
ABSTRACT
A demonstration of cellulose degrading bacterium from human gut changed our view that human cannot degrade the cellulose. However, investigation of cellulose degradation by human gut microbiota on molecular level has not been completed so far. We showed here, using cellobiose as a model that promoted the growth of human gut key members, such as Bacteroides ovatus (BO), to clarify the molecular mechanism. Our results showed that a new polysaccharide utilization locus (PUL) from BO was involved in the cellobiose capturing and degradation. Further, two new cellulases BACOVA_02626GH5 and BACOVA_02630GH5 on the cell surface performed the degradation of cellobiose into glucose were determined. The predicted structures of BACOVA_02626GH5 and BACOVA_02630GH5 were highly homologous with the cellulase from soil bacteria, and the catalytic residues were highly conservative with two glutamate residues. In murine experiment, we observed cellobiose reshaped the composition of gut microbiota and probably modified the metabolic function of bacteria. Taken together, our findings further highlight the evidence of cellulose can be degraded by human gut microbes and provide new insight in the field of investigation on cellulose.
Introduction
Cellulose is one of the major components of plant cell walls and a complex polysaccharide comprising backbone of D-glucopyranosyl residues linked via β1,4-glycosidic bond.Citation1 It is neither digested nor absorbed in the upper gut of mammals and is degraded only via a symbiotic association established between cellulolytic microorganisms and their host.Citation2–8 Increasing evidences have demonstrated that cellulose could be digested by the cellulolytic bacteria of ruminant herbivorous mammals and omnivorous mammals, including human.Citation2,Citation9–12 Despite their small number, cellulolytic bacteria play a crucial role in the cellulose degradation as their absence affecting the remainder of the microbial community.Citation13 On top of that, cellulolytic bacteria play a vital role in the provision of energy to the host and impact the health status.Citation14 Consequently, there is an increased interest for cellulose investigation.
To date, cellulolytic bacteria isolated from the large intestine or rumen of animals including bovine, horse, rabbit, tapir, capybara, rhinoceros, monkey, elephant, rat, pig and so on have been reported.Citation15–18 These cellulolytic bacteria were mainly Fibrobacter succinogenes subsp succinogenes, Fibrobacter succinogenes subsp elongatus, Fibrobacter succinogenes, Fibrobacter intestinalis and Ruminococcus flavefaciens. Cellulose breakdown in the human gut has been studied since from 19th century.Citation19 Up to now, cellulolytic bacteria isolated from human feces including Ruminococcus champanellensis, Ruminococcus spp., Bacteroides cellulosilyticus, Enterococcus spp., Eubacterium spp., Clostridium spp., Clostridium herbivorans and Bacillus cellulosae have been reported.Citation8,Citation11,Citation20–22 Despite there are dozens of cellulolytic bacteria have been isolated and identified from animals and humans, the studies of these strains have not been completed fully.Citation23 Among these strains, only the type strains of Fibrobacter and Ruminococcus from herbivorous mammals were further investigated.Citation24,Citation25 In addition to the cellulolytic bacteria from animals and humans, bacteria isolated from soil and termite, such as Pseudoalteromonas haloplanktis and Cytophaga hutchinsonii, can digest the cellulose into soluble sugars and biofuels.Citation26,Citation27 Taken together, although a large number of cellulolytic bacteria have been isolated, most of these bacteria are from animals or soil waiting for further study on molecular level. For human isolates, despite research on Ruminococcus champanellensis has made progress on the degradation of cellulose, we speculated that human gut may still has some other cellulases to confer cellulose degradation.
Genes encoding cellulases found in the genome of isolated bacteria from animals’ gut mainly belong to the glycoside hydrolases (GHs), such as GH5, GH8, GH9, GH44, GH45, GH48, GH51 and GH74 and families of endoglucanases, GH48 and GH9 families of cellobiohydrolases, and GH1 and GH3 families of β-glucosidases.Citation13,Citation23 In the human gut, GH5, GH8, GH9 and GH48 from Ruminococcus champanellensis are the only cellulases demonstrated so far.Citation25 Thus, research on the cellulases encoded by the genome of human gut microbes need to be paid more attention. Cellobiose as the smallest unit of cellulose has been well investigated in rumen and the termite gut.Citation28,Citation29 However, cellulases encoded by the genome of human gut microbes involved in the degradation of cellobiose remains unknown. Bacteroides ovatus (BO, ATCC 8483) isolated from human feces which belongs to the dominant phyla Bacteroidetes has been demonstrated to be capable of utilizing nearly all the major plant and host glycans including cellobiose and mixed-linkage beta-glucans.Citation30,Citation31 Hence, we explored the genes encoding cellulases to uncover the mechanism of cellobiose degradation by human gut key member BO on molecular level in this study.
To address, RNA-seq combined with molecular biology and biochemistry techniques were employed to discover the possible new enzymes. Meanwhile, we also analyzed the bioactivity of cellobiose on the composition of gut microbiota of mice by 16S rRNA sequencing. Our results may contribute to elucidate how cellulose is degraded by human gut microbiota and explore the significance of cellulose degradation in the human gut.
Results
Cellobiose promotes the growth of Bacteroides ovatus and probiotics
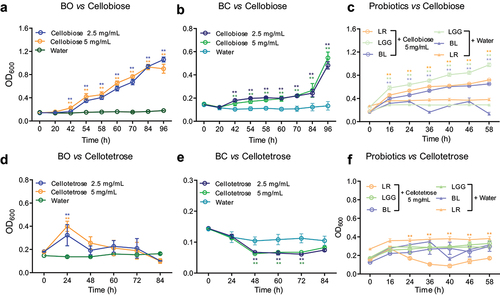
Previous data show BO can utilize cellobiose,Citation30 and Bacteroides cellulosilyticus (BC) is an adaptive forager with a strong emphasis on hemicellulose.Citation32 Hence, BO and BC were chosen for the current research. Firstly, the growth curve of BO and BC grown on either at 2.5 or 5 mg/mL cellobiose as the sole carbon source in MM medium were tested. Without cellobiose supplementation, BO and BC could not grow in MM medium (). In consonance with the previous data, our result showed BO grew well both on 2.5 mg/mL and 5 mg/mL cellobiose (). However, BC grew quite slowly, and the max OD600 value was almost only half of BO grown on cellobiose at 96 h (). In addition to BO and BC, bioactivity of cellobiose on probiotics including Lactobacillus reuteri (LR), Lactobacillus rhamnosus (LGG) and Bifidobacterium longum (BL) were also analyzed. Without cellobiose supplementation, LGG, LR and BL could not grow well in MCB medium (). Intriguingly, we observed that LGG, LR and BL grew well on 5 mg/mL cellobiose with max OD600 value around 1, 0.7 and 0.5, respectively, at 58 h (). Since these bacteria could utilize the cellobiose, we further would like to know whether BO, BC and probiotics could grow on cellotetrose or not? The results indicated that BO grew on 5 mg/mL cellotetrose with max OD600 value around 0.4 at 24 h (). However, BC and probiotics could not grow on cellotetrose (). In addition, we also tested the growth of BO and BC on cellotriose as well. The result showed that neither BO nor BC could grow on cellotriose (Supplementary Figure S1a, b). These results implicated that BO and probiotics preferred cellobiose as carbon source. For clarifying how cellobiose was degraded by these bacteria, BO was chosen for the further investigation.
Figure 1. Growth curves of HGM grown on cello-oligosaccharides were detected.
