
ABSTRACT
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis, an infectious disease with one of the highest morbidity and mortality rates worldwide. Leveraging its highly evolved repertoire of non-protein and protein virulence factors, Mtb invades through the airway, subverts host immunity, establishes its survival niche, and ultimately escapes in the setting of active disease to initiate another round of infection in a naive host. In this review, we will provide a concise synopsis of the infectious life cycle of Mtb and its clinical and epidemiologic significance. We will also take stock of its virulence factors and pathogenic mechanisms that modulate host immunity and facilitate its spread. Developing a greater understanding of the interface between Mtb virulence factors and host defences will enable progress toward improved vaccines and therapeutics to prevent and treat tuberculosis.
Introduction
Tuberculosis (TB) is a contagious infectious disease caused by the bacillus Mycobacterium tuberculosis (Mtb), a remarkably successful pathogen that primarily infects the lungs, leading to the classic syndrome of pulmonary TB. In addition, all other organs and tissues including the lymph nodes, brain, kidneys, and spine can be affected in a disorder called extrapulmonary TB [Citation1]. According to the World Health Organization (WHO) Global TB Report 2021 [Citation2], approximately 25% of the world’s population has immunologic evidence of prior infection with Mtb as determined by surveillance testing, and in 2020, 10 million people developed the active form of tuberculosis (ATB). TB represents one of the deadliest infections in the world, and, along with malaria and HIV/AIDS, has had the most significant socioeconomic impact on humanity [Citation3]. The most up-to-date mortality data indicate that in 2020, 1.4 million people died of TB, thus representing the second leading infectious cause of death globally after COVID-19 [Citation2,Citation4]. While many of these cases and deaths arise from primary tuberculosis occurring after an initial infection, latent TB infection (LTBI), in which the bacteria can remain quiescent for decades from the initial exposure without causing TB disease [Citation4–6], also accounts for a significant proportion of ATB. Indeed, 5–10% of individuals with LTBI subsequently develop ATB, and the risk of converting from latent to active disease increases substantially when individuals experience other medical conditions such as cancer, HIV/AIDS, diabetes, kidney failure, and viral coinfections such as with coronavirus. Children under 5 years of age are also at high risk of progression from LTBI to ATB [Citation7,Citation8].
For a microbe isolated and identified nearly 150 years ago that has caused immeasurable suffering and death, it is remarkable that an effective vaccine is not available. While vaccination with the Bacillus Calmette–Guérin (BCG) – the attenuated vaccine derived from M. bovis – protects against disseminated forms of TB in children, it does not prevent primary infection and does not provide consistent protection against infection in adults. Thus, the search for an improved vaccine able to prevent childhood and adult pulmonary TB remains critical [Citation9,Citation10]. Recent work demonstrating that intravenous vaccination of macaques with BCG afforded the animals a superior immune response and marked protection against subsequent airway infection with Mtb provides compelling evidence that the mammalian immune system can be harnessed to prevent Mtb infection [Citation11,Citation12].
As previously stated, a vast number of people demonstrate evidence of past or ongoing infection with Mtb. Currently, there are several methods of diagnosis, depending on whether active or latent TB is suspected. For diagnosis of LTBI or concern for ATB, current immunologic methods include the Mantoux Tuberculin skin test (TST) and whole-blood interferon-gamma (IFN-γ) release assays (IGRAs) [Citation13]. Both assays test for the presence of an amnestic cell mediated immune response. TST is an in vivo test performed through a subcutaneous injection of an antigen (tuberculin or purified protein derivative) in the lower part of the arm of the subject, who must return within 48–72 hours for determination of induration of the skin at the site of injection. While TST is an affordable, simple method to screen and detect the presence of cell mediated immunity to Mtb, it can also yield false-positive results in people vaccinated with BCG or false-negative results in immunosuppressed individuals [Citation8,Citation14,Citation15]. IGRAs are blood tests that measure levels of IFN-γ following in vitro stimulation of human lymphocytes with two Mtb virulence factors – the early secreting antigen target-6 (ESAT-6, EsxA) and culture filtrate protein-10 (CFP-10, EsxB). IGRAs demonstrate improved accuracy, sensitivity, and specificity in detecting Mtb infection, and in addition are not affected by prior BCG vaccination since the BCG strain lacks the EsxA and EsxB antigens and thus individuals vaccinated with BCG (but not infected with Mtb) should have no immunologic memory of exposure to EsxA and EsxB [Citation16,Citation17]. However, neither TST nor IGRAs can distinguish between ATB and LTBI or predict the risk of developing ATB in people with LTBI [Citation18,Citation19]. At the present time, treatment of individuals with LTBI (i.e. with no symptoms or clear signs of developing ATB) is recommended in order to reduce the risk of progression to ATB, especially in populations with a high risk of reactivation such as recent LTBI converters, immunocompromised patients living with HIV/AIDS, health care workers, and patients starting tumor necrosis factor-alpha (TNF-α) inhibitors [Citation20,Citation21]. Research in this area is vital to predict those who would most benefit from treatment (i.e. those with LTBI and a high risk of progression to ATB) and to identify those who have immunologic memory of disease but are not infected, and thus would not benefit from treatment.
To diagnose ATB, most of the world relies on established techniques such as sputum acid fast bacillus (AFB) staining and traditional culture techniques. These laborious and time-consuming assays are being replaced by more modern molecular tests. In particular, the GeneXpert nucleic acid-based test not only can detect Mtb in the sputum of patients with active disease but also can identify the presence of resistance to rifampicin, one of the essential first-line drugs for ATB treatment. Thus, where this test is available, patients with suspected ATB can receive a prompt molecular diagnosis and a more effective initial treatment for drug-resistant TB.
Antibiotics to treat TB have been available for over 50 years, but a major obstacle to successful therapy is that chemotherapy against TB requires long-term multidrug combinations and strict adherence to treatment. The current first-line chemotherapy for drug-susceptible TB involves a combination of rifampin (RIF), isoniazid (INH), pyrazinamide (PZA), and ethambutol (EMB) for the first 2 months, followed by 4 months of RIF and INH [Citation22–24]. Mechanisms of action of these drugs have been elucidated, demonstrating complex roles of drug activation and binding to targets in Mtb [Citation24]. TB can be cured over a 6-month period assuming the initial organism is drug susceptible and therapy is completed with adherence to a daily drug regimen. However, if these criteria are not met, i.e. if the initial organism is resistant to some of the drugs or if the patient is inconsistent with their therapy, cure can be significantly delayed. In fact, a significant threat to global TB control is the emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) TB, as MDR/XDR-TB treatments are more complicated, often requiring longer treatment regimens with second-line agents [Citation25]. MDR-TB is caused by Mtb strains resistant to INH/RIF and XDR-TB reflects resistance to INH/RIF plus any fluoroquinolone and at least one of the three second-line drugs, respectively [Citation26]. Although MDR and XDR-TB primarily emerge by the development of mutations in drug targets such as katG and/or inhA (INH resistance), rpoB (RIF resistance), pncA (PZA), and embCAB operon (EMB resistance) [Citation27–30], the host–pathogen dynamics underlying drug resistance are not fully understood, as mutations that confer antibiotic resistance in Mtb can evolve dynamically over time. Fortunately, owing to the recent development of several new classes of drugs against TB, therapeutic regimens for MDR and XDR-TB show promise [Citation31]. Nevertheless, further studies are needed to better understand the relationship between the development of drug resistance and virulence, as well as to expand our knowledge of the molecular basis of Mtb pathogenesis, thereby contributing to new treatments for TB.
Evolutionary and comparative analyses of Mtb strains have identified virulence factors essential for Mtb survival and propagation inside the host, as well candidate genes for the development of new vaccines and anti-TB drugs. Studies using Mtb transposon mutant libraries and mouse model populations such as The Collaborative Cross and Diversity Outbred (inbred and outbred mouse lines, respectively) have identified a large number of bacterial and host genes, in some cases dependent on host genotype, that impact Mtb survival in vivo [Citation32–34]. Mtb is currently divided into nine phylogeographically distinct lineages (L1-L9), widely distributed around the world [Citation35–37]. Paleomicrobiology studies have suggested that the interaction between mycobacteria and humans began approximately 73,000 years ago and that TB was carried out of Africa through migrations during the Neolithic period [Citation35,Citation38]. The successful, long-standing association between Mtb and humans may have helped shape the biology and epidemiology of TB, as evolutionary “modern” lineages appear to be correlated with less severe early inflammatory responses, more efficient transmission rates, and differences in the emergence of MDR/XDR-TB [Citation39–41].
The current review will summarize our knowledge of Mtb virulence factors, based on their molecular structures, host targets, and functions in pathogenesis. We have categorized them into two subgroups: non-protein and protein virulence factors. As the number of studies on the host–pathogen interactions in the context of Mtb is vast, it is outside the scope of this review to discuss all known virulence factors, and we regret omitting some topics due to space limitations. Before discussing the virulence factors themselves, we provide a brief overview of the life cycle of Mtb to offer broad context on their roles during infection. Our aim is to provide a framework to better understand the pathogenicity of one of the deadliest infectious agents in human history and highlight new potential targets for anti-TB therapy.
Overview of infection
Pulmonary TB is both the most common manifestation of TB and the mechanism of its transmission. Since Mtb can only survive and persist within living organisms – it does not appear to have an environmental niche [Citation35] – we consider the Mtb life cycle initiated when it reaches the airway and lung (). This first encounter between the airway and bacillus is also called primary infection.
Figure 1. Overview of Mtb infection. Mtb enters the human body through the airway where it engages the innate immune system within the alveolar space. Macrophages and dendritic cells ingest the bacteria, recruiting new cells and activating adaptive immunity. Together, the innate and adaptive immune systems collaborate to eradicate the bacteria or restrict its active replication within a granuloma. Active tuberculosis occurs either after primary infection or after reactivation due to immunodeficiency, leading to symptomatic disease and transmission to a new host to start a new infection cycle.
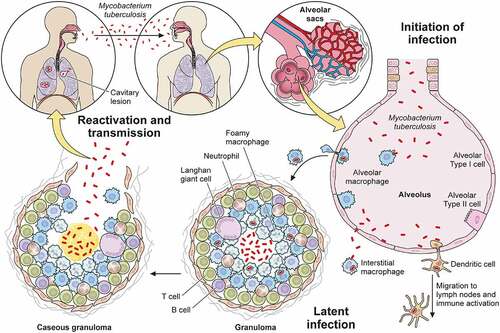
Mtb first enters through the nose or mouth, encounters cells in the upper airway, and in most cases passes into the distal lung to arrive in the alveolar space. To survive and establish infection, Mtb then invades beyond the mucosal or alveolar epithelium. As it flows through the upper and lower airways, Mtb can infect epithelial cells [Citation42] and microfold cells encountered in nasal associated mucosal tissue or bronchus-associated mucosal tissue [Citation43,Citation44] as an initial route to transit from the airway. Once the upper and larger distal airways are passed, the next site of interaction with the host occurs within the alveolus.
Because alveoli are continuously exposed to airborne particulates and pathogens, they also contain specialized innate immune cells known as alveolar macrophages that sample and engage with airborne antigens. In addition to alveolar macrophages, dendritic cells found within the interstitial space can also interact with airborne particles. Thus, during primary infection, Mtb that reaches the alveolus infects alveolar macrophages that reside in the alveolar space, as well as interstitial dendritic cells [Citation42,Citation45,Citation46]. As an alternative route of entry, type II alveolar epithelial cells can also be infected by Mtb [Citation47,Citation48]. These cells fail to control infection with associated high rates of cell death [Citation49]. Since alveolar epithelial cells vastly outnumber alveolar macrophages, this route may represent an important mechanism through which Mtb traverses the mucosa.
Mtb-infected alveolar macrophages and dendritic cells both serve as early reservoirs of infection and function to activate an adaptive immune response. Infected alveolar macrophages migrate from the alveolar sac into the interstitial space [Citation1]. In some circumstances, the infected macrophages will then reside within the interstitium [Citation50], and in other circumstances, infected macrophages in addition to infected dendritic cells migrate from the lung to draining lymph nodes to prime and activate T and B cells that function to limit progression of infection [Citation51–53]. Within the interstitium, resident interstitial macrophages engulf extracellular bacteria that escape initial phagocytosis or after their release from dying cells. Both types of infected macrophages – alveolar and interstitial – along with non-infected macrophages, inflammatory monocytes, neutrophils, and T cells recruited by the inflammation and tissue damage then form the characteristic TB granuloma. Whether this multicellular structure exists to limit or enhance Mtb infection is still unclear [Citation54,Citation55]. However, in most primary infections, infection is controlled, either through complete eradication of the bacteria leaving behind only immunologic memory of the interaction or through formation of a stable granuloma.
From the host’s perspective, a well-formed and stable granuloma limits the progression of infection and constrains any tissue damage to a small, well-circumscribed region. Most Mtb-infected individuals will contain the disease at this step and be asymptomatic. From the bacterial perspective, the granuloma permits the bacteria to maintain a state of dormancy and thus avoid clearance by the immune system [Citation56]. Thus, contained, Mtb may persist indefinitely. Aside from during autopsy or surgery where bacilli can be cultured from surreptitiously identified granuloma, this condition can currently only be diagnosed with TST or IGRA. Recently, some have questioned whether the vast numbers of individuals with LTBI, ~1/4 to 1/3 of the world’s population as estimated by the WHO, truly co-exist with viable organisms versus having cleared the infection and demonstrating an appropriate amnestic cell mediated immune response [Citation57,Citation58].
Notwithstanding the question of prevalence of “true” LTBI, some infected individuals proceed to active infection, either directly after the primary infection or after “reactivation” from latent infection, generally in the setting of immunosuppression [Citation59]. Often when this occurs, the structure of the granuloma changes and is associated with the presence of a material known as caseum which can facilitate more rapid Mtb growth. The central region of caseum can liquify, creating an even more favorable growth environment. Mtb can then spread beyond the lung to other parts of the body either via lymphatics [Citation60] or blood vessels [Citation61]. Ultimately, granuloma liquefaction, which is also associated with tissue destruction [Citation62], can facilitate the spread of tuberculosis to naive hosts through airborne transmission [Citation63,Citation64].
In the absence of treatment, ATB has a high mortality rate [Citation65]. During this phase, Mtb stimulates airway nociceptive neurons to produce a chronic, bloody cough that is one of the most characteristic symptoms of ATB disease. This persistent cough is also one vector of transmission that allows Mtb to escape and propagate infection [Citation63]. In this way, Mtb may renew its life cycle in a naive host.
Virulence factors
Considering the ability of Mtb to infect a variety of mammalian hosts [Citation66], manifest clinically in humans as both latent and active infection, and dwell in the air within motes of airborne particles to be transmitted to naive hosts, it comes as no surprise that it has evolved a variety of approaches to establish infection and survive within a variety of cells, tissues, and environments. Below we discuss the impact of these virulence traits, distinguishing between non-protein virulence factors such as lipids and sugars, and canonical proteinaceous virulence factors.
Non-protein virulence factors
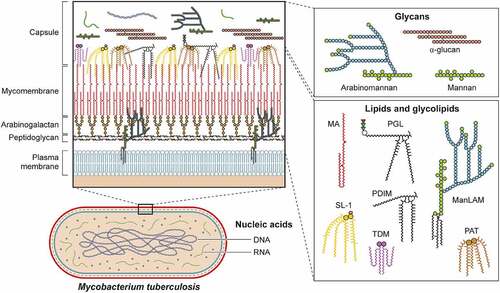
Though classically protein virulence factors have been the focus of microbiologists, organisms have evolved to leverage many other cellular components to facilitate their growth and survival within replicative niches. In this section, we discuss the functions of non-proteinaceous virulence factors organized by molecule class including lipids/glycolipids, glycans, nucleic acids, and metabolites. Many of these molecules are key components of the cell surface, and interact with host targets after secretion, shedding, or while attached to the bacterial surface. Indeed, mycobacterial and related species have a unique cell envelope with several components playing a role in virulence [Citation67,Citation68]. In addition to impacting the host through direct interactions, the structural rigidity of the mycobacterial cell envelope provides protection in the form of greater impermeability to antimicrobial molecules produced by the immune system the bacteria encounter in infection. Furthermore, the importance of the mycobacterial cell envelope to its survival in the host is highlighted by the fact that two of the first-line antibiotics to treat TB, isoniazid and ethambutol, target the biosynthesis of mycolic acids and arabinogalactan, arabinomannan, and lipoarabinomannan, respectively. The biosynthesis of cell surface molecules is discussed in great detail elsewhere [Citation69,Citation70], while here we focus on their roles in the context of host interactions [Citation69–72].
