
ABSTRACT
Streptococcus suis type 2 (SS2), a major emerging/re-emerging zoonotic pathogen found in humans and pigs, can cause severe clinical infections, and pose public health issues. Our previous studies recognized peptidyl-prolyl isomerase (PrsA) as a critical virulence factor promoting SS2 pathogenicity. PrsA contributed to cell death and operated as a pro-inflammatory effector. However, the molecular pathways through which PrsA contributes to cell death are poorly understood. Here in this study, we prepared the recombinant PrsA protein and found that pyroptosis and necroptosis were involved in cell death stimulated by PrsA. Specific pyroptosis and necroptosis signalling inhibitors could significantly alleviate the fatal effect. Cleaved caspase-1 and IL-1β in pyroptosis with phosphorylated MLKL proteins in necroptosis pathways, respectively, were activated after PrsA stimulation. Truncated protein fragments of enzymatic PPIase domain (PPI), N-terminal (NP), and C-terminal (PC) domains fused with PPIase, were expressed and purified. PrsA flanking N- or C-terminal but not enzymatic PPIase domain was found to be critical for PrsA function in inducing cell death and inflammation. Additionally, PrsA protein could be anchored on the cell surface to interact with host cells. However, Toll-like receptor 2 (TLR2) was not implicated in cell death and recognition of PrsA. PAMPs of PrsA could not promote TLR2 activation, and no rescued phenotypes of death were shown in cells blocking of TLR2 receptor or signal-transducing adaptor of MyD88. Overall, these data, for the first time, advanced our perspective on PrsA function and elucidated that PrsA-induced cell death requires its flanking N- or C-terminal domain but is dispensable for recognizing TLR2. Further efforts are still needed to explore the precise molecular mechanisms of PrsA-inducing cell death and, therefore, contribution to SS2 pathogenicity.
Introduction
Streptococcus suis is an important porcine bacterial pathogen and zoonotic agent that causes a variety of pathologies, including sudden death, septic shock, and meningitis among pigs and humans [Citation1,Citation2]. S. suis type 2 (SS2) is the most common serotype among the 29 reported S. suis serotypes based on serological reactivity differences against the antigenic capsular polysaccharide [Citation3]. Two large-scale human outbreaks of lethal SS2 infection occurred in China, with the novel symptom of streptococcal toxic shock-like syndrome (STSLS). It raises serious concerns regarding global public health. Moreover, these two sudden public health incidents challenged our understanding of sporadic severe S. suis infections in humans [Citation4].
Cell death is a conserved and intrinsic immune defence mechanism for cells that occurs in response to physical damage, active and genetically programmed pathways during normal development and physiology, and pathogen infection [Citation5,Citation6]. Many pathogenic microorganisms have been reported to induce eukaryotic cell death, by producing toxic products or secretion of virulence determinants that interact directly with key components of the death machinery [Citation7,Citation8]. Currently, the major proposed modalities of programmed cell death (PCD) with intricate types of machinery, including apoptosis, necroptosis, and pyroptosis, have been unveiled and well-defined [Citation9–11]. Apoptosis is a typical non-inflammatory programmed cell death pattern characterized by the activation of initiator caspases (caspase-2, −8, −9, and −10), as well as effector caspases (caspase-3, −6, and −7), to cleave cellular substrates [Citation12]. Apoptosis can be induced by either the intrinsic mitochondrial mechanism or the extrinsic receptor-mediated pathway. Morphologically, apoptotic cells are characterized as nuclei condensing with DNA fragmentation, membrane blebbing, cell shrinkage, mitochondrial permeability transition, and maintaining an intact plasma membrane [Citation13]. Necroptosis refers to a particularly intense form of programmed necrosis caused by stimulating death receptors with agonists or danger signals and can be discriminated from apoptosis by nuclear and organelle swelling, an increased cell volume, the plasma membrane collapses, and release of intracellular contents [Citation14]. Meanwhile, necroptosis exhibits caspase-independent release of inflammatory cellular contents and provokes mixed lineage kinase domain-like pseudokinase (MLKL) pores formation with the involvement of receptor-interacting protein kinase 1 and 3 (RIPK1 and RIPK3) signalling axis [Citation5,Citation6]. Pyroptosis is a pathway to cell death driven by plasma membrane pore formation based on the inflammasome mediated caspases (caspase-1, −11, or −4/5) activation and is inherently inflammatory accompanied by secretion of inflammatory IL-1β and IL-18, and featured with membrane rupture, cell lysis [Citation10,Citation15,Citation16]. The above three modes of PCD were initially considered to constitute mutually exclusive cellular states, whereas growing evidence has shown the extensive interplay and intricate crosstalk within pyroptosis, apoptosis, and necroptosis [Citation6,Citation9,Citation17]. Recently, PANoptosis, a novel, and highly interconnected cell death process, has been identified. PANoptosis is an inflammatory programmed cell death activated and controlled by a cytoplasmic multimeric protein complex called the PANoptosome, which can simultaneously induce pyroptosis, apoptosis, and necroptosis [Citation10,Citation18].
Peptidyl-prolyl isomerase, one of the ubiquitously distributed membrane-associated lipoproteins which prompt the cis/trans isomerization at peptidyl bonds preceding proline residues, is important for proteins secretion and maturation by handling protein folding [Citation19,Citation20]. PrsA, a parvulin-like peptidyl-prolyl isomerase, has been found as a key modulator for specific extracellular proteins secretion and contributes to bacterial infection and pathogenesis in many bacteria such as Bacillus subtilis, Staphylococcus aureus and Listeria monocytogenes [Citation21–26]. Recently, we identified a PrsA homolog which was a potential effector in the type IV-like secretion system in a highly pathogenic S. suis type 2 strain that can induce STSLS symptoms [Citation27]. The ΔprsA mutant was significantly attenuated in mice and prone to be cleared in macrophage or whole blood bactericidal environment [Citation28]. PrsA protein of SS2 was non-haemolytic, suggesting that PrsA may damage cellular integrity differently from the pore-forming cholesterol-dependent haemolysin as previously described [Citation29]. The present study aimed to investigate PrsA-induced cell death modes, activation signalling pathways, and PrsA structural domains that control cell death. TLR2 receptor-mediated PrsA recognition is also evaluated.