Cellobiose is degraded into glucose by Bacteroides ovatus
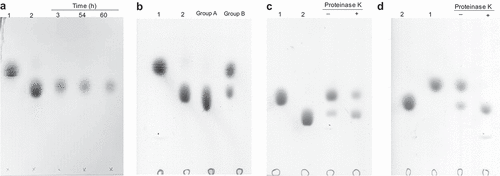
To explore whether the cellobiose could be degraded into glucose, the supernatants of BO grown on cellobiose were collected for further analysis by TLC. Unfortunately, glucose could not be detected from the supernatants (). Thus, we considered that the cellobiose might be transported into the cell for further digestion. Otherwise, the glucose creating from cellobiose might be utilized by BO quickly. To that end, BO was co-cultured with glucose (as group A) or cellobiose (as group B). Following, the cells were collected after 12 h or 72 h, respectively. After washing by HEPES-Na buffer for three times, the collected cells were then incubated with cellobiose, respectively, again for overnight at 37°C to detect the glucose by TLC. The results showed glucose could not be detected from the group A while it could be found from the group B (). This result suggested that enzymes which degraded the cellobiose into glucose only expressed in the presence of cellobiose, and these enzymes might be localized on the cell surface. Meanwhile, this result also demonstrated that cellobiose was not be transported into the cell for further digestion. To demonstrate our speculation, BO grown on cellobiose were collected. Following, the collected cells were treated with proteinase K for 6 h and 12 h, respectively. After removing the proteinase K, the cells were incubated with cellobiose again. The results indicated that cells treated with proteinase K for 6 h generated less glucose, and there was no glucose produced after cells treated with proteinase K for 12 h (). Briefly, the above results demonstrated that cellobiose was degraded into glucose by BO, and these results implicated that the enzymes might be localized on the cell surface. However, this speculation needed more data to confirm.
Figure 2. The degradation products of cellobiose were analyzed by TLC.
Two discrete PULs in the genome of Bacteroides ovatus are activated by cellobiose
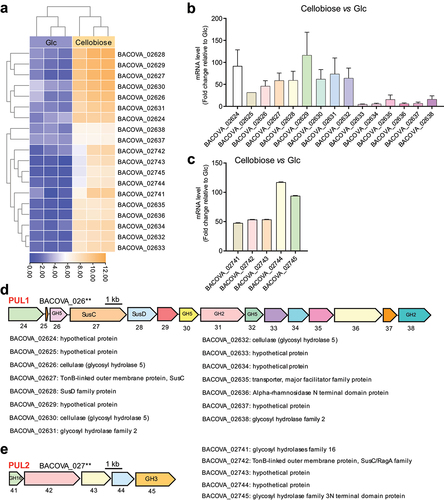
To explore the PULs which conferred the degradation of cellobiose, BO co-cultured with cellobiose were collected for RNA sequencing (RNA-seq). The prime sequencing results was showed in (Supplementary Table S1). Briefly, a total of 91 genes exhibited altered expression in response to cellobiose (Supplementary Figure S2a). Among the 91 genes, 19 genes significantly up-regulated compared with BO grown on glucose ( and Supplementary Figure S2b, c). The 19 up-regulated genes were then arranged into two discrete PULs including PUL1 (BACOVA_20624-BACOVA_02638) and PUL2 (BACOVA_02741-BACOVA_02745) which were further confirmed by RT-qPCR (). The schematics of each PUL was described subsequently based on the gene annotation of BO (GenBank: CP012938.1). PUL1 was clustered by fifteen genes including a pair of SusC/SusD, glycosidase hydrolysis (GH5 and GH2) and six hypothetical proteins (). PUL2 was clustered by five genes mainly including two glycosidase hydrolysis (GH16 and GH3) (). Intriguingly, we noted that PUL1 had never been reported till now. These results suggested that PUL1 and PUL2 might be involved in the degradation of cellobiose.
Figure 3. Polysaccharide utilization loci (PULs) were screened by RNA-seq and confirmed by RT-qPCR.
Discovery of two new cellulases to confer the degradation of cellobiose
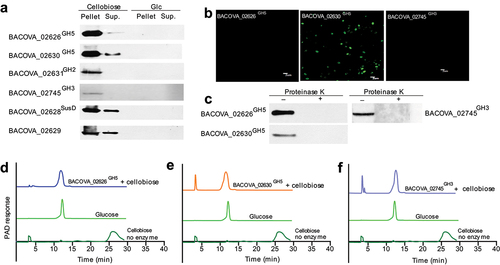
To determine which PUL mediated the degradation of cellobiose, the recombined proteins encoded by each PUL were purified and the corresponding polyclonal antibody were prepared. Results showed that BACOVA_02626GH5, BACOVA_02630GH5, BACOVA_02631GH2 and BACOV-A_02745GH3 specifically expressed in the presence of cellobiose while BACOVA_02632GH5, BACOVA_02638GH2 and BACOVA_02741GH16 expressed non-specificity which showed band in the presence of glucose (Glc) (, Supplementary Figure S3a). This result suggested that cellulases BACOVA_02626GH5 and BACOVA_02630GH5 and β-glucosidase BACOVA_02745GH3 might be involved in the degradation of cellobiose. Together with the results that cellobiose could be degraded into glucose detected by TLC, we next wondered to know whether BACOVA_02626GH5, BACOVA_02630GH5 and BACOVA_02745GH3 expressed on the cell surface or not. To that end, we analyzed the location of BACOVA_02626GH5 and BACOVA_02630GH5 by immunofluorescence and Western blotting. We observed that BACOVA_02630GH5 could be detected from the cell surface while BACOVA_02626GH5 and BACOVA_02745GH3 could not be found from the cell surface by immunofluorescence (). Meanwhile, we observed that BACOVA_02626GH5, BACOVA_02630GH5 and BACOVA_02745GH3 could not be detected in the presence of glucose (Supplementary Figure S3b). However, when the cell surface proteins were removed by proteinase K, BACOVA_02626GH5, BACOVA_02630GH5 and BACOVA_02745GH3 could not be tested by Western blotting (). Here, an interesting data was BACOVA_02626GH5 and BACOVA_02745GH3 could not be detected by immunofluorescence while Western blotting result showed they were possibly located on the cell surface. This probably suggested that the expression of BACOVA_02626GH5 and BACOVA_02745GH3 on the cell surface was pretty low. This phenomenon was also observed in our previous study.Citation33 Hence, cells treated by proteinase K following by Western blotting might be the gold standard to identify the surface enzymes as previous publications.Citation34,Citation35 In addition, we observed that surface glycan binding protein BACOVA_02628SusD and BACOVA_02629 were located on the cell surface (Supplementary Figure S3c). In contrast, BACOVA_02628SusD and BACOVA_02629 could not be tested in the presence of glucose (Supplementary Figure S3b). Taken together, the above results suggested that BACOVA_02626GH5, BACOVA_02630GH5 and BACOVA_02745GH3 were located on the cell surface of BO. To determine whether BACOVA_02626GH5, BACOVA_02630GH5 and BACOVA_02745GH3 conferred the degradation of cellobiose into glucose or not, we next analyzed the enzymatic activity. The recombinant proteins including BACOVA_02626GH5, BACOVA_02630GH5 and BACOVA_02745GH3 were incubated with cellobiose in vitro, respectively (Supplementary Figure S3d – f). The end products were analyzed by HPAEC-PAD. Intriguingly, we found that BACOVA_02626GH5, BACOVA_02630GH5 and BACOVA_02745GH3 could digest the cellobiose into glucose while glucose could not be detected in the absence of BACOVA_02626GH5, BACOVA_02630GH5 or BACOVA_02745GH3 (). Here, we did not analyze the enzymatic activity of BACOVA_02631GH2 because it was a sugar binding domain protein. Taken together, these results directly showed that cellobiose was degraded into glucose by BACOVA_02626GH5, BACOVA_02630GH5 and BACOVA_02745GH3 which expressed on the cell surface of BO. Notably, BACOVA_02745GH3 had been mentioned in the previous report with a poorly enzymatic activity on cellobiose.Citation31 To sum up, we might conclude that two new cellulases conferred the degradation of cellobiose into glucose were determined in this study.
Figure 4. Two new cellulases were determined.
