
ABSTRACT
Introduction
Albinism is a heterogenous disease with variable phenotypic and genotypic presentations. Diagnosis can be challenging and clinical evaluation strategies vary.
Areas covered
This review examines the phenotypic and genotypic characteristics of albinism and discusses evaluation strategies used to assess its variable clinical presentations. Additionally, it explores the challenges faced by clinicians in diagnosing albinism and issues related to genetic testing, interpretation and subsequent counseling of affected patients and their families. It is important to note that although current management of albinism is mainly supportive and focuses on optimizing vision, there are emerging therapies with promising translational benefit.
Expert opinion
Ongoing research in the field of albinism is benefiting from recent advances, particularly in retinal imaging and phenotyping. Similarly, access to advanced technologies like next-generation and long-read sequencing has improved diagnostic accuracy for previous cases with missing heritability. Deeper genotyping and phenotyping as well as multicentre collaborative approaches have allowed better understanding of genotype-phenotype correlations. There is still a need for more research on the psychosocial aspects of albinism. Encouraging involvement of patients and the public in determining research priorities in this area is essential for a better understanding of the psychosocial impact on individuals with albinism.
1. Introduction
Albinism is a heterogenous group of clinical disorders characterized by disrupted melanin synthesis or melanosome maturation during development causing cutaneous and/or ocular hypopigmentation [Citation1]. Albinism is historically divided into ocular albinism (OA) and oculocutaneous albinism (OCA). OA, characterized by X-linked inheritance, primarily presents with defects in the ocular and visual pathways. On the other hand, OCA follows an autosomal recessive inheritance pattern and manifests with both ocular and systemic abnormalities such as skin and hair hypopigmentation [Citation2]. A systematic review performed by Kromberg et al demonstrated wide variation in the prevalence of OCA, with an estimated prevalence of 1 in 4000 to 7000 in Africa and 1 in 12,000 to 15,000 in European countries [Citation3]. The prevalence of OA is about 1 in 60,000 males [Citation4].
In eyes of patients with albinism, pigment cells in the retinal pigment epithelium (RPE) which mainly contains eumelanin are affected [Citation5]. The increasing availability of genetic testing provides a possibility of a definitive molecular diagnosis and subtyping, as well as allows further understanding of these complex heterogenous eye diseases. Bakker et al recently proposed a new relationship between disease genes and function across various types of albinism, conceptualizing one retinal pigmentation pathway [Citation5]. This led to a new functional genetic classification of human albinism and facilitated a better understanding of its etiology.
Based on the aforementioned retinal pigmentation pathway hypotheses, several clinical and genetic subtypes of albinism exist. The first subtypes are syndromic forms of albinism such as Hermansky-Pudlak syndrome and Chediak-Higashi syndrome, which could pose systemic complications and morbidities as the general lysosome related organelles genesis and function are affected. The second subtype, OCA, affects only eye, skin and hair pigmentation [Citation5]. OCA1A is the most severe type with a complete lack of melanin production throughout life, with the milder forms of OCA1B, OCA2, OCA3 and OCA4 showing pigment accumulation over time [Citation6]. Other subtypes of OCA such as OCA5, OCA6, OCA7 and OCA8 have also been identified [Citation7]. The clinical phenotypes are not always distinguishable; therefore, it is important to utilize molecular diagnosis to identify the genetic mutations associated with the different types of OCA [Citation6].
Clinical manifestations of OA are usually limited to the eye because there is near normal melanin production but melanosome formation is affected [Citation5]. In addition to the primary ocular manifestations, OA has been observed to affect skin pigmentation at the microscopic level. Studies employing light and electron microscopy have revealed notable changes in the skin of patients with X-linked OA and female carries. These changes include the presence of abnormal or giant melanocytes and a general reduction in the number of melanocytes, regardless of size [Citation8]. Such findings suggest that the effects of OA extend beyond ocular symptoms, impacting melanocyte morphology and distribution in the skin. This also highlights the nuanced and systemic nature of OA, expanding our understanding of its dermatological implications [Citation8]. Finally, Foveal Hypoplasia, Optic Nerve Decussation defects and Anterior segment dysgenesis (FHONDA) resembles albinism remarkably with nystagmus, foveal hypoplasia and misrouting but without pigmentary abnormalities [Citation5].
This review aims to provide an update of the current understanding of the clinical and genotypic characteristics of albinism. With the current knowledge regarding albinism, we would like to discuss the research developments in the field that are likely to be important in the future.
2. Clinical characteristics of albinism
2.1. Cutaneous features
Cutaneous features of OCA which include reduced pigmentation of hair and skin are caused by reduction in tyrosinase function. Patients with OCA typically presents with reduced melanin pigment at birth. However, certain OCA subtypes such as OCA1B would result in gradual pigmentation with time due to the residual tyrosinase function. OCA2 is even milder in presentation, with cream-colored skin and blonde to red hair. OCA3, also known as red OCA, is associated with reddish hair and skin [Citation9]. Without adequate sun protection and sunscreens especially during outdoor activities, individuals with OCA have an increased risk of skin cancers [Citation1] which may include squamous cell carcinoma, basal cell carcinoma, melanoma and less commonly Merkel cell carcinoma, as well as sunburns and actinic damage [Citation10]. It is important to note that benign skin nevus from inappropriate sun exposure can also have cosmetic complications [Citation11].