Material and methods
Microbe and growth conditions
The SS2 pathogenic strain 05ZYH33 and its prsA mutant strain (ΔprsA) reported previously were used in this study [Citation28]. The recombinant Escherichia coli (E. coli) BL21 strain used for PrsA protein expression was constructed and maintained in our research laboratory. S. suis and E. coli strains were cultured in Brain Heart Infusion (BHI, OXOID, England) and Luria-Bertani Broth (Landbridge, Beijing) or Agar, respectively. Bacterial cultures were grown in a shaking incubator at 37°C with shaking at 150 rpm. Kanamycin (Sigma, UK) was added to the E. coli growth medium at a final concentration of 50 g/mL before IPTG (isopropyl – D-thiogalactoside) induction.
PrsA protein and truncated fragments expression and preparation
Three functional domains and truncated proteins, including enzymatic PPIase domain (PPI, 144-243aa), N-terminal (NP, 27-243aa), and C-terminal (PC, 144-333aa), were chosen as targeted fragments for recombinant protein expression. The primers for NP, PPI, and PC domains amplification and expression were listed in . Recombinant vectors were cloned in pET-30a, and protein purification was performed with a Ni-NTA column as previously described [Citation30]. IPTG (1 mM) was used to induce full-length PrsA, NP, PPI, and PC protein recombinant expression strains for 12 hours at 16 °C. Bacterial cells were isolated and ultrasonically treated. After centrifugation, supernatant soluble protein extracts were purified using Ni-NTA agarose chromatography (Novagen, Beijing, China). The purity of proteins was examined by SDS-PAGE and confirmed by western blotting with His-tag antibodies. Endotoxin removal and quantification of the purified recombinant proteins were carried out by the commercialized Endotoxin Removal and Chromogenic LAL Endotoxin Quantitative Kits (Chinese Horseshoe Crab Reagent Manufactory Co., China). After ultrafiltration and demineralization with a 10kDa hyperfiltration tube (Millipore, USA), the corresponding protein was then filtered by a 0.22-μm filter and determined by the BCA protein detection kit (Beyotime, China) and store at −80 for future use.
Table 1. Primers used in this study.
Cell culture and cytotoxicity determination
PrsA stimulation and cytotoxicity were assessed in RAW264.7 cells, cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum. Cells were incubated for a specific period at 37 °C with 5% CO2 after being treated with either recombinant PrsA or truncated PrsA proteins. Cell viability was analysed through fluorescent microscopic observation after staining with a LIVE/DEADTM Viability/Cytotoxicity Kit (Invitrogen, Eugene, OR) and quantified by a Lactate Dehydrogenase (LDH) Cytotoxicity Assay Kit (Beyotime, China) as previously described [Citation27]. The synchronized treatments of PBS and 0.2% Triton X-100 were conducted and served as negative and positive controls for basal and 100% cell death, respectively. Cytotoxicity variation in SS2 infection groups between the WT and ΔprsA strain was also determined with the same method at a multiplicity of infection (MOI) of 50 for 4hs.
Apoptosis detection
DAPI (4,’ 6-diamidino-2-phenylindole) (Sigma-Aldrich, USA) staining for fluorescence microscopy and DNA ladders detected by 1.5% agarose gel electrophoresis were used to identify nuclei condensation and fragmentation, respectively. Staurosporine (200 nM), an apoptosis inducer, was added and treated as a positive control. Genomes from different cell groups were extracted using DNA Ladder Extraction Kit (Beyotime, China). After PrsA stimulation, cells were washed and stained with an Annexin V-FITC apoptosis assay kit (Beyotime, China) or TUNNEL staining kit (Beyotime, China) to examine the early stages of apoptosis by flow cytometry (BD, USA) and fluorescence detection, respectively [Citation31].
Detection of pyroptosis and necroptosis
The cells were pre-incubated for 1 hour at 37°C with an irreversible inhibitor of Ac-YVAD-cmk (100 μM, Selleck, China) targeting the cysteine protease caspase-1, as previously described [Citation32] before being stimulated with PrsA protein (75 μg/ml) or infected with SS2 cells. Control groups consisted of cells treated with only recombinant proteins or SS2 cells, a particular inhibitor, Triton X-100, and PBS. The viability of the cells was assessed using the methods mentioned above. Small molecule inhibitors of Necrostatin-1 (Nec-1, 20 μM, Selleck, China) and Necrosulfonamide (NSA, 10 μM, Selleck, China) that functioned specifically to RIPK-1 and MLKL in the necroptosis pathway were used to detect necroptotic cell death [Citation33,Citation34].
Ultra-structural morphological observation
RAW264.7 cells were washed twice with PBS and fixed in 2.5% glutaraldehyde for 12 hours after PrsA protein stimulation. Samples for scanning and transmission electron microscopy were prepared as previously described [Citation28]. Scanning and transmission electron microscopic images were observed in Hitachi Model SU-8010 and H-7650 (Hitachi, Japan), respectively.
