [Citation40] extracted 70,000 parameters from large structural datasets by employing a learning algorithm that combines the discriminative structured-prediction and online passive-aggressive frameworks. They then built an RNA secondary structure prediction tool that they called ContextFold and analyzed its prediction quality. As a result, ContextFold was found to yield an error reduction of 50% over the published results at that time. However, one criticism of ContextFold is that it has been affected by overfitting, as evidenced by its poor accuracy in predicting secondary structures for RNA families not included in the training data [Citation56].
Table 3. RNA secondary and tertiary structure prediction tools that employs AI methods*.
A third approach to estimating parameters for RNA secondary structure prediction is the use of a combination of thermodynamic and structural data. Andronescu et al. [Citation55] used a parameter estimation algorithm, constraint generation, that can be trained rapidly on both thermodynamic and structural data. Their algorithm applies an iterative strategy in which the thermodynamic energy parameters are initially generated as the solution to a constrained optimization problem. The newly generated energy parameters are then utilized to update the optimization constraints, allowing the parameters to be better optimized in the succeeding iteration. When tested on biologically relevant RNA structures, the parameters learned using the constraint generation algorithm gave better accuracy compared to the CONTRAfold [Citation39] and thermodynamic energy parameters [Citation33]. Akiyama et al. [Citation43] developed MXfold, an RNA secondary structure prediction algorithm that combines thermodynamic and ML-based weighted approaches. They employed a max-margin framework, structured SVM, to train the scoring parameters of their fine-grained model. Their benchmark showed that MXfold had better prediction accuracy compared to some of the prediction algorithms available at the time. Encouraged by the success of MXfold, the same group built another algorithm, MXfold2, for predicting RNA structures using DL [Citation44]. This time around, they combined the thermodynamic energy parameter with folding scores, which were obtained using DL. This strategy proved useful in minimizing overfitting, and MXfold2 gave robust and accurate predictions of RNA secondary structures.
Besides parameter estimation, AI tools have been used to build the architecture and folding algorithms of RNA secondary structure prediction engines (). Such AI-based prediction engines accept an RNA sequence or a sequence alignment to produce an RNA secondary structure prediction. For instance, Bindewald and Shapiro [Citation42] built KNetFold that uses a hierarchical network of k-nearest neighbor classifiers to predict a consensus RNA secondary structure from an RNA sequence alignment. The network predicts if any two columns of an alignment correspond to a base pair. KNetFold was evaluated with a set of 49 RFAM database alignments and was shown to have higher prediction accuracy compared to a few prediction engines available at the time. Lu et al. [Citation38] employed a long short-term memory (LSTM) network to develop an RNA secondary structure prediction method. RNA sequences are usually truncated before their features can be employed in DL; their method negates the need for sequence truncation. They also used a free energy-based filter to prevent overfitting of the base pairs. The LSTM network-based method with energy-based filter substantially increased the accuracy of the RNA secondary structure prediction, with the Matthews correlation coefficient being 28% higher compared to three other algorithms. Singh et al. [Citation45] applied deep contextual learning in the prediction of Watson-Crick and non-Watson-Crick base pairs, as well as non-nested (pseudoknot) base pairs stabilized by tertiary interactions. They used a large database, bpRNA (>100,000 RNA sequences) [Citation57], which is suitable for deep learning but may not be reliable. To address this issue, they first trained their models of residual and LSTM networks using a non-redundant set of RNA sequences from bpRNA. Then, they further trained the models on base pairs extracted from high-resolution non-redundant RNA structures. The resulting method, SPOT-RNA, showed a significant improvement in predicting all types of base pairs. In a rather elaborate study, Wayment-Steele et al. [Citation41] established EternaBench, a benchmark dataset with >20,000 data, including those that were obtained from high-throughput RNA structure experiments. They then trained a multitask model using the different data types in EternaBench to improve RNA secondary structure prediction. Aside from secondary structure prediction, the model, which the authors named ‘EternaFold,’ was tasked to maximize the likelihood of chemical probing data and predict affinities of RNA molecules to proteins and small molecules. Gratifyingly, EternaFold gave best-of-class performance in multiple tasks for the EternaBench data and many other datasets from literature.
3.3. RNA three-dimensional (3D) structure prediction
Knowledge of the 3D structures of RNA molecules is valuable as it can increase our understanding of the mechanisms of RNA function and provide clues for the design of RNA-targeted drugs. Currently, there are several biophysical methods – X-ray crystallography, nuclear magnetic resonance spectroscopy, cryo-electron microscopy, and small-angle X-ray scattering – that are being used to determine the 3D structures of RNAs. However, only 3.2% of the structures deposited in the Protein Data Bank (as of February 2024) contain RNA, and the available structures are mostly for short motifs and enzymatic RNAs. The paucity of 3D structural information on RNA molecules is partly due to their dynamic nature, which makes solving their structures more challenging compared to proteins.
AI tools have been developed to fill the need for 3D structural information on RNA molecules (). Such tools are used either for directly predicting the tertiary structures of RNAs or for scoring and evaluating the quality of the predicted tertiary structures obtained from non-AI-based computational methods. One of the first learning-based direct methods for predicting RNA tertiary structure is the BAyesian network model of RNA using Circular distributions and maximum Likelihood Estimation [BARNACLE [Citation47]. What this method focuses on is the problem of conformational sampling, which is the generation of highly probable RNA conformations. By combining a dynamic Bayesian network with directional statistics, BARNACLE enables the efficient sampling of 3D RNA conformations that are compatible with the secondary structure information. Another direct method for RNA tertiary structure prediction that tackles the sampling problem is TreeFolder [Citation54], a conditional random fields (CRF) model that estimates the probability of an RNA conformation from its primary and secondary structures. The CRF model is trained on a set of RNAs with solved structures and overcomes the issue of overfitting by regularization of the model parameters. After model training, TreeFolder employs a tree-based sampling approach that can sample the conformations of two distant segments and the same approach as BARNACLE to differentiate between native structures and decoys.
In addition to sampling techniques, several groups created AI-based tools to identify structural RNA modules, which are the building blocks of RNA motifs comprised of stacked non-Watson-Crick base pairs [Citation58]. Cruz and Westhof [Citation52] developed RMDetect, a tool that can be used to search for RNA modules. RMDetect uses Bayesian network models, base pair probability prediction, and positional clustering to detect all possible variations of the tertiary interactions and base pairs. When tested on 1,444 ncRNAs alignments, RMDetect identified 141 known and 21 novel modules. Additionally, RMDetect can be used in conjunction with other tools to assemble RNA 3D models. Theis et al. [Citation51] developed a pipeline, metaRNAmodules, to map short modules onto RFAM alignments. Subsequently, using RMDetect, the module features are represented as a Bayesian network model in which all the bases of the module are interpreted as nodes and all base pairings as connections between modes. Zirbel et al. [Citation50] constructed probabilistic models based on a combination of stochastic context-free grammars and Markov random fields (SCFG/MRF) for sequence variability of RNA 3D motifs using atomic-resolution (<2.0 Å) structures. The SCFG technique is used to model nested pairs and insertions, while the MRF technique handles base triples and non-nested base pairs. RNA sequences are aligned to the SCFG/MRF models using the Java-based Alignment of RNA using 3D structure information (JAR3D) software.