Lipids and glycolipids
Many bacteria are known to shed components of their outer membrane or cell wall, oftentimes having the capacity to dramatically impact host biology. The best studied of these, the lipopolysaccharide (LPS) of gram-negative bacteria, mediates a vast array of host responses through its binding to pattern recognition receptors such as toll-like receptor 4 (TLR4) on the cell surface and caspase 4/11 in the cytoplasm [Citation73]. In a similar vein, mycobacterial lipid and glycan molecules can either be shed or potentially transported directly into the host via secretion or membrane vesicles [Citation74–76]. As noted above, such molecules may also serve to buttress the mycobacterial cell wall and/or create impenetrable barriers to host defences [Citation77]. Although these activities are not mutually exclusive, below we focus on the virulence functions of these complex molecules ().
Table 1. Non-protein virulence factors of Mtb.
Mycolic acid
Mycolic acids, long chain fatty acids ranging between (C60-C90), are the primary constituents of the inner leaflet of the outer membrane. Mycolic acids are covalently linked to the arabinogalactan layer or are present as free mycolic acid within the capsule () [Citation134,Citation135]. This region is also known as the mycomembrane for the abundance of this lipid species. In alveolar epithelial cells, mycolic acids inhibit TLR2 leading to a decrease in IL-8 production [Citation79]. Free mycolic acids can activate the DAP12-associated triggering receptor expressed on myeloid cells 2 (TREM2) on innate immune cells, which in turn increases MCP-1 production and recruitment of Mtb permissive macrophages [Citation80]. Overproduction of MCP-1 correlates with susceptibility to ATB [Citation136]. Thus, regulation of inflammation via IL-8 and MCP-1 is but one of several mechanisms by which mycolic acids influence host immunity [Citation137]. That Mtb can control both the abundance and structure of mycolic acids in response to its environment [Citation81] suggests that temporal and spatial control of mycolic acids allows Mtb to fine-tune the host response during various stages of infection.
Figure 2. Non-protein virulence factors of Mtb. Schematic of Mtb lipids, glycolipids, glycans, and nucleic acids that contribute to virulence. TDM: trehalose dimycolate, SL-1: sulfolipid-1, PGL: phenolic glycolipid, MA: mycolic acid, PAT: pentaacyl trehalose, PDIM: phthiocerol dimycocerosates, ManLAM: mannosylated lipoarabinomannan.
Trehalose monomycolate and trehalose dimycolate
Trehalose monomycolate (TMM) and trehalose dimycolate (TDM) are abundant glycolipids formed by the conjugation of mycolic acids to the disaccharide trehalose via ester bonds [Citation138,Citation139]. TMM is a precursor to mycolic acid and presents within the cell envelope where it can be shed or secreted by Mtb by an unknown mechanism [Citation75]. TDM is also known as cord factor for mediating the formation of long filaments or “cords” when Mtb is extracellular. Because the interaction of TMM is similar to TDM, here we focus on the virulence capacity of TDM, with the recognition that TMM has overlapping activities. Monocyte-Inducible C-type Lectin (Mincle), a pattern recognition receptor (PRR) present in macrophages and dendritic cells, binds to the trehalose motif of TDM [Citation96,Citation140]. This interaction triggers SH2-domain-containing inositol polyphosphate 5’ phosphatase 1 (SHP-1) and Fc gamma receptor IIB (FcγRIIB) leading to inhibition of phagosome arrest [Citation90–92]. Mincle also activates the PI3K-AKT-GSK3 signaling pathway leading to the production of TNF-α, IL-6, and MCP-1 with subsequent recruitment of monocytes and neutrophils [Citation141–143]. Notably, by modifying TDM via enzymatic cyclopropanation, Mtb can temporally modulate the host immune response during infection [Citation144,Citation145].
Phthiocerol dimycocerosates
Another major class of lipids with diverse immunoregulatory activities are the phthiocerol dimycocerosates (PDIM) [Citation78]. Briefly, these lipid species are present on the outer leaflet of the outer membrane and may be secreted or shed from mycobacteria in a similar manner as described previously. While several glycolipids are known to interact with host factors (), PDIM is thought to mask pathogen-associated molecular patterns (PAMPs) of Mtb, allowing the bacteria to evade recognition by TLRs [Citation146,Citation147]. After phagocytosis, PDIM is suggested to have a role in membrane rupture including both phagosome and mitochondria membranes [Citation148,Citation149]. Though damaged phagosomes signal the host cell to initiate degradative autophagy, a cell-intrinsic host defence, PDIM also inhibits MyD88 signaling, leading to a decrease in autophagy [Citation150,Citation151].
Phenolic glycolipids
Phenolic Glycolipid (PGL), the covalent linkage of PDIM to a phenolic moiety and one to 4 sugars, is another important component of the mycobacterial outer membrane [Citation152]. While some epidemiologic evidence links PGL production to more “hypervirulent” lineage 2 strains of Mtb [Citation85–87] owing to a disruption of polyketide synthase gene pks15/1 essential for PGL production in lineage 4 Mtb strains [Citation153,Citation154], recent work has identified “ancient” or “ancestral” subspecies within lineage 2 strains that also contain mutations in pks15/1 [Citation155]. PGL activates the cytosolic surveillance pathway and induces MCP-1 secretion (also known as CCL2) [Citation156,Citation157]. CCL2 in turn interacts with host chemokine receptor 2 (CCR2) and recruits Mtb permissive macrophages [Citation146]. In support of this model, a human genetic polymorphism that increases MCP-1 production is associated with increased likelihood of developing pulmonary tuberculosis [Citation136]. However, CCR2 knockout mice are not more susceptible to infection by a lineage 4 Mtb strain, which genetically lacks PGL, and while CCR2 knockout mice do display enhanced susceptibility to a lineage 2 strain (HN878), this outcome is PGL independent [Citation157]. PGL may also modulate the immune system by antagonizing the function of TLR2 [Citation158], or by inhibiting inflammatory cytokine activity by interfering with T-cell receptor signaling [Citation88].
Polyacyl trehaloses
The disaccharide trehalose is a precursor for multiple Mtb glycolipids. In addition to the covalent attachment of mycolic acids as discussed above, trehalose can be acylated in a variety of positions [Citation159]. Here, we concentrate on the role of diacyl trehalose (DAT), because of its greater implication in Mtb virulence. When dendritic cells are exposed to DAT, production of pro-inflammatory interleukin-12 (IL-12) is downregulated, and production of the anti-inflammatory cytokine IL-10 is increased [Citation160]. In the context of macrophage infection with Mtb, both DAT and triacyl trehalose (TAT) downregulate expression of inducible nitric oxide synthase (iNOS), leading to reduced nitric oxide (NO) production [Citation161]. Like TDM, acyl trehaloses bind cell surface Mincle receptors, triggering a signaling cascade that increases production of TNF-α and other cytokines [Citation110,Citation111]. In parallel, DAT may mask the interaction of PRRs with other lipid virulence factors [Citation162,Citation163]. DAT may also impact adaptive immune responses to Mtb as it inhibits T cell proliferation and cytokine production through protein kinase C (PKC) activation and inhibition of mitogen-activated protein kinase (MAPK) [Citation164,Citation165]. Because DAT, TDM, and other lipids are presented by the MHC class I CD1 protein to initiate T-cell activation [Citation110], the ability of DAT to inhibit T cell activities may prevent a more robust anti-Mtb adaptive immune response.
Sulfolipids
While sharing structural similarity to tetra acyl trehalose, sulfolipid-1 (SL-1) has a sulphate group at the 2’ carbon of trehalose (). SL-1 is one of the most abundant lipids in Mtb and represents approximately 1% of its dry weight [Citation166]. SL-1 does not have a classic virulence function, as Mtb strains lacking SL-1 are not attenuated in mice or guinea pigs in traditional infection models [Citation167,Citation168]. However, recent work has shown that SL-1 is sufficient to activate nociceptive neurons from mice and humans and to stimulate a cough reflex in guinea pigs [Citation63]. SL-1 production is reduced during latent infection and upregulated during active infection [Citation169]. Taken together, Mtb may regulate SL-1 production to facilitate another critical stage of infection, namely, transmission.
Phosphatidylinositol mannosides/Lipomannan/Mannosylated lipoarabinomannan
The most abundant glycolipids present in the mycobacterial cell envelope are phosphatidyl-myo-inositol mannosides (PIMs) [Citation134,Citation170–172]. Acyl phosphatidyl-myo-inositol dimannoside (AcPIM2) comprises most of the inner membrane in most mycobacteria species [Citation134]. Furthermore, AcPIM6, one of the other major PIM species [Citation173,Citation174], helps maintain cell envelope integrity [Citation175]. The structure of lipomannan (LM) represents the decoration of PIM species with multiple mannose sugars [Citation174]. Finally, lipoarabinomannan (LAM) is formed by the attachment of multiple arabinose residues to LM, and when LAM is capped by another mannan group, forms a structure known as ManLAM (). Through its engagement of the scavenger receptor, CD36, ManLAM inhibits nitric oxide production [Citation176]. Recognized by their mannose residues, PIMs, LM, LAM, and ManLAM interact with several C-type lectin receptors including mannose receptor (MR), dendritic cell immunoactivating receptor (DCAR), and dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN, also known as CD209) [Citation105,Citation177]. C-type lectin receptors initiate phagocytosis in macrophages, dendritic cells, and neutrophils [Citation178], and these interactions may facilitate mycobacterial uptake by alveolar macrophages to provide an early replicative niche. In its most well-characterized activity, ManLAM protects Mtb from degradation in the phagolysosome of macrophages by arresting the maturation and acidification of phagosomes [Citation179–182] through both inhibition of phosphoinositide-3-kinase [Citation182,Citation183] and its interaction with lipid rafts leading to delayed phagosome-lysosome fusion [Citation184]. Thus, these phosphatidylinositol containing glycolipids can not only engage host receptors to facilitate Mtb uptake into host cells but also prevent their degradation by cell intrinsic immune mechanisms.
Glycans
Peptidoglycan and arabinogalactan within the cell envelope play a critical role for Mtb infection and the overall impermeability of its cell envelope [Citation71,Citation185,Citation186]. The glycans found within the 40 nm capsular space (), namely mannan, arabinomannan, and α-glucan [Citation101,Citation119,Citation187–190], play important roles in Mtb pathogenesis. It should be noted that when investigators grow and prepare Mtb for animal infection experiments, to prevent bacterial clumping, Mtb is often grown in Tween-80, which acts as both a nutrient and surfactant. However, Tween-80 also disrupts the Mtb capsule, thus making interpretation of experiments that characterize the role of the Mtb capsule in initial infection challenging [Citation187,Citation191–194]. Mannan and arabinomannan share structural similarities to LM and LAM, respectively. These glycans similarly manipulate the host, and since they form a part of the bacterial capsule, they are more likely to interact with host cells prior to LAM and LM [Citation114–117,Citation195]. α-Glucan is the major capsular glycan identified during most experimental conditions [Citation189,Citation196]. Highlighting its role in virulence, an α-glucan deficient Mtb strain is highly attenuated in a mouse model of infection [Citation119] possibly owing to its inability to modulate innate and adaptive immunity [Citation120,Citation197].
Nucleic acids
Through its specialized Type VII secretion system called ESX-1, Mtb can rupture or damage the phagosome and enter the cytosol [Citation198]. Once that occurs, mycobacterial DNA has been proposed to enter the cytosol to interact with cyclic GMP-AMP synthase (cGAS) [Citation122–125] though other studies have suggested that host mitochondrial DNA also acts as a cGAS ligand [Citation199]. Subsequent activation of STING via cGAS-derived cGAMP results in downstream signaling that leads to robust production of interferon-beta (IFN-β), which is detrimental to the host in the context of Mtb infection [Citation200,Citation201], potentially through decreased macrophage metabolism [Citation202]. In parallel, Mtb secretes RNA into the host cytoplasm to induce IFN-β via the host RNA sensing pathway mediated by RIG-I and MAVS [Citation126]. While Mtb may benefit from inducing excess IFN-β, activation of STING also induces autophagy, a host defence that can facilitate bacterial clearance [Citation203].
Protein virulence factors
Since Mtb is an obligate intracellular pathogen, it comes as no surprise that it has dedicated a significant proportion of its proteome to proteins that enhance its survival. In this section, we focus on several categories of Mtb protein virulence factors that directly impact the host, taking a predominantly bacterial-centered view. We group virulence determinants into the following categories based on their functions in the context of their interaction with the host: (i) modifiers of TLR2 activity, (ii) regulators of cytokine production, (iii) disruptors of phagosome function, and modulators of (iv) autophagy, and (v) cell death. Wherever possible, we highlight virulence protein activities () but also provide a summary table of proteins with known virulence function ().
Figure 3. Protein virulence factors of Mtb. Schematic representation of Mtb interactions within infected macrophages highlighting (i) toll-like receptors, (ii) phagosome arrest and escape, (iii) autophagy subversion, (iv) cell death modulation, and (v) cytokine production. Each box contains representative Mtb virulence factors that engage host cell biologic processes detailed in the text.
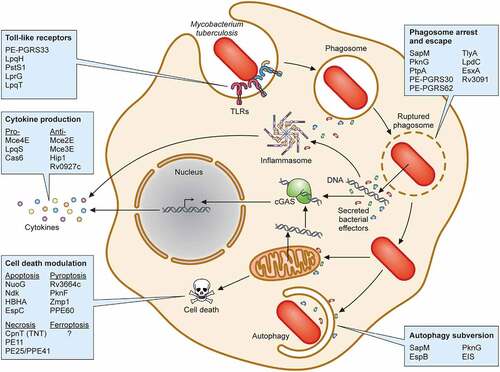
Table 2. Protein virulence factors of Mtb.
TLR2 activity modifiers
A myriad of host cell receptors can recognize Mtb cell wall ligands, including scavenger receptors, C-type lectin receptors, and TLRs [Citation313]. Among the TLRs, TLR2 plays a major role in detecting Mtb [Citation314,Citation315]. After TLR2 activation, the adapter protein myeloid differentiation primary response protein 88 (MyD88) is recruited and an intracellular signaling cascade is triggered [Citation316,Citation317]. Ultimately, this signaling pathway leads to the production of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-12, chemokines, and antimicrobial molecules (reactive oxygen intermediates (ROI) and reactive nitrogen intermediates (RNI)), which all play a role in controlling Mtb infection [Citation318,Citation319].
Considering its importance to the host, Mtb has developed multiple mechanisms to exploit TLR2 activation for its benefit. One mechanism involves leveraging the PE and PPE protein families, which are characterized by a conserved Pro-Glu and Pro-Pro-Glu motifs in their N-terminal region, respectively [Citation318]. Almost 10% of the Mtb genome is devoted to PE/PPE proteins [Citation320] and many members of these families interact with TLR2 to modulate the host innate immune response including PPE34, PE-PGRS11, PE-PGRS17, PE-PGRS33, PPE26, PPE57, and PPE60 [Citation220,Citation221,Citation223,Citation224,Citation226,Citation314]. The best characterized of these is PE-PGRS33, which interacts with TLR2 in a Ca2+-dependent manner [Citation206,Citation218] resulting in the secretion of pro-inflammatory cytokines and chemokines [Citation207,Citation321].
In addition to PE/PPE proteins, other Mtb virulence factors have been described as modulators of TLR2 signaling. Lipoproteins are both well-established TLR2 ligands and abundant components of the Mtb cell envelope. To that end, there are multiple Mtb lipoproteins that may engage and modulate TLR2 including LprG [Citation209], LpqH [Citation204], LpqT [Citation215] and PstS1 [Citation211]. Of note, the context of interaction may play an important role in the outcome of the response, such that free protein may cause a different response in the host than bacterial associated protein. Thus, when recombinant LprG [Citation209], LpqH [Citation204], and LpqT [Citation215] are added to macrophages, they activate pro-inflammatory cytokine secretion, inhibit antigen processing and IFN-γ-mediated MHC-II antigen presentation to T cells in a TLR-dependent manner. Likewise, PstS1 induces TNFα and IL-6 in primary human monocytes predominantly through TLR2 activation [Citation220]. However, experiments conducted using M. smegmatis expressing Mtb LpqT [Citation216] or LpqH [Citation322] yield conflicting results to those using purified proteins. For example, neither M. smegmatis expressing LpqT nor LpqH suppress macrophage MHC-II expression [Citation216,Citation322]. Both studies also showed that M. smegmatis expressing LpqT or LpqH decrease pro-inflammatory cytokine secretion [Citation216,Citation322], suggesting that they may act to dampen TLR2 activity. Taken together, it remains unclear how these and other Mtb lipoproteins impact TLR2 activity, and whether direct bacterial contact with innate immune cells impacts the TLR2 response. Indeed, it was demonstrated recently that phagosome membrane damage mediated by PDIM and ESX-1 are necessary to interfere with the host TLR2 response [Citation323], indicating that cytoplasmic access may be a critical requirement to impact the TLR2 signaling system.