Immunoblot analysis
Cell lysates were extracted and dissolved in a RIPA lysis buffer (Beyotime, China), then mixed with a 6× protein loading buffer before being run on a 12% SDS-PAGE. Proteins were then transferred to a PVDF membrane, blocked with 5% skim milk, washed with TBST, and incubated overnight at 4°C with the corresponding primary antibodies for the detection of LC3-B (Sigma, St. Louis, USA), caspase-1 (Proteintech, CHINA), IL-1β (Proteintech), GSDMD (Peoteintech), pMLKL (Proteintech), MyD88 (Cell signalling technology, USA) and TLR2 (Cell signalling technology). Actin was served and detected as an internal control with a β-actin specific antibody (Proteintech). The membrane was washed with TBST buffer and treated with the appropriate secondary antibody conjugated to horseradish peroxidase as previously described [Citation35]. Executed protein blots were developed by ECL detection reagents (GlpBio, USA) and visualized with a chemiluminescent imaging system (Thermofisher, USA).
Cytokines release
Quantitative real-time PCR (qPCR) analysis, as previously described [Citation27], was used to determine the expression of inflammatory cytokines TNFα, IL-6, and IL-1β in cells treated with PrsA truncated proteins at a concentration of 50 μg/ml for 4 hrs. The data was collected using an Agligent MX3000P qPCR equipment (Stratagene, USA), and the fold changes were assessed using a 2−ΔΔCt approach. The actin gene was employed as an internal control for normalization.
Confocal immunofluorescence assay
PrsA protein-induced targeted cell death was imaged by Confocal fluorescence microscopy. Briefly, RAW264.7 cells (2–2.5 × 105 cells) cultured in DMEM medium were incubated with PrsA protein (50 μg/ml) for 4 hours, then washed, and labelled with primary rabbit anti-PrsA antibodies, followed by Alexa Fluor 488 goat anti-rabbit Ab. DAPI and Alexa Fluor 568 phalloidin (Invitrogen) was used to stain cell nuclei and cytoplasmic actin. Samples were then analysed by the FV-1000 imaging system on an IX-81 confocal microscope (Olympus, Germany).
RNA interference
A small RNA interference strategy was used to knock down the expression level of TLR-2 and myeloid differentiation factor 88 (MyD88). Specifically, RAW264.7 macrophages cultured in 24-well plate were transfected with the commercially available siRNA molecules synthesized from GenePharma () by Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. After 12 hs siRNAs incubation with antibiotics free Opti-MEM, the percent interference efficacy was determined and confirmed by qPCR and western blotting analysis. Cells transfected with ineffective interference RNAs (siNC) were conducted as a negative control. Following TLR2 or MyD88 knocked-down assays, the cells were then washed and subjected to further cytotoxic PrsA stimulation with DMEM containing 10% FBS. The viability of the cells were determined as described above.
Statistical analysis
All the experimental results were expressed as mean ± square deviation. A two-tailed, unpaired Student’s t-test was used for statistical difference analysis. A P value of < 0.05 or < 0.01 was considered significant and highly significant, respectively. All cytotoxicity determination assays were repeated at least three times. The figures were depicted by GraphPad prism version-5.
Results
PrsA-induced cell death without activation of autophagy and apoptosis pathway
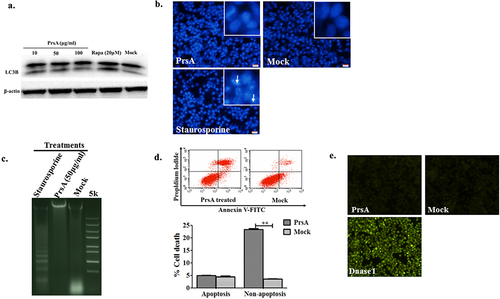
The activation of LC3B marker molecule within autophagosome, nuclear condensation and DNA fragmentation, and typical feature of membrane changes in apoptosis were evaluated after recombinant PrsA stimulation to determine whether the milder patterns of autophagy and apoptosis were involved in cell death or not. The results found that rapamycin (20 μM) can significantly induce cellular autophagy, while PrsA protein in different concentrations could not induce LC3B activation (). The cells induced by staurosporine (200 nM) showed typical apoptosis features such as nucleus pyknosis, dense staining and brightness, distortion of some nuclei, and aggregation or fragmentation of chromatin to form a crescent, splay, and petal shapes. However, no observed cellular changes and increased apoptotic cell ratio were shown in groups induced with PrsA in comparison to the negative control groups (). Tunnel staining for genomic DNA fragmentation confirmed the non-apoptotic result in cells upon PrsA stimulation ().
Figure 1. PrsA induced cell death without autophagy and early apoptosis pathway activation in RAW264.7. a. Autophagy activation detected by western blotting with LC3B molecules. Cells were stimulated with PrsA in different concentrations or Rapamycin for 4 hs. Whole-cell lysates were subjected to western blotting. b. Apoptosis identification by chromatin staining. Cells were stimulated with PrsA (50 μg/ml) for 4 hrs. Then, cells nuclei were stained with DAPI. Increased apoptotic cells with fragmented and brighter chromatin were indicated with white arrows in the positive control group treated with staurosporine (200 nM). Magnification, ×400. c. Agarose gel electrophoresis analysis of DNA fragmentation in cells genome treated as b. d & e. Apoptotic cell death determined by flow cytometric analysis and TUNNEL staining in RAW264.7 cells after PrsA protein stimulation. DNase I was conducted as positive control for TUNNEL assay. The asterisk of “**” indicated significant at P < 0.01.