Recently, two end-to-end DL-based approaches – DeepFoldRNA [Citation48] and E2Efold-3D [Citation49]—have been developed for de novo RNA 3D structure prediction from sequence alone. DeepFoldRNA uses a self-attention-based neural network architecture to predict geometric restraints with high accuracy and limited-memory Broyden-Fletcher-Goldfarb-Shanno minimization simulations to build the 3D RNA structures (). Testing of DeepFoldRNA on the RFAM dataset resulted in models with an average root-mean-square deviation (RMSD) of 2.68 Å, which is significantly more accurate than the non-AI-based SimRNA [Citation59] and FARFAR2 [Citation60] software (RMSD values equal to 19.37 and 21.07 Å, respectively). Moreover, DeepFoldRNA is 345 times and 4134 times faster than SimRNA and FARFAR2. E2Efold-3D adopts several strategies including: (1) a fully differentiable end-to-end deep learning model that directly produces the atomic coordinates of the RNA, (2) informative representation of the RNA sequence to address the problem of data scarcity, (3) secondary structure-assisted self-distillation, and (4) parameter-efficient backbone formulation. When tested on the RNA puzzle dataset, E2Efold-3D outperformed FARFAR2 by a large margin, with RMSD values of <4 Å for E2Efold-3D and 7.3 Å for FARFAR2.
Figure 2. (a) Workflow for the DeepFoldRNA method [Citation48]. (b) Model of Saccharomyces cerevisiae P-site tRNA generated by DeepFoldRNA (green) superimposed with the native structure (blue, PDB ID: 6Q8Y). L-BFGS, limited-memory Broyden-Fletcher-Goldfarb-Shanno; RMSD, root mean square deviation.
![Figure 2. (a) Workflow for the DeepFoldRNA method [Citation48]. (b) Model of Saccharomyces cerevisiae P-site tRNA generated by DeepFoldRNA (green) superimposed with the native structure (blue, PDB ID: 6Q8Y). L-BFGS, limited-memory Broyden-Fletcher-Goldfarb-Shanno; RMSD, root mean square deviation.](/cms/asset/73b8fc00-86c3-429b-b158-fb013f8dc4d9/iedc_a_2313455_f0002_oc.jpg)
One of the challenges in RNA 3D structure prediction is designing a scoring function that reliably distinguishes accurate structural models from less accurate ones. Li et al. [Citation53] developed two scoring functions, RNA3DCNN_MD and RNA3DCNN_MDMC, to respectively assess the quality of the local and global structural models of RNAs. The group treated each nucleotide and its surrounding atoms as a 3D image, which was fed into the 3D convolutional neural networks (CNNs), resulting in an RMSD-based nucleotide unfitness score that indicates the extent to which the nucleotide fits into its environment. A performance comparison revealed that RNA3DCNN performs similarly or better than four prior scoring functions. Townshend et al. [Citation46] designed Atomic Rotationally Equivariant Scorer (ARES), a deep neural network that enables the identification of accurate structural models. Whereas the first layer of the ARES network gathers only the 3D coordinates and chemical element type of each atom, the succeeding layers collect information across all atoms. This enables ARES to evaluate the accuracy of the global 3D model while also capturing the details of the motifs, modules, and interatomic interactions. Astonishingly, despite being trained on only 18 experimentally determined RNA structures, ARES significantly outperforms 3 earlier scoring functions and gives the best results in community-wide RNA structure prediction challenges.
3.4. Detection of small molecule binding sites in RNA targets
Following the identification of a structural motif on a disease-associated RNA and the determination of its 3D structure, the next step is to assess whether the RNA motif has an appropriate small molecule binding pocket. It has been argued that RNA motifs that form high-quality pockets are capable of binding small molecules with high specificity and potency [Citation2]. Thus, establishing that a putative RNA target contains a ligandable pocket can help in target prioritization and selection. At present, there are a few non-AI-based computational methods for identifying small molecule binding sites on RNAs [Citation61–63]. However, these methods still suffer some limitations, such as high false-positive rates and difficulty in extracting information on short-range interactions.
ML techniques have been used to improve the detection of binding sites on RNAs. For example, Su et al. [Citation64] developed RNAsite, an ML approach that comprises a sequence- and structure-based module. The features extracted from the two modules are fed into a random forest algorithm for training and testing. Evaluation of RNAsite showed that it is competitive with existing methods, despite it being trained on only 60 RNAs. Xie and Frank [Citation65] combined cavity mapping methods with ML classifiers to distinguish between ligandable and decoy RNA cavities. They used a distance-based cavity fingerprinting method that encodes the physicochemical composition and geometry of the cavities. They showed that the method can effectively distinguish ligandable cavities from decoys with an area under the curve of greater than 0.83.
Kozlovskii and Popov [Citation66] developed BiteNetN, a DL approach that could detect small molecule binding sites on both RNA and DNA structures. First, the nucleic acid structures are voxelized and split into grids to create 3D images. Then, the 3D images are fed into 3D CNNs, which give probability scores and the coordinates of the predicted binding sites. By not limiting the data to RNAs, they were able to improve the performance of BiteNetN compared to prior methods. Moreover, they showed that BiteNetN can correctly identify conformation-specific ligand binding sites on the HIV-1 transactivating response region (TAR), as well as on the conformational ensembles of the ATP aptamer. Jiang et al. [Citation67] also performed analyses on RNA and DNA structures to detect the binding sites on these macromolecules. They extracted descriptors from three features, namely sequence, structure, and preference. Then, they established a feature-based ensemble learning classifier to identify the binding sites using three ML algorithms (random forest, extreme gradient boosting, and light gradient boosting), as well as a template-based classifier using structural conservation. They showed that the resulting algorithm, Nucleic Acid Binding Site (NABS), which combines the two classifiers, outperforms existing methods and are effective in predicting the binding sites for both RNA and DNA.
Wang et al. [Citation68] employed a deep CNN approach, RLBind, to identify small molecule binding sites in RNAs. What distinguishes RLBind from the previous approaches is that it obtains global information from the full-length sequence and structural features of the RNA to capture long-range interactions. Furthermore, RLBind extracts local information from the limited neighbors of the targeted nucleotides. The authors posit that the combination of the global and local RNA information renders RLBind with improved accuracy in predicting small molecule binding sites in RNAs. The above-mentioned ML approaches can be used to generate RNA targets that are expected to bind small molecules, thereby limiting the possible target search space. However, it is worth noting that the RNA targets that have been characterized as having small molecule binding sites are best validated further.
4. Applications of AI in the identification of RNA-targeted small molecules
4.1. Virtual screening
The identification of RNA targets – disease-relevant RNA motifs with ligandable sites – is only half the battle. Finding small molecules that have high affinity and selectivity for the RNA target is, in our belief, the more difficult half of the drug discovery process. In many instances, extensive experimental or virtual high-throughput screening (HTS) of large chemical compound libraries is required to identify RNA binders that can modulate the function of the RNA target. Many experimental screening approaches have been developed specifically for RNA targets and have proven to be promising in identifying RNA binders/modulators [reviewed in Citation32,Citation69,Citation70]. Although experimental approaches tend to be more robust for identifying promising small molecules, virtual screening is an attractive alternative as it can greatly expand the accessible chemical space while also reducing time and cost.
Compared to the experimental screening approaches, examples of virtual screening against RNA targets are limited [Citation71–76], owing perhaps to inadequate accuracy of the virtual screening tools. In particular, the scoring functions in virtual screening tools usually fail to recover the correct 3D orientation and conformation (or ‘pose’) of a small molecule bound to an RNA [Citation77–80]. To address this issue, ML approaches have been applied in scoring for distinguishing between native and non-native ligand poses, since they can be trained to identify complex relationships with limited structural data. For example, Chhabra et al. [Citation81] used random forest to train a set of pose classifiers, RNAPosers, to estimate the relative ‘nativeness’ of RNA – ligand poses. The classifiers in RNAPosers were generated from pose fingerprints based on the pairwise distance between the heavy atoms in the RNA and those in the small molecule. Testing of RNAPosers revealed a recovery of native poses in up to 80% of the cases examined. Likewise, Stefaniak and Bujnicki [Citation82] developed an ML statistical scoring function, AnnapuRNA, for evaluating the structures of RNA – small molecule complexes generated by docking methods. AnnapuRNA specifically uses k-nearest neighbors and multilayer feed-forward artificial neural network models for scoring. When used to predict the binding mode of the BRX1354 small molecule to the FMN riboswitch, AnnapuRNA was able to find a near-native pose at a higher accuracy than an existing scoring method. Both RNAPosers and AnnapuRNA are freely available to the scientific community and can be utilized in virtual screening to identify near-native poses that are suitable for further binding affinity prediction.
