
ABSTRACT
Heartland virus (HRTV), an emerging tick-borne pathogenic bunyavirus, has been a concern since 2012, with an increasing incidence, expanding geographical distribution, and high pathogenicity in the United States. Infection from HRTV results in fever, thrombocytopenia, and leucopenia in humans, and in some cases, symptoms can progress to severe outcomes, including haemorrhagic disease, multi-organ failure, and even death. Currently, no vaccines or antiviral drugs are available for treatment of the HRTV disease. Moreover, little is known about HRTV-host interactions, viral replication mechanisms, pathogenesis and virulence, further hampering the development of vaccines and antiviral interventions. Here, we aimed to provide a brief review of HRTV epidemiology, molecular biology, pathogenesis and virulence on the basis of published article data to better understand this virus and provide clues for further study.
Introduction
Heartland virus (HRTV) is a novel tick-borne pathogenic bunyavirus and was identified in 2009 from two Missouri farmers who were hospitalized for a severe acute disease characterized by fever, fatigue, diarrhoea, thrombocytopenia, and leucopenia [Citation1]. HRTV is genetically related to the severe fever with thrombocytopenia syndrome virus (SFTSV), another highly pathogenic bunyavirus emerging in East Asia (including China and neighbouring countries) [Citation2]. According to the latest International Committee on Taxonomy of Viruses (ICTV) report, HRTV is classified into Bandavirus genus (Phenuiviridae family, Bunyavirales order), as the second representative species of bandaviruses following SFTSV [Citation3]. Due to expanding geographical distribution, increasing infection cases and the potential high pathogenicity, bandaviruses have posed threats to public health and raised more concerns. HRTV is regarded as a Category C Priority Pathogen by the National Institute of Allergy and Infectious Diseases (NIAID). However, to date, there are no specific antiviral drugs and vaccines to control the infections of HRTV and HRTV-related viruses. Despite ongoing researches, the interactions of HRTV-host and the pathogenesis of HRTV are largely unknown, hindering the development of prevention and therapies. Here, we summarize the recent progress on HRTV and provide some hints for further study.
Epidemiology
According to the Centers for Disease Control and Prevention (CDC), as of November 2022, more than 60 cases of HRTV infections have been reported in 14 states in the Midwestern, Northeastern, and Southern United States (https://www.cdc.gov/heartland-virus/statistics/index.html). The Lone Star tick (Amblyomma americanum) has been implicated as the main vector of HRTV, if not the only vector of the virus [Citation4–10], because most patients were exposed to ticks in the 2 weeks before illness onset, and HRTV could be isolated from the Lone Star tick [Citation11,Citation12]. In addition, Marvin et al. found that larval and nymphal Lone Star ticks can be infected with HRTV following immersion of HRTV suspensions [Citation6]. Moreover, transstadial transmission, nonviremic transmission, vertical transmission, and horizontal transmission of HRTV infection also occur under laboratory conditions, further demonstrating that the Lone Star tick is the vector of HRTV [Citation6]. Recently, the Asian longhorned tick (Haemaphysalis longicornis), a vector of SFTSV, has invaded several states in the United States and can transmit HRTV under laboratory conditions [Citation13,Citation14]. Thus, the Asian longhorned tick is likely a potential vector for HRTV transmission. Serological investigations indicated that many domestic and wild animals (white-tail deer, raccoons, horses, dogs, opossums, moose, and coyotes) may be potential reservoir hosts of HRTV and the geographic range of seropositive animals was almost consistent with that of the Lone Star tick [Citation15–17]. In addition, a retrospective survey showed that vertebrate animals with HRTV-specific neutralizing antibodies can be found in several states where no human infection cases were recorded, indicating that the geographic range of HRTV distribution is likely beyond where the cases occur [Citation15]. The modes of animal-to-human and human-to-human transmission in SFTSV infection should be watched out in HRTV study and HRTV disease monitoring [Citation18–20], although no cases have been reported to date.