OCA1 is the most common type of albinism in European or Western American population, whereas OCA2 (also known as yellow albinism) is the most common type of albinism in African Black OCA patients [Citation12]. OCA4 has clinical characteristics that are indistinguishable to OCA2 [Citation9] and its mutations have been reported in about 5–8% of German patients and 18% of Japanese patients with albinism [Citation13,Citation14]. The amount of cutaneous pigmentation in OCA4 can vary from minimal to near normal [Citation15]. Rarer subtypes of OCA, such as OCA5 presents with white skin and golden yellow hair, whereas OCA6 patients have light hair that darkens over time. OCA7 and OCA8 patients have mild skin and hair hypopigmentation. Skin hypopigmentation also imposes a significant psychosocial effect, especially on individuals with darker skin tone as the effect of skin hypopigmentation is more apparent [Citation16].
2.2. Ophthalmic features
All types of OCA and OA have similar ocular findings but can range in severity. Patients with albinism can have iris transillumination defects, foveal and optic disc hypoplasia and fundus hypopigmentation (); they often present with reduced vision and photophobia [Citation17–19]. Abnormal optic nerve fiber decussation at the optic chiasm (excessive crossing of temporal fibers at the chiasm) is also characteristic in albinism [Citation19].
Figure 1. Staging schemes in albinism. Iris transillumination. Anterior segment photos in albinism show various degrees of iris transillumination according to the grading scheme by Summers et al. [Citation17]. Adapted and reprinted with permission from Papageorgiou et al [Citation18].
![Figure 1. Staging schemes in albinism. Iris transillumination. Anterior segment photos in albinism show various degrees of iris transillumination according to the grading scheme by Summers et al. [Citation17]. Adapted and reprinted with permission from Papageorgiou et al [Citation18].](/cms/asset/0024bbec-728e-42d1-af7c-56eee94a9c49/ierl_a_2320117_f0001_oc.jpg)
2.2.1. Reduced visual acuity
Patients with albinism often have reduced visual acuity in varying degree. Visual acuity has been shown to be correlated with grade of foveal hypoplasia and measurement of photoreceptor thickness [Citation20]. Grading of foveal hypoplasia can also predict future visual acuity in young children with albinism [Citation19,Citation21]. Other factors which also correlate to poor visual acuity include complete albinism genotypes and the presence of nystagmus [Citation22]. Visual acuity usually ranges between −0.10 and 1.60 logMAR acuity, with median visual acuity tends to be 0.60 logMAR acuity [Citation23]. OCA1A tends to give poorer visual acuity with significant photophobia. Among different phenotypes of OCA, OCA3 usually gives better visual acuity [Citation6].
2.2.2. Iris transillumination defect
Iris transillumination defect occurs due to loss of iris pigment epithelium. This is reported to be present in 91–100% of individuals with albinism [Citation24,Citation25]. As a result, these individuals often complain of photophobia. In OCA1A, irises are usually light blue to pink and can be fully translucent. Pigments do not develop and amelanotic nevi may be present. As pigment may develop with time in OCA1B, blue irises may change to green or brown. In OCA3, hypopigmentation is usually not sufficient to alter ophthalmic development [Citation6]. Iris transillumination defect can be graded using classification systems for objectively documenting iris involvement [Citation24].
2.2.3. Refractive error
Patients with albinism often have hypermetropia and with-the rule astigmatism, with a smaller proportion of them with myopia [Citation26,Citation27]. Refractive errors have been found to correlate with axial length [Citation27]. As albinism can cause foveal hypoplasia-related nystagmus, it can lead to perception of smeared image motion of the retina [Citation27]. Amongst the different subtypes of OCA, OCA1A has the highest increase in astigmatism with age [Citation28].
2.2.4. Nystagmus
Nystagmus due to albinism has similar characteristics to other forms of infantile nystagmus, which includes conjugate horizontal nystagmus with an accelerating slow phase. A null zone if often present can be eccentric, resulting in abnormal head posture (AHP) [Citation29]. The nature of the nystagmus can be jerk or pendular in the horizontal plane. This usually develops within the first few weeks of life, with its amplitude diminishing with age [Citation30]. A small vertical component has however been previously documented [Citation29]. Periodic alternating nystagmus is also more common among patients with albinism as compared to other etiologies [Citation31]. Absence of nystagmus has been reported in up to 7.7% of individuals with albinism [Citation24].
2.2.5. Strabismus
The prevalence of strabismus is as high as 100% in OCA1 and esotropia is common among patients with albinism [Citation29]. Albinism can also lead to positive angle kappa and subsequent exotropia [Citation32].
2.2.6. Fundus hypopigmentation
Fundus hypopigmentation is present in more than 94% of individuals with albinism and is characterized by prominent choroidal vessels within the posterior pole as the result of reduced pigmentation of retinal pigment epithelium and choroid. The degree of fundus hypopigmentation can be graded objectively using a classification system [Citation24]. Patients with albinism and disease carriers typically have fundus appearances that are being described as ‘mud splattered’ due to the mixed patches of hypopigmentation and normal pigmentation. It will have the appearance of dark radial streaks against a bright background on fundus autofluorescence imaging. To enhance the diagnostic accuracy and assess the full extent of fundus hypopigmentation, ultra-widefield fundus imaging is recommended. Moreover, previous studies using Optos imaging may exhibit concentric macular rings, which may be a distinctive feature of foveal hypoplasia that supports the diagnosis of albinism [Citation33].