The application of graph neural networks (GNNs) in virtual screening of a chemical library against an RNA target was demonstrated in a recent paper by Haga et al. [Citation76]. In their study, the authors performed virtual screening against miR-21, a microRNA that has been implicated in the pathogenesis of interstitial lung disease, using three GNN architectures – graph convolutional networks, graph isomorphism networks, and graph attention networks. With the use of GNNs, they were able to extract relevant feature representations of the compound library and perform predictions on unseen compounds in less than a week, further proving that AI-based virtual screening can significantly reduce time and cost required for identifying RNA-binding small molecules. However, the authors also stress that GNNs and other neural networks require large amounts of experimentally derived training data, which is still very limited for RNA-binding small molecules.
The virtual screening methods described above are similar in the way that they require information about the binding site on the RNA target and the small molecule ligands. Oliver et al. [Citation83] deviated from this norm and developed a platform, RNAmigos, that creates augmented base pairing (i.e. both Watson-Crick and non-Watson-Crick) networks to depict RNA structures. These networks, built using graph convolutional neural networks, are used to predict a molecular fingerprint for a ligand. The fingerprint can then be used when searching for active binders within a chemical compound library. RNAmigos was trained on 773 RNA – small molecule complex structures (corresponding to 270 small molecules) obtained from the Protein Data Bank. When its performance was compared to a similar method, Inforna [Citation84], it was shown that RNAmigos outperformed Inforna in almost all ligand classes. However, a criticism of RNAmigos is that it posits that different RNA – small molecule complexes are equally likely to form [Citation85]. In reality, different RNA – small molecule complexes have varying likelihood of occurrence, and this simplification can cause bias in training.
4.2. Focused library design
Recently, the use of smaller chemical libraries is becoming increasingly prevalent due to the high resource requirements of chemical compound screening. Many pharmaceutical and biotech companies prefer curated collections that contain drug- or lead-like compounds without undesirable molecular features. By using curated chemical libraries, one can reduce the incidence of false positives and enhance downstream optimization performance, consequently saving R&D resources [Citation86].
Several initiatives to design RNA-focused chemical libraries have been made in recent years using empirical studies and ML algorithms. For instance, a group at Merck built an RNA-focused library of about 3,800 small molecules that is privileged for RNA targets compared with their previous compound libraries [Citation87]. Prior to building the RNA-focused library, they conducted HTS against 42 RNA targets using ~55,100 compounds and a compound screening technique, Automated Ligand Identification System (ALIS; ). With the use of molecular descriptors, naïve Bayes models were trained on binders versus non-binders to examine the specific features required for RNA binding. The model was then used to select promising small molecules from Merck’s compound collection, and the resulting RNA-focused library () was used in subsequent screenings against 32 of the initial RNA targets. The hit rate with the focused library was higher compared with the original library, which suggests that the ML model-informed RNA-focused library facilitated the discovery of novel RNA binders.
Figure 3. Chemical substructures enriched in selective binders in RNA-focused libraries. The Merck group adapted the (a) Automated ligand identification system as a method to screen small molecule libraries against RNAs [Citation87]. Using an ML approach and the results of their screening, they built an RNA-focused library and found (b) 12 substructures enriched in RNA binders. Likewise, the Schneekloth group built an RNA-focused library, ROBIN, from their previous (c) Small molecule microarray screening projects and identified (d) Ten substructures enriched in selective RNA binders [Citation88]. RP-HPLC, reversed-phase high-performance liquid chromatography.
![Figure 3. Chemical substructures enriched in selective binders in RNA-focused libraries. The Merck group adapted the (a) Automated ligand identification system as a method to screen small molecule libraries against RNAs [Citation87]. Using an ML approach and the results of their screening, they built an RNA-focused library and found (b) 12 substructures enriched in RNA binders. Likewise, the Schneekloth group built an RNA-focused library, ROBIN, from their previous (c) Small molecule microarray screening projects and identified (d) Ten substructures enriched in selective RNA binders [Citation88]. RP-HPLC, reversed-phase high-performance liquid chromatography.](/cms/asset/256e5079-279a-41ba-996b-ca34e50841bb/iedc_a_2313455_f0003_oc.jpg)
Another RNA-focused library that was recently generated is the Repository Of BInders to Nucleic acids (ROBIN) [Citation88]. The results of small molecule microarray screens () against 36 nucleic acid targets (27 RNA and 9 DNA) using 24,572 compounds were used to compile ROBIN, which is a collection of 2,003 RNA binders (). To classify RNA binders from FDA-approved drugs and protein binders, the group used the hits from the small molecule microarray screens and a least absolute shrinkage and selection operator (Lasso) regression and multilayer perceptron. This study revealed that RNA and protein binders may have similar drug-like properties and yet have distinct chemical features, e.g. van der Waals surface area, topological charge, aromaticity and nitrogen ring systems, sp3 character, and hydrogen bond acceptors.
A third example of an RNA-focused library is the Duke RNA-Targeted Library (DRTL), which was biased toward the physicochemical and structural properties of small molecules that are biologically active against non-ribosomal RNAs [Citation89]. The 804 DRTL compounds were selected by using a k-nearest neighbor algorithm and the molecular descriptors of the compounds in the RNA-targeted BIoactive ligaNd Database (R-BIND). The binding of DRTL compounds to four biologically relevant RNAs (HIV-1 TAR, HIV-2 TAR, and CAG and CUG repeats) was evaluated using fluorescence-based indicator displacement assays. The hit rates ranged from 0.9% to 3.1%, which are comparable to the hit rates of other RNA-focused libraries. In addition, the screens identified several RNA-binding small molecules with novel scaffolds, supporting the goal of expanding the chemical space for RNA targets.
5. Applications of AI in hit-to-lead optimization
5.1. Quantitative structure – activity/property relationships (QSAR/QSPR)
Due to the difficulty in obtaining the 3D structures of RNA – small molecule complexes, the rational design of RNA-targeted small molecules remains a challenge. To the best of our knowledge, the structure-guided design of small molecules that bind RNA was only attempted in a few studies [Citation90–93]. AI may be able to fill in the gap by providing predictive analytics that will aid the hit-to-lead optimization of small molecules. Specifically, ML strategies can be applied to model QSAR/QSPR for predicting the biological activity and pharmacokinetic properties (absorption, distribution, metabolism, excretion, and toxicity or ADMET) of small molecules. First, the structural characteristics of small molecules are usually converted into molecular descriptors. Then, QSAR/QSPR models are created to obtain the quantitative correlation between the biological activity and/or ADMET data derived from experiments and the molecular descriptors of the small molecules. It should be noted, though, that a large amount of high-quality data is paramount to achieving robust QSAR/QSPR models.
Recently, the Hargrove group published a series of studies on the use of QSAR/QSPR models for identifying small molecule properties that impact binding to RNA targets [Citation94,Citation95]. In the first study, they employed the screening data for amilorides against HIV RNA stem-loops obtained using a transactivator of transcription Tat) peptide displacement assay [Citation94]. First, they analyzed the data using agglomerative hierarchical clustering to categorize the small molecules based on their binding profiles. Then, they performed linear discriminant analysis using the groups derived from the clustering and the molecular descriptors of the amilorides. Next, QSAR models were constructed for every HIV sequence by linear regression, model searching using the method of exhaustion, and leave-one-out cross validation. The findings of the study suggest that optimizing amilorides for binding to the HIV-1 TAR stem-loop should focus on aromatic surface area and electrostatic properties, whereas optimizing amilorides for binding to the HIV-1 exonic splicing silencer of Vpr (ESSV) stem-loop should focus on molecular shape.