To identify the homologous PUL in the genome of other gut microbes, we blasted the amino acid sequence of PUL1 encoding protein involved in the cellobiose degradation process using National Center for Biotechnology Information (NCBI) database. The blast results are listed in Supplementary Table S2. We observed that PUL1 encoding protein involved in the cellobiose degradation mainly distributed in the Bacteroides spp. Meanwhile, we also analyzed BACOVA_02626GH5 and BACOVA_02630GH5 by molecular evolutionary genetic analysis 11 (MEGA11.0), respectively. Our results showed that BACOVA_02626GH5 and BACOVA_02630GH5 were highly homologous with the cellulase from Bacteroides spp. (Supplementary Figure S3g, h). To our surprised, we found that the amino acid sequence of BACOVA_02626GH5 and BACOVA_02630GH5 only have 17.1% identity (data not shown). In addition, we found BACOVA_02626GH5 and BACOVA_02630GH5 have no homology with β-glucanase and cellulase from probiotics, such as LR, LGG and BL (data not shown). Briefly, we found two new cellulases which homologues widely existed in Bacteroides spp. deconstructed the cellobiose into glucose.
The predicted structure of BACOVA_02626GH5 and BACOVA_02630GH5 is highly homologous with the cellulase from soil bacteria
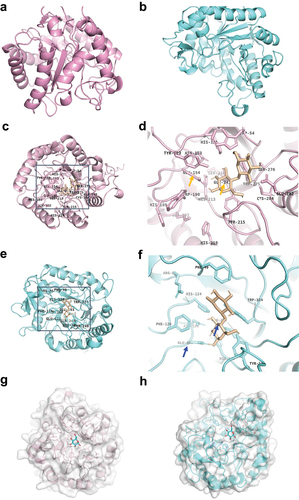
To understand the catalytic mechanism of BACOVA_02626GH5 and BACOVA_0–2630GH5 on cellobiose, their overall structure was predicted by Alphafold2, respectively. The carton structure of BACOVA_02626GH5 and BACOVA_02630GH5 were monomer shown by pink or blue with a pLDDT equal to 94.732 and 92.247, respectively (). Next, point-site and catalytic site of BACOVA_02626GH5 and BACOVA_02630GH5 were predicted based on the carton structure. For BACOVA_02626GH5, we found the catalytic pocket including Ser 276, Trp 54, Asn 153, Glu 154, Tyr 215, Glu 241, Cys 284, Glu 281 (). For BACOVA_02630GH5, the catalytic pocket was composed of Glu 293, Tpr 324, Asn 180, Phe 126, Arg 85, Tyr 248, Phe 49 (). In addition, we found the catalytic residues of BACOVA_02626GH5 were Glu 154 and Glu 241 (the orange arrow showed) while for BACOVA_02630GH5 was Glu 181 and Glu 293 (the blue arrow showed) (). This result was consisted with the data of sequence alignment of catalytic residues of BACOVA_02626GH5 and BACOVA_02630GH5. Amino acids residues of BACOVA_02626GH5 including Tyr 123, Trp 54, Trp 275, His 121, Asn 153, Glu 282 were used to fix the cellobiose (). For BACOVA_02630GH5, the amino acids residues, such as Arg 85, His 124, Asn 180, Trp 324, Tyr 333, were responsible for the fixation of cellobiose (). Although the homology of amino acids sequence of BACOVA_02626GH5 and BACOVA_02630GH5 were not high, we observed the predicted structure of BACOVA_02626GH5 and BACOVA_02630GH5 were high homologous to endoglucanase Cel5R (PDB ID 5I2U) and Cel5A from Pseudomonas stutzeri A1501 (PDB ID 4LX4) with a similarity more than 0.5 (Supplementary Figure S4a, b). On top of that, the catalytic residues of BACOVA_02626GH5 and BACOVA_02630GH5 were highly conservative with Cel5R and Cel5A (Supplementary Table S3). To explore the binding of cellobiose with the cellulase, docking was performed. As the results showed glucose bound BACOVA_02626GH5 and BACOVA_02630GH5, respectively (). The above results might supply the information for better understanding how the cellulase digest the β1,4 glucosidic bond. Together with the previous data, we might conclude that BACOVA_02626GH5 and BACOVA_02630GH5 were two cognate cellulase.
Figure 5. Structure biology of BACOVA_02626GH5 and BACOVA_02630GH5.
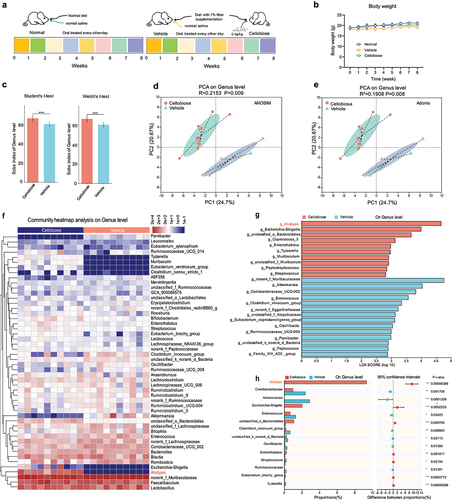
Cellobiose reshapes the composition of gut microbiota of mice feeding low fiber diet
To explore the function of cellobiose on the composition of gut microbiota of mice feeding with low fiber diet, mice were oral treated with cellobiose every other day for eight weeks (). We observed that the cellobiose improved body weight loss (). Next, the feces of mice in vehicle and cellobiose groups were analyzed by diversity sequencing based on the 16S rRNA. α diversity analysis using student’s t-test or Welch’s t-test on genus level showed a significantly difference (p < 0.01) (). PCA analysis showed distinct clustering of intestinal microbe communities of each group with a separation totally alongside PC1 at 24.74% based on the ANOSIM (p = 0.009) or Adonis (p = 0.008) on genus level (). Following, the differences of the relative abundance of the top 50 species between groups were visualized with heatmap on genus level. The results indicated that the relative abundance of Alistipes in the cellobiose group increased significantly compared with the vehicle group (p < 0.05) (). Next, LEfSe and two groups of comparison on genus level were analyzed. We found the abundance of Alistipes was enriched significantly after cellobiose treatment (). These results inferred that the composition of gut microbiota of mice feeding low fiber diet might be modified after cellobiose treatment. To verify these data, the third generation sequencing was performed. Alpha diversity and beta diversity analysis showed that cellobiose group was significantly different from the vehicle group (Supplementary Figure S5a-c). Heatmap and LEfSe analysis on species level showed that the abundance of both Bacteroides ovatus (BO) and Lactobacillus reuteri (LR) increased significantly (p < 0.05) in cellobiose group (Supplementary Figure S5d, e). In addition, we also found the abundance of Alistipes was enriched on genus level (Supplementary Figure S5e). Taken together, we might conclude that cellobiose reshaped the composition of gut microbiota of mice feeding low fiber diet and contributed to the proliferation of species including Lactobacillus reuteri and Bacteroides ovatus.
Figure 6. Function of cellobiose on the composition of gut microbiota was analyzed by diversity sequencing based on 16S rRNA.