Molecular virology
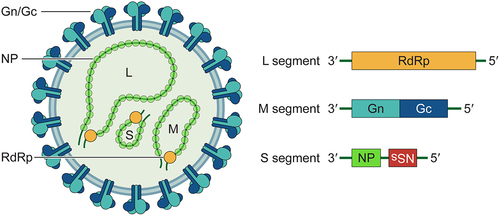
Similar to other members of Phenuiviridae family, the HRTV genome comprises three single-stranded negative-sense RNA segments designated as large (L), medium (M), and small (S). The L segment encodes the RNA-dependent RNA polymerase (RdRp), the M segment encodes the polyprotein precursor that is further cleaved into two glycoprotein subunits, Gn and Gc, and the S segment utilizes an ambisense coding strategy to encode the nucleoprotein (NP) and the nonstructural protein (NSs). The NSs proteins of multiple pathogenic bunyaviruses, including HRTV, have been considered as pivotal virulence factors likely contributing to the pathogenesis, which will be discussed in detail in the following section. NP wraps viral genomic RNA and interacts with RdRp to form ribonucleoprotein complexes (RNPs), which are core components of the virion. A schematic of the virion and genome segments is shown in .
Figure 1. Schematic diagram of the HRTV virion and genome organization. HRTV particles are enveloped with the Gn/Gc heterodimers incorporated on the surface. The tripartite single-stranded RNA genome (L, M and S segment) are packaged into ribonucleoproteins (RNPs) by nucleocapsid protein (NP) and viral RNA-dependent RNA polymerase (RdRp). The L segment encodes RdRp. The M segment encodes Gn and Gc. The S segment encodes NP and the nonstructural protein (NSs) in ambisense orientation.
Clinical manifestations and pathogenesis
The majority of reported hospitalized cases infected with HRTV presented with fever, fatigue, diarrhoea, headache, and myalgia [Citation1,Citation11,Citation12]. Laboratory abnormalities include leucopenia, thrombocytopenia, and elevated aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels [Citation1,Citation11,Citation12,Citation21,Citation22]. Most of the reported hospitalized patients recovered, but several deaths occurred. At necropsy, HRTV antigen was detected in multiple tissues, including the brain, liver, gallbladder, pancreas, heart, lung, large and small bowel, kidney, testes, bone marrow, lymph nodes, and spleen, accompanied by multiple organ failure and haemorrhage [Citation23,Citation24]. Hemophagocytosis was observed in most deaths, and at least two deaths were associated with secondary hemophagocytic lymphohistiocytosis (HLH), which has also been reported in cases of SFTSV infection [Citation11,Citation23–27]. Thus, hyperinflammation occurred in these patients. Despite these common features, the full spectrum of HRTV remains unknown.
Although HRTV-specific antibodies have been identified in domestic and wild animal populations near the residences of patients, clinical disease, viraemia and virus shedding were not observed, even if several animals were inoculated with HRTV under laboratory conditions [Citation15–17,Citation28,Citation29]. However, IFNAR-/- mice (interferon-α/β receptor knockout) intraperitoneally and Ag129 mice (interferon-α/β/γ receptor knockout) infected with HRTV showed detectable viraemia, associated illness, and death in a dose-dependent manner [Citation28,Citation30–32]. Euthanized IFNAR-/- mice and Ag129 mice both showed hepatic haemorrhage, enlarged pale spleen, and excess peritoneal fluid [Citation28,Citation31,Citation32]. Hematoxylin and eosin staining revealed neutrophilic infiltration in the liver of intraperitoneally infected IFNAR-/- mice [Citation31], but in the spleen, liver, and kidneys of Ag129 mice [Citation28,Citation33]. Moreover, HRTV antigens could be detected in the spleen, liver, and kidneys, consistent with the target organs infected with HRTV in humans [Citation28,Citation31,Citation33]. Signal transducer and activator of transcription 2 (STAT2) knockout hamsters, another immunodeficient animal model, were also susceptible to HRTV infection, and acute inflammation was detected in the spleen, lymph nodes, liver, and lungs [Citation34]. These data suggest that interferon (IFN) response is critical for controlling HRTV infection.