Regulators of cytokine production
While modulating TLR signaling is one approach that Mtb takes to impact the host response and later cytokine production, Mtb uses other effector proteins to manipulate signaling pathways important for appropriately timed and regulated cytokine production. By inducing anti-inflammatory cytokines or suppressing pro-inflammatory cytokines, Mtb can fine-tune the host innate immune response to enhance its virulence.
The mammalian cell entry (Mce) operons are critical both for initial phagocytosis and manipulation of host cell cytokine responses [Citation230,Citation324–326]. Within these operons, several Mce lipoproteins are known to manipulate cytokine production via inhibition of phosphorylation cascades. For instance, Mce2E, also known as LprL, inhibits the activation of ERK and JNK MAPK signaling pathways and K48 ubiquitination of eEF1A1. Mce2E has a MAPK-docking motif that directly binds to mitogen-activated protein kinase 1 (MAPK1 or ERK2) and mitogen-activated protein kinase 8 (MAPK8 or JNK1), inhibiting their activation and resulting in decreased production of TNF-α and IL-6 [Citation228]. Similarly, another Mce (Mce3E or LprM) binds to MAPK1, inhibiting its phosphorylation and thus reducing production of TNF-α and IL-6 [Citation229]. Thus, a variety of Mce proteins function to suppress innate immune signaling pathways.
In addition to members of the Mce families, other Mtb virulence proteins modulate cytokine production. The serine hydrolase, Hip1, inhibits the production of several pro-inflammatory cytokines such as IL-12, IL-6, and TNF-α in dendritic cells [Citation241]. Likewise, Rv0927c, a short-chain dehydrogenase/reductase (Rv0927c) also downregulates TNF-α, IL-1β, and IL-6 production, by preventing NF-kB activation [Citation300,Citation327]. In contrast, another lipoprotein, LpqS stimulates TNF-α and IL-12 production [Citation234]. Recently, in an unexpected discovery, an Mtb Type III CRISPR/Cas system protein, Cas6, was demonstrated not only to be important for virulence of Mtb in a mouse model of disease but also a secreted virulence factor that induces IL-6, IL-1β, and TNF-α production via an unknown mechanism [Citation239]. This study, among others, indicates that CRISPR/Cas is not only a bacterial defence against bacteriophages but can also function as a virulence factor. Overall, that a wide array of Mtb virulence factors can impact host cytokine production, and in some cases have opposing effects on the same cytokine (), suggests that fine-tuned control of both pro- and anti- inflammatory cytokines at various stages of infection is critical for Mtb virulence.
Phagosome maturation arrest and phagosome escape
After recognition of Mtb ligands by host cell receptors on professional phagocytes, bacteria are internalized by phagocytosis and subsequently located within early phagosomes. During their maturation process, phagosomes acidify to pH ≤5 and fuse with lysosomes, forming phagolysosomes. In the absence of any intervention, the fate of these internalized organisms is killing by host antimicrobial processes and digestion by phagolysosomal hydrolases. However, Mtb has evolved sophisticated strategies to avoid the degradative phagolysosomal environment by delaying or inhibiting phagosome-lysosome maturation, and by mediating phagosome rupture and escape into the cytosol.
Several Mtb proteins are involved in arresting phagosome maturation. First identified in 2000, Mtb SapM is a phosphatase involved in phagosome-lysosome inhibition [Citation244]. SapM dephosphorylates phosphatidylinositol 3-phosphate (PI3P) present in the phagosome membrane leading to inhibition of phagosome-lysosome fusion in the murine macrophage cell line RAW 264.7 [Citation245] and PMA-differentiated human THP-1 macrophages (hereafter named THP-1 cells) [Citation246]. The secreted protein PknG also inhibits phagosome-lysosome fusion favoring bacterial survival inside THP-1 cells through its interaction with GDP-bound Rab7L1 (Rab29 in mouse) on the Golgi complex, blocking its transition to active Rab7L1-GTP and, consequently, inhibiting Rab7L1-GTP recruitment to phagosomes [Citation256]. Inhibition of Rab7L1-GTP recruitment to phagosomes blocks the maturation of phagosomes as evidenced by impaired recruitment of other phagolysosomal markers like early endosome autoantigen 1 (EEA1), Rab7, and LAMP2 [Citation256]. PknG has also been implicated in host immunity impairment [Citation257], LTBI reactivation [Citation328], and autophagic flux inhibition [Citation258]. Of note, both SapM and PknG are exported by the SecA2 protein export system [Citation247] and then likely enter the cytoplasm through EsxA mediated phagosome disruption. Another Mtb phosphatase that interferes with phagosome maturation is PtpA, which blocks phagosome-lysosome fusion via dephosphorylation of VPS33B, a host protein involved in intracellular vesicle trafficking [Citation253]. PtpA also binds subunit H of the macrophage vacuolar-H+ATPase (V-ATPase) protein complex, inhibiting phagosome acidification [Citation253,Citation329,Citation330]. Thus, targeting of V-ATPase by PtpA hampers the proper function of several host proteins thereby inhibiting phagosome acidification and ultimately favoring Mtb infection in THP-1 macrophages [Citation253]. PtpA activity is regulated by protein tyrosine kinase A (PtkA) and protein kinase A (PknA) through phosphorylation [Citation331]. Like PknG, PtpA impacts other host cell pathways, including (i) modulating the host immune response by co-opting host ubiquitin [Citation254], (ii) regulating expression of host genes [Citation332], and (iii) blocking host cell apoptosis via GSK3 dephosphorylation [Citation333]. Thus, Mtb secreted virulence factors with enzymatic activities like SapM, PknG and PtpA can be multifunctional.
In this vein, considering their high prevalence in the Mtb genome, among the multiple functions of PE/PPE proteins in host cells, they also participate in arresting the maturation of Mtb-containing phagosomes [Citation250]. For example, during macrophage infection, an Mtb mutant lacking PE-PGRS30 demonstrated increased colocalization with LAMP-1 compared to wild-type Mtb, suggesting that PE-PGRS30 inhibits phagosome-lysosome fusion [Citation250]. This mutant was also attenuated in a mouse infection model, highlighting its importance to Mtb virulence. Furthermore, when J774 macrophages are infected with M. smegmatis expressing Mtb PE-PGRS62, phagosome maturation is blocked through inhibition of Rab7 and LAMP-1 recruitment [Citation261].
Other Mtb proteins involved in phagosome maturation arrest are TlyA and LpdC. TlyA impairs phagosome maturation by reducing recruitment of Rab5, Rab7, and EEA1 to the phagosome [Citation334]. Rather than block recruitment of Rab proteins (a common feature of virulence factors), Mtb LpdC leads to retention of coronin-1 on the phagosome membrane [Citation265]. Coronin-1 activates the Ca2+-dependent phosphatase calcineurin and calcineurin activity blocks fusion of phagosomes with lysosomes [Citation335], suggesting a mechanism of action of LpdC in phagosome maturation arrest via coronin-1 and calcineurin.
Another strategy to subvert the host immune response is to cause phagosome rupture and escape into the cytosol (termed cytosolic translocation). ESAT-6 (EsxA) has been implicated in Mtb translocation to the cytosol [Citation198,Citation336,Citation337]. EsxA is secreted as a heterodimer with CFP-10 (EsxB) through the ESX-1 Type 7 secretion system [Citation75]. The Mtb genome encodes for 5 unique Type 7 secretion systems (ESX-1 to ESX-5), and among these, ESX-1 is the most studied. Several components and substrates of the ESX-1 secretion system are Mtb virulence factors (). It should also be noted that other Mtb secreted proteins not directly secreted via the ESX-1 machine can access the cytoplasm via the ruptured phagosome [Citation305] or because the bacteria now reside in the cytoplasm.
As the canonical virulence factor of the ESX-1 secretion system, EsxA has been implicated in a variety of virulence activities [Citation248]. EsxA can bind the cell surface of epithelial cells via laminin [Citation338], M cells via scavenger receptor B1 [Citation43] and macrophages via β2-microglobulin [Citation339] and TLR2 [Citation340] to mediate activities like cell entry [Citation43,Citation338] and immunomodulation [Citation339,Citation340]. Once Mtb is internalized, EsxA facilitates phagosome rupture [Citation341] which results in activation of the cytosolic surveillance pathway and induction of IFN-β [Citation342]. Membrane binding and lysis occur when EsxA dissociates from EsxB under acidic conditions [Citation343] typically found in late phagosomes, in a process requiring EsxA Nα-acetylation [Citation344]. EsxA membranolytic activity is enhanced by PDIM [Citation345] and can be modulated by single amino acid substitutions at glutamine 5 of the EsxA N-terminus [Citation249]. Interestingly, mutants in two conserved residues in the unstructured C-terminal tail of EsxA which are required for phagosome rupture and virulence maintain the ability to lyse acidified liposomes, suggesting that virulence activities and membrane lysis can be decoupled [Citation346].
While the majority of focus has been on the impact of Mtb ESX-1 and EsxA on membrane rupture, another Mtb protein, Rv3091, was recently shown to allow phagosome escape of an avirulent organism, M. smegmatis [Citation268]. Rv3091 is homologous to patatin-like phospholipases (PLPs) that are essential virulence factors of Rickettsia typhi [Citation347,Citation348] and Legionella pneumophila [Citation349,Citation350]. Indeed, Mtb Rv3091 has PLP activity [Citation268], suggesting that it may serve a similar function in Mtb pathogenesis as for R. typhi and L. pneumophila, though studies in Mtb have yet to be reported.
Subversion of host autophagy
Autophagy is an evolutionarily conserved process mediating degradation of intracellular organelles and proteins during cell differentiation and stress. Autophagy can be harnessed by eukaryotic cells as a host defence against intracellular pathogens and in this context is termed xenophagy. During xenophagy, intracellular organisms are ensnared within autophagosomes that eventually fuse with lysosomes forming autolysosomes [Citation203,Citation351]. The first report of a role for autophagy in mycobacterial pathogenesis showed that autophagy is engaged in macrophages in the context of infection with BCG [Citation352]. Subsequent results demonstrated that the Mtb ESX-1 machine (which is absent in the BCG strain [Citation353]) is necessary for autophagy activation [Citation124]. Importantly, Mtb xenophagy requires the activity of a variety of host molecules including cGAS/STING [Citation122–125] and the ubiquitin ligases Parkin [Citation354] and Smurf1 [Citation355] that target Mtb for degradation via ubiquitination. Ubiquitination of Mtb or Mtb-containing phagosomes leads to recruitment of autophagy adaptors such as p62 and NBR1 [Citation354,Citation355] which then mediate autophago(lyso)some formation [Citation124,Citation354–357]. In response, Mtb has evolved multiple strategies to counteract autophagic clearance during infection [Citation351], some of which are discussed below.
Because the autophagy process requires the concerted activity of several proteins involved in vesicular trafficking, Mtb virulence factors that target the autophagy machinery overlap with those involved in phagosome maturation arrest. For example, the Mtb proteins SapM and PknG, discussed above, also function to modulate autophagy. SapM interacts with host Rab7 to inhibit autophagosome-lysosome fusion [Citation358]. PknG has the unusual activity of both enhancing autophagy induction but inhibiting autophagosome maturation [Citation258]. It enhances autophagy induction by binding to the pleckstrin-homology domain of AKT, preventing activation of the PI3K-AKT-mTOR pathway and resulting in autophagy induction [Citation258]. Counteracting this activity, PknG also blocks RAB14-GTP hydrolysis preventing Mtb-containing autophagosomes from maturing in autolysosomes, with the net effect of blocking autophagic flux and improving bacterial survival [Citation258]. Highlighting the importance of the PI3K/AKT/mTOR pathway, another Mtb virulence factor known as EIS disrupts autophagy in macrophages [Citation270,Citation271] by indirectly impacting this pathway [Citation271]. EIS increases acetylation of histone H3, which up-regulates IL-10 expression, and IL-10 then activates the PI3K/AKT/mTOR pathway, thus suppressing autophagy. Finally, EspB, an ESX-1–secreted protein, suppresses IFN-γ-induced autophagy in mouse macrophages in part through the downregulation of IFN-γ-receptor [Citation273]. Taken together, the autophagy pathway appears to be a high-value target for Mtb.
Modulation of cell death pathways
Multicellular organisms are privileged in their ability to sacrifice individual cells for the greater good of the host. There exist several routes to cell death, including apoptosis, pyroptosis, necrosis, and ferroptosis, and Mtb, as an intracellular pathogen, has evolved ways to modulate these pathways. Given their importance to both host and pathogen, these pathways have been extensively studied. Much like the case of cytokine regulation, examples exist of Mtb virulence factors that either inhibit or induce cell death. Here, we will discuss some representative virulence factors that demonstrate the breadth of proteins that target these essential pathways. An exhaustive discussion is, however, beyond the scope of this review.
Apoptosis, or programmed cell death, can be detrimental to Mtb pathogenesis by eliminating its intracellular niche [Citation359] and facilitating antigen presentation by cross-presentation [Citation360]. Various Mtb virulence factors inhibit apoptosis, such as NuoG [Citation279], Ndk [Citation361], PknE [Citation282], Rv3655c [Citation281], Rv3654c [Citation281], and Rv3033 [Citation284]. Macrophages infected with Mycobacterium kansasii expressing NuoG had reduced apoptosis whereas infection with Mtb lacking nuoG demonstrated increased apoptosis [Citation279]. NuoG was further shown to inhibit the extrinsic apoptosis pathway mediated by TNF-α via neutralization of NOX2-derived ROS [Citation362]. In addition to macrophages, NuoG also inhibits apoptosis in neutrophils and dendritic cells hampering Mtb cell-to-cell spread and delaying the onset of adaptive immunity [Citation280]. Like NuoG, Ndk inhibits apoptosis in macrophages via a mechanism dependent on the neutralization of NOX2 activity [Citation361]. In contrast to the inhibitory roles ascribed to NuoG and Ndk, several virulence factors have been identified that induce apoptosis, including the heparin-binding hemagglutinin antigen (HBHA) [Citation274] and the ESX-1-secreted substrate protein EspC [Citation278]. Whether inhibition or activation of apoptosis by Mtb is more favorable is not yet clear, and may depend on the cell type infected, the growth phase of the bacteria or local environmental conditions.
Another form of cell death that appears to mainly restrict Mtb growth is pyroptosis. Pyroptosis is a programmed necrosis that occurs primarily in myeloid cells such as macrophages [Citation363] that is triggered by the caspase 1/4/5/11-dependent pathway, resulting in inflammasome activation and, consequently, the release of IL-1β and IL-18 [Citation364]. The Mtb protein Rv3364c, a serine protease inhibitor, translocates into the host cytosol where it inhibits cathepsin G activity. This interaction hampers caspase-1 activation and prevents macrophage pyroptosis [Citation293]. More recently, it was found that the Mtb secreted effector protein PknF, a serine/threonine phosphokinase, inhibits the NLRP3 inflammasome dampening pyroptosis activation independently of the ESX-1 secretion system [Citation294]. Another Mtb protein, the zinc metalloprotease Zmp1 may also function to inhibit pyroptosis by inhibiting inflammasome activation [Citation291], though another study did not demonstrate a role for Zmp1 independent of the ESX-1 effector EsxA [Citation365]. More recently, Zmp1 was shown to bind to a mitochondrial respiratory chain complex 1 protein, GRIM-19 [Citation366]. Consistent with the early finding that Zmp1 inhibits pyroptosis [Citation291], loss of GRIM-19 or expression of Zmp1 leads to loss of mitochondrial membrane potential, increased mitochondrial reactive oxygen species and NLRP3 activation [Citation366]. Much like its variable impact on apoptosis, Mtb can also induce pyroptosis in host cells to enhance its survival. For example, Mtb PPE60 induces pyroptosis in human THP-1 cells [Citation225]. In addition, Mtb causes plasma membrane damage in an ESX-1-dependent manner, triggering pyroptosis in THP-1 cells [Citation367]. These results suggest that pyroptosis induction in macrophages by Mtb could be advantageous to the bacteria because it can facilitate Mtb infection of neighboring, uninfected cells. To that end, it is tempting to hypothesize that Mtb temporally regulates host cell pyroptosis (and possibly other cell death pathways) to first establish an intracellular niche for replication by inhibiting pyroptosis (and cell death), and then once that is achieved, to then escape and spread by activating it.