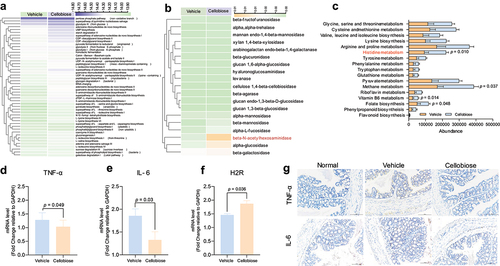
The metabolic function of bacteria may be modified and possibly contributes to inhibit the inflammation
To investigate the functional activities of bacteria, we used the PICRUSt1/2 and Tax4Fun tool to map the 16S rRNA sequence of the genes and pathways that these bacterial species might harbor. Unsupervised clustering analysis of 49 predicted metabolic MetaCyc pathways showed the greatest variability between the vehicle and cellobiose group. This result revealed that microbiome metabolic pathways were relatively specific to cellobiose (). Further, we predicted the enzymes related with the metabolism of carbohydrate by Tax4Fun tool. Unexpectedly, only β-N-acetylhexosaminidase, alpha-glucosidase and β-galactosidase showed difference compared with the vehicle group (). Besides, we also predicted the KEGG pathway by PICRUSt1. Metabolism related KEGG pathways including histidine, methane, Vitamin B6 and folate biosynthesis were significantly enriched after cellobiose treatment (). Intriguingly, β-N-acetylhexosaminidase and histidine, which was metabolized by LR to histamine, have been demonstrated to inhibit the inflammation of colon.Citation36,Citation37 Thus, the pre-inflammatory factors including TNF-α and IL-6 from colon tissues were analyzed. We found the mRNA expression level of TNF-α and IL-6 significantly decreased after cellobiose treatment (). Since the histidine metabolism pathway was enriched significantly, histamine H2 receptor (H2R) was also analyzed. The result showed the mRNA expression level of H2R increased significantly after cellobiose administration (). In addition, the protein level of TNF-α and IL-6 were detected by IHC, and the result indicated that IL-6 and TNF-α decreased after cellobiose treatment (). Taken together, these results implied that cellobiose might change the metabolic function of bacteria. However, it was not clear whether the change in metabolic function of bacteria related with the inflammation. The biological function of cellobiose on the host needs to be explored in the future.
Figure 7. Predication of metabolic function of bacteria and inflammatory factors detection.
Discussion
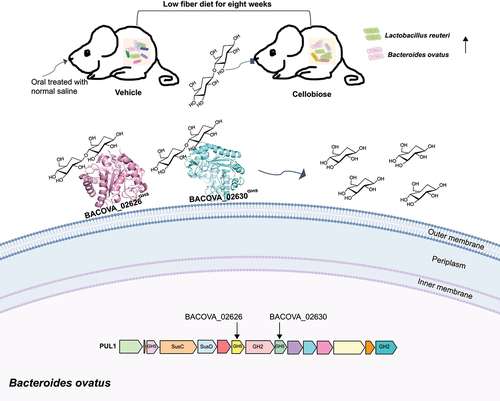
Cellulolytic bacteria are key participants in the breakdown process of cellulose.Citation23 Although cellulolytic bacteria have been widely isolated, they have not been well studied on molecular level. Cellobiose as the smallest unit of cellulose has been reported to promote the growth of human gut key member BO,Citation30 yet the underlying molecular mechanism is vague so far. Collectively, data present here uncovered how BO degraded cellobiose from diet into glucose, and the function of cellobiose on the composition of gut microbes of mice feeding low fiber diet was explored. As shown in , two new cell surface cognate cellulases, which structure showed homology with the cellulase from soil bacteria, conferred the degradation of cellobiose into glucose were clarified. In vivo test, we observed cellobiose reshaped the composition of gut microbiota and probably modified the metabolic function of bacteria. The current knowledge may shed considerable new light on plant cell wall hydrolysis by human gut microbiota and contributes to the research on cellulose.
Figure 8. Model of degradation of cellobiose by human gut BO. Two new cellulases on the cell surface conferred the degradation of cellobiose into glucose were determined. In vivo test, we observed that cellobiose reshaped the composition of gut microbiota, and the abundance of LR and BO were enriched significantly after eight weeks administration by gavage.
To date, at least three pathways including β-glucosidase (EC 3.2.1.21), cellobiose phosphorylases (EC 2.4.1.20) and 6-P-β-glucosidases (EC 3.2.1.86) have been reported to cleave the β1,4 linkage.Citation38–40 However, cellular localization analysis of cellulase has not been described on molecular level till now. The current results firstly uncovered two new cell surface cellulases in this study. Cellobiose as one of the most common intermediates, whose accumulation can inhibit the upstream cellulases. Timely removal of cellobiose is crucial for the entire cellulolytic process. Thus, we speculated that cellulases located on the cell surface may be beneficial to cleave cellobiose, and this is possibly the evolutionary needs. However, this needs more data to confirm. Notably, despite the amino acids sequence shared lower identity, the predicted structure showed BACOVA_02626GH5 and BACOVA_02630GH5 had similar structure with endoglucanase Cel5A (PDB ID 4LX4) and Cel5R (PDB ID 5I2U).Citation41,Citation42 The catalytic residues of BACOVA_02626GH5, BACOVA_02630GH5, Cel5A and Cel5R were highly conservative with two glutamate residues. This might support the result that BACOVA_02626GH5 and BACOVA_02630GH5 were endoglucanase and further implicated that these cellulases might evolve from soil bacteria. However, enzyme activity analysis showed that BACOVA_02626GH5 and BACOVA_02630GH5 exhibited exocellulases activity to make two glucose units. In fact, exocellulases are scarce among bacterial cellulolytic systems. Up to now, there were only a few reported cases mainly attributed to the cellulosomal bacteria affiliated to the Clostridiales.Citation43 Exo-cellulases are particularly scarce in anaerobic gut metagenomic datasets, with limited number of cellobiohydrolases observed in several rumen microbiomes, marine and termite gutsCitation29,Citation44–47. BACOVA_02745GH3 with lower catalytic activity was the only β-glucosidases from human gut microbiota reported so far.Citation45 In spite of this, we consider that the exocellulases are still believed to be necessary for microbes to effectively extract energy.
In vivo test, we found our result was consistent with the previous report that Alistipes increased significantly after cellulose treatment.Citation48 Although Alistipes is equipped with enzymes to degrade cellulose, cellulases encoded by Alistipes are not clear so far. Histamine, which is converted by histidine, has been reported to modulate intestinal immune response and inhibit colonic inflammation.Citation37 In this study, we firstly observed that LR, which secretes histamine significantly, was increased after cellobiose treatment compared with vehicle group. Secondly, we found the KEGG pathway of histidine metabolism was enriched significantly in cellobiose group. Thirdly, we showed that the mRNA expression level of colonic H2R was increased significantly. Fourthly, we observed that the proinflammatory factors, such as IL-6 and TNF-α, were decreased after cellobiose treatment. Combined these results together, we might conclude that cellobiose might have the bioactivity to inhibit colonic inflammation. However, we were not clear the underly molecular mechanism why the abundance of LR was increased after cellobiose treatment. We speculated that it was possibly that LR competed cellobiose with BO in vivo. Despite the abundance of LR was increased significantly, the amino acid sequence of currently reported cellulase and β-glucanase from LR have no identity with BACOVA_02626GH5 and BACOVA_02630GH5. This result implicated that gut microbes might harbor cellulases beyond our imagination. We may need to pay more attention on this field to explore more cellulases from human gut microbes in the future, especially for cellulases generating cellobiose or cello-oligosaccharide. Overall, this study provides critical evidence to support the phenomenon that cellulose can be degraded into glucose in the human gut.