Pyroptosis and necroptosis involved in cell death after PrsA induction
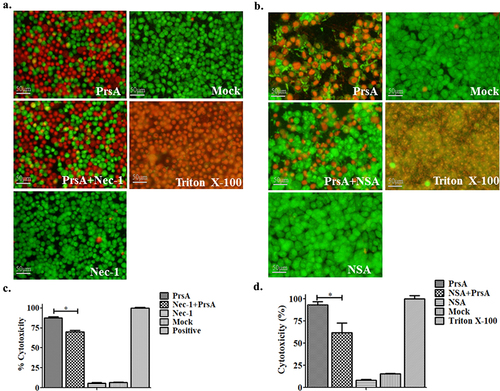
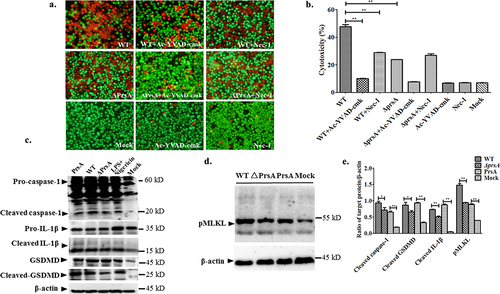
Small molecule inhibitors targeting critical signal pathways of pyroptosis and necroptosis were used to examine the possible effect of these inhibitors after recombinant PrsA stimulation. The results of pyroptosis and necroptosis involved in cell death after PrsA induction showed that 82.9% of the cell death prompted by PrsA can be significantly reduced to 27.3% in the group with preliminary incubation of the irreversible inhibitor of Ac-YVAD-cmk targeting the cysteine protease caspase-1 (P < 0.01) (), which was important for inflammasome activation. These results demonstrated the caspase-1 signalling pathway mediated in PrsA cytotoxic effect. Similar but weaker inhibition of cell death rate induced by PrsA proteins with Nec-1 and NSA inhibitors, which blocked specific to the RIPK-1 and MLKL in the necroptosis pathway as shown in ().
Figure 2. Pyroptosis was involved in cell death induced by the PrsA protein. a & b. Cytotoxicity determination of PrsA at different concentrations in RAW264.7 cells detected by fluorescent observation of live (green) and dead (red) and LDH quantification kit after 1 h or 2hs incubation. c. Fluorescent observation of RAW264.7 cells treated with PrsA only (75 μg/ml) or PrsA with a cell-permeable, selective, and irreversible inhibitor of the cysteine protease caspase-1 (Ac-YVAD-cmk, 100 μM) for 4 hs. Bar scales were 50 μm. d. Quantitative detection of cytotoxicity in cells subjected with or without Ac-YVAD-cmk inhibitor by an LDH release measurement kit. Cellular lysis buffer of triton X-100 (0.2%) was treated and considered 100% death for cytotoxicity calculation. DMEM medium treatments were used as a negative control. The asterisk of “**” indicated significant at P < 0.01.
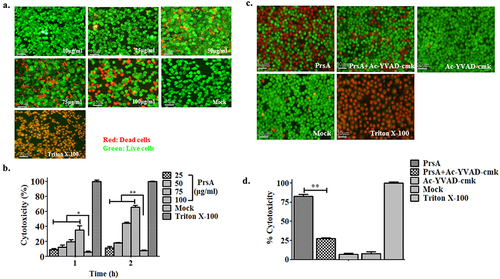
Figure 3. PrsA-induced cell death partially by necroptosis signalling pathway. a & b. Fluorescent observation of live (green) and dead (red) RAW264.7 or HEp-2 cells treated with PrsA only (75 μg/ml) or PrsA with specific inhibitor Nec-1 (20 μM) and NSA (10 μM) for 4 hs targeted RIPK-1 and MLKL, respectively, in necroptosis pathway. Bar scales indicated 50 μm. c & d. Evaluation of cytotoxicity with LDH release assay kit. RAW264.7 or HEp-2 cells were treated as panels a & b.
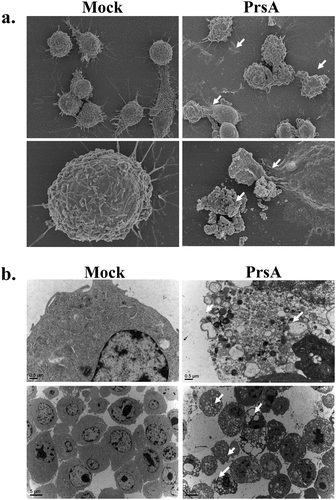
Scanning and transmission electron microscopy were used to observe the ultra-structural morphological changes among samples of RAW264.7 cells stimulated by PrsA. Typically, normal cells without PrsA treatment were intact and regular in shape with a smooth and flat surface and boundary of the cell membrane, clear mitochondrial and endoplasmic reticulum structure, and a dense and uniform nucleus. Whereas the cells in the PrsA stimulation group were morphologically characterized by non-apoptotic cell death with rupture membrane, cell degeneration and vacuolization, organelles swelling, and the release of cellular contents and was accompanied by cell fragmentation ().
Figure 4. Electronic microscopy observation.
S. suis infection assays of the WT and ΔprsA strains were conducted to verify the putative lethal function of PrsA effector to host cell death. The prsA gene mutation would lead to a significant decrease in cell cytotoxicity induced by ΔprsA strain (P < 0.01). The Ac-YVAD-cmk and Nec-1 inhibitors explicitly targeted to the pyroptosis and necroptosis pathways could alleviate the cytotoxic effect caused by the WT strain, respectively (P < 0.01) (). Furthermore, after PrsA stimulation, there was an observed activation of critical cell signalling molecules such as caspase-1, GSDMD and IL-1β in pyroptosis, and pMLKL in the necroptosis pathway. Differential expression levels of cleaved caspase-1, IL-1β, and pMLKL between the WT and prsA strains of SS2 infection groups were determined using western blotting assay (). Significant decreases of cleaved caspase-1, IL-1β, and pMLKL were observed in the PrsA deficient strain of SS2.
Figure 5. Confirmation of lethal function and potential cell death patterns activated by PrsA in RAW264.7 cells infected with SS2.