Studies have demonstrated that induction of necrosis, an uncontrolled cell death, favors the survival of Mtb in the host cell [Citation368,Citation369]. CpnT, an outer membrane channel protein in Mtb, also secretes its C-terminal domain, known as tuberculosis necrotizing toxin (TNT) [Citation285,Citation286]. Once it reaches the host cytoplasm through the combined activities of Mtb ESX-1, ESX-2, ESX-4, and ESX-5 Type VII secretion systems [Citation370,Citation371], TNT hydrolyzes NAD+ and induces macrophage necroptosis with associated mitochondrial depolarization [Citation285,Citation372]. Curiously, while CpnT/TNT are sufficient to induce necroptosis in mouse and human macrophages, CpnT is not required for Mtb pathogenesis in C57BL/6 mice [Citation286]. Instead, it has been suggested that CpnT can participate in Mtb dissemination and reactivation in vivo [Citation286]. Another possible explanation is that Mtb uses redundant pathways to induce necrosis or necroptosis in the host [Citation75]. To that end, it has been reported that PE11 [Citation288–290] and the protein complex PE25/PPE41 [Citation373] induce necrosis in the host by unknown mechanisms.
It has also been demonstrated that ferroptosis, a type of regulated necrosis dependent on free iron and lipid peroxides accumulation, which is modulated by Mtb showed that ferroptosis induction favors Mtb replication in the host [Citation374]; however, no studies have described Mtb virulence factors responsible for inducing this cell death pathway yet.
Outlook
In 2015, the WHO launched the “End TB Strategy,” an ambitious plan to eliminate TB incidence by 90% and mortality by 95% by the year 2035 [Citation375]. However, the extended duration required for anti-TB chemotherapy and the emergence of MDR/XDR-TB pose significant challenges toward achieving those goals. Until tests with greater sensitivity and specificity to identify those with ATB or LTBI that will progress to ATB become available, TST and IGRA are the primary immunologic tests for exposure to Mtb. In addition, the 100-year-old BCG vaccine remains the only licensed TB vaccine [Citation376]. One hope is that the study of Mtb virulence mechanisms may provide new avenues for the development of anti-TB therapies and vaccines that will revolutionize the prevention, diagnosis, and treatment of TB to achieve the targets of the WHO End TB strategy.
Although improved diagnostics and chemotherapy are crucial to achieve the milestones proposed by the End TB Strategy, a better vaccine is urgently needed to prevent TB-dependent morbidity and mortality, especially in vulnerable populations at high risk of developing ATB. Currently, about 16 TB vaccine candidates are at various clinical trial phases, including live-attenuated Mtb, killed Mtb, viral vectored and protein/adjuvant-based candidates [Citation377]. A novel vaccine candidate M72/AS01E showed promising results in a Phase 2b trial after 3 years of follow-up in adults from South Africa, Kenya, and Zambia, with a vaccine efficacy in preventing ATB of 50%, the first time in a century that a vaccine against TB has had significant efficacy [Citation378]. Another vaccine candidate, MTBVAC, is based on an attenuated Mtb clinical isolate genetically engineered to stably delete the phoP and fadD26 genes, thus interfering with transcription, synthesis, and/or secretion of multiple virulence factors, including ESAT-6 and PDIM. In contrast to BCG, however, MTBVAC conserves most of the T cell epitopes described for TB including ESAT-6 and CFP-10 [Citation379,Citation380]. Preclinical studies in small animal models and BCG-naive adult rhesus macaques showed that MTBVAC is safe, with high protection against Mtb challenge when compared to BCG [Citation379,Citation381]. MTBVAC has successfully entered a multi-center Phase 3 efficacy trial in newborn babies in South Africa, Madagascar, and Senegal [Citation382]. Finally, after the remarkable success of the mRNA-containing lipid-particle SARS-CoV-2 vaccines, this promising platform has been adapted to TB vaccines with the intent to deliver mRNA molecules encoding Mtb surface molecules into host cells using lipid nanoparticles. Indeed, clinical trials of an mRNA vaccine for TB are set to begin this year, according to a BioNTech Press Release [Citation383]. Leveraging advances in our understanding of Mtb virulence and pathogenesis with new delivery and adjuvant technologies has the potential to dramatically alter the trajectory of TB prevention.
Newer TB antigen-based skin tests (TBSTs) have been developed to measure the immune response to Mtb specific antigens, many of which are also Mtb virulence factors. Emerging evidence suggests that TBSTs tests are cost-effective, accurate, offer similar specificity to IGRA, and may provide more reliable results in immunosuppressive patients, such as people living with HIV [Citation384]. Regarding IGRAs, other mycobacterial virulence factors can also be used to improve their specificity and sensitivity, including EspC (an ESX-1 substrate), EspF (an ESX-1 associated protein), and Rv2348-B (unknown function), to identify individuals that do not react to ESAT-6 or CFP-10 [Citation385]. Finally, as discussed above, immunologic-based tests cannot definitively identify individuals infected with living Mtb. Recently, novel glycan-based dyes [Citation386] and positron-emission-based probes [Citation387] that take advantage of the unique components of the mycobacterial cell wall have been developed that can visualize living Mtb in vitro or in vivo, respectively. These or other compounds targeting either bacterial growth-specific or host inflammation-specific processes [Citation388] hold promise as cutting-edge diagnostics to reduce diagnostic uncertainty and shorten treatment delays associated with our currently available tools.
Despite recent advances in TB drug development, additional antibiotics and shorter treatment regimens are necessary to prevent the emergence of drug-resistant strains. While traditional approaches toward developing directly acting antimicrobials have yielded several new classes of therapeutics [Citation31,Citation389], targeting bacterial virulence pathways and their host responses holds promise as the next frontier of Mtb drug development [Citation390]. Thus, targeting critical virulence lipids [Citation391] or virulence factors [Citation392] could yield unique therapeutics not identified by traditional methods. Alternatively, whether by enhancing autophagy [Citation393], reversing Mtb-induced blockade of phagosome maturation [Citation394] or balancing the host inflammatory response [Citation395], developing approaches that direct the host immune system to overcome its “fundamental immunodeficiency” [Citation396] that licenses Mtb to successfully replicate will help end the world’s longest-lasting endemic pandemic.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Pai M, Behr MA, Dowdy D, et al. Tuberculosis. Nat Rev Dis Primers. 2016;2:16076.
- World Health Organization. Global tuberculosis report 2021. 2021.
- Bourzac K. Infectious disease: beating the big three. Nature. 2014;507:S4–29.
- World Health Organization. Tuberculosis. 2021.
- Cohen A, Mathiasen VD, Schon T, et al. The global prevalence of latent tuberculosis: a systematic review and meta-analysis. Eur Respir J. 2019;54:1900655.
- Houben RM, Dodd PJ. The global burden of latent tuberculosis infection: a re-estimation using mathematical modelling. PLOS Med. 2016;13:e1002152.
- Chee CBE, Reves R, Zhang Y, et al. Latent tuberculosis infection: opportunities and challenges. Respirology. 2018;23:893–900.
- Gong W, Wu X. Differential diagnosis of latent tuberculosis infection and active tuberculosis: a key to a successful tuberculosis control strategy. Front Microbiol. 2021;12:745592.
- Fatima S, Kumari A, Das G, et al. Tuberculosis vaccine: a journey from BCG to present. Life Sci. 2020;252:117594.
- Dockrell HM, Smith SG. What have we learnt about BCG vaccination in the last 20 years? Front Immunol. 2017;8:1134.
- Darrah PA, Zeppa JJ, Maiello P, et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature. 2020;577:95–102.
- Irvine EB, O’Neil A, Darrah PA, et al. Robust IgM responses following intravenous vaccination with Bacille Calmette-Guerin associate with prevention of Mycobacterium tuberculosis infection in macaques. Nat Immunol. 2021;22:1515–1523.
- Pai M, Behr M. Latent mycobacterium tuberculosis infection and interferon-gamma release assays. Microbiol Spectr. 2016;4. DOI:10.1128/microbiolspec.TBTB2-0023-2016
- McShane H. Tuberculosis vaccines: beyond bacille Calmette-Guerin. Philos Trans R Soc Lond B Biol Sci. 2011;366:2782–2789.
- Farhat M, Greenaway C, Pai M, et al. False-positive tuberculin skin tests: what is the absolute effect of BCG and non-tuberculous mycobacteria? Int J Tuberc Lung Dis. 2006;10:1192–1204.
- Takwoingi Y, Whitworth H, Rees-Roberts M, et al. Interferon gamma release assays for Diagnostic Evaluation of Active tuberculosis (IDEA): test accuracy study and economic evaluation. Health Technol Assess. 2019;23:1–152.
- Pai M, Denkinger CM, Kik SV, et al. Gamma interferon release assays for detection of Mycobacterium tuberculosis infection. Clin Microbiol Rev. 2014;27:3–20.
- Carranza C, Pedraza-Sanchez S, de Oyarzabal-Mendez E, et al. Diagnosis for latent tuberculosis infection: new alternatives. Front Immunol. 2020;11:2006.
- Munoz L, Stagg HR, Abubakar I. Diagnosis and management of latent tuberculosis infection: table 1. Cold Spring Harb Perspect Med. 2015;5:a017830.
- Huaman MA, Sterling TR. Treatment of latent tuberculosis infection-an update. Clin Chest Med. 2019;40:839–848.
- Marx FM, Cohen T, Menzies NA, et al. Cost-effectiveness of post-treatment follow-up examinations and secondary prevention of tuberculosis in a high-incidence setting: a model-based analysis. Lancet Glob Health. 2020;8:e1223–33.
- Dela Cruz CS, Lyons PG, Pasnick S, et al. Treatment of drug-susceptible tuberculosis. Ann Am Thorac Soc. 2016;13:2060–2063.
- Nahid P, Dorman SE, Alipanah N, et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: treatment of drug-susceptible tuberculosis. Clin Infect Dis. 2016;63:e147–95.
- Peloquin CA, Davies GR. The treatment of tuberculosis. Clin Pharmacol Ther. 2021;110:1455–1466.
- Ramachandran G, Swaminathan S. Safety and tolerability profile of second-line anti-tuberculosis medications. Drug Saf. 2015;38:253–269.
- Quenard F, Fournier PE, Drancourt M, et al. Role of second-line injectable antituberculosis drugs in the treatment of MDR/XDR tuberculosis. Int J Antimicrob Agents. 2017;50:252–254.
- Heym B, Alzari PM, Honore N, et al. Missense mutations in the catalase-peroxidase gene, katG, are associated with isoniazid resistance in Mycobacterium tuberculosis. Mol Microbiol. 1995;15:235–245.
- Telenti A, Imboden P, Marchesi F, et al. Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet. 1993;341:647–650.
- Scorpio A, Lindholm-Levy P, Heifets L, et al. Characterization of pncA mutations in pyrazinamide-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1997;41:540–543.
- Parsons LM, Salfinger M, Clobridge A, et al. Phenotypic and molecular characterization of Mycobacterium tuberculosis isolates resistant to both isoniazid and ethambutol. Antimicrob Agents Chemother. 2005;49:2218–2225.
- Conradie F, Diacon AH, Ngubane N, et al. Treatment of highly drug-resistant pulmonary tuberculosis. N Engl J Med. 2020;382:893–902.
- Joshi SM, Pandey AK, Capite N, et al. Characterization of mycobacterial virulence genes through genetic interaction mapping. Proc Natl Acad Sci U S A. 2006;103:11760–11765.
- Smith CM, Baker RE, Proulx MK, et al. Host-pathogen genetic interactions underlie tuberculosis susceptibility in genetically diverse mice. Elife. 2022;11. DOI:10.7554/eLife.74419.
- Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A. 2003;100:12989–12994.
- Comas I, Coscolla M, Luo T, et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45:1176–1182.
- Coscolla M, Gagneux S, Menardo F, et al. Phylogenomics of Mycobacterium africanum reveals a new lineage and a complex evolutionary history. Microb Genom. 2021;7. DOI:10.1099/mgen.0.000477.
- Coll F, McNerney R, Guerra-Assuncao JA, et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat Commun. 2014;5:4812.
- Cardona PJ, Catala M, Prats C. Origin of tuberculosis in the Paleolithic predicts unprecedented population growth and female resistance. Sci Rep. 2020;10:42.
- Moller M, Hoal EG. Current findings, challenges and novel approaches in human genetic susceptibility to tuberculosis. Tuberculosis (Edinb). 2010;90:71–83.
- Portevin D, Gagneux S, Comas I, et al. Human macrophage responses to clinical isolates from the Mycobacterium tuberculosis complex discriminate between ancient and modern lineages. PLOS Pathog. 2011;7:e1001307.
- Napier G, Campino S, Merid Y, et al. Robust barcoding and identification of Mycobacterium tuberculosis lineages for epidemiological and clinical studies. Genome Med. 2020;12:114.
- Rodrigues TS, Conti BJ, Fraga-Silva TFC, et al. Interplay between alveolar epithelial and dendritic cells and Mycobacterium tuberculosis. J Leukocyte Biol. 2020;108:1139–1156.
- Khan HS, Nair VR, Ruhl CR, et al. Identification of scavenger receptor B1 as the airway microfold cell receptor for Mycobacterium tuberculosis. Elife. 2020;9. DOI:10.7554/eLife.52551.
- Nair VR, Franco LH, Zacharia VM, et al. Microfold cells actively translocate mycobacterium tuberculosis to initiate infection. Cell Rep. 2016;16:1253–1258.
- Killick KE, Ni Cheallaigh C, O’Farrelly C, et al. Receptor-mediated recognition of mycobacterial pathogens. Cell Microbiol. 2013;15:1484–1495.
- Cohen SB, Gern BH, Delahaye JL, et al. Alveolar macrophages provide an early mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe. 2018;24:439–46 e4.
- Bermudez LE, Goodman J. Mycobacterium tuberculosis invades and replicates within type II alveolar cells. Infect Immun. 1996;64:1400–1406.
- Hernandez-Pando R, Jeyanathan M, Mengistu G, et al. Persistence of DNA from Mycobacterium tuberculosis in superficially normal lung tissue during latent infection. Lancet. 2000;356:2133–2138.
- Danelishvili L, McGarvey J, Li YJ, et al. Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell Microbiol. 2003;5:649–660.
- Lovey A, Verma S, Kaipilyawar V, et al. Early alveolar macrophage response and IL-1R-dependent T cell priming determine transmissibility of Mycobacterium tuberculosis strains. Nat Commun. 2022;13:884.
- Urdahl KB, Shafiani S, Ernst JD. Initiation and regulation of T-cell responses in tuberculosis. Mucosal Immunol. 2011;4:288–293.
- Shaler CR, Horvath C, Lai R, et al. Understanding delayed T-cell priming, lung recruitment, and airway luminal T-cell responses in host defense against pulmonary tuberculosis. Clin Dev Immunol. 2012;2012:628293.
- Carpenter SM, Lu LL. Leveraging antibody, B cell and Fc receptor interactions to understand heterogeneous immune responses in tuberculosis. Front Immunol. 2022;13:830482.
- Cronan MR. In the thick of it: formation of the tuberculous granuloma and its effects on host and therapeutic responses. Front Immunol. 2022;13:820134.
- Cohen SB, Gern BH, Urdahl KB. The tuberculous granuloma and preexisting immunity. Annu Rev Immunol. 2022;40:589–614.
- Park HD, Guinn KM, Harrell MI, et al. Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol Microbiol. 2003;48:833–843.
- Behr MA, Kaufmann E, Duffin J, et al. Latent tuberculosis: two centuries of confusion. Am J Respir Crit Care Med. 2021;204:142–148.
- Behr MA, Edelstein PH, Ramakrishnan L. Is Mycobacterium tuberculosis infection life long? BMJ. 2019;367:l5770.
- Zumla A, Raviglione M, Hafner R, et al. Tuberculosis. N Engl J Med. 2013;368:745–755.
- Lerner TR, Queval CJ, Lai RP, et al. Mycobacterium tuberculosis cords within lymphatic endothelial cells to evade host immunity. JCI Insight. 2020;5. DOI:10.1172/jci.insight.136937.
- Barr DA, Schutz C, Balfour A, et al. Serial measurement of M. tuberculosis in blood from critically-ill patients with HIV-associated tuberculosis. EBioMedicine. 2022;78:103949.
- Ong CW, Elkington PT, Brilha S, et al. Neutrophil-Derived MMP-8 drives AMPK-dependent matrix destruction in human pulmonary tuberculosis. PLOS Pathog. 2015;11:e1004917.
- Ruhl CR, Pasko BL, Khan HS, et al. Mycobacterium tuberculosis Sulfolipid-1 activates nociceptive neurons and induces cough. Cell. 2020;181:293–305 e11.
- Shiloh MU. Mechanisms of mycobacterial transmission: how does Mycobacterium tuberculosis enter and escape from the human host. Future Microbiol. 2016;11:1503–1506.
- Haas MK, Belknap RW. Updates in the treatment of active and latent tuberculosis. Semin Respir Crit Care Med. 2018;39:297–309.
- Kock R, Michel AL, Yeboah-Manu D, et al. Zoonotic tuberculosis - the changing landscape. Int J Infect Dis. 2021;113 Suppl 1:S68–72.
- Zuber B, Chami M, Houssin C, et al. Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J Bacteriol. 2008;190:5672–5680.