Fundus hypopigmentation usually occurs to lesser degree in OA than in OCA [Citation34]. Fundus hypopigmentation of varying degree can be explained using animal studies that uncrossed fibers located in the temporal periphery of the retina is more susceptible to changes in retinal pigmentation [Citation35]. The localized differences in melanin concentration in the RPE also affects retinal development to a varying degree [Citation36].
2.2.7. Foveal hypoplasia
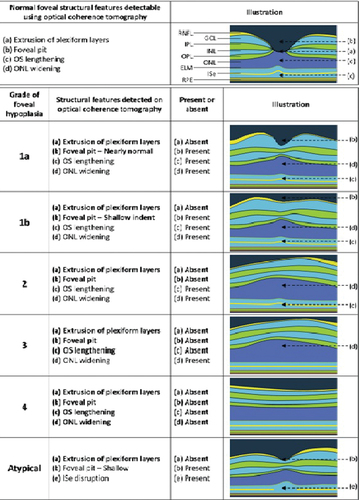
Foveal hypoplasia occurs where there is complete or partial failure of foveal pit formation and cone photoreceptor specialization. It has been postulated that lack of tyrosinase activity and DOPA results in increased mitosis and cell death during retinal development [Citation37]. On fundoscopy, foveal hypoplasia is characterized by blunting of foveal reflex [Citation1]. On optical coherence tomography (OCT), there is continuation of inner retinal layers posterior to the foveola and reduced cone photoreceptor specialization [Citation19]. Foveal hypoplasia has been reported to be 94–100% of individuals with albinism [Citation23]. There is a classification grading system that can be used for diagnosis and predicting future visual acuity in preverbal children with albinism () [Citation19,Citation21]. In non-syndromic OCA, foveal hypoplasia ranges from grades 1 to 4 [Citation23]. In Hermansky-Pudlak syndrome and GPR143 variants, foveal hypoplasia ranges from grades 3 to 4. FHONDA is caused by SLC38A8 variants and is inherited in an autosomal recessive manner. In biallelic pathogenic SLC38A8 variants, pigmentation is normal [Citation23]. However, severe grades of foveal hypoplasia are reported (only grade 3 and grade 4) [Citation24,Citation38].
Figure 2. Schematic drawing of the leicester grading system for Foveal Hypoplasia using OCT, shows a normal fovea, followed by typical and atypical grades of foveal hypoplasia. The normal fovea features outward displacement, termed extrusion, of the plexiform layers.
2.2.8. Optic nerve abnormalities
Albinism has been associated with optic nerve abnormalities such as small cup-to-disc ratio, elongation in the horizontal plane, thinning of temporal peripapillary retinal nerve fiber layer and oblique cup with situs inversus [Citation39,Citation40]. Fundus examination may also reveal abnormal retinal blood vessel patterns such as wide exit angles from the optic nerve head and vessels that encroach upon the central macular area [Citation41].
2.2.9. Chiasmal misrouting
Albinism can cause abnormal decussation of temporal fibers onto the contralateral hemisphere [Citation42]. Chiasmal misrouting can be diagnosed using visual evoked potentials (VEPs) () [Citation43] and it has been reported in 84–100% of individuals with albinism [Citation24,Citation29]. Another important differential for asymmetric VEPs is FHONDA () [Citation44].
Figure 3. Multichannel VEPs to investigate intracranial visual pathway dysfunction (a and b).
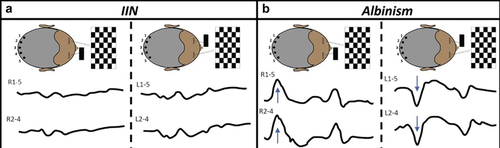
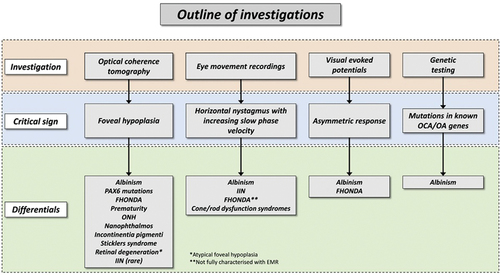
Figure 4. In evaluating patients with suspected albinism, the outline of investigations, characteristic sign that should be elicited with each investigative modality and differential diagnoses are shown.
3. Genetic classification of albinism
OA is inherited in an X-linked manner, whereas OCA is inherited in an autosomal recessive manner.
GPR143 variants cause OA. In those with pathogenic GPR143 variants, only ocular hypopigmentation is present. Female carriers show mild patchy features of the disease, with fundus showing ‘mud splattered’ features.
shows the classification of non-syndromic oculocutaneous and ocular albinism based on gene affected
Table 1. Classification of non-syndromic subtypes of albinism by gene.
There are also syndromic OCAs which includes Hermansky-Pudlak syndrome and Chediak-Higashi syndrome which are all inherited in an autosomal recessive manner.
shows the genes involved in different forms of syndromic OCAs.
Table 2. Genes involved in different syndromic OCAs.
