
1. Introduction
Drug design and development (DDD) is a lengthy and time-consuming process. The process can take up to 14–15 years, and the cost ranges from US$944 million to US$2,826 million (2019 estimate), and these costs keep rising [Citation1]. For several decades, computers have played a key role in DDD. The application of an extensive group of computational or in silico tools are referred to as computer-aided drug design (CADD), which have been used in academia and industry for several years [Citation2]. CADD is an interdisciplinary field and has become one of the pillars in the pharmaceutical discovery pipelines. Current significant technological advances that are transforming CADD into computationally-driven drug discovery (CDDD) include but are not limited to [Citation3]: large (106–107) compound libraries in stock; ultra-large (1010–1015) on-demand, and generative virtual libraries (1023–1060); advances in structure-based and artificial intelligence (AI)-based virtual screening of the large and ultra-large spaces; de novo design; free energy perturbation and AI-based QSAR/QSPR modeling to enhance potency and selectivity; data-driven computational tools to predict absorption, distribution, metabolism, excretion, and bioactivity, and toxicology. The latter predictions are now routinely used in multiparameter hit-to-lead optimization. However, any computational model – regardless of its accuracy and applicability – will never ensure that all the predictions are correct. Strictly speaking, exact prediction of an experimental value is not possible as any experimental determination also has a degree of uncertainty. Computational predictions require experimental validation with appropriate and robust in vitro and in vivo assays. In turn, the experimental data can be fed back into the models to improve the quality [Citation3,Citation4]. In addition, as discussed in Section 2, AI plays a key role in synthesis planning and computational approaches that are used in the wet lab as part of the DDD automatization process.
There are several benefits of CADD and CDDD versus traditional DD and DDD that typically include experimentally driven high-throughput screening (HTS). For example, in silico tools provide a more cost-effective approach than traditional DD; computational tools offer faster and more efficient access to large and ultra-large chemical spaces, thus facilitating intellectual property novelty; hit-to-lead optimization/identification driven by computational tools is faster than traditional methods that typically require custom synthesis [Citation3]. As discussed in Section 2, incorporating AI and machine learning (ML) methods into CADD heavily shifts traditional hypothesis-driven research into a data-driven discovery [Citation5]. In any case, the influential role of CADD/CDDD can be exemplified by the more than 70 drugs approved for clinical use discovered with the aid of computational approaches [Citation6].
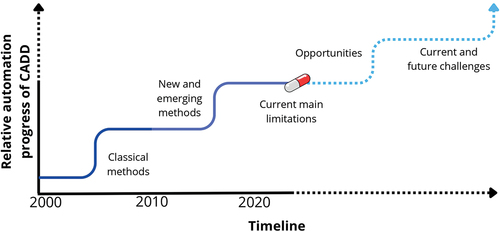
The impressive role of current in silico tools and methodologies in DDD impacts the expectations (and hype). It poses questions like: ‘What is the plausibility that all drugs will be designed by computers by the end of the decade?,’ or equivalent interrogations. This Editorial aims to provide the author’s opinion on this plausibility. The opinion is based on their experience and very recent literature from various research groups from different settings – academia and industry – that have reflected on this topic. outlines the coverage of the Editorial. The X-axis of the figure represents a timeline from 2020 to present, and the Y-axis depicts the evolution of the automatization processes in CADD relative to main decades since the classical CADD approaches to the new and emerging methods. The figure also outlines recent opportunities and challenges that are discussed in the following sections.
Figure 1. Timeline of automation of CADD. The X-axis represents the timeline of the most representative periods in CADD, and Y-axis illustrates the relative progress of automation in CADD. The continue line illustrates the events before 2024, while the dotted line illustrates the future events that CADD faces. Color coding is used to illustrate past (marine blue), emerging (purple), and future (cyan) events. Finally, the pill represents the current progress around automation in CADD.
2. New methods and technologies in CADD
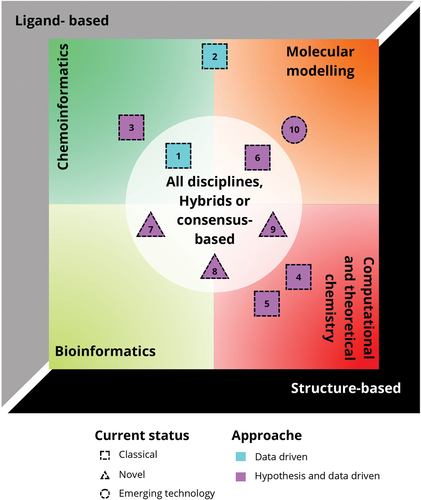
CADD comprises several well-established theoretical disciplines such as molecular modeling, bioinformatics, chemoinformatics, data mining and visualization, among others [Citation7]. As recently and extensively reviewed, many structure- and ligand-based DD methods have been used in CADD for several decades [Citation8]. Common examples include molecular docking and dynamics, pharmacophore modeling, QSAR/QSPR modeling, and similarity searching, among several others. Recently, the data-driven approaches of AI, including ML, have been integrated into CADD reshaping the contribution of computers in DD [Citation5]. For example, as reviewed by Chang et al., AI/ML has been incorporated in property prediction, molecular docking, de novo design, and compound retrosynthesis (useful to synthesize and obtain physical samples of generated libraries), among many other approaches [Citation9]. Niazi also published a recent comprehensive review of the application of AI/ML in DDD, clinical testing, and manufacturing [Citation10]. shows the different classifications of CADD schematically, its conceptual basis, and its integration into the DD pipeline.