PrsA flanking N- and C-terminal responsible for PrsA function in inducing cell death and inflammation
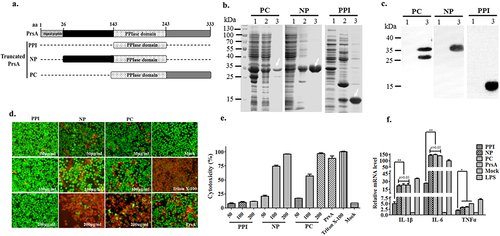
In order to identify PrsA’s essential functional domains that cause cell death, three predicted and potentially truncated segments, the enzymatic PPIase domain (PPI), N-terminal (NP), and C-terminal (PC) domains fused with PPIase (), were expressed as targeted fragments for cytotoxicity evaluation. The results showed that these three truncated PPI, NP, and PC proteins were successfully expressed and purified in a soluble form (). The proteins were identified by western blotting with a His tag (). Cell viability assays showed that different concentrations of enzymatic PPIase domain protein PPI did not induce cell death with a basal cytotoxic effect compared to the control group (P > 0.05). However, in RAW264.7 cells, the NP or PC truncated protein containing PPIase can cause cell death in a concentration gradient dependent manner (). The quantitative detection results of LDH tests were consistent with fluorescence observation (). Furthermore, significant activation of the inflammatory response induced by NP or PC truncated proteins was observed in RAW264.7, as shown by an increase in the expression of IL-1β, IL-6, and TNFα cytokines (). Their proinflammatory effects were close to complete PrsA group, while the PPI domain induced subtle cytokines release. All these data confirmed that PrsA flanking N- and C-terminal but single enzymatic PPIase domain were mainly responsible for PrsA function in causing cell death and inflammation.
Figure 6. PrsA flanking N- and C-terminal but not enzymatic PPIase domains were essential for PrsA function in inducing cell death and inflammation. a. Amino acid regions of three fragments of PrsA truncated proteins of PPIase, NP, and PC. b. Prokaryotic expression and purification of truncated PrsA proteins by SDS-PAGE. Lane 1, recombinant E. coli cell lysate from cultures before IPTG induction; Lane 2, e. coli cell lysate from IPTG-induced cultures; Lane 3, purified PrsA proteins. c. Western blotting of Lane 1 and Lane 3 of panel B using anti-His monoclonal antibody. d. Fluorescent observation of live (green) and dead (red) RAW264.7 cells treated with three truncated PrsA proteins PPI, NP, and PC at different concentrations, respectively. PrsA (200 μg/ml) and DMEM medium treatments were used as positive and negative controls. Cellular lysis buffer of triton X-100 (0.2%) was treated and considered 100% death for cytotoxicity calculation. The bar scales indicated were 50 μm. e. Evaluation of cytotoxicity induced by PrsA truncated proteins PPI, NP, and PC with LDH concentration determination kit. f. Relative mRNA levels of proinflammatory cytokines IL-1β, IL-6, and TNF-α, as determined by qPCR. The results were presented as fold induction relative to non-infected cells against β-actin gene expression using 2−Ct method. RAW264.7 cells were stimulated by 50 μg/ml of PrsA proteins, PPI, NP and PC for 4 hs. Lipopolysaccharide (LPS) was used at 400 ng/ml as a positive control. Asterisks of “*” and “**” indicated significant at P < 0.05 or P < 0.01, respectively.
PrsA-induced cell death independent of TLR2/MyD88 pathway activation
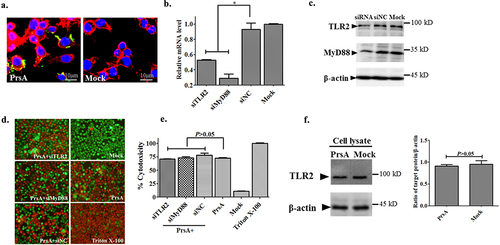
Cellular colocalization analysis by immunofluorescence microscopy indicated that the PrsA protein of SS2 could interact with host cells, mainly anchored on the cell surface, though partially endocytosed into the cytoplasm (). RNA interference assays were used to examine PrsA’s lethal function when the TLR2 signal pathway was blocked or knocked down to show the possible role of the recognition receptor TLR2 in cell death pathways induced by the probable lipoprotein PrsA. The results showed that the siRNA molecules’ specific to the TLR2 and the critical downstream adapter of MyD88 was effective and downregulated about 45% to 70% of the expression of the corresponding genes in RAW264.7 cells, respectively (), as compared to the negative control siRNA groups. The interference efficacy of corresponding proteins was confirmed by western blotting analysis (). Surprisingly, the knocking down of TLR2 or MyD88 could still not compensate for the cell-death-inducing effect caused by the PrsA protein. No significant differences in cytotoxicity and cell morphology changes were observed in PrsA stimulation groups among specific treatments of siTLR2, siMyD88, and siNC molecules (). Furthermore, there were no detected expression changes of TLR2 in the cell lysates after the PrsA protein stimulus as compared with the PBS control group after western blotting analysis ().
Figure 7. TLR2/MyD88 pathway was not involved in cell death induced by PrsA. a. Immunofluorescence observation of RAW264.7 cells and PrsA protein interaction. PrsA proteins were labelled with rabbit anti-PrsA poly-Ab with a second antibody Alexa Fluor 488 goat anti-rabbit Ab. Cell nuclei and cytoplasmic actin were stained with DAPI and Alexa Fluor 568 phalloidin. b. qPCR analysis of tlr2 and myd88 genes expression level in RAW264.7 after RNA interference. c. Western blotting analysis of targeted proteins expression after RNA interference. The siRNA and siNC meant specific small RNA interference molecules of TLR2, MyD88, and negative control treatment. d. Fluorescent observation of cell viability between normal RAW264.7 cells and TLR2 or MyD88 molecule downregulated cells after cytotoxic PrsA (200 μg/ml) treatment. Triton X-100 (0.2%) and DMEM medium treatments were used as positive and negative controls. e. Cytotoxicity determination with LDH release detection from the supernatants of RAW264.7 cells treated as panel d. f. Western blotting analysis of TLR2 activation after PrsA stimulation for 4hs in RAW264.7 cells as compared with PBS control. Densitometric difference of TLR2 was also analysed. The asterisk of “*” indicated significant at P < 0.05.