The IFN response is initiated by the recognition of viral infections through pattern recognition receptors (PRRs) [Citation35]. Previous studies by us and others have shown that SFTSV can be recognized by retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), especially RIG-I and several Toll-like receptors (TLRs) [Citation36–40]. Upon recognition, PRRs interact with the common adaptors (such as MAVS and TRIF) and subsequently recruit the crucial kinases such as TBK1/IKKε and IKKα/IKKβ/IKKγ to direct the activation of the transcription factors IRF3 and NF-κB, respectively. Activated transcription factors are translocated to the nucleus to induce IFN and pro-inflammatory cytokine production [Citation35,Citation41]. The synthesized type I and III IFNs are secreted and bind to their receptors on the cell surface, resulting in the phosphorylation of STAT2 and STAT1, which then heterodimerize and interact with IFN regulatory factor 9 (IRF9) to form an IFN-stimulated gene factor 3 (ISGF3) transcription complex. ISGF3 translocates to the nucleus to trigger the expression of more than 300 IFN-stimulated genes (ISGs), which are considered to interfere with multiple steps of viral infection [Citation41]. Therein, moloney leukaemia virus 10 protein (MOV10) has been recently identified as a potent host restriction factor against the infection and pathogenicity of bandaviruses including HRTV. Although the expression of MOV10 seemed to be only weakly up-regulated by IFNs, being recognized as a weak ISG, this host factor can be strongly induced during infections with bandaviruses, such as SFTSV and HRTV [Citation42]. Moreover, interestingly, a significant induction of MOV10 expression was observed at the early stage of HRTV infection [Citation42], suggesting that MOV10 is also likely a virus-stimulated gene (VSG) upon viral infection. Furthermore, MOV10 imposes significant restriction on bandavirus replication by interacting with the viral NPs to disable RNP formation [Citation42]. Importantly, MOV10 knockdown in a mouse model enhanced viral replication and pathogenicity, substantiating its restrictive activity against bandaviruses [Citation42]. Many ISGs have been reported to restrict bunyaviruses. For example, Myxovirus resistance protein A (MxA) exerts suppressive effects against several bunyaviruses including La Crosse virus (LACV) [Citation43–45], Crimean-Congo haemorrhagic fever virus (CCHFV) [Citation46], Dugbe virus (DUGV) [Citation47], and Rift Valley fever virus (RVFV) [Citation48]. Recently, our team found that MxA also suppresses SFTSV infection by interacting with the NP and disrupting the NP-RdRp interaction [Citation49]. Also, IFN-induced transmembrane proteins (IFITMs) inhibits the infection of RVFV, Hantaan virus (HTNV) and SFTSV [Citation50–52]. But the role of these or other ISGs in HRTV infection remains undetermined. As mentioned above, IFN system-deficient animals are highly susceptible to HRTV and present pathological manifestations, indicating the pivotal antiviral roles of the IFN system and the downstream ISG effectors (such as MOV10) in HRTV infection [Citation28,Citation30,Citation31,Citation34]. Conversely, viruses evolve strategies to escape the IFN antiviral response, which would contribute to efficient viral replication and hence pathogenesis. Thus, the immune evasion of HRTV from the IFN system could be a pivotal aspect of the viral pathogenesis (as discussed below).
Host factors and HRTV infection
Upon HRTV infection, several host factors were reported to modulate the viral infection. (I) Complications and physical status. A heart transplant recipient infected with HRTV presented severe myalgia and spontaneous retroperitoneal bleed except for common symptoms [Citation53]. In addition, an elderly man with rheumatoid arthritis was infected with HRTV, subsequently developed HLH and died [Citation25]. SFTS patients combined with comorbidities including diabetes mellitus, chronic viral hepatitis, central neurological complications, coinfections with bacteria or fungi, etc. were at higher risk of death [Citation54–57]. Thus, preexisting complications are likely important factors for HRTV and HRTV related-bandavirus infection and pathogenesis. This knowledge might allow clinical physicians to predict a severe course in advance and provide timely clinical intervention and treatment. (II) Tick salivary factors. Recently, Erin S et al. found that the group of A129 mice infected with HRTV in the presence of tick salivary gland extract (SGE) showed more severe disease outcome and higher viral load compared to the group infected with HRTV only [Citation32]. That means, SGE could enhance HRTV infection and pathogenicity in A129 mice. This is also the evidence that tick bites are the main route of HRTV transmission. Identification of the specific salivary factors responsible for transmission of HRTV or other tick-borne viruses is necessary for analyses of the interactions between pathogen, host, and vector. (III) Innate immunity. As mentioned above, HRTV infection triggered the expression of MOV10 (as both an ISG and a VSG) and in turn, MOV10 could restrict HRTV replication probably via disrupting the formation of RNP [Citation42]. IFN receptor deficiency and STAT1/STAT2-KO animals are more susceptible to HRTV infection and can mimic human clinical outcomes compared to their immunocompetent counterparts, further confirming IFN system-directed innate immunity is a key limit factor [Citation28,Citation30–34,Citation58]. (IV) Autophagy. Autophagy, a conserved intracellular homoeostatic process, is emerging as a critical player in viral infection. One hand, autophagy can restrict viral infection by multiple strategies; on the other hand, the autophagic process is exploited by some viruses for their own benefit. We found that HRTV likely induce autophagy and conversely, modulates autophagy to promote its own replication [Citation59], as further discussed below. (V) Age and sex. According to investigation of Heartland virus disease, the majority of patients infected with HRTV were older males [Citation11,Citation12], consistent with outcome of SFTSV infection [Citation60]. It could be due to the differences of immune response and exposure frequency to viral vectors in outdoor activities. Therefore, the complex interactions of the virus with multiple host factors could significantly contribute to the viral pathogenesis and spread.