4. Current evaluation strategies and management of patients with albinism
Due to the heterogeneity of symptoms and signs of OCA and OA, a multidisciplinary team approach involving Ophthalmologist and Dermatologist is required in assessing and managing patients with albinism. Children with albinism should undergo cycloplegic refraction to ensure prompt correction of refractive error. Careful assessment of nystagmus is also essential, noting its plane, axis of oscillation, conjugacy, frequency and amplitude [Citation49]. Modern video-based eye movement recordings can help in analyzing the nystagmus waveform and characteristics [Citation50]. AHP when null zone is not in the primary position can be objectively measured using torticollometer, Harms’ wall and orthopedic goniometer [Citation51]. Accurate measurement of deviation using prism cover tests is also crucial as patients with albinism often have strabismus. Other investigations include VEPs, which would show misrouting of visual pathway with abnormally high number of temporal fibers decussating at the optic chiasm. The International Society for Clinical Electrophysiology of Vision standard recommended multichannel ‘onset-offset’ pattern VEP presented with a field of 30 degrees to record chiasmal misrouting [Citation52]. OCT is another valuable tool to assess the degree of foveal hypoplasia, which is graded using the Leicester Grading System [Citation19].
Differentiating subtypes of albinism based on clinical presentation alone is challenging due to overlapping clinical features. Thus, genetic testing is helpful in confirming clinical suspicion, obtaining an accurate diagnosis and can aid in discussing prognosis and genetic counseling. This includes the differences in prognosis between syndromic forms compared to non-syndromic forms and searching for systemic phenotypes in syndromic OCA. Prior to genetic testing, a detailed family history should be taken, with attention to relatives with clinical manifestations of OCA and OA [Citation2]. Molecular genetic testing approaches can include a combination of targeted testing (multigene panel and chromosomal microarray) and comprehensive genomic testing (exome sequencing or genome sequencing). The genetic information on causative pathologic variants is important for carrier testing of family member, prenatal diagnosis and genetic counseling. Issues related to genetic counseling that need to be explored further include family planning, DNA banking and prenatal testing [Citation53]. summarized the aforementioned investigations, critical signs and the respective differential diagnoses when evaluating patients with suspected albinism [Citation44].
One of the challenges in obtaining a genetic diagnosis of albinism has been due to missing heritability or incomplete genetic diagnosis (where only one pathogenic TYR variant is identified). However, work by Gronskov et al. highlighted that a pathogenic haplotype (the allele p.S192Y-p.R402Q in cis, (also referred to as ‘cis-YQ’)) is a significant contributor to the disease and is common in Europeans [Citation54]. They suggest that these variants in combination may have an additive effect. This has been verified by other groups [Citation55]. Michaud et al. suggested that a promoter variant had a significant role in the pathogenesis of TYR associated albinism [Citation56], however a recent study by Loftus et al. did not find a difference in TYR mRNA expression in primary melanocytes between the different promoter genotypes [Citation57]. To identify the pathogenic haplotype, phasing of TYR variants is recommended. This is a scenario where long-read sequencing could help resolve whether the pathogenic haplotype is present. If there is still missing heritability, investigating for structural variants is recommended [Citation57]. Interestingly in carriers with albinism, phenotypic manifestations have also been reported [Citation58].
Recent advances in genetics offer the possibility of precise diagnosis. This includes the application of adeno-associated virus-based gene therapy for OA1, which showed positive outcomes in animal models [Citation59]. Gene editing of TYR gene with the CRISPR-associated nuclease protein (CRISPR/Cas9) system was also demonstrated on animal level by Song et al [Citation60]. More recently, patient-derived stem cell model for studying eye conditions related to OCA was developed. This is believed to be an important step forward in understanding albinism and testing potential therapies to treat it [Citation61]. Investigating albinism in some animal models may be limited as they lack fovea [Citation62].
Management of albinism at present usually includes supportive treatment aimed at optimizing vision, managing and addressing clinical manifestations especially photophobia, protecting retina from ultraviolet rays and reducing related complications such as skin cancers. Correction of refractive error using spectacles and/or contact lenses to reduce the risk of amblyopia is essential. When considering choice of contact lenses, soft contact lenses were associated with worse visual acuity compared to rigid gas permeable lenses and spectacles in a randomized controlled trial [Citation63]. Patients with albinism may have nystagmus which could cause misalignment of the toric soft contact lenses resulting in blurred vision. Some patients with albinism also do not find refractive correction helpful as their vision was limited by foveal hypoplasia [Citation64]. In cases of poor vision, low visual aids are helpful. Nystagmus, strabismus and AHP can be managed by prism glasses or surgery. The Anderson-Kestanbaum procedure is an effective surgical approach to move null point to correct AHP. However, there may be a higher risk of non-optimal success after surgery among patients with albinism due to poor fusion secondary to lower visual acuity and nystagmus [Citation65]. Tinted glasses or contact lenses can help to improve glare [Citation44]. Individuals with albinism should be advised to wear sun protective clothing and apply sunscreens in outdoors. Psychosocial and educational support should also be provided to individuals with albinism [Citation2].
5. Conclusion
Albinism which includes OCA and OA has heterogenous clinical manifestations and there is increasing understanding of its genetic variants. Treatment for albinism is focused on managing associated clinical features and complications. This includes the use of better optical aids for low vision with potential to improve quality of life. With an improved understanding of its pathophysiology, there is evidence from pre-clinical trials of novel therapies which directly address the molecular errors of albinism, in particular with the biochemical pathways in melanin biosynthesis. There are also emerging gene therapies that shows clinical success on animal models which have the potential for translational benefit for patients with albinism.