- Peterson EJ, Bailo R, Rothchild AC, et al. Path-seq identifies an essential mycolate remodeling program for mycobacterial host adaptation. Mol Syst Biol. 2019;15:e8584.
- Batt SM, Minnikin DE, Besra GS. The thick waxy coat of mycobacteria, a protective layer against antibiotics and the host’s immune system. Biochem J. 2020;477:1983–2006.
- Rahlwes KC, Sparks IL, Morita YS. Cell walls and membranes of actinobacteria. Subcell Biochem. 2019;92:417–469.
- Dulberger CL, Rubin EJ, Boutte CC. The mycobacterial cell envelope - a moving target. Nat Rev Microbiol. 2020;18:47–59.
- Jackson M, Stevens CM, Zhang L, et al. Transporters involved in the biogenesis and functionalization of the mycobacterial cell envelope. Chem Rev. 2021;121:5124–5157.
- Zamyatina A, Heine H. Lipopolysaccharide recognition in the crossroads of TLR4 and Caspase-4/11 mediated inflammatory pathways. Front Immunol. 2020;11:585146.
- Layre E. Trafficking of mycobacterium tuberculosis envelope components and release within extracellular vesicles: host-pathogen interactions beyond the wall. Front Immunol. 2020;11:1230.
- Augenstreich J, Briken V. Host cell targets of released lipid and secreted protein effectors of mycobacterium tuberculosis. Front Cell Infect Microbiol. 2020;10:595029.
- Prados-Rosales R, Carreno LJ, Batista-Gonzalez A, et al. Mycobacterial membrane vesicles administered systemically in mice induce a protective immune response to surface compartments of Mycobacterium tuberculosis. MBio. 2014;5: e01921-14. DOI:10.1128/mBio.01921-14.
- Siegrist MS, Bertozzi CR. Mycobacterial lipid logic. Cell Host Microbe. 2014;15:1–2.
- Rens C, Chao JD, Sexton DL, et al. Roles for phthiocerol dimycocerosate lipids in Mycobacterium tuberculosis pathogenesis. Microbiology (Reading). 2021;167. DOI:10.1099/mic.0.001042
- Sequeira PC, Senaratne RH, Riley LW. Inhibition of toll-like receptor 2 (TLR-2)-mediated response in human alveolar epithelial cells by mycolic acids and Mycobacterium tuberculosis mce1 operon mutant. Pathog Dis. 2014;70:132–140.
- Iizasa E, Chuma Y, Uematsu T, et al. TREM2 is a receptor for non-glycosylated mycolic acids of mycobacteria that limits anti-mycobacterial macrophage activation. Nat Commun. 2021;12:2299.
- Sharma NK, Rathor N, Sinha R, et al. Expression of mycolic acid in response to stress and association with differential clinical manifestations of tuberculosis. Int J Mycobacteriol. 2019;8:237–243.
- Buter J, Cheng TY, Ghanem M, et al. Mycobacterium tuberculosis releases an antacid that remodels phagosomes. Nat Chem Biol. 2019;15:889–899.
- Ghanem M, Dube JY, Wang J, et al. Heterologous production of 1-Tuberculosinyladenosine in mycobacterium kansasii models pathoevolution towards the transcellular lifestyle of mycobacterium tuberculosis. MBio. 2020;11. DOI:10.1128/mBio.02645-20.
- Buter J, Heijnen D, Wan IC, et al. Stereoselective synthesis of 1-tuberculosinyl adenosine; a virulence factor of mycobacterium tuberculosis. J Org Chem. 2016;81:6686–6696.
- Sinsimer D, Huet G, Manca C, et al. The phenolic glycolipid of Mycobacterium tuberculosis differentially modulates the early host cytokine response but does not in itself confer hypervirulence. Infect Immun. 2008;76:3027–3036.
- Constant P, Perez E, Malaga W, et al. Role of the pks15/1 gene in the biosynthesis of phenolglycolipids in the Mycobacterium tuberculosis complex. Evidence that all strains synthesize glycosylated p-hydroxybenzoic methyl esters and that strains devoid of phenolglycolipids harbor a frameshift mutation in the pks15/1 gene. J Biol Chem. 2002;277:38148–38158.
- Reed MB, Domenech P, Manca C, et al. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. 2004;431:84–87.
- Dagur PK, Sharma B, Upadhyay R, et al. Phenolic-glycolipid-1 and lipoarabinomannan preferentially modulate TCR- and CD28-triggered proximal biochemical events, leading to T-cell unresponsiveness in mycobacterial diseases. Lipids Health Dis. 2012;11:119.
- Mishra M, Adhyapak P, Dadhich R, et al. Dynamic remodeling of the host cell membrane by virulent mycobacterial sulfoglycolipid-1. Sci Rep. 2019;9:12844.
- Patin EC, Geffken AC, Willcocks S, et al. Trehalose dimycolate interferes with FcgammaR-mediated phagosome maturation through Mincle, SHP-1 and FcgammaRIIB signalling. PLoS ONE. 2017;12:e0174973.
- Bowker N, Salie M, Schurz H, et al. Polymorphisms in the pattern recognition receptor mincle gene (CLEC4E) and association with tuberculosis. Lung. 2016;194:763–767.
- Huber A, Kallerup RS, Korsholm KS, et al. Trehalose diester glycolipids are superior to the monoesters in binding to Mincle, activation of macrophages in vitro and adjuvant activity in vivo. Innate Immun. 2016;22:405–418.
- Ishikawa E, Ishikawa T, Morita YS, et al. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med. 2009;206:2879–2888.
- Miyake Y, Toyonaga K, Mori D, et al. C-type lectin MCL is an FcRgamma-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity. 2013;38:1050–1062.
- Walton EM, Cronan MR, Cambier CJ, et al. Cyclopropane modification of trehalose dimycolate drives granuloma angiogenesis and mycobacterial growth through VEGF signaling. Cell Host Microbe. 2018;24:514–25 e6.
- Feinberg H, Jegouzo SA, Rowntree TJ, et al. Mechanism for recognition of an unusual mycobacterial glycolipid by the macrophage receptor mincle. J Biol Chem. 2013;288:28457–28465.
- Feinberg H, Rambaruth ND, Jegouzo SA, et al. Binding sites for acylated trehalose analogs of glycolipid ligands on an extended carbohydrate recognition domain of the macrophage receptor mincle. J Biol Chem. 2016;291:21222–21233.
- Furukawa A, Kamishikiryo J, Mori D, et al. Structural analysis for glycolipid recognition by the C-type lectins Mincle and MCL. Proc Natl Acad Sci U S A. 2013;110:17438–17443.
- Doz E, Rose S, Nigou J, et al. Acylation determines the toll-like receptor (TLR)-dependent positive versus TLR2-, mannose receptor-, and SIGNR1-independent negative regulation of pro-inflammatory cytokines by mycobacterial lipomannan. J Biol Chem. 2007;282:26014–26025.
- Puissegur MP, Lay G, Gilleron M, et al. Mycobacterial lipomannan induces granuloma macrophage fusion via a TLR2-dependent, ADAM9- and beta 1 integrin-mediated pathway. J Immunol. 2007;178:3161–3169.
- Kalscheuer R, Palacios A, Anso I, et al. The Mycobacterium tuberculosis capsule: a cell structure with key implications in pathogenesis. Biochem J. 2019;476:1995–2016.
- Yuan C, Qu ZL, Tang XL, et al. Mycobacterium tuberculosis mannose-capped lipoarabinomannan induces IL-10-producing B cells and hinders CD4(+)Th1 immunity. iScience. 2019;11:13–30.
- Maeda N, Nigou J, Herrmann JL, et al. The cell surface receptor DC-SIGN discriminates between Mycobacterium species through selective recognition of the mannose caps on lipoarabinomannan. J Biol Chem. 2003;278:5513–5516.
- Stoop EJ, Mishra AK, Driessen NN, et al. Mannan core branching of lipo(arabino)mannan is required for mycobacterial virulence in the context of innate immunity. Cell Microbiol. 2013;15:2093–2108.
- Toyonaga K, Torigoe S, Motomura Y, et al. C-Type lectin receptor DCAR recognizes mycobacterial phosphatidyl-inositol mannosides to promote a Th1 response during infection. Immunity. 2016;45:1245–1257.
- Lugo-Villarino G, Troegeler A, Balboa L, et al. The C-Type lectin receptor DC-SIGN has an anti-inflammatory role in human M(IL-4) macrophages in response to mycobacterium tuberculosis. Front Immunol. 2018;9:1123.
- Driessen NN, Ummels R, Maaskant JJ, et al. Role of phosphatidylinositol mannosides in the interaction between mycobacteria and DC-SIGN. Infect Immun. 2009;77:4538–4547.
- Doz E, Rose S, Court N, et al. Mycobacterial phosphatidylinositol mannosides negatively regulate host Toll-like receptor 4, MyD88-dependent proinflammatory cytokines, and TRIF-dependent co-stimulatory molecule expression. J Biol Chem. 2009;284:23187–23196.
- Torrelles JB, Azad AK, Schlesinger LS. Fine discrimination in the recognition of individual species of phosphatidyl-myo-inositol mannosides from Mycobacterium tuberculosis by C-type lectin pattern recognition receptors. J Immunol. 2006;177:1805–1816.
- Reijneveld JF, Holzheimer M, Young DC, et al. Synthetic mycobacterial diacyl trehaloses reveal differential recognition by human T cell receptors and the C-type lectin Mincle. Sci Rep. 2021;11:1.
- Holzheimer M, Reijneveld JF, Ramnarine AK, et al. Asymmetric total synthesis of mycobacterial diacyl trehaloses demonstrates a role for lipid structure in immunogenicity. ACS Chem Biol. 2020;15:1835–1841.
- Daffe M, Lacave C, Laneelle MA, et al. Polyphthienoyl trehalose, glycolipids specific for virulent strains of the tubercle bacillus. Eur J Biochem. 1988;172:579–584.
- Decout A, Silva-Gomes S, Drocourt D, et al. Rational design of adjuvants targeting the C-type lectin Mincle. Proc Natl Acad Sci U S A. 2017;114:2675–2680.
- Glatman-Freedman A, Casadevall A, Dai Z, et al. Antigenic evidence of prevalence and diversity of Mycobacterium tuberculosis arabinomannan. J Clin Microbiol. 2004;42:3225–3231.
- Kang PB, Azad AK, Torrelles JB, et al. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J Exp Med. 2005;202:987–999.
- Rajaram MVS, Arnett E, Azad AK, et al. M. tuberculosis-initiated human mannose receptor signaling regulates macrophage recognition and vesicle trafficking by FcRgamma-Chain, Grb2, and SHP-1. Cell Rep. 2017;21:126–140.
- Taylor ME, Drickamer K. Structural requirements for high affinity binding of complex ligands by the macrophage mannose receptor. J Biol Chem. 1993;268:399–404.
- Ishida E, Corrigan DT, Malonis RJ, et al. Monoclonal antibodies from humans with Mycobacterium tuberculosis exposure or latent infection recognize distinct arabinomannan epitopes. Commun Biol. 2021;4:1181.
- Koliwer-Brandl H, Syson K, van de Weerd R, et al. Metabolic network for the biosynthesis of intra- and extracellular alpha-glucans required for virulence of mycobacterium tuberculosis. PLOS Pathog. 2016;12:e1005768.
- Geurtsen J, Chedammi S, Mesters J, et al. Identification of mycobacterial alpha-glucan as a novel ligand for DC-SIGN: involvement of mycobacterial capsular polysaccharides in host immune modulation. J Immunol. 2009;183:5221–5231.
- Agrawal P, Gupta P, Swaminathan K, et al. Alpha-Glucan pathway as a novel Mtb drug target: structural insights and cues for polypharmcological targeting of GlgB and GlgE. Curr Med Chem. 2014;21:4074–4084.
- Wassermann R, Gulen MF, Sala C, et al. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe. 2015;17:799–810.
- Watson RO, Bell SL, MacDuff DA, et al. The cytosolic sensor cGAS detects mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe. 2015;17:811–819.
- Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012;150:803–815.
- Collins AC, Cai H, Li T, et al. Cyclic GMP-AMP synthase is an innate immune DNA sensor for mycobacterium tuberculosis. Cell Host Microbe. 2015;17:820–828.
- Cheng Y, Schorey JS. Mycobacterium tuberculosis-induced IFN-beta production requires cytosolic DNA and RNA sensing pathways. J Exp Med. 2018;215:2919–2935.
- Chang DPS, Guan XL. Metabolic versatility of mycobacterium tuberculosis during infection and dormancy. Metabolites. 2021;11:11.
- Ehrt S, Schnappinger D, Rhee KY. Metabolic principles of persistence and pathogenicity in Mycobacterium tuberculosis. Nat Rev Microbiol. 2018;16:496–507.
- Eoh H, Wang Z, Layre E, et al. Metabolic anticipation in Mycobacterium tuberculosis. Nat Microbiol. 2017;2:17084.
- Fineran P, Lloyd-Evans E, Lack NA, et al. Pathogenic mycobacteria achieve cellular persistence by inhibiting the Niemann-Pick Type C disease cellular pathway. Wellcome Open Res. 2016;1:18.
- Baek SH, Li AH, Sassetti CM. Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol. 2011;9:e1001065.
- Lau SK, Lam CW, Curreem SO, et al. Identification of specific metabolites in culture supernatant of Mycobacterium tuberculosis using metabolomics: exploration of potential biomarkers. Emerg Microbes Infect. 2015;4:e6.
- Collins JM, Walker DI, Jones DP, et al. High-resolution plasma metabolomics analysis to detect Mycobacterium tuberculosis-associated metabolites that distinguish active pulmonary tuberculosis in humans. PLoS ONE. 2018;13:e0205398.
- Bansal-Mutalik R, Nikaido H. Mycobacterial outer membrane is a lipid bilayer and the inner membrane is unusually rich in diacyl phosphatidylinositol dimannosides. Proc Natl Acad Sci U S A. 2014;111:4958–4963.
- Ojha AK, Baughn AD, Sambandan D, et al. Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol Microbiol. 2008;69:164–174.
- Flores-Villanueva PO, Ruiz-Morales JA, Song CH, et al. A functional promoter polymorphism in monocyte chemoattractant protein-1 is associated with increased susceptibility to pulmonary tuberculosis. J Exp Med. 2005;202:1649–1658.
- Moopanar K, Mvubu NE. Lineage-specific differences in lipid metabolism and its impact on clinical strains of Mycobacterium tuberculosis. Microb Pathog. 2020;146:104250.
- Fujita Y, Naka T, Doi T, et al. Direct molecular mass determination of trehalose monomycolate from 11 species of mycobacteria by MALDI-TOF mass spectrometry. Microbiology (Reading). 2005;151:1443–1452.
- Fujita Y, Naka T, McNeil MR, et al. Intact molecular characterization of cord factor (trehalose 6,6’-dimycolate) from nine species of mycobacteria by MALDI-TOF mass spectrometry. Microbiology (Reading). 2005;151:3403–3416.
- Matsunaga I, Moody DB. Mincle is a long sought receptor for mycobacterial cord factor. J Exp Med. 2009;206:2865–2868.
- Lee WB, Kang JS, Yan JJ, et al. Neutrophils promote mycobacterial trehalose dimycolate-induced lung inflammation via the mincle pathway. PLOS Pathog. 2012;8:e1002614.
- Hansen M, Peltier J, Killy B, et al. Macrophage phosphoproteome analysis reveals MINCLE-dependent and -independent mycobacterial cord factor signaling. Mol Cell Proteomics. 2019;18:669–685.
- Wells CA, Salvage-Jones JA, Li X, et al. The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J Immunol. 2008;180:7404–7413.
- Rao V, Fujiwara N, Porcelli SA, et al. Mycobacterium tuberculosis controls host innate immune activation through cyclopropane modification of a glycolipid effector molecule. J Exp Med. 2005;201:535–543.
- Rao V, Gao F, Chen B, et al. Trans-cyclopropanation of mycolic acids on trehalose dimycolate suppresses Mycobacterium tuberculosis -induced inflammation and virulence. J Clin Invest. 2006;116:1660–1667.
- Cambier CJ, Takaki KK, Larson RP, et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature. 2014;505:218–222.
- Day TA, Mittler JE, Nixon MR, et al. Mycobacterium tuberculosis strains lacking surface lipid phthiocerol dimycocerosate are susceptible to killing by an early innate host response. Infect Immun. 2014;82:5214–5222.
- Augenstreich J, Arbues A, Simeone R, et al. ESX-1 and phthiocerol dimycocerosates of Mycobacterium tuberculosis act in concert to cause phagosomal rupture and host cell apoptosis. Cell Microbiol. 2017;19:e12726.