In the second QSAR study by the Hargrove group, they used only the HIV-1 TAR element against several small molecules having diverse scaffolds to determine whether their model can be applied to the prediction of ligands with varying architectures [Citation95]. The protonation and tautomerization states for each small molecule were first evaluated, followed by the calculation of 435 molecular descriptors. In cases where a small molecule has several potential conformations, the descriptors were determined as the Boltzmann-weighted average of the conformations. The descriptors that were found identical in value for more than 80% of the compounds were deleted. This degree of care in calculating and selecting the molecular descriptors is seldom exercised but is a prerequisite for robust QSAR models. The experimental data, namely kinetic rate constants and binding affinities, were measured using surface plasmon resonance (SPR). Representative data splitting was performed using the Kennard – Stone algorithm, which ensured that representative small molecules are selected to achieve uniform representation of the descriptor space. Lasso was applied to further reduce the dimension of the data and extract the most important descriptors. The QSAR models were developed using multiple linear regression, and their predictive performance was confirmed by comparing to ensemble tree methods. The experimental data obtained from SPR were satisfactorily explained by the models. Furthermore, the results of the binding and kinetics predictions of 12 untested compounds were very precise, demonstrating the effectiveness of multiple linear regression models as tools for small molecule optimization.
5.2. Binding activity prediction
Currently, there are several biophysical methods for measuring the binding affinity of small molecules to RNA targets [Citation32]. These methods do not only provide binding affinities but also other information about the binding event and, as such, are helpful in hit-to-lead optimization. However, conducting biophysical experiments to assess binding may not always be straightforward. As an alternative to experimental measurements, ML models were developed to predict the binding of small molecules to RNA.
Szulc et al. [Citation96] combined molecular fingerprints (or Structural Interaction Fingerprints; SIFts) with ML techniques to predict the binding activity of small molecules to RNA targets. First, they prepared a benchmark set based on a literature search for small molecules with known binding for six RNA targets. Next, they docked the ligands to the targets and calculated the SIFts for the top-scored poses. Then, they evaluated the performance of 15 ML algorithms into which the SIFts for each RNA – ligand complex and binding activity were fed. From this evaluation, they found that CatBoost, extremely randomized trees, and light gradient boosting models performed best in predicting ligand binding. Finally, they applied explainable AI on one of the RNA targets they studied, HIV-1 TAR, to identify the positive interactions and quantify their effect on binding. Recently, Krishnan et al. [Citation97] developed a more quantitative model for RNA – small molecule binding affinity prediction that can be applied on multiple RNA targets. Using the large database of RNA-small molecule interactions that they previously curated [Citation98], they developed multivariate linear regression models to predict the binding affinities (log-scale dissociation constant) of small molecule for six RNA subtypes. Evaluation of the models revealed that their models outperformed existing QSAR and classification models. Finally, they performed Shapley Additive explanations (SHAP) analysis to quantify the contributions of the features to the model prediction.
5.3. De novo design of RNA-targeted small molecules
The process of generating new small molecule compounds that best satisfy a desired pharmacological profile is known as de novo molecular design. The major difference between virtual screening and de novo design is that, prior to evaluation, the structures of the small molecules are known in the former and unknown in the latter. Even though some virtual screening libraries contain over a billion compounds, these libraries still only account for a tiny fraction of the vast chemical space (estimated to be 1060–10100) [Citation99]. By employing de novo design, we can expect to explore the chemical space more efficiently and obtain small molecules with higher potency and selectivity for RNA targets than afforded by brute force virtual screening of enormous chemical libraries.
The potential of de novo design in generating small molecules with optimal properties against RNA targets was demonstrated in a study performed by Grimberg et al. [Citation100]. The study was divided into two parts: (1) a computational part that employs ML approaches to design new small molecule inhibitors that bind the RNA hairpin 91 in the ribosomal peptidyl transfer center of Mycobacterium tuberculosis (), and (2) an experimental part where the designed inhibitors were synthesized and evaluated. First, the group trained a Lasso regression model on a dataset containing the molecular descriptors (as features) of 791 small molecules having the 2-phenylthiazole moiety and their binding affinities for hairpin 91. The model enabled the identification of features that are significant predictors of RNA binding affinity. Second, they used a decision tree model to classify the small molecules as binding or non-binding and determine the important descriptors for RNA binding. Third, they analyzed the unique geometrical patterns in the small molecule structures using CNN on the simplified molecular input line entry system (SMILES) representation and images of the small molecules. Based on the insights obtained from these approaches, the group designed ten molecules that contain 2-phenylthiazole, four of which were found to be potent inhibitors of the RNA target ().
Figure 4. Application of AI in the optimization of small molecules that target (a) RNA hairpin 91 (red) in the ribosomal peptidyl transfer center of mycobacterium tuberculosis (PDB ID: 5O61). (b) Secondary structure of hairpin 91. (c) Chemical structures, ribosome activity, and inhibitory concentration (IC50) of the small molecules that were designed based on the insights obtained from the machine and deep learning approaches [Citation100].
![Figure 4. Application of AI in the optimization of small molecules that target (a) RNA hairpin 91 (red) in the ribosomal peptidyl transfer center of mycobacterium tuberculosis (PDB ID: 5O61). (b) Secondary structure of hairpin 91. (c) Chemical structures, ribosome activity, and inhibitory concentration (IC50) of the small molecules that were designed based on the insights obtained from the machine and deep learning approaches [Citation100].](/cms/asset/a45a3a48-7360-44e9-a66e-ab9d16e139e1/iedc_a_2313455_f0004_oc.jpg)
6. Conclusion
In this review article, we described how AI is helping advance the field of RNA-targeted small molecule drug discovery. We provided examples of the different applications of AI in the identification of therapeutically-compelling RNA motifs, RNA structure prediction, screening for small molecules that modulate RNA activity, and hit-to-lead optimization. We anticipate the discovery of more RNA-targeted small molecule drugs, particularly for diseases with unmet medical needs, as more advances in experimental technologies are made and big data is efficiently harnessed with AI.
7. Expert opinion
AI has seen exponential growth in various sectors as a result of advances in computer hardware, such as graphical processing units, which have made parallel computing faster and enabled the handling of massive amounts of data. This growth is becoming more apparent in the pharmaceutical industry, where a number of successful applications of ML to protein-targeted small molecule drug discovery have already been reported [Citation5,Citation6]. Although efforts to proactively target the RNA with small molecules [Citation101] began at around the same time as ML gained prominence at the beginning of the millennium, interest has only risen in the past few years. This rise in interest has already led to the rapid accumulation of promising data on RNA targets and RNA – small molecule interactions. We anticipate that the accumulation of data will, in turn, trigger increased applications of AI in RNA-targeted small molecule drug discovery.
As we highlighted above, target identification is the stage of RNA-targeted small molecule drug discovery where most AI applications have been made. We expect that, with the exponentially increasing amount of multi-omics data that provide multidimensional views of disease pathology, the application of AI in target identification will continue to grow. In particular, we foresee AI-driven multi-omics analysis as being a driving force for the development of precision medicine against cancer. For example, the identification of cancer-related mutations on mRNAs and ncRNAs, elucidation of their function, and prediction of small molecule drug targets, which are important for developing precision medicine, may benefit from AI-driven multi-omics analysis [Citation102].