6. Expert opinion
Albinism is a condition that can cause significant health, social, educational and financial consequences for the person as well as for the family and community affected. Patients with albinism also have a high risk of skin cancer and can affect vision to varying extent, causing significant morbidity and mortality. Epidemiological data which have shown higher prevalence of albinism in the African subcontinent have been mostly published many years ago and are now outdated. New and better research studies on prevalence rates is therefore essential as the prevalence rates especially in the African subcontinent may be underestimated. With a better understanding of the epidemiology of albinism around the world, effort can then be focused on improving related health services as well as increasing awareness and acceptance of the condition in that area [Citation66].
As there are no effective therapies for the ophthalmic manifestations, more research is required to improve diagnostic techniques. Foveal abnormalities have been well described in albinism [Citation19]. Kuht et al. performed a cross-sectional study who identified foveal hypoplasia in 32.1% of OCA carriers using OCT. With better retinal imaging technologies, it may improve phenotyping of subtle ocular characteristics of patients or carriers with albinism [Citation58]. Handheld OCT has also allowed clinicians to image and characterize foveal findings in children with poor cooperation where other clinical diagnostic modalities are difficult to perform [Citation67]. Improvement in genetic testing techniques has also allowed us to classify different non-syndromic OCA and to better differentiate the significant phenotypic overlap of albinism with other disorders.
Albinism is one of the conditions commonly associated with nystagmus, which gives negative psychosocial and functional consequences. Therefore, there has been increased emphasis on individualized interventions based on patient age, function, needs and concerns. Studies regarding this topic to date were limited by small sample sizes, and there is no randomized controlled trial comparing different interventions to treat nystagmus [Citation68]. As treatment of albinism is mainly supportive, more patient-centered research should be encouraged, in particular studies of quality-of-life measures. As nystagmus is one of the phenotypically visible characteristics of albinism, affected patients are often stigmatized. With optimal refractive correction, individuals with albinism often attain satisfactory visual function. In addition, albinos perform equally well in intelligence tests as compared to control subjects [Citation69]. This means that improved parental and teacher understanding as well as early recognition of any specific educational problems is of paramount importance to support children with albinism and to improve their quality of life [Citation70]. The extent of the psychosocial challenges needs to be better understood and should be the main focus of future research in this field [Citation10].
6.1. Five-year view
Albinism is a heterogenous clinical condition and in the next five years, the hope is that advances in genotyping and phenotyping using modern imaging and genetic sequencing techniques will help to identify groups of patients that would need more support. A more personalized and stratified approach in looking after patients affected by the condition should be developed. Whilst using state-of-the-art technologies might improve the diagnostic perspective and clinical understanding of the disease, knowing what matters most for each individual patient who suffers from albinism and its associated complications should be the aim of future research. Patient and public involvement should be encouraged in setting research focus in the field of albinism. It is also important to determine a cost-effective pathway to screen and to identify children with albinism, especially in areas and countries with higher prevalence rates such as in Africa. When the prevalence of albinism is underreported and there is general lack of awareness of the condition in regions such as Africa, it is inevitable that the special needs of many individuals with albinism will not be met. Education of the importance of skin protection and eye care is necessary among patients with albinism. Spreading awareness and knowledge about this condition can also help eliminate the stigma and prejudice associated with the disease. Through the provision of adequate care to patients with albinism, the disease course can be changed and comorbidities can be treated when present.
Article highlights
Albinism has variable syndromic and non-syndromic presentations of cutaneous and/or ocular hypopigmentation caused by disruption of melanin synthesis and melanosome development.
In regions where ultraviolet radiation risk is elevated, cutaneous hypopigmentation in albinism increases the susceptibility to skin cancer, constituting a substantial burden of its morbidity and mortality.
Clinically stratifying ophthalmic manifestations of albinism, in particular objectively grading foveal hypoplasia using optical coherence tomography can help with estimating future vision in preverbal children and predicting likely genetic etiologies linked with foveal hypoplasia.
Improvement of diagnostic modalities and genetic testing techniques has allowed more accurate classification of the variable clinical phenotypic presentations of albinism.
The routine use of next-generation sequencing panels in clinical genetics workup has highlighted the challenges in variant interpretation and the role of common variants in development of albinism.
Current treatment strategies are aimed at addressing manifestations of the disease and optimizing visual potential. There are no curative treatments available, although early-stage clinical trials are underway.
More robust epidemiological data is needed in areas where albinism is more common and research should focus on improving knowledge and healthcare access for patients and family members affected by albinism.
Declaration of interests
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
I Gottlob and MG Thomas provided senior leadership for this manuscript. CS Chean drafted the paper. I Gottlob and MG Thomas made substantial contributions to study conception, design and data collection. CS Chean, Z Tu, MG Thomas and I Gottlob edited the draft paper and approved the final version submitted for publication.
Additional information
Funding
References
- Neveu MM, Padhy SK, Ramamurthy S, et al. Ophthalmological manifestations of oculocutaneous and ocular albinism: Current perspectives. Clin Ophthalmol. 2022;16:1569–1587. doi: 10.2147/OPTH.S329282
- MG T, Zippin J, BP B. Oculocutaneous albinism and ocular albinism overview. Adam MP, Mirzaa GM, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.