- Astarie-Dequeker C, Le Guyader L, Malaga W, et al. Phthiocerol dimycocerosates of M. tuberculosis participate in macrophage invasion by inducing changes in the organization of plasma membrane lipids. PLOS Pathog. 2009;5:e1000289.
- Bah A, Sanicas M, Nigou J, et al. The lipid virulence factors of mycobacterium tuberculosis exert multilayered control over autophagy-related pathways in infected human macrophages. Cells. 2020;9:666.
- Quigley J, Hughitt VK, Velikovsky CA, et al. The cell wall lipid PDIM contributes to phagosomal escape and host cell exit of mycobacterium tuberculosis. MBio. 2017;8. DOI:10.1128/mBio.00148-17
- Barnes DD, Lundahl MLE, Lavelle EC, et al. The emergence of phenolic glycans as virulence factors in mycobacterium tuberculosis. ACS Chem Biol. 2017;12:1969–1979.
- Brites D, Gagneux S. The nature and evolution of genomic diversity in the mycobacterium tuberculosis complex. Adv Exp Med Biol. 2017;1019:1–26.
- Gagneux S. Host-pathogen coevolution in human tuberculosis. Philos Trans R Soc Lond B Biol Sci. 2012;367:850–859.
- Carey AF, Wang X, Cicchetti N, et al. Multiplexed strain phenotyping defines consequences of genetic diversity in mycobacterium tuberculosis for infection and vaccination outcomes. mSystems. 2022;7:e0011022.
- Cambier CJ, O’Leary SM, O’Sullivan MP, et al. Phenolic glycolipid facilitates mycobacterial escape from microbicidal tissue-resident macrophages. Immunity. 2017;47:552–65 e4.
- Dunlap MD, Howard N, Das S, et al. A novel role for C-C motif chemokine receptor 2 during infection with hypervirulent Mycobacterium tuberculosis. Mucosal Immunol. 2018;11:1727–1742.
- Elsaidi HR, Lowary TL. Inhibition of cytokine release by mycobacterium tuberculosis phenolic glycolipid analogues. Chembiochem. 2014;15:1176–1182.
- Purdy GE, Hsu FF. Complete characterization of polyacyltrehaloses from mycobacterium tuberculosis H37Rv biofilm cultures by multiple-stage linear ion-trap mass spectrometry reveals a new tetraacyltrehalose family. Biochemistry. 2021;60:381–397.
- Magallanes-Puebla A, Espinosa-Cueto P, Lopez-Marin LM, et al. Mycobacterial glycolipid Di-O-acyl trehalose promotes a tolerogenic profile in dendritic cells. PLoS ONE. 2018;13:e0207202.
- Espinosa-Cueto P, Escalera-Zamudio M, Magallanes-Puebla A, et al. Mycobacterial glycolipids di-O-acylated trehalose and tri-O-acylated trehalose downregulate inducible nitric oxide synthase and nitric oxide production in macrophages. BMC Immunol. 2015;16:38.
- Passemar C, Arbues A, Malaga W, et al. Multiple deletions in the polyketide synthase gene repertoire of Mycobacterium tuberculosis reveal functional overlap of cell envelope lipids in host-pathogen interactions. Cell Microbiol. 2014;16:195–213.
- Rousseau C, Neyrolles O, Bordat Y, et al. Deficiency in mycolipenate- and mycosanoate-derived acyltrehaloses enhances early interactions of Mycobacterium tuberculosis with host cells. Cell Microbiol. 2003;5:405–415.
- Palma-Nicolas JP, Hernandez-Pando R, Segura E, et al. Mycobacterial di-O-acyl trehalose inhibits Th-1 cytokine gene expression in murine cells by down-modulation of MAPK signaling. Immunobiology. 2010;215:143–152.
- Saavedra R, Segura E, Leyva R, et al. Mycobacterial di-O-acyl-trehalose inhibits mitogen- and antigen-induced proliferation of murine T cells in vitro. Clin Diagn Lab Immunol. 2001;8:1081–1088.
- Goren MB. Sulfolipid I of Mycobacterium tuberculosis, strain H37Rv. I. Purification and properties. Biochim Biophys Acta. 1970;210:116–126.
- Gilmore SA, Schelle MW, Holsclaw CM, et al. Sulfolipid-1 biosynthesis restricts Mycobacterium tuberculosis growth in human macrophages. ACS Chem Biol. 2012;7:863–870.
- Rousseau C, Turner OC, Rush E, et al. Sulfolipid deficiency does not affect the virulence of Mycobacterium tuberculosis H37Rv in mice and guinea pigs. Infect Immun. 2003;71:4684–4690.
- Rodriguez JE, Ramirez AS, Salas LP, et al. Transcription of genes involved in sulfolipid and polyacyltrehalose biosynthesis of Mycobacterium tuberculosis in experimental latent tuberculosis infection. PLoS ONE. 2013;8:e58378.
- Morita YS, Fukuda T, Sena CB, et al. Inositol lipid metabolism in mycobacteria: biosynthesis and regulatory mechanisms. Biochim Biophys Acta. 2011;1810:630–641.
- Gilleron M, Ronet C, Mempel M, et al. Acylation state of the phosphatidylinositol mannosides from Mycobacterium bovis bacillus Calmette Guerin and ability to induce granuloma and recruit natural killer T cells. J Biol Chem. 2001;276:34896–34904.
- Mishra AK, Driessen NN, Appelmelk BJ, et al. Lipoarabinomannan and related glycoconjugates: structure, biogenesis and role in Mycobacterium tuberculosis physiology and host-pathogen interaction. FEMS Microbiol Rev. 2011;35:1126–1157.
- Morita YS, Sena CB, Waller RF, et al. PimE is a polyprenol-phosphate-mannose-dependent mannosyltransferase that transfers the fifth mannose of phosphatidylinositol mannoside in mycobacteria. J Biol Chem. 2006;281:25143–25155.
- Crellin PK, Kovacevic S, Martin KL, et al. Mutations in pimE restore lipoarabinomannan synthesis and growth in a Mycobacterium smegmatis lpqW mutant. J Bacteriol. 2008;190:3690–3699.
- Eagen WJ, Baumoel LR, Osman SH, et al. Deletion of PimE mannosyltransferase results in increased copper sensitivity in Mycobacterium smegmatis. FEMS Microbiol Lett. 2018;365. DOI:10.1093/femsle/fny025
- Jozefowski S, Sobota A, Pawlowski A, et al. Mycobacterium tuberculosis lipoarabinomannan enhances LPS-induced TNF-alpha production and inhibits NO secretion by engaging scavenger receptors. Microb Pathog. 2011;50:350–359.
- Zheng RB, Jegouzo SAF, Joe M, et al. Insights into interactions of mycobacteria with the host innate immune system from a novel array of synthetic mycobacterial glycans. ACS Chem Biol. 2017;12:2990–3002.
- Li K, Underhill DM. C-type lectin receptors in phagocytosis. Curr Top Microbiol Immunol. 2020;429:1–18.
- Torrelles JB, Knaup R, Kolareth A, et al. Identification of Mycobacterium tuberculosis clinical isolates with altered phagocytosis by human macrophages due to a truncated lipoarabinomannan. J Biol Chem. 2008;283:31417–31428.
- Birch HL, Alderwick LJ, Appelmelk BJ, et al. A truncated lipoglycan from mycobacteria with altered immunological properties. Proc Natl Acad Sci U S A. 2010;107:2634–2639.
- Nakayama H, Kurihara H, Morita YS, et al. Lipoarabinomannan binding to lactosylceramide in lipid rafts is essential for the phagocytosis of mycobacteria by human neutrophils. Sci Signal. 2016;9:ra101.
- Fratti RA, Chua J, Vergne I, et al. Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci U S A. 2003;100:5437–5442.
- Hawkins PT, Stephens LR, Suire S, et al. PI3K signaling in neutrophils. Curr Top Microbiol Immunol. 2010;346:183–202.
- Dell’Angelica EC, Mullins C, Caplan S, et al. Lysosome-related organelles. FASEB J. 2000;14:1265–1278.
- Sani M, Houben EN, Geurtsen J, et al. Direct visualization by cryo-EM of the mycobacterial capsular layer: a labile structure containing ESX-1-secreted proteins. PLOS Pathog. 2010;6:e1000794.
- Angala SK, Belardinelli JM, Huc-Claustre E, et al. The cell envelope glycoconjugates of Mycobacterium tuberculosis. Crit Rev Biochem Mol Biol. 2014;49:361–399.
- Barnes DD, Lundahl MLE, Lavelle EC, Scanlan, EM, et al. The emergence of phenolic glycans as virulence factors in Mycobacterium tuberculosis. ACS Chem Biol. 2017;12 8 :1969–1979.
- Rashid AM, Batey SF, Syson K, et al. Assembly of alpha-Glucan by GlgE and GlgB in Mycobacteria and Streptomycetes. Biochemistry. 2016;55:3270–3284.
- Schwebach JR, Glatman-Freedman A, Gunther-Cummins L, et al. Glucan is a component of the Mycobacterium tuberculosis surface that is expressed in vitro and in vivo. Infect Immun. 2002;70:2566–2575.
- Lemassu A, Daffe M. Structural features of the exocellular polysaccharides of Mycobacterium tuberculosis. Biochem J. 1994;297(Pt 2):351–357.
- Venkataswamy MM, Goldberg MF, Baena A, et al. In vitro culture medium influences the vaccine efficacy of Mycobacterium bovis BCG. Vaccine. 2012;30:1038–1049.
- Prados-Rosales R, Carreno LJ, Weinrick B, et al. The type of growth medium affects the presence of a mycobacterial capsule and is associated with differences in protective efficacy of BCG vaccination against mycobacterium tuberculosis. J Infect Dis. 2016;214:426–437.
- Stokes RW, Norris-Jones R, Brooks DE, et al. The glycan-rich outer layer of the cell wall of Mycobacterium tuberculosis acts as an antiphagocytic capsule limiting the association of the bacterium with macrophages. Infect Immun. 2004;72:5676–5686.
- Ortalo-Magne A, Lemassu A, Laneelle MA, et al. Identification of the surface-exposed lipids on the cell envelopes of Mycobacterium tuberculosis and other mycobacterial species. J Bacteriol. 1996;178:456–461.
- Selvaraj P, Jawahar MS, Rajeswari DN, et al. Role of mannose binding lectin gene variants on its protein levels and macrophage phagocytosis with live Mycobacterium tuberculosis in pulmonary tuberculosis. FEMS Immunol Med Microbiol. 2006;46:433–437.
- Yang L, Sinha T, Carlson TK, et al. Changes in the major cell envelope components of Mycobacterium tuberculosis during in vitro growth. Glycobiology. 2013;23:926–934.
- Gagliardi MC, Lemassu A, Teloni R, et al. Cell wall-associated alpha-glucan is instrumental for Mycobacterium tuberculosis to block CD1 molecule expression and disable the function of dendritic cell derived from infected monocyte. Cell Microbiol. 2007;9:2081–2092.
- Houben D, Demangel C, van Ingen J, et al. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol. 2012;14:1287–1298.
- Wiens KE, Ernst JD. The mechanism for type I interferon induction by mycobacterium tuberculosis is bacterial strain-dependent. PLOS Pathog. 2016;12:e1005809.
- Zhang L, Jiang X, Pfau D, et al. Type I interferon signaling mediates Mycobacterium tuberculosis-induced macrophage death. J Exp Med. 2021;218. DOI:10.1084/jem.20200887
- Moreira-Teixeira L, Mayer-Barber K, Sher A, et al. Type I interferons in tuberculosis: foe and occasionally friend. J Exp Med. 2018;215:1273–1285.
- Olson GS, Murray TA, Jahn AN, et al. Type I interferon decreases macrophage energy metabolism during mycobacterial infection. Cell Rep. 2021;35:109195.
- Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2018;14:243–251.
- Gehring AJ, Rojas RE, Canaday DH, et al. The Mycobacterium tuberculosis 19-kilodalton lipoprotein inhibits gamma interferon-regulated HLA-DR and Fc gamma R1 on human macrophages through Toll-like receptor 2. Infect Immun. 2003;71:4487–4497.
- Becker K, Sander P. Mycobacterium tuberculosis lipoproteins in virulence and immunity - fighting with a double-edged sword. FEBS Lett. 2016;590:3800–3819.
- Palucci I, Camassa S, Cascioferro A, et al. PE_PGRS33 contributes to mycobacterium tuberculosis entry in macrophages through interaction with TLR2. PLoS ONE. 2016;11:e0150800.
- Basu S, Pathak SK, Banerjee A, et al. Execution of macrophage apoptosis by PE_PGRS33 of Mycobacterium tuberculosis is mediated by Toll-like receptor 2-dependent release of tumor necrosis factor-alpha. J Biol Chem. 2007;282:1039–1050.
- Aguilar-Lopez BA, Correa F, Moreno-Altamirano MMB, et al. LprG and PE_PGRS33 Mycobacterium tuberculosis virulence factors induce differential mitochondrial dynamics in macrophages. Scand J Immunol. 2019;89:e12728.
- Gehring AJ, Dobos KM, Belisle JT, et al. Mycobacterium tuberculosis LprG (Rv1411c): a novel TLR-2 ligand that inhibits human macrophage class II MHC antigen processing. J Immunol. 2004;173:2660–2668.
- Alonso H, Parra J, Malaga W, et al. Protein O-mannosylation deficiency increases LprG-associated lipoarabinomannan release by Mycobacterium tuberculosis and enhances the TLR2-associated inflammatory response. Sci Rep. 2017;7:7913.
- Jung SB, Yang CS, Lee JS, et al. The mycobacterial 38-kilodalton glycolipoprotein antigen activates the mitogen-activated protein kinase pathway and release of proinflammatory cytokines through Toll-like receptors 2 and 4 in human monocytes. Infect Immun. 2006;74:2686–2696.
- Byun EH, Kim WS, Kim JS, et al. Mycobacterium tuberculosis Rv0577, a novel TLR2 agonist, induces maturation of dendritic cells and drives Th1 immune response. FASEB J. 2012;26:2695–2711.
- Buchko GW, Kim H, Myler PJ, et al. Chemical shift assignments for Rv0577, a putative glyoxalase associated with virulence from Mycobacterium tuberculosis. Biomol NMR Assign. 2012;6:43–46.
- Huard RC, Chitale S, Leung M, et al. The Mycobacterium tuberculosis complex-restricted gene cfp32 encodes an expressed protein that is detectable in tuberculosis patients and is positively correlated with pulmonary interleukin-10. Infect Immun. 2003;71:6871–6883.
- Su H, Zhu S, Zhu L, et al. Recombinant lipoprotein Rv1016c derived from mycobacterium tuberculosis is a TLR-2 ligand that induces macrophages apoptosis and inhibits MHC II antigen processing. Front Cell Infect Microbiol. 2016;6:147.
- Li F, Feng L, Jin C, et al. LpqT improves mycobacteria survival in macrophages by inhibiting TLR2 mediated inflammatory cytokine expression and cell apoptosis. Tuberculosis (Edinb). 2018;111:57–66.
- Su H, Peng B, Zhang Z, et al. The Mycobacterium tuberculosis glycoprotein Rv1016c protein inhibits dendritic cell maturation, and impairs Th1/Th17 responses during mycobacteria infection. Mol Immunol. 2019;109:58–70.
- Yeruva VC, Kulkarni A, Khandelwal R, et al. The PE_PGRS proteins of mycobacterium tuberculosis are Ca(2+) binding mediators of host-pathogen interaction. Biochemistry. 2016;55:4675–4687.
- Xie Y, Zhou Y, Liu S, et al. PE_PGRS: vital proteins in promoting mycobacterial survival and modulating host immunity and metabolism. Cell Microbiol. 2021;23:e13290.
- Bansal K, Sinha AY, Ghorpade DS, et al. Src homology 3-interacting domain of Rv1917c of Mycobacterium tuberculosis induces selective maturation of human dendritic cells by regulating PI3K-MAPK-NF-kappaB signaling and drives Th2 immune responses. J Biol Chem. 2010;285:36511–36522.
- Su H, Kong C, Zhu L, et al. PPE26 induces TLR2-dependent activation of macrophages and drives Th1-type T-cell immunity by triggering the cross-talk of multiple pathways involved in the host response. Oncotarget. 2015;6:38517–38537.
- Mi Y, Bao L, Gu D, et al. Mycobacterium tuberculosis PPE25 and PPE26 proteins expressed in Mycobacterium smegmatis modulate cytokine secretion in mouse macrophages and enhance mycobacterial survival. Res Microbiol. 2017;168:234–243.
- Xu Y, Yang E, Huang Q, et al. PPE57 induces activation of macrophages and drives Th1-type immune responses through TLR2. J Mol Med (Berl). 2015;93:645–662.