Virulence factors
Previous studies by us and others have suggested that SFTSV NSs, as a crucial virulence factor, hijacks multiple host factors to inclusion bodies (IB) formed by NSs itself to interfere with the host antiviral responses, contributing to the viral pathogenesis [Citation61–69]. Based on these studies, the NSs-induced cytoplasmic IBs in cells infected by SFTSV and other related bunyaviruses were proposed as a virally built “jail” which can sequestrate critical host signalling molecules and thus spatially interfere with the corresponding biological processes [Citation63,Citation65,Citation66]. Although HRTV NSs share ~ 60% amino acid sequence identity with SFTSV NSs, HRTV NSs do not induce noticeable IB “jail” formation [Citation70]. Interestingly, HRTV NSs also act as a main virulence factor of HRTV and maintain the capacity to antagonize or usurp host responses via similar but differentiated mechanisms.
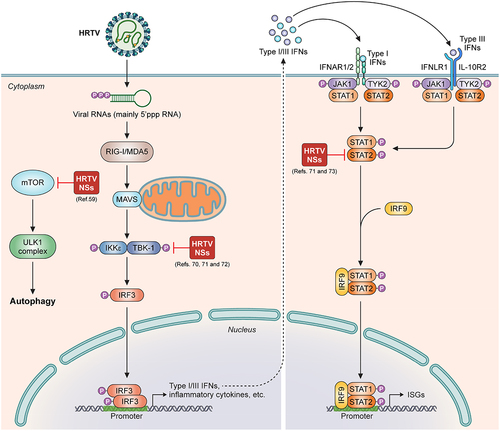
HRTV NSs antagonize IFN induction (Citation70,Citation71). Reporter gene assay results showed that HRTV NSs inhibited IFN-β promoter activation and expression of IFNB1 and RANTES at the level of kinase TBK1 (Citation70,Citation71). Consistently, the phosphorylation and nuclear translocation of IRF3 can be strongly blocked by HRTV NSs (Citation70,Citation71). Mechanistically, HRTV NSs interacts with kinase TBK1 and hence dose-dependently hinders TBK1-IRF3 association, thus disabling IRF3 activation (Citation70,Citation71). This mechanism is different from that of SFTSV NSs sequestering the TBK1-IRF3 complex into its IBs (Citation63). The scaffold dimerization domain (SDD) of TBK1 and two conserved amino acids at positions 21 and 23 of HRTV NSs have been shown to be required for the TBK1-HRTV NSs interaction (Citation70,Citation72).
HRTV NSs inhibits type I and III IFN signalling (Citation71,Citation73). HRTV NSs suppresses IFN-β- and IFN-λ-mediated antiviral responses by targeting STAT2 and specifically blocking STAT2 actions (but not STAT1), including its phosphorylation and nuclear translocation (Citation71,Citation73). This mechanism is different from the SFTSV NSs disruption of both STAT2 and STAT1 nuclear translocation (Citation65). In addition, the DNA-binding domain (DBD) and linker domain (LD) of STAT2 are required for HRTV NSs – STAT2 interaction (Citation73). In contrast, only DBD is required for the SFTSV NSs – STAT2 interaction (Citation65). These findings highlight the functional conservativeness and mechanism divergence of these homologous viral NSs proteins in antagonizing host antiviral responses. Of note, in the context of HRTV infection, STAT1 phosphorylation and nuclear accumulation also could be inhibited, suggesting an additional viral strategy to antagonize IFN signalling besides that directed by HRTV NSs. However, the further mechanism underlying HRTV infection-mediated inhibition to STAT1 remains to be elucidated.