- Kromberg JGR, Flynn KA, Kerr RA. Determining a worldwide prevalence of oculocutaneous albinism: a systematic review. Invest Ophthalmol Vis Sci. 2023;64(10):14. doi: 10.1167/iovs.64.10.14
- Tsang SH, Sharma T. X-linked Ocular Albinism. Adv Exp Med Biol. 2018;1085:49–52.
- Bakker R, Wagstaff EL, Kruijt CC, et al. The retinal pigmentation pathway in human albinism: not so black and white. Prog Retin Eye Res. Prog Retin Eye Res. 2022;91:101091
- Gronskov K, Ek J, Brondum-Nielsen KOA. Oculocutaneous albinism. Orphanet J Rare Dis. 2007;2(1):43–43. doi: 10.1186/1750-1172-2-43
- Kausar T, Bhatti MA, Ali M, et al. OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24. Clin Genet. 2013;84(1):91–93. doi: 10.1111/cge.12019
- O’Donnell FEJ, Green WR, Fleischman JA, Hambrick GW. X-linked ocular albinism in blacks. Ocular Albinism Cum Pigmento Arch Ophthalmol. 1978;96(7):1189–1192. doi: 10.1001/archopht.1978.03910060023005
- Summers CG. Albinism: classification, clinical characteristics, and recent findings. Optom Vis Sci. 2009;86(6):659–662. doi: 10.1097/OPX.0b013e3181a5254c
- Ma EZ, Zhou AE, Hoegler KM, et al. Oculocutaneous albinism: epidemiology, genetics, skin manifestation, and psychosocial issues. Arch Dermatol Res. 2023;315(2):107–116. doi: 10.1007/s00403-022-02335-1
- Dessinioti C, Stratigos AJ, Rigopoulos D, et al. A review of genetic disorders of hypopigmentation: lessons learned from the biology of melanocytes. Exp Dermatol. 2009;18(9):741–749. doi: 10.1111/j.1600-0625.2009.00896.x
- Oetting WS, King RA. Molecular basis of albinism: mutations and polymorphisms of pigmentation genes associated with albinism. Hum Mutat. 1999;13(2):99–115. doi: 10.1002/(SICI)1098-1004(1999)13:2<99:AID-HUMU2>3.0.CO;2-C
- Rundshagen U, Zuhlke C, Opitz S, et al. Mutations in the MATP gene in five German patients affected by oculocutaneous albinism type 4. Hum Mutat. 2004;23(2):106–110. doi: 10.1002/humu.10311
- Inagaki K, Suzuki T, Shimizu H, et al. Oculocutaneous albinism type 4 is one of the most common types of albinism in Japan. Am J Hum Genet. 2004;74(3):466–471. doi: 10.1086/382195
- Hayashi M, Suzuki T. Oculocutaneous albinism type 4. Adam MP, Feldman J, Mirzaa GM, et al., editors. Seattle (WA): University of Washington, Seattle; 1993. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.
- Anshelevich EE, Mosojane KI, Kenosi L, et al. Factors affecting quality of life for people living with albinism in Botswana. Dermatol Clin. 2021;39(1):129–145. doi: 10.1016/j.det.2020.08.012
- Summers CG, Knobloch WH, Witkop CJJ, et al. Hermansky-pudlak syndrome. Ophthalmic Findings Ophthalmology. 1988;95(4):545–554. doi: 10.1016/S0161-6420(88)33152-0
- Papageorgiou E, McLean RJ, Gottlob I. Nystagmus in childhood. Pediatr Neonatol. 2014;55(5):341–351. doi: 10.1016/j.pedneo.2014.02.007
- Thomas MG, Kumar A, Mohammad S, et al. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmol. 2011;118(8):1653–1660. doi: 10.1016/j.ophtha.2011.01.028
- Mohammad S, Gottlob I, Kumar A, et al. The functional significance of foveal abnormalities in albinism measured using spectral-domain optical coherence tomography. Ophthalmol. 2011;118(8):1645–1652. doi: 10.1016/j.ophtha.2011.01.037
- Rufai SR, Thomas MG, Purohit R, et al. Can structural grading of foveal hypoplasia predict future vision in infantile nystagmus? A longitudinal study. Ophthalmol. 2020;127(4):492–500. doi: 10.1016/j.ophtha.2019.10.037
- Dumitrescu AV, Tran J, Pfeifer W, et al. Clinical albinism score, presence of nystagmus and optic nerves defects are correlated with visual outcome in patients with oculocutaneous albinism. Ophthalmic Genet. 2021;42(5):539–552. doi: 10.1080/13816810.2021.1933544
- Kuht HJ, Maconachie GDE, Han J, et al. Genotypic and phenotypic spectrum of Foveal Hypoplasia: a multicenter study. Ophthalmol. 2022;129(6):708–718. doi: 10.1016/j.ophtha.2022.02.010
- Kruijt CC, Gradstein L, Bergen AA, et al. The Phenotypic and Mutational Spectrum of the FHONDA Syndrome and oculocutaneous albinism: similarities and differences. Invest Ophthalmol Vis Sci. 2022;63(1):19. doi: 10.1167/iovs.63.1.19
- Sheth V, Gottlob I, Mohammad S, et al. Diagnostic potential of iris cross-sectional imaging in albinism using optical coherence tomography. Ophthalmol. 2013;120(10):2082–2090. doi: 10.1016/j.ophtha.2013.03.018
- Yahalom C, Tzur V, Blumenfeld A, et al. Refractive profile in oculocutaneous albinism and its correlation with final visual outcome. Br J Ophthalmol. 2012;96(4):537–539. doi: 10.1136/bjophthalmol-2011-300072
- Wildsoet CF, Oswald PJ, Clark S. Albinism: its implications for refractive development. Invest Ophthalmol Vis Sci. 2000;41(1):1–7.