- Su H, Zhang Z, Liu Z, et al. Mycobacterium tuberculosis PPE60 antigen drives Th1/Th17 responses via Toll-like receptor 2-dependent maturation of dendritic cells. J Biol Chem. 2018;293:10287–10302.
- Gong Z, Kuang Z, Li H, et al. Regulation of host cell pyroptosis and cytokines production by Mycobacterium tuberculosis effector PPE60 requires LUBAC mediated NF-kappaB signaling. Cell Immunol. 2019;335:41–50.
- Bansal K, Elluru SR, Narayana Y, et al. PE_PGRS antigens of Mycobacterium tuberculosis induce maturation and activation of human dendritic cells. J Immunol. 2010;184:3495–3504.
- Chaturvedi R, Bansal K, Narayana Y, et al. The multifunctional PE_PGRS11 protein from Mycobacterium tuberculosis plays a role in regulating resistance to oxidative stress. J Biol Chem. 2010;285:30389–30403.
- Qiang L, Wang J, Zhang Y, et al. Mycobacterium tuberculosis Mce2E suppresses the macrophage innate immune response and promotes epithelial cell proliferation. Cell Mol Immunol. 2019;16:380–391.
- Li J, Chai QY, Zhang Y, et al. Mycobacterium tuberculosis Mce3E suppresses host innate immune responses by targeting ERK1/2 signaling. J Immunol. 2015;194:3756–3767.
- Zhang F, Xie JP. Mammalian cell entry gene family of Mycobacterium tuberculosis. Mol Cell Biochem. 2011;352:1–10.
- Pasricha R, Saini NK, Rathor N, et al. The Mycobacterium tuberculosis recombinant LprN protein of mce4 operon induces Th-1 type response deleterious to protection in mice. Pathog Dis. 2014;72:188–196.
- Tufariello JM, Mi K, Xu J, et al. Deletion of the Mycobacterium tuberculosis resuscitation-promoting factor Rv1009 gene results in delayed reactivation from chronic tuberculosis. Infect Immun. 2006;74:2985–2995.
- Kim J-S, Kim WS, Choi H-G, et al. Mycobacterium tuberculosis RpfB drives Th1-type T cell immunity via a TLR4-dependent activation of dendritic cells. J Leukocyte Biol. 2013;94(4):733–749. DOI:10.1189/jlb.0912435
- Sakthi S, Narayanan S. The lpqS knockout mutant of Mycobacterium tuberculosis is attenuated in macrophages. Microbiol Res. 2013;168:407–414.
- Sala C, Odermatt NT, Soler-Arnedo P, et al. EspL is essential for virulence and stabilizes EspE, EspF and EspH levels in Mycobacterium tuberculosis. PLOS Pathog. 2018;14:e1007491.
- Sha S, Shi X, Deng G, et al. Mycobacterium tuberculosis Rv1987 induces Th2 immune responses and enhances Mycobacterium smegmatis survival in mice. Microbiol Res. 2017;197:74–80.
- Sha S, Shi Y, Tang Y, et al. Mycobacterium tuberculosis Rv1987 protein induces M2 polarization of macrophages through activating the PI3K/Akt1/mTOR signaling pathway. Immunol Cell Biol. 2021;99:570–585.
- Kim WS, Kim JS, Cha SB, et al. Mycobacterium tuberculosis PE27 activates dendritic cells and contributes to Th1-polarized memory immune responses during in vivo infection. Immunobiology. 2016;221:440–453.
- Jiao J, Zheng N, Wei W, et al. M. tuberculosis CRISPR/Cas proteins are secreted virulence factors that trigger cellular immune responses. Virulence. 2021;12:3032–3044.
- Yang F, Xu L, Liang L, et al. The involvement of mycobacterium type III-A CRISPR-Cas system in oxidative stress. Front Microbiol. 2021;12:774492.
- Madan-Lala R, Sia JK, King R, et al. Mycobacterium tuberculosis impairs dendritic cell functions through the serine hydrolase Hip1. J Immunol. 2014;192:4263–4272.
- Rengarajan J, Murphy E, Park A, et al. Mycobacterium tuberculosis Rv2224c modulates innate immune responses. Proc Natl Acad Sci U S A. 2008;105:264–269.
- Madan-Lala R, Peixoto KV, Re F, et al. Mycobacterium tuberculosis Hip1 dampens macrophage proinflammatory responses by limiting toll-like receptor 2 activation. Infect Immun. 2011;79:4828–4838.
- Saleh MT, Belisle JT. Secretion of an acid phosphatase (SapM) by Mycobacterium tuberculosis that is similar to eukaryotic acid phosphatases. J Bacteriol. 2000;182:6850–6853.
- Vergne I, Chua J, Lee HH, et al. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2005;102:4033–4038.
- Puri RV, Reddy PV, Tyagi AK. Secreted acid phosphatase (SapM) of Mycobacterium tuberculosis is indispensable for arresting phagosomal maturation and growth of the pathogen in guinea pig tissues. PLoS ONE. 2013;8:e70514.
- Zulauf KE, Sullivan JT, Braunstein M. The SecA2 pathway of Mycobacterium tuberculosis exports effectors that work in concert to arrest phagosome and autophagosome maturation. PLOS Pathog. 2018;14:e1007011.
- Bao Y, Wang L, Sun J. A small protein but with diverse roles: a review of EsxA in mycobacterium–host interaction. Cells. 2021;10:10.
- Zhang Q, Wang D, Jiang G, et al. EsxA membrane-permeabilizing activity plays a key role in mycobacterial cytosolic translocation and virulence: effects of single-residue mutations at glutamine 5. Sci Rep. 2016;6:32618.
- Iantomasi R, Sali M, Cascioferro A, et al. PE_PGRS30 is required for the full virulence of Mycobacterium tuberculosis. Cell Microbiol. 2012;14:356–367.
- Chatrath S, Gupta VK, Dixit A, et al. PE_PGRS30 of Mycobacterium tuberculosis mediates suppression of proinflammatory immune response in macrophages through its PGRS and PE domains. Microbes Infect. 2016;18:536–542.
- Sullivan JT, Young EF, McCann JR, et al. The Mycobacterium tuberculosis SecA2 system subverts phagosome maturation to promote growth in macrophages. Infect Immun. 2012;80:996–1006.
- Wong D, Bach H, Sun J, et al. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H±ATPase to inhibit phagosome acidification. Proc Natl Acad Sci U S A. 2011;108:19371–19376.
- Wang J, Li BX, Ge PP, et al. Mycobacterium tuberculosis suppresses innate immunity by coopting the host ubiquitin system. Nat Immunol. 2015;16:237–245.
- Bach H, Papavinasasundaram KG, Wong D, et al. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe. 2008;3:316–322.
- Pradhan G, Shrivastva R, Mukhopadhyay S. Mycobacterial PknG targets the Rab7l1 signaling pathway to inhibit phagosome-lysosome fusion. J Immunol. 2018;201:1421–1433.
- Wang J, Ge P, Lei Z, et al. Mycobacterium tuberculosis protein kinase G acts as an unusual ubiquitinating enzyme to impair host immunity. EMBO Rep. 2021;22:e52175.
- Ge P, Lei Z, Yu Y, et al. M. tuberculosis PknG manipulates host autophagy flux to promote pathogen intracellular survival. Autophagy. 2021;18(3):576-594. doi:10.1080/15548627.2021.1938912.
- Sannigrahi A, Nandi I, Chall S, et al. Conformational switch driven membrane pore formation by mycobacterium secretory protein MPT63 induces macrophage cell death. ACS Chem Biol. 2019;14:1601–1610.
- Siromolot AA, Oliinyk OS, Kolibo DV, et al. Mycobacterium tuberculosis antigens MPT63 and MPT83 increase phagocytic activity of murine peritoneal macrophages. Ukr Biochem J. 2016;88:62–70.
- Thi EP, Hong CJ, Sanghera G, et al. Identification of the Mycobacterium tuberculosis protein PE-PGRS62 as a novel effector that functions to block phagosome maturation and inhibit iNOS expression. Cell Microbiol. 2013;15:795–808.
- Long Q, Xiang X, Yin Q, et al. PE_PGRS62 promotes the survival of Mycobacterium smegmatis within macrophages via disrupting ER stress-mediated apoptosis. J Cell Physiol. 2019;234:19774–19784.
- Mittal E, Kumar S, Rahman A, et al. Modulation of phagolysosome maturation by bacterial tlyA gene product. J Biosci. 2014;39:821–834.
- Rahman MA, Sobia P, Dwivedi VP, et al. Mycobacterium tuberculosis TlyA protein negatively regulates T helper (Th) 1 and Th17 differentiation and promotes tuberculosis pathogenesis. J Biol Chem. 2015;290:14407–14417.
- Deghmane AE, Soualhine H, Bach H, et al. Lipoamide dehydrogenase mediates retention of coronin-1 on BCG vacuoles, leading to arrest in phagosome maturation. J Cell Sci. 2007;120:2796–2806.
- Heo DR, Shin SJ, Kim WS, et al. Mycobacterium tuberculosislpdC, Rv0462, induces dendritic cell maturation and Th1 polarization. Biochem Biophys Res Commun. 2011;411:642–647.
- Sun J, Wang X, Lau A, et al. Mycobacterial nucleoside diphosphate kinase blocks phagosome maturation in murine RAW 264.7 macrophages. PLoS ONE. 2010;5:e8769.
- Cui Z, Dang G, Song N, et al. Rv3091, an extracellular patatin-like phospholipase in mycobacterium tuberculosis, prolongs intracellular survival of recombinant mycolicibacterium smegmatis by mediating phagosomal escape. Front Microbiol. 2020;11:2204.
- Mehra A, Zahra A, Thompson V, et al. Mycobacterium tuberculosis type VII secreted effector EsxH targets host ESCRT to impair trafficking. PLOS Pathog. 2013;9:e1003734.
- Shin DM, Jeon BY, Lee HM, et al. Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLOS Pathog. 2010;6:e1001230.
- Duan L, Yi M, Chen J, et al. Mycobacterium tuberculosis EIS gene inhibits macrophage autophagy through up-regulation of IL-10 by increasing the acetylation of histone H3. Biochem Biophys Res Commun. 2016;473:1229–1234.
- Kim KH, An DR, Song J, et al. Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc Natl Acad Sci U S A. 2012;109:7729–7734.
- Huang D, Bao L. Mycobacterium tuberculosis EspB protein suppresses interferon-gamma-induced autophagy in murine macrophages. J Microbiol Immunol Infect. 2016;49:859–865.
- Choi JA, Lim YJ, Cho SN, et al. Mycobacterial HBHA induces endoplasmic reticulum stress-mediated apoptosis through the generation of reactive oxygen species and cytosolic Ca2+ in murine macrophage RAW 264.7 cells. Cell Death Dis. 2013;4:e957.
- Lanfranconi MP, Arabolaza A, Gramajo H, et al. Insights into the evolutionary history of the virulent factor HBHA of Mycobacterium tuberculosis. Arch Microbiol. 2021;203:2171–2182.
- Menozzi FD, Rouse JH, Alavi M, et al. Identification of a heparin-binding hemagglutinin present in mycobacteria. J Exp Med. 1996;184:993–1001.
- Pethe K, Alonso S, Biet F, et al. The heparin-binding haemagglutinin of M. tuberculosis is required for extrapulmonary dissemination. Nature. 2001;412:190–194.
- Guo Q, Bi J, Wang H, et al. Mycobacterium tuberculosis ESX-1-secreted substrate protein EspC promotes mycobacterial survival through endoplasmic reticulum stress-mediated apoptosis. Emerg Microbes Infect. 2021;10:19–36.
- Velmurugan K, Chen B, Miller JL, et al. Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLOS Pathog. 2007;3:e110.
- Blomgran R, Desvignes L, Briken V, et al. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe. 2012;11:81–90.
- Danelishvili L, Yamazaki Y, Selker J, et al. Secreted Mycobacterium tuberculosis Rv3654c and Rv3655c proteins participate in the suppression of macrophage apoptosis. PLoS ONE. 2010;5:e10474.
- Kumar D, Narayanan S. pknE, a serine/threonine kinase of Mycobacterium tuberculosis modulates multiple apoptotic paradigms. Infect Genet Evol. 2012;12:737–747.
- Jayakumar D, Jacobs WR Jr., Narayanan S. Protein kinase E of Mycobacterium tuberculosis has a role in the nitric oxide stress response and apoptosis in a human macrophage model of infection. Cell Microbiol. 2008;10:365–374.
- Zhang W, Lu Q, Dong Y, et al. Rv3033, as an emerging anti-apoptosis factor, facilitates mycobacteria survival via inhibiting macrophage intrinsic apoptosis. Front Immunol. 2018;9:2136.
- Sun J, Siroy A, Lokareddy RK, et al. The tuberculosis necrotizing toxin kills macrophages by hydrolyzing NAD. Nat Struct Mol Biol. 2015;22:672–678.
- Danilchanka O, Sun J, Pavlenok M, et al. An outer membrane channel protein of Mycobacterium tuberculosis with exotoxin activity. Proc Natl Acad Sci U S A. 2014;111:6750–6755.
- Chen W, Bao Y, Chen X, et al. Mycobacterium tuberculosis PE25/PPE41 protein complex induces activation and maturation of dendritic cells and drives Th2-biased immune responses. Med Microbiol Immunol. 2016;205:119–131.
- Singh P, Rao RN, Reddy JR, et al. PE11, a PE/PPE family protein of Mycobacterium tuberculosis is involved in cell wall remodeling and virulence. Sci Rep. 2016;6:21624.
- Rastogi S, Singh AK, Pant G, et al. Down-regulation of PE11, a cell wall associated esterase, enhances the biofilm growth of Mycobacterium tuberculosis and reduces cell wall virulence lipid levels. Microbiology (Reading). 2017;163:52–61.
- Deng W, Zeng J, Xiang X, et al. PE11 (Rv1169c) selectively alters fatty acid components of Mycobacterium smegmatis and host cell interleukin-6 level accompanied with cell death. Front Microbiol. 2015;6:613.
- Master SS, Rampini SK, Davis AS, et al. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe. 2008;3:224–232.
- Vemula MH, Medisetti R, Ganji R, et al. Mycobacterium tuberculosis zinc metalloprotease-1 assists mycobacterial dissemination in zebrafish. Front Microbiol. 2016;7:1347.
- Danelishvili L, Everman JL, McNamara MJ, et al. Inhibition of the plasma-membrane-associated serine protease Cathepsin G by mycobacterium tuberculosis Rv3364c suppresses Caspase-1 and pyroptosis in macrophages. Front Microbiol. 2011;2:281.
- Rastogi S, Ellinwood S, Augenstreich J, et al. Mycobacterium tuberculosis inhibits the NLRP3 inflammasome activation via its phosphokinase PknF. PLOS Pathog. 2021;17:e1009712.
- Sethi D, Mahajan S, Singh C, et al. Lipoprotein LprI of mycobacterium tuberculosis acts as a lysozyme inhibitor. J Biol Chem. 2016;291:2938–2953.
- Vazquez Reyes S, Ray S, Aguilera J, et al. Development of an in vitro membrane model to study the function of EsxAB Heterodimer and establish the role of EsxB in membrane permeabilizing activity of mycobacterium tuberculosis. Pathogens. 2020;9:1015.
- Welin A, Bjornsdottir H, Winther M, et al. CFP-10 from Mycobacterium tuberculosis selectively activates human neutrophils through a pertussis toxin-sensitive chemotactic receptor. Infect Immun. 2015;83:205–213.
- Mubin N, Pahari S, Owais M, et al. Mycobacterium tuberculosis host cell interaction: role of latency associated protein Acr-1 in differential modulation of macrophages. PLoS ONE. 2018;13:e0206459.
- Siddiqui KF, Amir M, Gurram RK, et al. Latency-associated protein Acr1 impairs dendritic cell maturation and functionality: a possible mechanism of immune evasion by Mycobacterium tuberculosis. J Infect Dis. 2014;209:1436–1445.
- Beaulieu AM, Rath P, Imhof M, et al. Genome-wide screen for Mycobacterium tuberculosis genes that regulate host immunity. PLoS ONE. 2010;5:e15120.
- Deng G, Zhang F, Yang S, et al. Mycobacterium tuberculosis Rv0431 expressed in Mycobacterium smegmatis, a potentially mannosylated protein, mediated the immune evasion of RAW 264.7 macrophages. Microb Pathog. 2016;100:285–292.
- Rath P, Huang C, Wang T, et al. Genetic regulation of vesiculogenesis and immunomodulation in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2013;110:E4790–7.
- Georgieva M, Sia JK, Bizzell E, et al. Mycobacterium tuberculosis GroEL2 modulates dendritic cell responses. Infect Immun. 2018;86. DOI:10.1128/IAI.00387-17
- Li H, Dang G, Liu H, et al. Characterization of a novel Mycobacterium tuberculosis serine protease (Rv3194c) activity and pathogenicity. Tuberculosis (Edinb). 2019;119:101880.