HRTV NSs has the potential to induce pro-viral autophagy. We previously showed that SFTSV infection triggers autophagy and SFTSV NSs expression induces the conversion of LC3‐I to LC3‐II (Citation59). Mechanistically, SFTSV NSs interacts with mechanistic target of rapamycin (mTOR) and sequesters mTOR into inclusion bodies to impair the inhibitory effect of mTOR on unc‐51‐like kinase 1 (ULK1) and then stimulate autophagy to bolster SFTSV infection (Citation59). By using SFTSV reverse genetic minigenome system, we found that autophagy bolsters SFTSV replication and propagation by promoting the activity of SFTSV replication/transcription machinery RNP (Citation59). In the same study, we found that HRTV NSs also targets mTOR by the protein-protein interaction in extended analyses. Furthermore, it was demonstrated that HRTV NSs inhibits the ULK1 phosphorylation at Ser 757 and enhances the conversion of LC3‐I to LC3‐II as well. Thus, HRTV is likely able to regulate autophagy by the NSs protein for its own benefit via a similar mechanism. The study was overall focused on SFTSV NSs and many details in HRTV NSs manipulation of the host autophagy need further investigations. Recently, Liu et al. found that SFTSV NSs interacts with vimentin to block Beclin1-vimentin complex formation and promote autophagy in an IB-independent manner (Citation74). In addition, Yan et al. reported that SFTSV NP interacts with BECN1 and disrupts BECN1-BCL2 association, thus triggering autophagy flux to promote viral assembly and exocytosis (Citation75). These observations suggest that SFTSV can use various strategies to induce autophagy and enhance viral infection and pathogenicity. Further investigations are required to determine whether HRTV also use the similar mechanisms to trigger autophagy.
In addition, Taniguchi et al. reported the construction of NSs-knockout HRTV using a reverse genetics system [Citation33]. The authors further confirmed that HRTV NSs is an important virulence factor in cellular and animal infection experiments with mutant viruses. Compared with wild-type HRTV, NSs-knockout HRTV could induce higher levels of IFN-β and ISGs in infected cells and was less pathogenic in Ag129 mice [Citation33].
Here, we present a summarized model of the HRTV NSs actions (). As for other proteins encoded by HRTV, there are very limited reports regarding their potential involvement in viral regulations of host responses. Virus entry into host cells is the first step for infection. Glycoproteins (GPs) constitute the molecular machinery for the virus entry and thus largely determine the host tissue tropism and should be intimately linked to the pathogenicity and virulence. Currently, the mechanism by which HRTV penetrates host cells, and in particular, the identity of host factors that may interact with the glycoproteins to facilitate entry, remain elusive. However, several host factors including the C-type lectins [Citation76,Citation77], nonmuscle myosin heavy chain IIA [Citation78], and C-C motif chemokine receptor 2 [Citation79] have been identified as possible entry factors for SFTSV infection. It will be merited to test whether they may be also implicated in HRTV invasion into host cells. Additionally, Zhang et al. found that HRTV and HRTV-related bandaviruses including SFTSV and Guertu virus (GTV) glycoproteins might activate the host cell’s unfolded-protein response to favour their replication; however, the underlying mechanism remains to be determined [Citation80]. There is no report concerning any potential roles of the HRTV NP in viral perturbation of host biological processes. Of note, in our previous studies on the IFN antagonism by HRTV NSs, it was repeatedly suggested that, at least, NP has no evident effects on the IFN responses [Citation70,Citation73]. However, as aforementioned, the study by Yan et al. revealed a role of SFTSV NP in the induction of autophagy. It is possible that HRTV NP may have a similar effect. Overall, further investigation is needed to explore whether and how HRTV-encoded proteins aside from NSs may regulate host responses to facilitate the viral replication and/or aggravate the pathogenicity.
Figure 2. The model of HRTV NSs interfering with host antiviral responses as a virulence factor. HRTV NSs targets to TBK1 and hinders the association of TBK1 with IRF3, impairing IFN induction (middle). HRTV NSs interacts with STAT2 and dampens STAT2 activities to antagonize both type I and III IFN signalling (right). Additionally, HRTV NSs likely binds to mTOR to induce autophagy for its own benefit (left).