Discussion
Pathogens can facilitate infection by the exportation or secretion of diverse virulence factors to the extracellular environment [Citation36]. PrsA is ubiquitously distributed in Gram-positive bacteria and functions as a chaperone for processing variable periplasmic proteins and extracellular secretion factors to maturation. The post-translocation essence of this protein has foldase activity mediated in the folding and stability of secreted effectors as well as contributing to virulence regulation [Citation22,Citation37–39]. The surface-anchored and secreted protein of PrsA in S. suis type 2 is found pro-inflammatory and cytotoxic to host cells [Citation27]. PrsA itself is also a virulence factor that can mediate SS2 infection and pathogenesis by modulating the secretion and translocation of suilysin and other important adhesion molecules, such as GAPDH and enolase [Citation30]. However, the cytotoxic function of PrsA in SS2 and its mechanism leading to cell death are not clearly understood.
In this study, we combine multiple molecular activation and detection techniques to explore the patterns of cell death with possible activation pathways and the probable function domains of PrsA leading to cell death. With a range of cell death modes investigation, it was found that the PrsA protein of SS2 could participate in cell death mainly through pyroptosis. The lethal effect of PrsA on RAW264.7 cells can be markedly alleviated shown in , with the preincubation of Ac-YVAD-cmk inhibitor specific to inflammasome activation. PrsA also mediated cell death accompanied by the participation of the necroptosis pathway. Inhibitors targeting the critical RIPK-1 and MLKL molecules in necroptosis can reduce the cell death rate induced by PrsA to a certain extent in , though the effect is not as obvious as that of Ac-YVAD-cmk. Electron microscopic observation of morphological changes in dead cells has been considered one of the gold standards for determining the manner of cell death. The typical phenotypic changes of cell membrane rupture, the release of cytoplasmic contents, and severe inflammatory reaction were reflected in the results of electron microscope observation (). All these features were consistent with the morphological characteristics of pyroptosis and necroptosis. The cell death patterns of pyroptosis and necroptosis induced by PrsA were confirmed in SS2 microbial infection tests. The critical inhibitors specific to the pyroptosis and necroptosis signalling pathways could relieve the lethal cytotoxicity stimulated by the WT strain of SS2. The prsA gene deletion would lead to a significant decrease in cell cytotoxicity as compared to the WT strain (). This may be owing to the down-regulation and translocation of the secretion level of suilysin with other virulence factors when the prsA gene was mutant [Citation28]. The cytotoxic suilysin itself was found to be essential for NLRP3 inflammasome activation and triggering cytokine storm contributes to STSLS in SS2 [Citation35]. Furthermore, the secreted peptidyl-prolyl cis, trans-isomerase of PrsA in H. pylori, was found to induce gastric Th17 response, promote proinflammatory response, and execute gastric epithelial cell apoptosis [Citation40,Citation41]. So the inflammatory effect triggered by PrsA may help to induce cell death [Citation16,Citation42].
After the expression and purification of PrsA truncated proteins, it was revealed that the PPIase enzymatic domain is not directly involved in cell death and pro-inflammatory effect. PrsA flanking N- or C-terminal domain of SS2 was indispensable for inducing cell death, as shown in . Similar findings were observed in the research of PrsA functional domains investigation in B. subtilis and L. monocytogenes [Citation23,Citation43,Citation44]. The enzymatic activity of the PPI domain within PrsA in B. subtilis may be dispensable for the proper folding of multiple repertoires of secreted proteins in vivo. In L. monocytogenes, the PrsA2 flanking N- and C-terminal domains were critical for protein chaperone functions as well as for the complementation of haemolytic activity, phospholipase activity, and plaque formation. While PPIase activity within PrsA2 was insufficient to restore numerous in vitro defects, the PPIase activity appeared to enhance PrsA2 function, contributed to full bacterial virulence, and mediated in optimal resistance to β-lactam antibiotics [Citation43,Citation45]. The accessory PPI domain of SS2 itself participated in mediating subtle inflammation but contributed to inducing substantial cytokines expression with chimeric proteins of NP and PC domains. The difference in lethal effects among variable PrsA proteins may due to their divergent capability to induce inflammation response.
PrsA is a membrane-exposed lipoprotein that modulates several secreted proteins in the microenvironment within the cytoplasmic membrane-cell wall interface at a post-translocational folding level [Citation22,Citation46,Citation47]. It is well known that the pathogen-associated molecular patterns (PAMPs) of a typical microbial ligand of lipoproteins can be recognized and activated by the host innate immune receptor of TLR2 expressed on the cell surface and then interact with TLR1 or TLR6 to form heterodimers [Citation48,Citation49]. The receptor clusters would recruit adaptors with mainly MyD88 molecule to initiate downstream defensive immune responses, including the secretion of pro-inflammatory cytokines [Citation50]. Protein cell interaction analysis illustrated that the probable surface anchored essence of PrsA in RAW264.7 and prompted us to speculate if the TLR2 signalling pathway played a role in cell death that was stimulated by PrsA. However, the comprehensive experimental results in , contrary to our expectation, confirmed that TLR2/MyD88 pathway cannot be activated and was not involved in the lethal effect induced by the PrsA protein.
Based on our findings, we clarified the cell death modes of pyroptosis and necroptosis induced by prsA factor in SS2 and examined the indispensable lethal domains of N- or C-termini fused with PPIase within PrsA. These would help to provide novel insights for PrsA biological functions research. Future studies should focus on the investigation of the individual recognizing and interacting proteins as well as the precise mechanisms upon PrsA induction within the cell host. The putative chaperone function with the foldase substrates analysis and the relationship between cell death and inflammatory effects still need further investigations.