It is now widely known that RNA molecules adopt an ensemble of conformations in response to certain physiological signals to fine-tune and expand their functionality [Citation103]. However, compared to single static structures, dynamic RNA ensembles are more challenging to characterize both with experiments (usually with nuclear magnetic resonance or NMR spectroscopy) and traditional computational methods. Molecular dynamics (MD) simulations can be used to capture RNA structural dynamics with unparalleled spatiotemporal resolution and give insights into the relationship between the structure and function of the RNA [Citation104]. The outcome of MD simulations is largely dependent on the underlying model, the force field, which defines the relationship between the simulated RNA’s exact geometry and potential energy [Citation104]. This poses another challenge since force fields for RNA are still not accurate enough to generate ensembles consistent with NMR experimental data [Citation105]. In the future, applying ML approaches in the refinement of the dihedral angles, such that they better match the experimental data, will likely improve the RNA force fields and, by extension, the accuracy of MD simulations.
Compared to our understanding of the structures of RNA motifs, we know less about the physicochemical properties and enriched features that influence RNA binding and selectivity. Several groups have described a number of properties and features enriched in RNA-binding small molecules [Citation84,Citation87,Citation95,Citation106–111]. Commendably, the Schneekloth group has gone as far as to publish all the results of their small molecule microarray screening against diverse RNA targets [Citation88]. Data sharing initiatives like this, as well as precompetitive collaborations, will accelerate the development of effective AI models that could further our understanding of the desirable chemotypes and properties. Likewise, the sharing of algorithms through code hosting tools, such as GitHub and SourceForge, will enable comparative benchmarking of newly developed algorithms.
One of the latest trends in the pharmaceutical industry that is likely to make an impact in RNA-targeted small molecule drug discovery is generative AI. In this technique, a corpus of existing small molecule compounds is used to train a DL model, which usually encodes a high-dimensional representation, such as smiles, to a low-dimensional representation to generate novel compounds [Citation112]. The advantage of using generative models is that one can rapidly search a large chemical space to identify promising drug candidates. However, as with de novo design, whether the output of the generative models is significant will only be known after synthesis and biological evaluation of the novel compounds.
The previous ML-based QSAR models described above are based on data from in vitro experiments that investigate the binding of small molecules to an RNA target. Perhaps due to the newness of this field, there is still very little information on the biological activity of small molecules that bind RNAs. To fully realize the usefulness of QSAR models, more experimental data from biological systems must be generated to train the models on the bioactivity features. This will promote the rational, function-based design of RNA-targeted small molecules.
It is worth mentioning here that AI can also be divided into two systems based on their underlying principles and approaches: (1) rule-based systems, which rely on explicit rules and knowledge to make decisions or provide expert advice, and (2) data-driven systems, which are dependent on vast datasets and use ML algorithms to learn patterns and relationships and make predictions based on the data. In the preceding sections, we touched mainly on the applications of data-driven AI simply because of the lack of literature on the use of rule-based AI in the field. We surmise that the applications of rule-based AI are more pervasive than evident.
Because rule-based AI algorithms are fast and extremely thorough, they are ideal for processes that are time-sensitive and where errors cannot be tolerated. For instance, rule-based systems can be used for interpreting the results of HTS and selecting the hit compounds based on the criteria provided by the programmer. While rule-based AI is simpler to implement and promotes precision, its scope is limited, and its intelligence is restricted to the set of rules set by the programmer. We have seen a transition from rule-based AI to data-driven AI in drug discovery [Citation113] to the point that the former is rarely discussed in the context of RNA-targeted small molecule drug discovery. It is likely that rule-based systems are not being reported to protect intellectual property, while ML tools are released to the public for beta testing.
Both rule-based and data-driven systems have their strengths and weaknesses. When selecting the appropriate AI system to be used in the drug discovery project or a particular process, such strengths and weaknesses should be considered. In addition, the use of a hybrid system, a combination of rule-based and data-driven systems, is also a possibility to be explored [Citation114]. To be more specific, rule-based AI can be embedded into neural networks or employed for pre-processing of the training dataset for ML predictions. On the other hand, data-driven AI can be used to extract the rules to be applied in rule-based AI. This synergy between the two AI systems will open up new opportunities to accelerate the discovery of RNA-targeted small molecules.
The latest surveys regarding the use of data for AI applications in healthcare and medicine revealed significant ethical challenges [Citation115]. Concrete measures to ensure data privacy and ownership, fairness, accountability, algorithmic transparency, and liability must be set and properly implemented. Tackling the ethical issues even at this early stage of the RNA-targeted drug discovery is critical for preventing the risks and problems that will impede the adoption and spread of AI use in the field.
On a final note, for AI to be more widely adopted in this field, it is crucial that an AI-friendly culture be promoted from top down. Senior leadership would have to make bold decisions and allocate both financial and non-financial resources for AI adoption. Since data scientists who understand the concepts of RNA-targeted small molecule drug discovery are few and in demand, the training of in-house scientists on the use of AI tools may be necessary. This could be made possible by partnering with academic institutions where AI innovation thrives. We believe that the next five years will be an exciting and fast-paced chapter in the history of RNA-targeted small molecule drug discovery once organizations figure out how to better conquer the growing mountains of data through AI.
Article highlights
Artificial intelligence (AI) tools can be used for generating hypotheses on the role of RNAs in disease.
Recent machine learning and deep learning approaches have shown improved accuracy in predicting RNA secondary and tertiary structures, as well as in detecting small molecule binding sites on RNAs.
Applications of AI-based approaches in virtual screening against RNA targets and the design of RNA-focused libraries have emerged and are expected to boost the discovery of novel RNA binders/modulators.
Although the applications are still limited, it is clear that AI will be integral to quantitative structure – activity/property relationship modeling and de novo drug design.
AI approaches to improve the accuracy of molecular dynamics simulations and that combine rule-based and data-driven systems, data and algorithm sharing, and the generation vast amounts of high-quality data will accelerate the discovery of novel RNA-targeted small molecule drugs.
Declaration of interest
EC Morishita is a senior investigator at Veritas In Silico, Inc. S Nakamura is the scientific founder and CEO of Veritas In Silico, Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Ratni H, Ebeling M, Baird J, et al. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem. 2018;61(15):6501–6517. doi: 10.1021/acs.jmedchem.8b00741
- Warner KD, Hajdin CE, Weeks KM. Principles for targeting RNA with drug-like small molecules. Nat Rev Drug Discov. 2018;17(8):547–558. doi: 10.1038/nrd.2018.93
- ENCODE Project Consortium, Birney E, Stamatoyannopoulos JA, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816.
- Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–874. doi: 10.1038/nrg3074
- Vamathevan J, Clark D, Czodrowski P, et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov. 2019;18(6):463–477. doi: 10.1038/s41573-019-0024-5
- Jiménez-Luna J, Grisoni F, Weskamp N, et al. Artificial intelligence in drug discovery: recent advances and future perspectives. Expert Opin Drug Discov. 2021;16(9):949–959. doi: 10.1080/17460441.2021.1909567
- Garber K. Drugging RNA. Nat Biotechnol. 2023;41(6):745–749. doi: 10.1038/s41587-023-01790-z
- Jiménez-Luna J, Grisoni F, Schneider G. Drug discovery with explainable artificial intelligence. Nat Mach Intell. 2020;2(10):573–584. doi: 10.1038/s42256-020-00236-4
- Hughes JP, Rees S, Kalindjian SB, et al. Principles of early drug discovery. Br J Pharmacol. 2011;162(6):1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x
- Bagewadi S, Bobic T, Hofmann-Apitius M, et al. Detecting miRNA mentions and relations in biomedical literature. F1000Res. 2014;3:205. doi: 10.12688/f1000research.4591.2
- Jiang Q, Wang Y, Hao Y, et al. miR2disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37(Database issue):D98–D104. doi: 10.1093/nar/gkn714
- Naeem H, Küffner R, Csaba G, et al. miRsel: automated extraction of associations between microRnas and genes from the biomedical literature. BMC Bioinf. 2010;11(1):135. doi: 10.1186/1471-2105-11-135
- Lamurias A, Clarke LA, Couto FM, et al. Extracting microRNA-gene relations from biomedical literature using distant supervision. PLoS One. 2017;12(3):e0171929. doi: 10.1371/journal.pone.0171929
- Joppich M, Weber C, Zimmer R. Using context-sensitive text mining to identify miRnas in different stages of atherosclerosis. Thromb Haemost. 2019;119(8):1247–1264. doi: 10.1055/s-0039-1693165
- Carlevaro-Fita J, Lanzós A, Feuerbach L, et al. Cancer LncRNA census reveals evidence for deep functional conservation of long noncoding RNAs in tumorigenesis. Commun Biol. 2020;3(1):56. doi: 10.1038/s42003-019-0741-7
- Xu F, Wang Y, Ling Y, et al. dbDEMC 3.0: functional exploration of differentially expressed miRnas in cancers of human and model organisms. Int J Genomics Proteomics. 2022;20(3):446–454. doi: 10.1016/j.gpb.2022.04.006
- Zhou B, Ji B, Liu K, et al. EVLncRNAs 2.0: an updated database of manually curated functional long non-coding RNAs validated by low-throughput experiments. Nucleic Acids Res. 2021;49(D1):D86–D91. doi: 10.1093/nar/gkaa1076
- Cui C, Zong B, Fan R, et al. HMDD v4.0: a database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2024;52(D1):D1327–D1332. doi: 10.1093/nar/gkad717
- Gao Y, Shang S, Guo S, et al. Lnc2Cancer 3.0: an updated resource for experimentally supported lncRna/circrna cancer associations and web tools based on RNA-seq and scRNA-seq data. Nucleic Acids Res. 2021;49(D1):D1251–D1258. doi: 10.1093/nar/gkaa1006
- Bao Z, Yang Z, Huang Z, et al. LncRNADisease 2.0: an updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019;47(D1):D1034–D1037. doi: 10.1093/nar/gky905
- Yang Y, Wang D, Miao Y, et al. lncRNASNP v3: an updated database for functional variants in long non-coding RNAs. Nucleic Acids Res. 2023;51(D1):D192–D198. doi: 10.1093/nar/gkac981
- Jiang Q, Wang Y, Hao Y, et al. miR2disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37(Database):D98–D104. doi: 10.1093/nar/gkn714
- Xie B, Ding Q, Han H, et al. miRcancer: a microRNA-cancer association database constructed by text mining on literature. Bioinformatics. 2013;29(5):638–644. doi: 10.1093/bioinformatics/btt014
- Chen J, Lin J, Hu Y, et al. Rnadisease v4.0: an updated resource of RNA-associated diseases, providing RNA-disease analysis, enrichment and prediction. Nucleic Acids Res. 2023;51(D1):D1397–D1404. doi: 10.1093/nar/gkac814
- Li J, Han L, Roebuck P, et al. TANRIC: an interactive open platform to explore the function of lncRnas in cancer. Cancer Res. 2015;75(18):3728–3737. doi: 10.1158/0008-5472.CAN-15-0273
- Zhao T, Xu J, Liu L, et al. Identification of cancer-related lncRnas through integrating genome, regulome and transcriptome features. Mol Biosyst. 2015;11(1):126–136. doi: 10.1039/C4MB00478G
- Zhang X, Wang J, Li J, et al. CRlncRC: a machine learning-based method for cancer-related long noncoding RNA identification using integrated features. BMC Med Genomics. 2018;11(Suppl 6):120. doi: 10.1186/s12920-018-0436-9
- Khalid R, Naveed H, Khalid Z. Computational prediction of disease related lncRnas using machine learning. Sci Rep. 2023;13(1):806. doi: 10.1038/s41598-023-27680-7
- Ning S, Zhang J, Wang P, et al. Lnc2Cancer: a manually curated database of experimentally supported lncRnas associated with various human cancers. Nucleic Acids Res. 2016;44(D1):D980–D985. doi: 10.1093/nar/gkv1094
- Chen G, Wang Z, Wang D, et al. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013;41(D1):D983–D986. doi: 10.1093/nar/gks1099
- Ning L, Cui T, Zheng B, et al. MNDR v3.0: mammal ncRNA-disease repository with increased coverage and annotation. Nucleic Acids Res. 2021;49(D1):D160–D164. doi: 10.1093/nar/gkaa707
- Morishita EC. Discovery of RNA-targeted small molecules through the merging of experimental and computational technologies. Expert Opin Drug Discov. 2023;18(2):207–226. doi: 10.1080/17460441.2022.2134852
- Xia T, Santa Lucia J Jr, Burkard ME, et al. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry. 1998;37(42):14719–14735. doi: 10.1021/bi9809425
- Mathews DH, Sabina J, Zuker M, et al. Expanded sequence dependence of thermodynamic parameters provides improved prediction of RNA secondary structure. J Mol Biol. 1999;288(5):911–940. doi: 10.1006/jmbi.1999.2700
- Mathews DH, Disney MD, Childs JL, et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci U S A. 2004;101(19):7287–7292. doi: 10.1073/pnas.0401799101
- Lu ZJ, Turner DH, Mathews DH. A set of nearest-neighbor parameters for predicting the enthalpy change of RNA secondary structure formation. Nucleic Acids Res. 2006;34(17):4912–4924. doi: 10.1093/nar/gkl472
- Dawson WK, Shino A, Kawai G, et al. Developing an updated strategy for estimating the free-energy parameters in RNA duplexes. Int J Mol Sci. 2021;22(18):9708. doi: 10.3390/ijms22189708
- Lu W, Tang Y, Wu H, et al. Predicting RNA secondary structure via adaptive deep recurrent neural networks with energy-based filter. BMC Bioinf. 2019;20(Suppl 25):684. doi: 10.1186/s12859-019-3258-7
- Do CB, Woods DA, Batzoglou S. Contrafold: RNA secondary structure prediction without physics-based models. Bioinformatics. 2006;22(14):e90–e98. doi: 10.1093/bioinformatics/btl246
- Zakov S, Goldberg Y, Elhadad M, et al. Rich parameterization improves RNA structure prediction. J Comput Biol. 2011;18(11):1525–1542. doi: 10.1089/cmb.2011.0184
- Wayment-Steele HK, Kladwang W, Strom AI, et al. RNA secondary structure packages evaluated and improved by high-throughput experiments. Nat Methods. 2022;19(10):1234–1242. doi: 10.1038/s41592-022-01605-0
- Bindewald E, Shapiro BA. RNA secondary structure prediction from sequence alignments using a network of k-nearest neighbor classifiers. RNA. 2006;12(3):342–352. doi: 10.1261/rna.2164906
- Akiyama M, Sato Y, Sakakibara Y. A max-margin training of RNA secondary structure prediction integrated with the thermodynamic model. J Bioinform Comput Biol. 2018;16(6):1840025. doi: 10.1142/S0219720018400255
- Sato K, Akiyama M, Sakakibara Y. RNA secondary structure prediction using deep learning with thermodynamic integration. Nat Commun. 2021;12(1):941. doi: 10.1038/s41467-021-21194-4
- Singh J, Hanson J, Paliwal K, et al. RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning. Nat Commun. 2019;10(1):5407. doi: 10.1038/s41467-019-13395-9
- Townshend RJL, Eismann S, Watkins AM, et al. Geometric deep learning of RNA structure. Science. 2021;373(6558):1047–1051. doi: 10.1126/science.abe5650