Conclusion
In recent years, HRTV has become one of the representative high-pathogenic bunyaviruses, posing a significant threat to public health. According to the latest statistics, the human disease caused by HRTV infection could lead to estimated case mortality rates of 5–10 %. Although some aspects of the pathogenesis and virulence have been explored as stated, the HRTV-host interactions, HRTV infection mechanisms, HRTV animal reservoirs and transmission cycle remain largely unknown, which severely impedes the development of vaccines and antiviral interventions. Thus, both basic and applied researches are urgently needed to expedite the development of prevention and treatment strategies against HRTV infection.
Several critical gaps and questions need to be addressed in the future. 1) The life cycle and interactions of HRTV with host cells are largely inferred from knowledge of other bunyaviruses. More studies on HRTV-host interactions are needed to understand the life cycle and host factors that regulate virus infection. 2) On the other hand, the pathogenesis and virulence of several other bunyaviruses, especially SFTSV, have been investigated more comprehensively. Comparative studies are merited to determine the similarities/differences and to obtain a quick view of the virology of HRTV, in light of the knowledge of other bunyaviruses. 3) A more appropriate animal model is urgently required. Various animal models of SFTSV have been investigated [Citation58], including the aged ferret model, which can fully recapitulate the clinical manifestations of human infections [Citation81]. Animal models suitable for SFTSV are references for studying HRTV. 4) Additional clinical data and investigations are required to depict the full spectrum and pathogenesis of the HRTV disease. 5) Vaccines and drugs used to control HRTV infection need to be explored and developed as soon as possible. Several promising SFTSV candidate vaccines and anti-SFTSV drugs have been reported [Citation82] and similar strategies can be used to test their inhibitory activity against HRTV.
Authors’ contributions
Conceptualization: YJN. Resources: YJN, HLW, and FD. Funding acquisition: YJN, HLW, KF, and SL. Writing-original draft: KF, YJN, HYZ, BBU, and SL. Writing-review and editing: YJN. All authors have read and approved the final manuscript.
Acknowledgements
The work in our lab was supported by the National Key Research and Development Program of China (2022YFC2303300, to YJN and HLW), Youth Innovation Promotion Association of Chinese Academy of Sciences (2020333, to YJN), the National Natural Science Foundation of China (32170171, to YJN), the Open Research Fund Program of the State Key Laboratory of Virology of China (2022IOV004, to KF), and the Science and Technology Program of Guangzhou, China (202102020057, to SL). We thank the National Virus Resource Center (Wuhan Institute of Virology) for virus resources and the Core Facility and Technical Support of Wuhan Institute of Virology for technical assistance in our study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data Availability statement
Data sharing is not applicable to this article as no new data were created or analysed in this study.
Additional information
Funding
References
- McMullan LK, Folk SM, Kelly AJ, et al. A new phlebovirus associated with severe febrile illness in Missouri. N Engl J Med. 2012;367(9):834–10. doi: 10.1056/NEJMoa1203378
- Yu XJ, Liang MF, Zhang SY, et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med. 2011;364(16):1523–1532. doi: 10.1056/NEJMoa1010095
- Kuhn JH, Adkins S, Alkhovsky SV, et al. 2022 taxonomic update of phylum negarnaviricota (Riboviria: orthornavirae), including the large orders bunyavirales and Mononegavirales. Arch Virol. 2022;167(12):2857–2906. doi: 10.1007/s00705-022-05546-z
- Savage HM, Godsey MS, Lambert A, et al. First detection of heartland virus (bunyaviridae: phlebovirus) from field collected arthropods. Am J Trop Med Hyg. 2013;89(3):445–452. doi: 10.4269/ajtmh.13-0209