Material and methods
Bacterial strains and growth conditions
Bacterial strains used in the current research are listed in (Supplementary Table S4). Bacteroides spp. were routinely cultured under anaerobic conditions at 37°C using an anaerobic cabinet (Whitley A25 Workstation; Don Whitley, 80% N2, 10% H2, 10% CO2) in brain-heart infusion (BHI) supplement with 10% sterile defibrillated sheep blood or minimal medium (MM) (Supplementary Table S5) containing an appropriate carbon source plus 1.2 mg/mL porcine hematin (Sigma-Aldrich) as previously described.Citation34 Lactobacillus rhamnosus GG (LGG), Lactobacillus reuteri (LR) and Bifidobacterium longum (BL) were routinely cultured under anaerobic conditions at 37°C in MRS medium or Medium for colon bacteria (MCB) (Supplementary Table S6).Citation49 For growth curve, the turbidity at OD600 was around 0.2. Cultures supplement with either 2.5 mg/mL or 5 mg/mL cellobiose, cellotetrose or cellotrise were grown at 37°C in anaerobic incubator for a couple of days, and the OD600 value was recorded at different time points by Micro plates Reader. Notably, all the bacteria used in the present study were identified by amplifying the full length of 16S rRNA and sequencing before to use.Citation50
Thin layer chromatography (TLC)
TLC experiments were done as described previously.Citation51 Briefly, glucose and cellobiose were used as standards. TLC Silica gel 60 F254 (Merck) was used for detecting the glucose. And 1-butanol/ethanol/water (5: 3: 2, v/v/v) was employed as mobile phase, and concentrated sulfuric acid/ethanol (1.9: 40, v/v) was served as staining solution. One microliter sample was spotted on the TLC silicone plate, which were developed upright in a TLC tank using mobile phase. After development, the plates were dried by a blow dryer and sprayed with staining solution. Subsequently, the plates were visualized for 1–2 min at high temperature by hot air gun.
Transcriptomic analysis by RNA sequencing (RNA-seq)
BO was cultured in triplet using MM supplemented with 5 mg/mL carbon source, glucose or cellobiose, as described above. Cells were harvested at mid-log phase. Total RNA was extracted by customer and checked for a RIN number to inspect RNA integrity by an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, US). Qualified total RNA was further purified by RNA Clean XP Kit (Cat A63987, Beckman Coulter, Inc. Kraemer Boulevard Brea, CA, USA) and RNase-Free DNase Set (Cat#79254, QIAGEN, GmBH, Germany). RNA-seq libraries were prepared using the Script Seq Complete kit from Epicentre. Samples were paired-end sequenced on an Illumina HiSeq 4000 instrument at Shanghai Biotechnology Corporation (SHBIO). Analysis of the RNA-seq results were performed exactly as the description.Citation52 Differential gene expression analysis was performed with the DeSeq2 package.Citation53
Quantitative RT-PCR (RT-qPCR)
Comparison of the levels of transcription of the PULs were performed by RT-qPCR as previous described.Citation53 BO was cultured in 5 mL of MM containing 5 mg/mL carbon source as described above. Triplicate bacterial cultures were harvested at mid-log phase (OD600 ∼ 0.6) and placed in RNA protect (Qiagen), then purified with RNeasy kit (Qiagen). RNA purity was assessed spectrophotometrically (NanoDrop 2000), and 1 μg of RNA was used immediately for reverse transcription using M-MLV. RT-qPCR was performed in a 96-well plate ABI VIIA7 system with SYBR Premix Ex Taq (Takara) according to manufacturer’s instructions. Reactions were carried out in 14 μL, including 0.4 μL forward primer and reverse primer, respectively, 0.4 μL ROX Reference Dye II (Takara), 7 μL SYBR® Premix Ex Taq II (2 ×) (Tli RNaseH Plus), Bulk (Takara), 1 μL reverse transcript cDNA and 4.8 μL ddH2O. Reaction conditions were same with previous publication.Citation54 Data were normalized to 16S rRNA transcript levels, and a change in expression level was calculated as a fold change relative to glucose cultures. All the primers are listed in (Supplementary Table S7).
Cloning and protein purification
The open-reading frames encoding mature forms of the proteins used in the study were amplified from the genomic DNA of BO by PCR using appropriate primers (Supplementary Table S7), which introduced BamHI/XhoI and NdeI/XhoI sites to the flanks of the target genes. Notably, the signal peptide was deleted. The amplified DNA was cloned into the expression vector pET28a and following transformed into Tuner (DE3) competent cells for protein expression (Supplementary Table S8). The recombinant proteins contain either N- or C-terminal His 6-tag. Tuner (DE3) contained vector were cultured in LB broth containing kanamycin (50 µg/mL) at 37°C. Cells were grown to mid-log phase (OD600 ~0.6), at which point isopropyl β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 0.2–0.5 mM, and the cultures were shaking (200 rpm) for a further 16 h at 16°C. The cells were harvested by centrifugation, sonicated and His 6-tagged recombinant protein were purified. Buffer for protein purification used in the present research is listed in Supplementary Table S9. Site-directed mutants were made using the Quikchange kit (Agilent), using primers shown in (Supplementary Table S7).
Polyclonal antibody preparation
All animal protocols were approved by Institute Animal Care and Use Committee at Shanghai Institute of Materia Medica (Ref. number 2019-02-YY-08). The polyclonal antibodies (Supplementary Table S10) were prepared following published procedures.Citation55 600 μg/mL purified recombinant proteins were mixed with an equal volume of Freund’s complete adjuvant (Sigma-Aldrich, St. Louis, MO, USA). Subsequently, 100 μg proteins in the adjuvant were injected subcutaneously into 6-week-old BALAB/c mice. Two weeks after the first injection, the mice were immunized twice at 1-week interval, using same amount of protein. Blood was collected at 7 days after the last injection and placed at room temperature for 1 h. The supernatant was collected via centrifugation at 2200 g for 20 min.
Protein cellar localization
Cellular localization of protein was determined by immunofluorescence and Western blotting as described previously.Citation56 In brief, BO cultures were grown overnight (OD600 ~0.6) in 5 mL MM containing 5 mg/mL cellobiose as final concentration. Cells were collected by centrifugation at 5,000 g for 10 min and resuspended in 2 mL of PBS. Proteinase K (0.5 mg/mL final concentration) was added to 1 mL of the suspension and the other half left untreated as control. Both samples were incubated at 37°C overnight followed by centrifugation at 5000 g for 10 min to collect cells. To eliminate residual proteinase K activity, cell pellets were resuspended in 1 mL of 1.5 M trichloroacetic acid and incubated on ice for 30 min. Precipitated mixtures were then centrifuged at 5000 g for 10 min and washed twice in 1 mL of ice-cold acetone (99.8%). The resulting pellets were allowed to dry in a 40°C heat block for 5 min and dissolved in 250 μL of Laemmli buffer. Samples were heated for 5 min at 98°C and mixed by pipetting several times before loading in 8–12% SDS-PAGE gels. Electrophoresed proteins were transferred to nitrocellulose membranes by Western blotting followed by immunochemical detection using primary mouse polyclonal antibodies (Eurogentec) generated against various proteins and secondary goat anti-mouse antibodies (Santa Cruz Biotechnology) (Supplementary Table S10).
Immunofluorescence microscopy
BO suspensions of mid-exponential phase cells (0.5 mL in PBS, produced as for Proteinase K treatment above) were fixed with an equal volume of 2 × formalin (9% formaldehyde in PBS), and rocked for 90 min at 25°C in Eppendorf tubes. The cells were then pelleted by centrifugation for 3 min at 8000 g and washed twice with 1 mL of 50 mM HEPES-Na buffer (pH 7.5). The bacterial cell pellet was resuspended in 1 mL of blocking solution (2% goat serum, 0.02% NaN3 in PBS) and incubated at 4°C for 16 h. After incubation, cells were centrifuged again at 8,000 g and the supernatants were discarded. For labeling, the bacteria were incubated with 0.5 mL of primary mouse IgG (1/500 dilution of IgG in 5% BSA powder in HEPES-Na buffer (pH 7.5), blocking solution) for 2 h at 25°C. The cells were then pelleted, washed in 1 mL of HEPES-Na buffer (pH 7.5) and resuspended in 0.4 mL goat anti-mouse IgG Alexa-Fluor 488 (Sigma-Aldrich), diluted 1/500 in blocking solution and incubated 1 h at 25°C in the dark. The cells were again pelleted, washed with 1 mL HEPES-Na buffer (pH 7.5), resuspended in 1 mL of 50 mM HEPES-Na buffer (pH 7.5) containing ProLong Gold antifade reagent (Life Technologies). Aliquots of labeled bacterial cells were mounted onto glass slides and stained by gram staining and secured with coverslips finally. Fluorescence was visualized using a laser scanning confocal microscope FV1000 (Olympus FV1000-SIM, Japan). Alexa-Fluor 488-labeled bacteria were visualized under a UV view and compared with bright-field phase-contrast of the same image.