- Schweigert A, Lunos S, Connett J, et al. Changes in refractive errors in albinism: a longitudinal study over the first decade of life. J AAPOS. 2018;22(6):462–466. doi: 10.1016/j.jaapos.2018.08.005
- Kumar A, Gottlob I, McLean RJ, et al. Clinical and oculomotor characteristics of albinism compared to FRMD7 associated infantile nystagmus. Invest Ophthalmol Vis Sci. 2011;52(5):2306–2313. doi: 10.1167/iovs.10-5685
- Hertle RW. Albinism: particular attention to the ocular motor system. Middle East Afr J Ophthalmol. 2013;20(3):248–255. doi: 10.4103/0974-9233.114804
- Abadi RV. Motor and sensory characteristics of infantile nystagmus. Br J Ophthalmol. 2002;86(10):1152–1160. doi: 10.1136/bjo.86.10.1152
- Brodsky MC, Fray KJ. Positive angle kappa: a sign of albinism in patients with congenital nystagmus. Am J Ophthalmol. 2004;137(4):625–629. doi: 10.1016/j.ajo.2003.11.066
- Ramtohul P, Comet A, Denis D. Multimodal imaging correlation of the concentric macular rings sign in foveal hypoplasia: a distinctive henle fiber layer geometry. Ophthalmol Retina. 2020;4(9):946–953. doi: 10.1016/j.oret.2020.03.022
- Marti A, Lasseaux E, Ezzedine K. Lessons of a day hospital: comprehensive assessment of patients with albinism in a European setting. Pigment Cell Melanoma Res. 2018;31(2):318–329. doi: 10.1111/pcmr.12651
- Colello RJ, Guillery RW. The early development of retinal ganglion cells with uncrossed axons in the mouse: retinal position and axonal course. Development. 1990;108(3):515–523. doi: 10.1242/dev.108.3.515
- Gimenez E, Lavado A, Jeffery G, et al. Regional abnormalities in retinal development are associated with local ocular hypopigmentation. J Comp Neurol. 2005;485(4):338–347. doi: 10.1002/cne.20495
- Wilk MA, Curcio CA, Brilliant MH, et al. Author response: relationship between foveal cone specialization and pit morphology in albinism. Invest Ophthalmol Vis Sci. 2014;55(9):5923–15470. doi: 10.1167/iovs.14-15470
- Kuht HJ, Han J, Maconachie GDE, et al. SLC38A8 mutations result in arrested retinal development with loss of cone photoreceptor specialization. Hum Mol Genet. 2020;29(18):2989–3002. doi: 10.1093/hmg/ddaa166
- Mohammad S, Gottlob I, Sheth V, et al. Characterization of abnormal optic nerve head morphology in albinism using optical coherence tomography. Invest Ophthalmol Vis Sci. 2015;56(8):4611–4618. doi: 10.1167/iovs.15-16856
- Chong GT, Farsiu S, Freedman SF, et al. Abnormal foveal morphology in ocular albinism imaged with spectral-domain optical coherence tomography. Arch Ophthalmol. 2009;127(1):37–44. doi: 10.1001/archophthalmol.2008.550
- Neveu MM, Holder GE, Sloper JJ, et al. Optic chiasm formation in humans is independent of foveal development. Eur J Neurosci. 2005;22(7):1825–1829. doi: 10.1111/j.1460-9568.2005.04364.x
- Hoffmann MB, Lorenz B, Morland AB, et al. Misrouting of the optic nerves in albinism: estimation of the extent with visual evoked potentials. Invest Ophthalmol Vis Sci. 2005;46(10):3892–3898. doi: 10.1167/iovs.05-0491
- Thomas MG, Maconachie GD, Sheth V, et al. Development and clinical utility of a novel diagnostic nystagmus gene panel using targeted next-generation sequencing. Eur J Hum Genet. 2017;25(6):725–734. doi: 10.1038/ejhg.2017.44
- Liu S, Kuht HJ, Moon EH, et al. Current and emerging treatments for albinism. Surv Ophthalmol. 2021;66(2):362–377. doi: 10.1016/j.survophthal.2020.10.007
- Okamura K, Suzuki T. Current landscape of Oculocutaneous Albinism in Japan. Pigment Cell Melanoma Res. 2021;34(2):190–203. doi: 10.1111/pcmr.12927
- Wei AH, Zang DJ, Zhang Z, et al. Exome sequencing identifies SLC24A5 as a candidate gene for nonsyndromic oculocutaneous albinism. J Invest Dermatol. 2013 Jul 1;133(7):1834–40. doi: 10.1038/jid.2013.49
- Grønskov K, Dooley CM, Østergaard E, et al. Mutations in c10orf11, a melanocyte-differentiation gene, cause autosomal-recessive albinism. Am J Hum Genet. 2013 Mar 7;92(3):415–21. doi: 10.1016/j.ajhg.2013.01.006
- Pennamen P, Tingaud-Sequeira A, Gazova I, et al. Dopachrome tautomerase variants in patients with oculocutaneous albinism. Genet Med. 2021 Mar 1;23(3):479–87. doi: 10.1038/s41436-020-00997-8
- Self JE, Dunn MJ, Erichsen JT, et al. Management of nystagmus in children: a review of the literature and current practice in UK specialist services. Eye (Lond). 2020;34(9):1515–1534. doi: 10.1038/s41433-019-0741-3