- Stamm CE, Pasko BL, Chaisavaneeyakorn S, et al. Screening mycobacterium tuberculosis secreted proteins identifies Mpt64 as a eukaryotic membrane-binding bacterial effector. mSphere. 2019;4. DOI:10.1128/mSphere.00354-19.
- Wang Q, Liu S, Tang Y, et al. MPT64 protein from Mycobacterium tuberculosis inhibits apoptosis of macrophages through NF-kB-miRNA21-Bcl-2 pathway. PLoS ONE. 2014;9:e100949.
- Sharma K, Gupta M, Pathak M, et al. Transcriptional control of the mycobacterial embCAB operon by PknH through a regulatory protein, EmbR, in vivo. J Bacteriol. 2006;188:2936–2944.
- Mata E, Farrell D, Ma R, et al. Independent genomic polymorphisms in the PknH serine threonine kinase locus during evolution of the Mycobacterium tuberculosis Complex affect virulence and host preference. PLOS Pathog. 2020;16:e1009061.
- Na I, Dai H, Li H, et al. Computational prediction and validation of specific EmbR binding site on PknH. Synth Syst Biotechnol. 2021;6:429–436.
- Shariq M, Quadir N, Sharma N, et al. Mycobacterium tuberculosis RipA Dampens TLR4-mediated host protective response using a multi-pronged approach involving autophagy, apoptosis, metabolic repurposing, and immune modulation. Front Immunol. 2021;12:636644.
- Gallant J, Heunis T, Beltran C, et al. PPE38-secretion-dependent proteins of M. tuberculosis alter NF-kB signalling and inflammatory responses in macrophages. Front Immunol. 2021;12:702359.
- Ates LS, Dippenaar A, Ummels R, et al. Mutations in ppe38 block PE_PGRS secretion and increase virulence of Mycobacterium tuberculosis. Nat Microbiol. 2018;3:181–188.
- Stamm CE, Collins AC, Shiloh MU. Sensing of Mycobacterium tuberculosis and consequences to both host and bacillus. Immunol Rev. 2015;264:204–219.
- Basu J, Shin DM, Jo EK. Mycobacterial signaling through toll-like receptors. Front Cell Infect Microbiol. 2012;2:145.
- Yu X, Zeng J, Xie J. Navigating through the maze of TLR2 mediated signaling network for better mycobacterium infection control. Biochimie. 2014;102:1–8.
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511.
- Underhill DM, Ozinsky A, Smith KD, et al. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A. 1999;96:14459–14463.
- Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–625.
- Songane M, Kleinnijenhuis J, Netea MG, et al. The role of autophagy in host defence against Mycobacterium tuberculosis infection. Tuberculosis (Edinb). 2012;92:388–396.
- Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544.
- Zumbo A, Palucci I, Cascioferro A, et al. Functional dissection of protein domains involved in the immunomodulatory properties of PE_PGRS33 of Mycobacterium tuberculosis. Pathog Dis. 2013;69:232–239.
- Post FA, Manca C, Neyrolles O, et al. Mycobacterium tuberculosis 19-kilodalton lipoprotein inhibits Mycobacterium smegmatis-induced cytokine production by human macrophages in vitro. Infect Immun. 2001;69:1433–1439.
- Hinman AE, Jani C, Pringle SC, et al. Mycobacterium tuberculosis canonical virulence factors interfere with a late component of the TLR2 response. Elife. 2021;10. DOI:10.7554/eLife.73984.
- Zhang Y, Li J, Li B, et al. Mycobacterium tuberculosis Mce3C promotes mycobacteria entry into macrophages through activation of β2 integrin-mediated signalling pathway. Cell Microbiol. 2018;20:20.
- Ahmad S, El-Shazly S, Mustafa AS, et al. The six mammalian cell entry proteins (Mce3a-F) encoded by the mce3 operon are expressed during in vitro growth of Mycobacterium tuberculosis. Scand J Immunol. 2005;62:16–24.
- Ahmad S, El-Shazly S, Mustafa AS, et al. Mammalian cell-entry proteins encoded by the mce3 operon of Mycobacterium tuberculosis are expressed during natural infection in humans. Scand J Immunol. 2004;60:382–391.
- Xia A, Li X, Quan J, et al. Mycobacterium tuberculosis Rv0927c inhibits NF-kappaB pathway by downregulating the phosphorylation level of IkappaBalpha and enhances mycobacterial survival. Front Immunol. 2021;12:721370.
- Khan MZ, Nandicoori VK. Deletion of pknG abates reactivation of latent mycobacterium tuberculosis in mice. Antimicrob Agents Chemother. 2021;65. DOI:10.1128/AAC.02095-20
- Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8:917–929.
- Hackam DJ, Rotstein OD, Zhang WJ, et al. Regulation of phagosomal acidification. Differential targeting of Na+/H+ exchangers, Na+/K±ATPases, and vacuolar-type H±atpases. J Biol Chem. 1997;272:29810–29820.
- Zhou P, Li W, Wong D, et al. Phosphorylation control of protein tyrosine phosphatase a activity in Mycobacterium tuberculosis. FEBS Lett. 2015;589:326–331.
- Wang J, Ge P, Qiang L, et al. The mycobacterial phosphatase PtpA regulates the expression of host genes and promotes cell proliferation. Nat Commun. 2017;8:244.
- Poirier V, Bach H, Av-Gay Y. Mycobacterium tuberculosis promotes anti-apoptotic activity of the macrophage by PtpA protein-dependent dephosphorylation of host GSK3alpha. J Biol Chem. 2014;289:29376–29385.
- Mittal E, Skowyra ML, Uwase G, et al. Mycobacterium tuberculosis type VII secretion system effectors differentially impact the ESCRT endomembrane damage response. MBio. 2018;9. DOI:10.1128/mBio.01765-18.
- Jayachandran R, Sundaramurthy V, Combaluzier B, et al. Survival of mycobacteria in macrophages is mediated by coronin 1-dependent activation of calcineurin. Cell. 2007;130:37–50.
- van der Wel N, Hava D, Houben D, et al. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. 2007;129:1287–1298.
- Simeone R, Bobard A, Lippmann J, et al. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLOS Pathog. 2012;8:e1002507.
- Kinhikar AG, Verma I, Chandra D, et al. Potential role for ESAT6 in dissemination of M. tuberculosis via human lung epithelial cells. Mol Microbiol. 2010;75:92–106.
- Sreejit G, Ahmed A, Parveen N, et al. The ESAT-6 protein of Mycobacterium tuberculosis interacts with beta-2-microglobulin (beta2m) affecting antigen presentation function of macrophage. PLOS Pathog. 2014;10:e1004446.
- Pathak SK, Basu S, Basu KK, et al. Direct extracellular interaction between the early secreted antigen ESAT-6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nat Immunol. 2007;8:610–618.
- Hsu T, Hingley-Wilson SM, Chen B, et al. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci U S A. 2003;100:12420–12425.
- Stanley SA, Johndrow JE, Manzanillo P, et al. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol. 2007;178:3143–3152.
- de Jonge MI, Pehau-Arnaudet G, Fretz MM, et al. ESAT-6 from Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic conditions and exhibits membrane-lysing activity. J Bacteriol. 2007;189:6028–6034.
- Aguilera J, Karki CB, Li L, et al. N (alpha)-Acetylation of the virulence factor EsxA is required for mycobacterial cytosolic translocation and virulence. J Biol Chem. 2020;295:5785–5794.
- Augenstreich J, Haanappel E, Sayes F, et al. Phthiocerol Dimycocerosates from mycobacterium tuberculosis increase the membrane activity of bacterial effectors and host receptors. Front Cell Infect Microbiol. 2020;10:420.
- Osman MM, Shanahan JK, Chu F, et al. The C terminus of the mycobacterium ESX-1 secretion system substrate ESAT-6 is required for phagosomal membrane damage and virulence. Proc Natl Acad Sci U S A. 2022;119:e2122161119.
- Rahman MS, Ammerman NC, Sears KT, et al. Functional characterization of a phospholipase A(2) homolog from Rickettsia typhi. J Bacteriol. 2010;192:3294–3303.
- Rahman MS, Gillespie JJ, Kaur SJ, et al. Rickettsia typhi possesses phospholipase A2 enzymes that are involved in infection of host cells. PLOS Pathog. 2013;9:e1003399.
- Ku B, Lee KH, Park WS, et al. VipD of Legionella pneumophila targets activated Rab5 and Rab22 to interfere with endosomal trafficking in macrophages. PLOS Pathog. 2012;8:e1003082.
- Gaspar AH, Machner MP. VipD is a Rab5-activated phospholipase A1 that protects Legionella pneumophila from endosomal fusion. Proc Natl Acad Sci U S A. 2014;111:4560–4565.
- Shariq M, Quadir N, Alam A, et al. The exploitation of host autophagy and ubiquitin machinery by Mycobacterium tuberculosis in shaping immune responses and host defense during infection. Autophagy. 2022;1–21. DOI:10.1080/15548627.2021.2021495.
- Gutierrez MG, Master SS, Singh SB, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766.
- Mahairas GG, Sabo PJ, Hickey MJ, et al. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J Bacteriol. 1996;178:1274–1282.
- Manzanillo PS, Ayres JS, Watson RO, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–516.
- Franco LH, Nair VR, Scharn CR, et al. The ubiquitin ligase Smurf1 functions in selective autophagy of mycobacterium tuberculosis and anti-tuberculous host defense. Cell Host Microbe. 2017;22:421–423.
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335.
- Chai Q, Wang X, Qiang L, et al. A Mycobacterium tuberculosis surface protein recruits ubiquitin to trigger host xenophagy. Nat Commun. 2019;10:1973.
- Hu D, Wu J, Wang W, et al. Autophagy regulation revealed by SapM-induced block of autophagosome-lysosome fusion via binding RAB7. Biochem Biophys Res Commun. 2015;461:401–407.
- Behar SM, Briken V. Apoptosis inhibition by intracellular bacteria and its consequence on host immunity. Curr Opin Immunol. 2019;60:103–110.
- Schaible UE, Winau F, Sieling PA, et al. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat Med. 2003;9:1039–1046.
- Sun J, Singh V, Lau A, et al. Mycobacterium tuberculosis nucleoside diphosphate kinase inactivates small GTPases leading to evasion of innate immunity. PLOS Pathog. 2013;9:e1003499.
- Miller JL, Velmurugan K, Cowan MJ, et al. The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PLOS Pathog. 2010;6:e1000864.
- Boise LH, Collins CM. Salmonella-induced cell death: apoptosis, necrosis or programmed cell death? Trends Microbiol. 2001;9:64–67.
- Rastogi S, Briken V. Interaction of mycobacteria with host cell inflammasomes. Front Immunol. 2022;13:791136.
- Wong KW, Jacobs WR Jr. Critical role for NLRP3 in necrotic death triggered by Mycobacterium tuberculosis. Cell Microbiol. 2011;13:1371–1384.
- Kurane T, Matsunaga T, Ida T, et al. GRIM-19 is a target of mycobacterial Zn(2+) metalloprotease 1 and indispensable for NLRP3 inflammasome activation. FASEB J. 2022;36:e22096.
- Beckwith KS, Beckwith MS, Ullmann S, et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat Commun. 2020;11:2270.
- Behar SM, Divangahi M, Remold HG. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol. 2010;8:668–674.
- Srinivasan L, Ahlbrand S, Briken V. Interaction of Mycobacterium tuberculosis with host cell death pathways. Cold Spring Harb Perspect Med. 2014;4:a022459.
- Pajuelo D, Tak U, Zhang L, et al. Toxin secretion and trafficking by Mycobacterium tuberculosis. Nat Commun. 2021;12:6592.
- Izquierdo Lafuente B, Ummels R, Kuijl C, et al. Mycobacterium tuberculosis Toxin CpnT is an ESX-5 substrate and requires three type VII secretion systems for intracellular secretion. MBio. 2021;12. DOI:10.1128/mBio.02983-20
- Pajuelo D, Gonzalez-Juarbe N, Tak U, et al. NAD(+) depletion triggers macrophage necroptosis, a cell death pathway exploited by mycobacterium tuberculosis. Cell Rep. 2018;24:429–440.
- Tundup S, Mohareer K, Hasnain SE. Mycobacterium tuberculosis PE25/PPE41 protein complex induces necrosis in macrophages: role in virulence and disease reactivation? FEBS Open Bio. 2014;4:822–828.
- Amaral EP, Costa DL, Namasivayam S, et al. A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J Exp Med. 2019;216:556–570.
- World Health Organization. The end TB strategy. 2015.
- Cobelens F, Suri RK, Helinski M, et al. Accelerating research and development of new vaccines against tuberculosis: a global roadmap. Lancet Infect Dis. 2022;22:e108–20.
- Bendre AD, Peters PJ, Kumar J. Tuberculosis: past, present and future of the treatment and drug discovery research. Curr Res Pharmacol Drug Discov. 2021;2:100037.
- Tait DR, Hatherill M, Van Der Meeren O, et al. Final analysis of a trial of M72/AS01E vaccine to prevent tuberculosis. N Engl J Med. 2019;381:2429–2439.
- Arbues A, Aguilo JI, Gonzalo-Asensio J, et al. Construction, characterization and preclinical evaluation of MTBVAC, the first live-attenuated M. tuberculosis-based vaccine to enter clinical trials. Vaccine. 2013;31:4867–4873.
- Gonzalo-Asensio J, Marinova D, Martin C, et al. MTBVAC: attenuating the human pathogen of tuberculosis (TB) toward a promising vaccine against the TB epidemic. Front Immunol. 2017;8:1803.
- White AD, Sibley L, Sarfas C, et al. MTBVAC vaccination protects rhesus macaques against aerosol challenge with M. tuberculosis and induces immune signatures analogous to those observed in clinical studies. NPJ Vaccines. 2021;6:4.
- Martin C, Marinova D, Aguilo N, et al. MTBVAC, a live TB vaccine poised to initiate efficacy trials 100 years after BCG. Vaccine. 2021;39:7277–7285.
- BioNTech. BioNtech provides update on plans to develop sustainable solutions to address infectious diseases on the African continent. 2021.
- World Health Organization. Rapid communication: tB antigen-based skin tests for the diagnosis of TB infection. (WHO/UCN/TB/2022.1). 2022.
- Villar-Hernandez R, Blauenfeldt T, Garcia-Garcia E, et al. Diagnostic benefits of adding EspC, EspF and Rv2348-B to the QuantiFERON Gold In-tube antigen combination. Sci Rep. 2020;10:13234.
- Kamariza M, Keyser SGL, Utz A, et al. Toward point-of-care detection of mycobacterium tuberculosis: a brighter solvatochromic probe detects mycobacteria within minutes. JACS Au. 2021;1:1368–1379.
- Rundell SR, Wagar ZL, Meints LM, et al. Deoxyfluoro-d-trehalose (FDTre) analogues as potential PET probes for imaging mycobacterial infection. Org Biomol Chem. 2016;14:8598–8609.
- Ordonez AA, Sellmyer MA, Gowrishankar G, et al. Molecular imaging of bacterial infections: overcoming the barriers to clinical translation. Sci Transl Med. 2019;11:11.
- Gomez GB, Siapka M, Conradie F, et al. Cost-effectiveness of bedaquiline, pretomanid and linezolid for treatment of extensively drug-resistant tuberculosis in South Africa, Georgia and the Philippines. BMJ Open. 2021;11:e051521.
- Huszar S, Chibale K, Singh V. The quest for the holy grail: new antitubercular chemical entities, targets and strategies. Drug Discov Today. 2020;25:772–780.
- Modak B, Girkar S, Narayan R, et al. Mycobacterial membranes as actionable targets for lipid-centric therapy in tuberculosis. J Med Chem. 2022;65:3046–3065.
- Fernandez-Soto P, Casulli J, Solano-Castro D, et al. Discovery of uncompetitive inhibitors of SapM that compromise intracellular survival of Mycobacterium tuberculosis. Sci Rep. 2021;11:7667.
- Silwal P, Paik S, Kim JK, et al. Regulatory mechanisms of autophagy-targeted antimicrobial therapeutics against mycobacterial infection. Front Cell Infect Microbiol. 2021;11:633360.
- Rankine-Wilson LI, Shapira T, Sao Emani C, et al. From infection niche to therapeutic target: the intracellular lifestyle of Mycobacterium tuberculosis. Microbiology (Reading). 2021;167. DOI:10.1099/mic.0.001041
- Krug S, Parveen S, Bishai WR. Host-directed therapies: modulating inflammation to treat tuberculosis. Front Immunol. 2021;12:660916.
- Nathan C. Kunkel lecture: fundamental immunodeficiency and its correction. J Exp Med. 2017;214:2175–2191.