HPAEC-PAD
Degradation products were detected by High-performance anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD) described as previously.Citation50 In brief, an analytical CARBOPACTM PA-100 anion-exchange column (Dionex) equipped with a guard column was used. The system had a loop size of 100 mL and was run at a flow rate of 1 mL/min. Carbohydrates were detected by pulsed amperometric detection (PAD) with electrode settings of E1 = + 0.05, E2 = + 0.6 and E3 = − 0.6. The elution conditions were 0–10 min, 100 mM NaOH; 10–25 min, 100 mM NaOH with a 0–75 mM sodium acetate gradient; and 25–35 min, 100 mM NaOH containing 500 mM sodium acetate, respectively. The standards used to identify the chromatographic peaks was glucose.
Protein structure predication
The predicted structure was performed by Alphafold2 based on the whole amino acids sequence of BACOVA_02626 and BACOVA_02630.Citation57 Here, three protein database including BFD, Uniref9 and Mgnify were selected for multiple sequence alignment.Citation58–61 Following, the monomer structure was predicted based on the inference of Alphafold2 to construct the 3D model and energy optimized.Citation58 After the predication of protein structure was finished, the possible small molecular binding site and amino acids were predicted based on the point-site.Citation62 Combined with the predicted binding site, the binding pattern of micromolecule and protein was predicted by Molecular docking algorithm (AutoDock Vina).Citation63 The most reasonable binding state for small molecules was calculated by OnionNet-SFCT.Citation64
Animal test
All animal protocols were approved by Institute Animal Care and Use Committee at Shanghai Institute of Materia Medica (Ref. number 2019-02-YY-08). Thirty 6-weeks-old specific pathogen free (SPF) male C57BL/6j mice were purchased from Shanghai Experimental Animal Research Center, China. After one weeks of adaptive feeding, mice were divided randomly into three groups named as normal, vehicle and cellobiose, respectively. For normal group, mice were treated with normal diet while mice in vehicle and cellobiose groups were feeding by 1% fiber supplementation diet (normally with 5% fiber supplementation). Mice in normal and vehicle group were treated with 200 µL normal saline by gavage while mice in Cellobiose group were oral treated cellobiose at dose 0.4 g/kg every other day. After eight weeks, all mice were sacrificed and fecal samples and colon tissue were collected for further analysis.
Tissue histology
Colon tissues were fixed in 10% neutral buffered-formalin, paraffin embedded, cut into 3–5 μm sections for H&E staining. TNF-α and IL-6 were detected by immunohistochemistry (IHC).
16S rRNA gene sequencing
For vehicle and cellobiose groups, the V3–V4 region of the bacterial 16S rRNA gene was amplified by PCR with barcode-indexed primers (338F and 806 R), using FastPfu Polymerase to analyze the composition of gut microbiota. Amplicons were then purified by gel extraction (AxyPrep DNA GelExtraction Kit, Axygen Biosciences, Union City, California, USA) and were quantified using QuantiFluor-ST (Promega, USA). The purified amplicons were pooled in equimolar concentrations, and paired-end sequencing was performed using an Illumina MiSeq instrument (Illumina, San Diego, California, USA).
Microbial analysis
The 16S rRNA sequencing data were processed using the Quantitative Insights to Microbial Ecology Platform (V.1.9.1). Sequencing reads were demultiplexed and filtered. Operational taxonomic units (OTUs) were picked at 97% similarity cutoff, and the identified taxonomy was then aligned using the Greengenes database (V.13.8). Chimeric sequences were identified and deleted. OTUs with numbers of sequences < 0.005% of the total number of sequences were removed from the OTUs table. After filtering, an average of 65,307 reads per sample was obtained. In addition, rarefaction was performed on the OTUs table to prevent methodological artifacts arising from varying sequencing depth. In efforts to dissect possible species for OTUs, we performed Mega BLAST search to align the reads of OTUs against reference sequences in the National Center for Biotechnology Information (NCBI) 16S rRNA database. All the data analysis were finished on line (https://cloud.majorbio.com/). Analysis software are listed in Supplementary Table S11.
Statistical analysis
GraphPad Prism 9.0 software (San Diego, CA, USA) was used to perform all the statistical analyses and made all the pictures. Image combination was performed by Adobe Illustrator 2020 software. All data are shown as means ± SD. Using unpaired two-tailed Student’s t-test for comparison or one-way ANOVA for multiple comparisons to measure p-value of the difference between groups. In addition, quantitative data were accorded with normal distribution and homogeneity of variance for ANOVA analysis.
Data statement
The data that support the findings of this study are available in Sequence Read Archive database (accession number PRJNA878605 for Transcriptomics and PRJNA877289 for 16S rRNA gene sequencing).
Author contributions
All authors have reviewed the final version of the manuscript and approved it for publication. This original manuscript has neither been published nor considering for publication in elsewhere. Among them, M.L. took the responsibility for all the experiments, design, data analysis and manuscript writing. Y.W. performed the Western blotting, qPCR, protein purification and antibody preparation. C.G. contributed to the preparation of antibody. S.W., Y.B., and L.Z. performed the protein structure predication and data analysis. K.D. supervised the whole project and revised the manuscript. All authors read and approved the final manuscript.
Supplemental Material
Download Zip (41.5 MB)Acknowledgments
We take the chance to thank Zhongshan Municipal Bureau of Science and Technology for their funding support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2227434
Additional information
Funding
References
- Golisch B, Lei Z, Tamura K, Brumer H. Configured for the human gut microbiota: molecular mechanisms of dietary β‑glucan utilization. ACS Chem Biol. 2021;16(11):2087–18. doi:10.1021/acschembio.1c00563.
- Macy JM, Farrand JR, Montgomery L. Cellulolytic and non-cellulolytic bacteria in rat gastrointestinal tracts. Appl Environ Microbiol. 1982;44(6):1428–1434. doi:10.1128/aem.44.6.1428-1434.1982.
- Varel VH, Fryda SJ, Robinson IM. Cellulolytic bacteria from pig large intestine. Appl Environ Microbiol. 1984;47(1):219–221. doi:10.1128/aem.47.1.219-221.1984.
- Varel VH, Pond WG. Characteristics of a new cellulolytic Clostridium sp. isolated from pig intestinal tract. Appl Environ Microbiol. 1992;58(5):1645–1649. doi:10.1128/aem.58.5.1645-1649.1992.
- Robert C, Del’Homme C, Bernalier-Donadille A. Interspecies H2 transfer in cellulose degradation between fibrolytic bacteria and H2-utilizing microorganisms from the human colon. FEMS Microbiol Lett. 2001;205(2):209–214. doi:10.1016/S0378-1097(01)00467-0.
- Robert C, Chassard C, Lawson PA, Bernalier-Donadille A. Bacteroides cellulosilyticus sp. nov., a cellulolytic bacterium from the human gut microbial community. Int J Syst Evol Microbiol. 2007;57(7):1516–1520. doi:10.1099/ijs.0.64998-0.
- Chassard C, Delmas E, Robert C, Bernalier- Donadille A. The cellulose-degrading microbial community of the human gut varies according to the presence or absence of methanogens. FEMS Microbiol Ecol. 2010;74(1):205–213. Available from. doi:10.1111/j.1574-6941.2010.00941.x.
- Chassard C, Delmas E, Robert C, Lawson PA, Bernalier-Donadille A. Ruminococcus champanellensis sp. nov., a cellulose-degrading bacterium from human gut microbiota. Int J Syst Evol Microbiol. 2012;62(1):138–143. doi:10.1099/ijs.0.027375-0.