- Frellsen J, Moltke I, Thiim M, et al. A probabilistic model of RNA conformational space. PLoS Comput Biol. 2009;5(6):e1000406. doi: 10.1371/journal.pcbi.1000406
- Pearce R, Omenn GS, Zhang Y. De novo RNA tertiary structure prediction at atomic resolution using geometric potentials from deep learning. bioRxiv Prepint. 2022. doi: 10.1101/2022.05.15.491755
- Shen T, Hu Z, Peng Z, et al. E2Efold-3D: End-to-end deep learning method for accurate de novo RNA 3D structure prediction. arXiv Preprint. 2022. doi: 10.48550/arXiv.2207.01586
- Zirbel CG, Roll J, Sweeney BA, et al. Identifying novel sequence variants of RNA 3D motifs. Nucleic Acids Res. 2015;43(15):7504–7520. doi: 10.1093/nar/gkv651
- Theis C, Höner Zu Siederdissen C, Hofacker IL, et al. Automatic identification of RNA 3D modules with discriminative power in RNA structural alignments. Nucleic Acids Res. 2013;41(22):9999–10009. doi: 10.1093/nar/gkt795
- Cruz JA, Westhof E. Sequence-based identification of 3D structural modules in RNA with RMDetect. Nat Methods. 2011;8(6):513–521. doi: 10.1038/nmeth.1603
- Li J, Zu W, Wang J, et al. RNA3DCNN: local and global quality assessments of RNA 3D structures using 3D deep convolutional neural networks. PLoS Comput Biol. 2018;14(11):e1006514. doi: 10.1371/journal.pcbi.1006514
- Wang Z, Xu J. A conditional random fields method for RNA sequence–structure relationships modeling and conformation sampling. Bioinformatics. 2011;27(13):i102–i110. doi: 10.1093/bioinformatics/btr232
- Andronescu M, Condon A, Hoos HH, et al. Efficient parameter estimation for RNA secondary structure prediction. Bioinformatics. 2007;23(13):i19–i28. doi: 10.1093/bioinformatics/btm223
- Rivas E, Lang R, Eddy SR. A range of complex probabilistic models for RNA secondary structure prediction that includes the nearest-neighbor model and more. RNA. 2012;18(2):193–212. doi: 10.1261/rna.030049.111
- Danaee P, Rouches M, Wiley M, et al. bpRNA: large-scale automated annotation and analysis of RNA secondary structure. Nucleic Acids Res. 2018;46(11):5381–5394. doi: 10.1093/nar/gky285
- Leontis NB, Westhof E. Analysis of RNA motifs. Curr Opin Struct Biol. Curr Opin Struct Biol. 2003;13(3):300–308. doi: 10.1016/S0959-440X(03)00076-9
- Boniecki M, Lach G, Dawson WK, et al. SimRNA: a course-grained method for RNA folding simulations and 3D structure prediction. Nucleic Acids Res. 2016;44(7):e63. doi: 10.1093/nar/gkv1479
- Watkins AM, Rangan R, Das R. FARFAR2: Improved de novo Rosetta prediction of complex global RNA folds. Structure. 2020;28(8):963–976. doi: 10.1016/j.str.2020.05.011
- Zeng P, Li J, Ma W, et al. Rsite: a computational method to identify the functional sites of noncoding RNAs. Sci Rep. 2015;5(1):9179. doi: 10.1038/srep09179
- Zeng P, Cui Q. Rsite2: an efficient computational method to predict the functional sites of noncoding RNAs. Sci Rep. 2016;6(1):19016. doi: 10.1038/srep19016
- Wang K, Jian Y, Wang H, et al. Rbind: computational network method to predict RNA binding sites. Bioinformatics. 2018;34(18):3131–3136. doi: 10.1093/bioinformatics/bty345
- Su H, Peng Z, Yang J, et al. Recognition of small molecule–RNA binding sites using RNA sequence and structure. Bioinformatics. 2021;37(1):36–42. doi: 10.1093/bioinformatics/btaa1092
- Xie J, Frank AT. Mining for ligandable cavities in RNA. ACS Med Chem Lett. 2021;12(6):928–934. doi: 10.1021/acsmedchemlett.1c00068
- Kozlovskii I, Popov P. Structure-based deep learning for binding site detection in nucleic acid macromolecules. NAR Genom Bioinform. 2021;3(4):lqab111. doi: 10.1093/nargab/lqab111
- Jiang Z, Xiao SR, Liu R. Dissecting and predicting different types of binding sites in nucleic acids based on structural information. Brief Bioinform. 2022;23(1):bbab411. doi: 10.1093/bib/bbab411
- Wang K, Zhou R, Wu Y, et al. Rlbind: a deep learning method to predict RNA–ligand binding sites. Brief Bioinform. 2023;24(1):bbac486. doi: 10.1093/bib/bbac486
- Haniff HS, Knerr L, Chen JL, et al. Target-directed approaches for screening small molecules against RNA targets. SLAS Discov. 2020;25(8):869–894. doi: 10.1177/2472555220922802
- Childs-Disney JL, Yang X, Gibaut QMR, et al. Targeting RNA structures with small molecules. Nat Rev Drug Discov. 2022;21(10):736–762. doi: 10.1038/s41573-022-00521-4
- Daldrop P, Reyes FE, Robinson DA, et al. Novel ligands for a purine riboswitch discovered by RNA-ligand docking. Chem Biol. 2011;18(3):324–335. doi: 10.1016/j.chembiol.2010.12.020
- Stelzer AC, Frank AT, Kratz JD, et al. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat Chem Biol. 2011;7(8):553–559. doi: 10.1038/nchembio.596
- Ganser LR, Lee J, Rangadurai A, et al. High-performance virtual screening by targeting a high-resolution RNA dynamic ensemble. Nat Struct Mol Biol. 2018;25(5):425–434. doi: 10.1038/s41594-018-0062-4
- Kallert E, Fischer TR, Schneider S, et al. Protein-based virtual screening tools applied for RNA-ligand docking identify new binders of the preQ1-riboswitch. J Chem Inf Model. 2022;62(17):4134–4148. doi: 10.1021/acs.jcim.2c00751
- Rocca R, Polerà N, Juli G, et al. Hit identification of novel small molecules interfering with MALAT1 triplex by a structure-based virtual screening. Arch Pharm (Weinheim). 2023;356(8):e2300134. doi: 10.1002/ardp.202300134
- Haga CL, Yang XD, Gheit IS, et al. Graph neural networks for the identification of novel inhibitors of a small RNA. SLAS Discov. 2023;28(8):402–409. doi: 10.1016/j.slasd.2023.10.002
- Pfeffer P, Gohlke H. DrugScoreRNA—knowledge-based scoring function to predict RNA-ligand interactions. J Chem Inf Model. 2007;47(5):1868–1876. doi: 10.1021/ci700134p
- Chen L, Calin GA, Zhang S. Novel insights of structure-based modeling for RNA-targeted drug discovery. J Chem Inf Model. 2012;52(10):2741–2753. doi: 10.1021/ci300320t
- Philips A, Milanowska K, Lach G, et al. LigandRNA: computational predictor of RNA-ligand interactions. RNA. 2013;19(12):1605–1616. doi: 10.1261/rna.039834.113
- Yan Z, Wang J. SPA-LN: a scoring function of ligand-nucleic acid interactions via optimizing both specificity and affinity. Nucleic Acids Res. 2017;45(12):e110. doi: 10.1093/nar/gkx255
- Chhabra S, Xie J, Frank AT. Rnaposers: machine leaning classifiers for ribonucleic acid–ligand poses. J Phys Chem B. 2020;124(22):4436–4445. doi: 10.1021/acs.jpcb.0c02322
- Stefaniak F, Bujnicki JM. AnnapuRNA: a scoring function for predicting small molecule binding poses. PLoS Comput Biol. 2021;17(2):e1008309. doi: 10.1371/journal.pcbi.1008309
- Oliver C, Mallet V, Gendron RS, et al. Augmented base pairing networks encode RNA–small molecule binding preferences. Nucleic Acids Res. 2020;48(14):7690–7699. doi: 10.1093/nar/gkaa583
- Disney MD, Winkelsas AM, Velagapudi SP, et al. Inforna 2.0: a platform for the sequence-based design of small molecules targeting structured RNAs. ACS Chem Biol. 2016;11(6):1720–1728. doi: 10.1021/acschembio.6b00001
- Zhou Y, Jiang Y, Chen SJ. RNA-ligand molecular docking: advances and challenges. Wiley Interdiscip Rev Comput Mol Sci. 2022;12(3):e1571. doi: 10.1002/wcms.1571
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53(7):2719–2740. doi: 10.1021/jm901137j
- Rizvi NF, Santa Maria JP Jr, Nahvi A, et al. Targeting RNA with small molecules: identification of selective, RNA-binding small molecules occupying drug-like chemical space. SLAS Discov. 2020;25(4):384–396. doi: 10.1177/2472555219885373
- Yazdani K, Jordan D, Yang M, et al. Machine learning informs RNA-binding chemical space. Angew Chem Int Ed Engl. 2023;62(11):e202211358. doi: 10.1002/anie.202211358
- Wicks SL, Morgan BS, Wilson AW, et al. Probing bioactive chemical space to discover RNA-targeted small molecules. bioRxiv preprint. 2023.