Figure 2. Different classifications of CADD and its role in modern DD. Classification method-based: Ligand-based, structure-based, and hybrids or consensus-based; classification theoretical discipline-based: chemoinformatics, molecular modeling, bioinformatics, or computational and theoretical chemistry; classification approach-based: data driven and hypothesis driven; Representative examples of methods used: (1) QSAR/QSPR modeling, (2) similarity searching, (3) chemical space analysis, (4) molecular docking, (5) molecular dynamics, (6) pharmacophore modeling, (7) machine or deep learning, (8) AI-QSAR, (9) generative or de novo design, and (10) quantum computing.
New synthesis technologies have emerged, facilitating and optimizing the generation of new small molecules and natural product derivatives, allowing the efficient generation of new compound libraries with high structural diversity [Citation11], and new testing technologies (e.g. high-throughput screening robots) that increase the velocity to manipulate and evaluate complete chemical libraries to specific endpoints, for example, from the inhibition/activation of a protein to the phenotypic changes in a 3D cell culture. However, despite these significant advances, large speed and quality accelerators are still expected in CADD and CDDD over the next five years.
2.1. Main limitations
A vast amount of data is being generated at an unprecedented pace. However, there is still an imbalance in the data that limits its use in CADD and CDDD. For instance, a report from 2022 indicates that only 11% of biologically validated targets for treating diseases have an equal number of active and inactive reported compounds. This suggests a systematic imbalance in public data [Citation12]. In this context, data imbalance could be explained because previously high-impact peer-reviewed journals have prioritized the publication of highly active molecules vs. publishing datasets with low or no activity against specific biological targets. However, in light of new AI technologies, which need to be built from active and inactive data, this has been a systematic error that must be addressed as soon as possible. To this end, some strategies to deal with unbalanced chemical data as emerged, such as decoy-generator tools and AI strategies to deal with unbalanced data sets [Citation12].
In addition to the quantity of data and its balance, another major limitation is the quality of public data. This is because previous reports demonstrate low reproducibility and/or significant variability in the available data due to using non-standardized protocols between different laboratories [Citation12].
On the other hand, the automated development of drugs brings with it ethical problems that must be resolved and legislated. For example, who will be responsible if an automatically generated drug generates side effects? Who will have access to personal data? How to ensure that automatic design is not applied to bad practices, for example to the design of chemical/biological weapons? And how do we ensure that automated drug design does not have biases about population types and gender? Thus, a critical step before the complete automation of drug design will be to generate policies, ethical guides, and strict regulations on using new methods applicable to DD and DDD. In this regard, a key step to do before the complete automatization of CADD and CDDD is to establish legal precedents around the AI intellectual property of models used to discover, design, repurpose, and optimize new drugs. This important step could generate legal, ethical, operative, and economic benefits to the pharmaceutic industry, like ethical drug design directed to specific genders or populations, improving the quality, accuracy, and clinical efficacy of their medical products, according to the emerging precision medicine paradigm.
3. Challenges and opportunities
A series of grand challenges that CADD faces have recently been discussed [Citation7]. Briefly, these challenges can be roughly organized into six areas, namely, chemical and biological spaces, methodology challenges, communication between research teams and groups (e.g. Human interaction), data sharing and scientific dissemination, and education and training. One of the most recent methodology challenges and opportunities is enhancing the interpretation of AI-models: explainable-AI (ML) [Citation5,Citation9]. Additionally, methodology challenges and opportunities are de novo molecular design, pairing CADD with click chemistry e.g. using building blocks, to expand the active chemical space with novel scaffolds [Citation13]; enhancement of semi-synthetic natural products [Citation13]; improvement of poly-pharmacology and rational drug repurposing [Citation7]; efficient capture and analysis of post-marketing data (e.g. side effects) [Citation7]; and integration of conventional DD data and -omic data (e.g. proteomics, metabolomics, lipidomics, genomics, etc.) [Citation10].