- Julliand V, de Vaux A, Millet L, Fonty G. Identification of Ruminococcus flavefaciens as the predominant cellulolytic bacterial species of the equine cecum. Appl Environ Microbiol. 1999;65(8):3738–3741. doi:10.1128/AEM.65.8.3738-3741.1999.
- Katole SB, Das A, Agarwal N, Prakash B, Saha SK, Saini M, Sharma AK. Influence of work on nutrient utilisation in semicaptive Asian elephants (Elephas maximus). J Appl Anim Res. 2014;42(4):380–388. doi:10.1080/09712119.2013.875904.
- Varel VH, Tanner RS, Woese CR. Clostridium herbivorans sp. nov., a cellulolytic anaerobe from the pig intestine. Int J Syst Bacteriol. 1995;45(3):490–494. doi:10.1099/00207713-45-3-490.
- Shinkai T, Kobayashi Y. Localization of ruminal cellulolytic bacteria on plant fibrous materials as determined by fluorescence in situ hybridization and real-time PCR. Appl Environ Microbiol. 2007;73(5):1646–1652. doi:10.1128/AEM.01896-06.
- Flint HJ, Scott KP, Duncan SH, Louis P, Forano E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes. 2012;3(4):289–306. doi:10.4161/gmic.19897.
- Li H, Qu J, Li T, Wirth S, Zhang Y, Zhao X, Li X. Diet simplification selects for high gut microbial diversity and strong fermenting ability in high-altitude pikas. Appl Microbiol Biotechnol. 2018;105(15):6739–6751. doi:10.1007/s00253-018-9097-z.
- Montgomery L, Macy JM. Characterization of rat cecum cellulolytic bacteria. Appl Environ Microbiol. 1982;44(6):1435–1443. doi:10.1128/aem.44.6.1435-1443.1982.
- Bryant MP, Robinson IM, Chu H. Observations on the nutrition of Bacteroides succinogenes-A ruminal cellulolytic bacterium. J Dairy Sci. 1959;42(11):1831–1847. doi:10.3168/jds.S0022-0302(59)90815-X.
- Neumann AP, McCormick CA, Suen G. Fibrobacter communities in the gastrointestinal tracts of diverse hindgut-fermenting herbivores are distinct from those of the rumen. Environ Microbiol. 2017;19(9):3768–3783. doi:10.1111/1462-2920.13878.
- Amann RI, Lin C, Key R, Montgomery L, Stahl DA. Diversity Among Fibrobacter isolates: towards a phylogenetic classification. Syst Appl Microbiol. 1992;15(1):23–31. doi:10.1016/S0723-2020(11)80133-5.
- Cummings JH, Macfarlane GT. The control and consequences of bacterial fermentation in the human colon. J Appl Bacteriol. 1991;70(6):443–459. doi:10.1111/j.1365-2672.1991.tb02739.x.
- Wedekind KJ, Mansfield HR, Montgomery L. Enumeration and isolation of cellulolytic and hemicellulolytic bacteria from human feces. Appl Environ Microbiol. 1988;54(6):1530–1535. doi:10.1128/aem.54.6.1530-1535.1988.
- Chassard C, Delmas E, Robert C, Bernalier-Donadille A. The cellulose-degrading microbial community of the human gut varies according to the presence or absence of methanogens. FEMS Microbiol Ecol. 2010;74(1):205–213. doi:10.1111/j.1574-6941.2010.00941.x.
- Robert C, Bernalier-Donadille A. The cellulolytic microflora of the human colon: evidence of microcrystalline cellulose-degrading bacteria in methane-excreting subjects. FEMS Microbiol Ecol. 2003;46(1):81–89. doi:10.1016/S0168-6496(03)00207-1.
- Froidurot A, Julliand V. Cellulolytic bacteria in the large intestine of mammals. Gut Microbes. 2022;14(1):2031694. doi:10.1080/19490976.2022.2031694.
- Yeoman CJ, Fields CJ, Lepercq P, Ruiz P, Forano E, White BA, Mosoni P. In Vivo Competitions between Fibrobacter succinogenes, Ruminococcus flavefaciens, and Ruminococcus albus in a Gnotobiotic Sheep Model Revealed by Multi-Omic Analyses. mBio. 2021;12(2):e03533–3520. 10.1128/mBio.03533-20.
- Moraïs S, David YB, Bensoussan L, Duncan SH, Koropatkin NM, Martens EC, Flint HJ, Bayer EA. Cellulose degradation in the human gut: ruminococcus champanellensis expands the cellulosome paradigm. Environ Microbiol. 2016;18(2):542–556. doi:10.1111/1462-2920.13047.
- Violot S, Aghajari N, Czjzek M, Feller G, Sonan GK, Gouet P, Gerday C, Haser R, Receveur-Bréchot V. Structure of a full length psychrophilic cellulase from Pseudoalteromonas haloplanktis revealed by X-ray diffraction and small angle X-ray scattering. J Mol Biol. 2005;348(5):1211–1224. doi:10.1016/j.jmb.20-05.03.026.
- Zhu Y, Han L, Hefferon KL, Silvaggi NR, Wilson DB, McBride MJ, Spormann AM. Periplasmic cytophaga hutchinsonii endoglucanases are required for use of crystalline cellulose as the sole source of carbon and energy. Appl Environ Microbiol. 2016;82(15):4835–4845. doi:10.1128/AEM.01298-16.
- Cao J, Deng Q, Gao D, He B, Yin S, Qian L, Wang J, Wang Q. A novel bifunctional glucanase exhibiting high production of glucose and cellobiose from rumen bacterium. Int J Biol Macromol. 2021;173:136–145. doi:10.1016/j.ijbiomac.2021.01.113.
- Liu N, Li H, Chevrette MG, Zhang L, Cao L, Zhou H, Zhou X, Zhou Z, Pope PB, Currie CR, et al. Functional metagenomics reveals abundant polysaccharide degrading gene clusters and cellobiose utilization pathways within gut microbiota of a wood-feeding higher termite. Isme J. 2019;13(1):104–117. doi:10.1038/s41396-018-0255-1.
- Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, McNulty NP, Abbott DW, Henrissat B, Gilbert HJ, Bolam DN, et al. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 2011;9(12):e1001221. doi:10.1371/journal.pbi-o.1001221.
- Tamura K, Hemsworth GR, Déjean G, Rogers TE, Pudlo NA, Urs K, Jain N, Davies JG, Martens EC, Brumer H. Molecular mechanism by which prominent human gut Bacteroidetes utilize mixed-linkage beta-glucans, major health-promoting cereal polysaccharides. Cell Rep. 2017;21(2):417–430. doi:10.1016/j.celrep.2017.09.049.
- Grondin JM, Tamura K, Déjean G, Abbott DW, Brumer H, O’Toole G. Polysaccharide utilization loci: fueling microbial communities. J Bacteriol. 2017;199(15):e00860–16. doi:10.1128/J-B.00860-16.
- Li M, Li S, Guo X, Guo C, Wang Y, Du Z, Zhang Z, Xie C, Ding K. Discrete genetic loci in human gut Bacteroides thetaiotaomicron confer pectin metabolism. Carbohydr Polym. 2021;272:118534. doi:10.1016/j.carbpol.2021.1-18534.
- Ndeh D, Rogowski A, Cartmell A, Luis AS, Baslé A, Gray J, Venditto I, Briggs J, Zhang X, Labourel A, et al. Complex pectin metabolism by gut bacteria reveals novel catalytic functions. Nature. 2017;544:65–70. doi:10.1038/nature21725.