- Vicens Q, Mondragón E, Reyes FE, et al. Structure–activity relationship of flavin analogues that target the flavin Mononucleotide Riboswitch. ACS Chem Biol. 2018;13(10):2908–2919. doi: 10.1021/acschembio.8b00533
- Connelly CM, Numata T, Boer RE, et al. Synthetic ligands for PreQ1 riboswitches provide structural and mechanistic insights into targeting RNA tertiary structure. Nat Commun. 2019;10(1):1501. doi: 10.1038/s41467-019-09493-3
- Menichelli E, Lam BJ, Wang Y, et al. Discovery of small molecules that target a tertiary-structured RNA. Proc Natl Acad Sci U S A. 2022;119(48):e2213117119. doi: 10.1073/pnas.2213117119
- Shino A, Otsu M, Imai K, et al. Probing RNA–small molecule interactions using biophysical and computational approaches. ACS Chem Biol. 2023;18(11):2368–2376. doi: 10.1021/acschembio.3c00287
- Patwardhan NN, Cai Z, Juru AU, et al. Driving factors in amiloride recognition of HIV RNA targets. Org Biomol Chem. 2019;17(42):9313–9320. doi: 10.1039/C9OB01702J
- Cai Z, Zafferani M, Akande OM, et al. Quantitative structure-activity relationship (QSAR) study predicts small-molecule binding to RNA structure. J Med Chem. 2022;65(10):7262–7277. doi: 10.1021/acs.jmedchem.2c00254
- Szulc NA, Mackiewicz Z, Bujnicki JM, et al. Structural interaction fingerprints and machine learning for predicting and explaining binding of small molecule ligands to RNA. Brief Bioinform. 2023;24(4):bbad187. doi: 10.1093/bib/bbad187
- Krishnan SR, Roy A, Gromiha MM. Reliable method for predicting the binding affinity of RNA-small molecule interactions using machine learning. Brief Bioinform. 2024;25(2):bbae002. doi: 10.1093/bib/bbae002
- Krishnan SR, Roy A, Gromiha MM. R-SIM: a database of binding affinities for RNA-small molecule interactions. J Mol Biol. 2022;435(14):167914. doi: 10.1016/j.jmb.2022.167914
- Dobson CM. Chemical space and biology. Nature. 2004;432(7019):824–828. doi: 10.1038/nature03192
- Grimberg H, Tiwari VS, Tam B, et al. Machine learning approaches to optimize small-molecule inhibitors for RNA targeting. J Chemoinform. 2022;14(1):4. doi: 10.1186/s13321-022-00583-x
- Nakamura S, Ueno H, inventors; Takeda Pharmaceutical Company Limited, assignee. Method of screening compound regulating the translation of specific mRNA. WIPO (PCT) patent W02006054788A1.
- Rezayi S, Kalhori SRN, Saeedi S. Effectiveness of artificial intelligence for personalized medicine in neoplasms: a systematic review. Biomed Res Int. 2022;2022:7842566. doi: 10.1155/2022/7842566
- Ganser LR, Kelly ML, Herschlag D, et al. The roles of structural dynamics in the cellular functions of RNAs. Nat Rev Mol Cell Biol. 2019;20(8):474–489. doi: 10.1038/s41580-019-0136-0
- Šponer J, Bussi G, Krepl M, et al. RNA structural dynamics as captured by molecular simulations: a comprehensive overview. Chem Rev. 2018;118(8):4177–4338. doi: 10.1021/acs.chemrev.7b00427
- Bergonzo C, Henriksen NM, Roe DR, et al. Highly sampled tetranucleotide and tetraloop motifs enable evaluation of common RNA force fields. RNA. 2015;21(9):1578–1590. doi: 10.1261/rna.051102.115
- Tran T, Disney MD. Identifying the preferred RNA motifs and chemotypes that interact by probing millions of combinations. Nat Commun. 2012;3(1):1125. doi: 10.1038/ncomms2119
- Morgan BS, Forte JE, Culver RN, et al. Discovery of key physicochemical, structural, and spatial properties of RNA-targeted bioactive ligands. Angew Chem Int Ed Engl. 2017;56(43):13498–13502. doi: 10.1002/anie.201707641
- Morgan BS, Sanaba BG, Donlic A, et al. R-BIND: and interactive database for exploring and developing RNA-targeted chemical probes. ACS Chem Biol. 2019;14(12):2691–2700. doi: 10.1021/acschembio.9b00631
- Haniff HS, Knerr L, Liu X, et al. Design of a small molecule that stimulates vascular endothelial growth factor a enabled by screening RNA fold–small molecule interactions. Nat Chem. 2020;12(10):952–961. doi: 10.1038/s41557-020-0514-4
- Padroni G, Patwardhan NN, Schapira M, et al. Systematic analysis of the interactions driving small molecule-RNA recognition. RSC Med Chem. 2020;11(7):802–813. doi: 10.1039/D0MD00167H
- Donlic A, Swanson EG, Chiu LY, et al. R-BIND 2.0: an updated database of bioactive RNA-targeting small molecules and associated RNA secondary structures. ACS Chem Biol. 2022;17(6):1556–1566. doi: 10.1021/acschembio.2c00224
- Walters WP, Murcko M. Assessing the impact of generative AI on medicinal chemistry. Nat Biotechnol. 2020;38(2):143–145. doi: 10.1038/s41587-020-0418-2
- Mervin L, Genheden S, Engkvist O. AI for drug design: from explicit rules to deep learning. Artif Intell Life Sci. 2022;2:100041. doi: 10.1016/j.ailsci.2022.100041
- Kierner S, Kucharski J, Kerner Z. Taxonomy of hybrid architectures involving rule-based reasoning and machine learning in clinical decisions: a scoping review. J Biomed Inform. 2023;144:104428. doi: 10.1016/j.jbi.2023.104428
- Vayena E, Blasimme A, Cohen IG. Machine learning in medicine: addressing ethical challenges. PLoS Med. 2018;15(11):e1002689. doi: 10.1371/journal.pmed.1002689