Authors’ Contribution
XWJ and MY: Conceptualization, methodology, formal analysis, writing – review and editing; GJY and LXZ: formal analysis, data curation, writing – review and editing; AS, DBZ and LHZ: formal analysis and data curation. All authors have read and agreed to the published version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data Availability statement
The authors confirmed that all data supporting the findings of this study are available within the article.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Segura M. Streptococcus suis research: progress and challenges. Pathogens. 2020;9(9):707. doi: 10.3390/pathogens9090707
- Segura M, Fittipaldi N, Calzas C, et al. Critical Streptococcus suis virulence factors: are they all really critical? Trends Microbiol. 2017;25(7):585–13. doi: 10.1016/j.tim.2017.02.005
- Xia X, Qin W, Zhu H, et al. How Streptococcus suis serotype 2 attempts to avoid attack by host immune defenses. J Microbiol Immunol Infect. 2019;52(4):516–525. doi: 10.1016/j.jmii.2019.03.003
- Haas B, Grenier D. Understanding the virulence of Streptococcus suis: a veterinary, medical, and economic challenge. Med Mal Infect. 2018;48(3):159–166. doi: 10.1016/j.medmal.2017.10.001
- Yan G, Elbadawi M, Efferth T. Multiple cell death modalities and their key features (review). World Acad Sci J. 2020. doi: 10.3892/wasj.2020.40
- Ashida H, Mimuro H, Ogawa M, et al. Cell death and infection: a double-edged sword for host and pathogen survival. J Cell Bio. 2011;195(6):931–942. doi: 10.1083/jcb.201108081
- Labbé K, Saleh M. Cell death in the host response to infection. Cell Death Diff. 2008;15(9):1339–1349. doi: 10.1038/cdd.2008.91
- Stephenson HN, Herzig A, Zychlinsky A. Beyond the grave: When is cell death critical for immunity to infection? Curr Opin Immunol. 2016;38:59–66. doi: 10.1016/j.coi.2015.11.004
- Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106–1121. doi: 10.1038/s41423-020-00630-3
- Wang Y, Kanneganti TD. From pyroptosis, apoptosis and necroptosis to PANoptosis: a mechanistic compendium of programmed cell death pathways. Computat Struct Biotechnol J. 2021;19:4641–4657. doi: 10.1016/j.csbj.2021.07.038
- Tang D, Kang R, Berghe TV, et al. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347–364. doi: 10.1038/s41422-019-0164-5
- Fink Susan L, Apoptosis CBT. Pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005
- Krysko DV, Vanden Berghe T, D’Herde K, et al. Apoptosis and necrosis: detection, discrimination and phagocytosis. Methods. 2008;44(3):205–221. doi: 10.1016/j.ymeth.2007.12.001
- Wu W, Liu P, Li J. Necroptosis: an emerging form of programmed cell death. Crit Rev Oncol Hematol. 2012;82(3):249–258. doi: 10.1016/j.critrevonc.2011.08.004
- Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity. 2019;50(6):1352–1364. doi: 10.1016/j.immuni.2019.05.020
- Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nature Rev Microbiol. 2009;7(2):99–109. doi: 10.1038/nrmicro2070
- Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Diff. 2019;26(1):99–114. doi: 10.1038/s41418-018-0212-6
- Place DE, Lee S, Kanneganti TD. Panoptosis in microbial infection. Curr Opin Microbiol. 2021;59:42–49. doi: 10.1016/j.mib.2020.07.012
- Schmidpeter PAM, Schmid FX. Prolyl isomerization and its catalysis in protein folding and protein function. J Mol Biol. 2015;427(7):1609–1631. doi: 10.1016/j.jmb.2015.01.023
- Schiene-Fischer C. Multidomain peptidyl prolyl cis/trans isomerases. Biochim Biophys Acta Gen Subj. 2015;1850(10):2005–2016. doi: 10.1016/j.bbagen.2014.11.012
- Ünal Can M, Steinert M. Microbial peptidyl-prolyl cis/trans isomerases (PPIases): virulence factors and potential alternative drug targets. Microbiol Mol Biol Rev. 2014;78(3):544–571. doi: 10.1128/MMBR.00015-14
- Sarvas M, Harwood CR, Bron S, et al. Post-translocational folding of secretory proteins in gram-positive bacteria. Biochim Biophys Acta, Mol Cell Res. 2004;1694:311–327. doi: 10.1016/j.bbamcr.2004.04.009
- Vitikainen M, Lappalainen I, Seppala R, et al. Structure-function analysis of PrsA reveals roles for the parvulin-like and flanking N- and C-terminal domains in protein folding and secretion in Bacillus subtilis. J Biol Chem. 2004;279(18):19302–19314. doi: 10.1074/jbc.M400861200
- Alonzo F 3rd, Port GC, Cao M, et al. The posttranslocation chaperone PrsA2 contributes to multiple facets of Listeria monocytogenes pathogenesis. Infect Immun. 2009;77(7):2612–2623. doi: 10.1128/IAI.00280-09
- Leuzzi R, Serino L, Scarselli M, et al. Ng-MIP, a surface-exposed lipoprotein of Neisseria gonorrhoeae, has a peptidyl-prolyl cis/trans isomerase (PPIase) activity and is involved in persistence in macrophages. Mol Microbiol. 2005;58(3):669–681. doi: 10.1111/j.1365-2958.2005.04859.x
- Guo L, Wu T, Hu W, et al. Phenotypic characterization of the foldase homologue PrsA in Streptococcus mutans. Mol Oral Microbiol. 2013;28:154–165. doi: 10.1111/omi.12014