- Luis AS, Briggs J, Zhang X, Farnell B, Ndeh D, Labourel A, Baslé A, Cartmell A, Terrapon N, Stott K, et al. Dietary pectic glycans are degraded by coordinated enzyme pathways in human colonic Bacteroides. Nature Microbiol. 2018;3(2):210–219. doi:10.1038/s41564-017-0079-1.
- Qian K, Chen S, Wang J, Sheng K, Wang Y, Zhang M. A β-N-acetylhexosaminidase Amuc_2109 from Akkermansia muciniphila protects against dextran sulfate sodium-induced colitis in mice by enhancing intestinal barrier and modulating gut microbiota. Food Funct. 2022;13(4):2216–2227. doi:10.1039/d1fo04094d.
- Gao C, Major A, Rendon D, Lugo M, Jackson V, Shi Z, Mori-Akiyama Y, Versalovic J. Histamine H2 Receptor-Mediated Suppression of Intestinal Inflammation by Probiotic Lactobacillus reuteri. mBio. 2015;6(6):e01358–1315. doi:10.1128/mBio.01358-15.
- Alexander JK. Characteristics of cellobiose phosphorylase. J Bacteriol. 1961;81(6):903–910. doi:10.1128/jb.81.6.903-910.1961.
- Romano AH, Brino G, Peterkofsky A, Reizer J. Regulation of β-Galactoside transport and accumulation in heterofermentative lactic acid bacteria. J Bacteriol. 1987;169(12):5589–5596. doi:10.1128/jb.169.12.5589-5596.1987.
- Thompson J, Gentry-Weeks CR, Nguyen NY, Folk JE, Robrish SA. Purification from Fusobacterium mortiferum ATCC 25557 of a 6-phosphoryl-O-a-D-glucopyranosyl: 6-phosphoglucohydrolase that hydrolyzes maltose 6-phosphate and related phospho-a-D-glucosides. J Bacteriol. 1995;177(9):2505–2512. doi:10.1128/jb.177.9.2505-2512.1995.
- Garg R, Srivastava R, Brahma V, Verma L, Karthikeyan S, Sahni G. Biochemical and structural characterization of a novel halotolerant cellulase from soil metagenome. Sci Rep. 2016;6:39634. doi:10.1038/srep39634.
- Dutoit R, Delsaute M, Collet L, Wauven CV, Elder DV, Berlemont R, Richel A, Galleni M, Bauvois C. Crystal structure determination of Pseudomonas stutzeri A1501 endoglucanase Cel5A: the search for a molecular basis for glycosynthesis in GH5_5 enzymes. Acta Crystallogr D Struct Biol. 2019;75(Pt 6):605–615. doi:10.1107/S2059798319007113.
- Lombard V, Ramulu HG, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(D1):D490–D495. doi:10.1093/nar/gkt1178.
- Svartström O, Alneberg J, Terrapon N, Lombard V, Bruijn I, Malmsten J, Dalin AM, Muller EE, Shah P, Wilmes P, et al. Ninety-nine de novo assembled genomes from the moose (Alces alces) rumen microbiome provide new insights into microbial plant biomass degradation. Isme J. 2017;11(11):2538–2551. doi:10.1038/ismej.2017.108.
- Stewart RD, Auffret MD, Warr A, Wiser AH, Press MO, Langford KW, Liachko I, Snelling TG, Dewhurst RG, Walker AW. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat Commun. 2018;9(1):870. doi:10.1038/s41467-018-03317-6.
- Watanabe H, Tokuda G. Cellulolytic systems in insects. Annu Rev Entomol. 2010;55:609–632. doi:10.1146/annurev-ento-112408-085319.
- Kern M, McGeehan JE, Streeter SD, Martin RNA, Besser K, Elias L, Eborall W, Malyon GP, Payne CM, Himmel ME, et al. Structural characterization of a unique marine animal family 7 cellobiohydrolase suggests a mechanism of cellulase salt tolerance. Proc Natl Acad Sci U S A. 2013;110(25):10189–10194. doi:10.1073/pnas.1301502110.
- Fischer F, Romero R, Hellhund A, Linne U, Bertrams W, Pinkenburg O, Eldin HS, Binder K, Jacob R, Walker A, et al. Dietary cellulose induces anti-inflammatory immunity and transcriptional programs via maturation of the intestinal microbiota. Gut Microbes. 2022;12(1):1–17. doi:10.1080/19490976.2020.1829962.
- Meulen RV, Makras L, Verbrugghe K, Adriany T, Vuyst LD. In vitro kinetic analysis of oligofructose consumption by Bacteroides and Bifidobacterium spp. indicates different degradation mechanisms. Appl Environ Microbiol. 2006;72(2):1006–1012. doi:10.1128/AEM.72.2.1006-1012.2006.
- Wilson KH, Blitchington RB, Greene RC. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol. 1990;28(9):1942–1946. doi:10.1128/jcm.28.9.1942-1946.1990.
- Rakoff-Nahoum S, Coyne MJ, Comstock LE. An ecological network of polysaccharide utilization among human intestinal symbionts. Curr Biol. 2014;24(1):40–49. doi:10.1016/j.cub.2013.10.077.
- Leth ML, Ejby M, Workman C, Ewald DA, Pedersen SS, Sternberg C, Bahl ML, Licht TR, Aachmann FL, Westereng B, et al. Differential bacterial capture and transport preferences facilitate co-growth on dietary xylan in the human gut. Nature Microbiol. 2018;3(5):570–580. doi:10.1038/s41564-018-0132-8.
- Love ML, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8.
- Luis AS, Briggs J, Zhang XY, Farnell B, Ndeh D, Labourel A, Baslé A, Cartmell A, Terrapon N, Stott K, et al. Dietary pectic glycans are degraded by coordinated enzyme pathways in human colonic Bacteroides. Nature Microbiol. 2018;3(2):210–219. doi:10.1038/s41564-017-0079-1.
- Laverde D, Wobser D, Romero-Saavedra F, Hogendorf W, Marel G, Berthold M, Kropec A, Codee J, Huebner J, Miyaji EN. Synthetic teichoic acid conjugate vaccine against nosocomial Gram-positive bacteria. PLoS One. 2014;9(10):e110953. doi:10.1371/journal.pone.01109-53.
- Rogowski A, Briggs JA, Mortimer JC, Tryfona T, Terrapon N, Lowe EC, Baslé A, Morland C, Day AM, Zheng H, et al. Glycan complexity dictates microbial resource allocation in the large intestine. Nat Commun. 2015;6:7481. doi:10.1038/ncomms8481.
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–589. doi:10.1038/s41586-021-03819-2.
- Steinegger M, Mirdita M, Söding J. Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nat Methods. 2019;16(7):603–606. doi:10.1038/s41592-019-0437-4.
- Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23(10):1282–1288. doi:10.1093/bioinformatics/btm098.
- Mitchell AL, Almeida A, Beracochea M, Boland M, Burgin J, Cochrane G, Crusoe MR, Kale V, Potter SC, Richardson LJ. Mgnify: the microbiome analysis resource in 2020. Nucleic Acids Res. 2020;48(D1):D570–D578. doi:10.1093/nar/gkz1035.
- Zhang Y, Liu H, Yang S, Luo R, Chen HF. Well-balanced force field ff 03 CMAP for folded and disordered proteins. J Chem Theory Comput. 2019;15(12):6769–6780. doi:10.1021/acs.jctc.9b00623.
- Li Z, Yan X, Wei Q, Gao X, Wang S, Cui S. PointSite: a point cloud segmentation tool for identification of protein ligand binding atoms. J Chem Inf Model. 2022;62(11):2835–2845. doi:10.1021/acs.jcim.1c01512.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334.
- Zheng L, Meng J, Jiang K, Lan H, Wang Z, Lin M, Li W, Guo H, Wei Y, Mu Y. Improving protein–ligand docking and screening accuracies by incorporating a scoring function correction term. Brief Bioinform. 2022;23(3). bbac051. doi:10.1093/bib/bbac051.