- Clark R, Blundell J, Dunn MJ, et al. The potential and value of objective eye tracking in the ophthalmology clinic. Eye (Lond). 2019;33(8):1200–1202. doi: 10.1038/s41433-019-0417-z
- Tyedmers M, Roper-Hall G. The harms tangent screen test. Am Orthopt J. 2006;56(1):175–179. doi: 10.3368/aoj.56.1.175
- Odom JV, Bach M, Brigell M, et al. ISCEV standard for clinical visual evoked potentials: (2016 update). Doc Ophthalmol. 2016;133(1):1–9. doi: 10.1007/s10633-016-9553-y
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Comm Genet. 2022;13(4):389–397. doi: 10.1007/s12687-022-00601-3
- Gronskov K, Jespersgaard C, Bruun GH, et al. A pathogenic haplotype, common in Europeans, causes autosomal recessive albinism and uncovers missing heritability in OCA1. Sci Rep. 2019;9(1):645–018–37272–37275. doi: 10.1038/s41598-018-37272-5
- Loftus SK, Gillis MF, Lundh L, et al. Haplotype-based analysis resolves missing heritability in oculocutaneous albinism type 1B. Am J Hum Genet. 2023;110(7):1123–1137. doi: 10.1016/j.ajhg.2023.05.012
- Michaud V, Lasseaux E, Green DJ, et al. The contribution of common regulatory and protein-coding TYR variants to the genetic architecture of albinism. Nat Commun. 2022;13(1):3939. doi: 10.1038/s41467-022-31392-3
- Loftus SK, Lundh L, Watkins-Chow DE, et al. A custom capture sequence approach for oculocutaneous albinism identifies structural variant alleles at the OCA2 locus. Hum Mutat. 2021;42(10):1239–1253. doi: 10.1002/humu.24257
- Kuht HJ, Thomas MG, McLean RJ, et al. Abnormal foveal morphology in carriers of oculocutaneous albinism. Br J Ophthalmol. 2023;107(8):1202–1208. doi: 10.1136/bjophthalmol-2020-318192
- Surace EM, Domenici L, Cortese K, et al. Amelioration of both functional and morphological abnormalities in the retina of a mouse model of ocular albinism following AAV-mediated gene transfer. Mol Ther. 2005;12(4):652–658. doi: 10.1016/j.ymthe.2005.06.001
- Song Y, Zhang Y, Chen M, et al. Functional validation of the albinism-associated tyrosinase T373K SNP by CRISPR/Cas9-mediated homology-directed repair (HDR) in rabbits. EBioMedicine. 2018;36:517–525. doi: 10.1016/j.ebiom.2018.09.041
- George A, Sharma R, Pfister T, et al. In vitro disease modeling of oculocutaneous albinism type 1 and 2 using human induced pluripotent stem cell-derived retinal pigment epithelium. Stem Cell Rep. 2022;17(1):173–186. doi: 10.1016/j.stemcr.2021.11.016
- van Beest E, Mukherjee S, Kirchberger L, et al. A fovea-like representation of space in mouse visual cortex. SSRN Electron J. 2019. doi:10.2139/ssrn.3441090
- Jayaramachandran P, Proudlock FA, Odedra N, et al. A randomized controlled trial comparing soft contact lens and rigid gas-permeable lens wearing in infantile nystagmus. Ophthalmol. 2014;121(9):1827–1836. doi: 10.1016/j.ophtha.2014.03.007
- Jhetam S, Mashige KP. Effects of spectacles and telescopes on visual function in students with oculocutaneous albinism. Afr Health Sci. 2020;20(2):758–767. doi: 10.4314/ahs.v20i2.28
- Villegas VM, Diaz L, Emanuelli A, et al. Visual acuity and nystagmus following strabismus surgery in patients with oculocutaneous albinism. P R Health Sci J. 2010;29(4):391–393.
- Human Rights Council thirty-first session agenda item 3 promotion and protection of all human rights, civil, political, economic, social and cultural rights, including the right to development report of the Independent expert on the enjoyment of human rights by persons with albinism note by the secretariat.
- Lee H, Sheth V, Bibi M, et al. Potential of handheld optical coherence tomography to determine cause of infantile nystagmus in children by using foveal morphology. Ophthalmol. 2013 Dec 1;120(12):2714–24. doi: 10.1016/j.ophtha.2013.07.018
- Penix K, Swanson MW, DeCarlo DK. Nystagmus in pediatric patients: interventions and patient-focused perspectives. Clin Ophthalmol. 2015;9:1527–1536. doi: 10.2147/OPTH.S62786
- Biswas S, IC L. Oculocutaneous albinism. Arch Dis Child. 1999;80(6):565–569. doi: 10.1136/adc.80.6.565
- Fulcher T, O’Keefe M, Bowell R, et al. Intellectual and educational attainment in albinism. J Pediatr Ophthalmol Strabismus. 1995;32(6):368–372. doi: 10.3928/0191-3913-19951101-09