- Jiang X, Yang Y, Zhou J, et al. Roles of the putative type IV-like secretion System key component VirD4 and PrsA in pathogenesis of Streptococcus suis type 2. Front Cell Infect Microbiol. 2016;6:172. doi: 10.3389/fcimb.2016.00172
- Liu H, Fu H, Jiang X, et al. PrsA contributes to Streptococcus suis serotype 2 pathogenicity by modulating secretion of selected virulence factors. Vet Microbiol. 2019;236:108375. doi: 10.1016/j.vetmic.2019.07.027
- Heuck AP, Moe PC, Johnson BB. The cholesterol-dependent cytolysin family of gram-positive bacterial toxins. In: Harris J, editor. Cholesterol binding and cholesterol transport proteins: structure and function in health and disease. DordrechtNetherlands: Springer Netherlands; 2010. pp. 551–577. doi: 10.1007/978-90-481-8622-8_20
- Jiang X, Yang Y, Zhou J, et al. Peptidyl isomerase PrsA is surface-associated on Streptococcus suis and offers cross-protection against serotype 9 strain. FEMS Microbiol Lett. 2019;366(2): doi: 10.1093/femsle/fnz002
- Wallberg F, Tenev T, Meier P. Analysis of apoptosis and necroptosis by fluorescence-activated cell sorting. Cold Spring Harb Protoc. 2016;2016:db prot087387. doi: 10.1101/pdb.prot087387
- Goldmann O, Sastalla I, Wos-Oxley M, et al. Streptococcus pyogenes induces oncosis in macrophages through the activation of an inflammatory programmed cell death pathway. Cell Microbiol. 2009;11(1):138–155. doi: 10.1111/j.1462-5822.2008.01245.x
- Cao L, Mu W. Necrostatin-1 and necroptosis inhibition: pathophysiology and therapeutic implications. Pharmacol Res. 2021;163:105297. doi: 10.1016/j.phrs.2020.105297
- de Almagro MC, Vucic D, de Almagro MC. Necroptosis: pathway diversity and characteristics. Semin Cell Dev Biol. 2015;39:56–62. doi: 10.1016/j.semcdb.2015.02.002
- Lin L, Xu L, Lv W, et al. An NLRP3 inflammasome-triggered cytokine storm contributes to streptococcal toxic shock-like syndrome (STSLS). PLOS Pathogens. 2019;15(6):e1007795. doi: 10.1371/journal.ppat.1007795
- Dal Peraro M, van der Goot FG. Pore-forming toxins: ancient, but never really out of fashion. Nat Rev Microbiol. 2016;14(2):77–92. doi: 10.1038/nrmicro.2015.3
- Henderson B, Allan E, Coates Anthony RM. Stress wars: the direct role of host and bacterial molecular chaperones in bacterial infection. Infect Immun. 2006;74(7):3693–3706. doi: 10.1128/IAI.01882-05
- Cahoon LA, Freitag NE. Listeria monocytogenes virulence factor secretion: don’t leave the cell without a chaperone. Front Cell Infect Microbiol. 2014;4:13. doi: 10.3389/fcimb.2014.00013
- Ernst K, Schnell L, Barth H. Host cell chaperones Hsp70/Hsp90 and peptidyl-prolyl cis/trans isomerases are required for the membrane translocation of bacterial adp-ribosylating toxins. In: Barth H, editor. Uptake and trafficking of protein toxins. Cham: Springer International Publishing; 2017. pp. 163–198. doi: 10.1007/82_2016_14
- Basak C, Pathak SK, Bhattacharyya A, et al. The secreted peptidyl prolyl cis, trans-isomerase HP0175 of Helicobacter pylori induces apoptosis of gastric epithelial cells in a TLR4- and apoptosis signal-regulating kinase 1-dependent manner. J Immunol. 2005;174(9):5672–5680. doi: 10.4049/jimmunol.174.9.5672
- Kundu M, Henderson B, editor. Moonlighting Cell Stress Proteins in Microbial Infections. Dordrecht: Springer; 2013. p. 81–91. doi: 10.1007/978-94-007-6787-4_5
- Karki R, Kanneganti TD. The ‘cytokine storm’: molecular mechanisms and therapeutic prospects. Trends Immunol. 2021;42(8):681–705. doi: 10.1016/j.it.2021.06.001
- Alonzo F 3rd, Xayarath B, Whisstock JC, et al. Functional analysis of the Listeria monocytogenes secretion chaperone PrsA2 and its multiple contributions to bacterial virulence. Mol Microbiol. 2011;80(6):1530–1548. doi: 10.1111/j.1365-2958.2011.07665.x
- Hyyrylainen HL, Marciniak BC, Dahncke K, et al. Penicillin-binding protein folding is dependent on the PrsA peptidyl-prolyl cis-trans isomerase in Bacillus subtilis. Mol Microbiol. 2010;77(1):108–127. doi: 10.1111/j.1365-2958.2010.07188.x
- Jousselin A, Renzoni A, Andrey Diego O, et al. The posttranslocational chaperone lipoprotein prsa is involved in both glycopeptide and oxacillin resistance in staphylococcus aureus. Antimicrob Agents Chemother. 2012;56(7):3629–3640. doi: 10.1128/AAC.06264-11
- Lin M-H, Li C-C, Shu J-C, et al. Exoproteome profiling reveals the involvement of the foldase prsa in the cell surface properties and pathogenesis of Staphylococcus aureus. Proteomics. 2018;18(5–6):1700195. doi: 10.1002/pmic.201700195
- Wiemels Richard E, Cech Stephanie M, Meyer Nikki M, et al. An Intracellular peptidyl-prolyl cis/trans isomerase is required for folding and activity of the staphylococcus aureus secreted virulence factor nuclease. J Bacteriol. 2016;199:e00453–16. doi: 10.1128/JB.00453-16
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34(5):637–650. doi: 10.1016/j.immuni.2011.05.006
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863
- Kumar S, Ingle H, Prasad DV, et al. Recognition of bacterial infection by innate immune sensors. Crit Rev Microbiol. 2013;39:229–246. doi: 10.3109/1040841X.2012.706249