4. Expert opinion
There is no doubt that computers play a pivotal role in the rationalization and development of drugs. In addition, computational approaches help rationalize the active compounds’ biological activity, including drug candidates and approved drugs, at the molecular level. Despite the many benefits, in silico tools still need to improve and be better incorporated into the drug discovery pipeline, thus enhancing the impact of the computational tools to identify hits and move them forward into preclinical and clinical development. There are critical specific challenges to address. One of them is the generation of high-quality data and the generation of an improvement of existing algorithms to process it. Data storage and maintenance can become a significant challenge, particularly for public sources. However, the effort is justified since it is well-known that public repositories of compounds annotated with biological activity, such as PubChem and ChEMBL, are vital to collaboration and global initiatives to foster decision-driven approaches such as AI/ML. Also, it is necessary to create models that better predict absorption, distribution, metabolism, excretion, bioactivity, and toxicology in order to prevent side effects. Experts from areas related to ‘big data’ and supercomputing are also required to be incorporated into DD, as are professionals in artificial, physical, and mathematical intelligence. Furthermore, new programs for training medicinal chemists and pharmacologists must allow the training of future chemists in these areas. Emerging technologies such as quantum computing [Citation14,Citation15] are anticipated to continue playing a pivotal role in selecting and identifying hits, their development, and rational drug repurposing.
The effectiveness of computer-based tools in drug discovery can be improved by enhancing communication and collaboration between academia and industry and establishing solid global initiatives. The scientific community should implement proper protocols to ensure data privacy, intellectual property rights, and regulatory compliance. Industry-academia partnerships and global initiatives are crucial in making DD more accessible [Citation5]. At the same time, pharmaceutical companies will be the ones that will be able to collect clinical data more efficiently and thereby improve their predictive algorithms and their ability to study their pharmaceutical innovations more rapidly [Citation10].
In summary, the authors believe that it is implausible for computers to design all drugs by the end of the decade. However, computers can increase the likelihood of designing faster, safer, cost-effective, and more successful therapeutics. Indeed, computers will play increasingly prominent roles in drug discovery.
Abbreviations
ADMET, absorption, distribution, metabolism, excretion, and toxicology; AI, artificial intelligence; CADD, computer-aided drug design; CDDD, computationally driven drug discovery; DD, drug design; DDD, drug design and development; HTS, high-throughput screening; ML, machine learning; QSAR/QSPR: quantitative structure-activity/property-relationships.
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
A reviewer of this manuscript has disclosed that they are employed by Charles River Laboratories. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Simoens S, Huys I. R&D costs of new medicines: a landscape analysis. Front Med. 2021;8:760762. doi: 10.3389/fmed.2021.760762
- Bassani D, Moro S. Past, present, and future perspectives on computer-aided drug design Methodologies. Molecules. 2023;28(9):3906. doi: 10.3390/molecules28093906
- Sadybekov AV, Katritch V. Computational approaches streamlining drug discovery. Nature. 2023;616(7958):673–685.
- Martinez-Mayorga K, Rosas-Jiménez JG, Gonzalez-Ponce K, et al. The pursuit of accurate predictive models of the bioactivity of small molecules. Chem Sci. 2024;1(6):1938–1952. doi: 10.1039/D3SC05534E
- Niazi SK, Mariam Z. Computer-aided drug design and drug discovery: a prospective analysis. Pharmaceuticals. 2023;17(1):22. doi: 10.3390/ph17010022
- Sabe VT, Ntombela T, Jhamba LA, et al. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: a review. Eur J Med Chem. 2021;224:113705. doi: 10.1016/j.ejmech.2021.113705
- Medina-Franco JL. Grand challenges of computer-aided drug design: the road ahead. Front Drug Discov. 2021;1:728581. doi: 10.3389/fddsv.2021.728551
- Azad I, Khan T, Ahmad N, et al. Updates on drug designing approach through computational strategies: a review. Future Sci OA. 2023;9(5):FSO862. doi: 10.2144/fsoa-2022-0085
- Chang Y, Hawkins BA, Du JJ, et al. A guide to in silico drug design. Pharmaceutics. 2022;15(1):49. doi: 10.3390/pharmaceutics15010049
- Niazi SK. The coming of age of AI/ML in drug discovery, development, clinical testing, and manufacturing: the FDA perspectives. Drug Des Devel Ther. 2023;17:2691–2725. doi: 10.2147/DDDT.S424991
- Gesmundo N, Dykstra K, Douthwaite JL, et al. Miniaturization of popular reactions from the medicinal chemists’ toolbox for ultrahigh-throughput experimentation. Nat Synth. 2023;2(11):1082–1091. doi: 10.1038/s44160-023-00351-1
- López-López E, Fernández-de Gortari E, Medina-Franco JL. Yes SIR! On the structure–inactivity relationships in drug discovery. Drug Discovery Today. 2022;27(8):2353–2362.
- Gao L, Shaabani S, Reyes Romero A, et al. “Chemistry at the speed of sound”: automated 1536-well nanoscale synthesis of 16 scaffolds in parallel. Green Chem. 2023;25:1380–1394. doi: 10.1039/D2GC04312B
- Blunt NS, Camps J, Crawford O, et al. Perspective on the current state-of-the-art of quantum computing for drug discovery applications. J Chem Theory Comput. 2022;18(12):7001–7023. doi: 10.1021/acs.jctc.2c00574
- Cova T, Vitorino C, Ferreira M, et al. Artificial intelligence and quantum computing as the next pharma disruptors. Methods Mol Biol. 2022;2390:321–347.