- Savage HM, Godsey MS Jr., Panella NA, et al. Surveillance for heartland virus (bunyaviridae: phlebovirus) in Missouri during 2013: first detection of virus in adults of amblyomma americanum (Acari: ixodidae). J Med Entomol. 2016;53(3):607–612. doi: 10.1093/jme/tjw028
- Godsey MS, Savage HM, Burkhalter KL, et al. Transmission of heartland virus (bunyaviridae: phlebovirus) by experimentally infected amblyomma americanum (Acari: ixodidae). J Med Entomol. 2016;53(5):1226–1233. doi: 10.1093/jme/tjw080
- Tuten HC, Burkhalter KL, Noel KR, et al. Heartland virus in humans and ticks, Illinois, USA, 2018–2019. Emerg Infect Dis. 2020;26(7):1548–1552. doi: 10.3201/eid2607.200110
- Newman BC, Sutton WB, Moncayo AC, et al. Heartland virus in Lone star ticks, Alabama, USA. Emerg Infect Dis. 2020;26(8):1954–1956. doi: 10.3201/eid2608.200494
- Romer Y, Adcock K, Wei Z, et al. Isolation of heartland virus from Lone star ticks, Georgia, USA, 2019. Emerg Infect Dis. 2022;28(4):786–792. doi: 10.3201/eid2804.211540
- Aziati ID, Jnr DM, Antia A, et al. Prevalence of bourbon and heartland viruses in field collected ticks at an environmental field station in st. Louis County, Missouri, USA. Ticks Tick Borne Dis. 2023;14(1):102080. doi: 10.1016/j.ttbdis.2022.102080
- Brault AC, Savage HM, Duggal NK, et al. Heartland virus epidemiology, vector association, and disease potential. Viruses. 2018;10(9):498. doi: 10.3390/v10090498
- Staples JE, Pastula DM, Panella AJ, et al. Investigation of heartland virus disease throughout the United States, 2013–2017. Open Forum Inf Dis. 2020;7(5):ofaa125. doi: 10.1093/ofid/ofaa125
- Cumbie AN, Trimble RN, Eastwood G. Pathogen spillover to an invasive tick species: first detection of Bourbon virus in haemaphysalis longicornis in the United States. Pathogens. 2022;11(4):11. doi: 10.3390/pathogens11040454
- Raney WR, Perry JB, Hermance ME. Transovarial transmission of Heartland virus by invasive Asian longhorned ticks under laboratory conditions. Emerg Infect Dis. 2022;28(3):726–729. doi: 10.3201/eid2803.210973
- Riemersma KK, Komar N. Heartland virus neutralizing antibodies in vertebrate wildlife, United States, 2009–2014. Emerg Infect Dis. 2015;21(10):1830–1833. doi: 10.3201/eid2110.150380
- Bosco-Lauth AM, Panella NA, Root JJ, et al. Serological investigation of heartland virus (bunyaviridae: phlebovirus) exposure in wild and domestic animals adjacent to human case sites in Missouri 2012–2013. Am J Trop Med Hyg. 2015;92(6):1163–1167. doi: 10.4269/ajtmh.14-0702
- Clarke LL, Ruder MG, Mead DG, et al. Heartland virus exposure in white-tailed deer in the Southeastern United States, 2001–2015. Am J Trop Med Hyg. 2018;99(5):1346–1349. doi: 10.4269/ajtmh.18-0555
- Yamanaka A, Kirino Y, Fujimoto S, et al. Direct transmission of severe fever with thrombocytopenia syndrome virus from domestic cat to veterinary personnel. Emerg Infect Dis. 2020;26(12):2994–2998. doi: 10.3201/eid2612.191513
- Tang X, Wu W, Wang H, et al. Human-to-human transmission of severe fever with thrombocytopenia syndrome bunyavirus through contact with infectious blood. J Infect Dis. 2013;207(5):736–739. doi: 10.1093/infdis/jis748
- Fang X, Hu J, Peng Z, et al. Epidemiological and clinical characteristics of severe fever with thrombocytopenia syndrome bunyavirus human-to-human transmission. PLOS Negl Trop Dis. 2021;15(4):e0009037. doi: 10.1371/journal.pntd.0009037
- Pastula DM, Turabelidze G, Yates KF, et al. Notes from the field: heartland virus disease - United States, 2012-2013. MMWR Morb Mortal Wkly Rep. 2014;63(12):270–271.
- Decker MD, Morton CT, Moncayo AC. One confirmed and 2 suspected cases of heartland virus disease. Clin Infect Dis. 2020;71(12):3237–3240. doi: 10.1093/cid/ciaa647
- Muehlenbachs A, Fata CR, Lambert AJ, et al. Heartland virus-associated death in tennessee. Clin Infect Dis. 2014;59(6):845–850. doi: 10.1093/cid/ciu434
- Fill MA, Compton ML, EC M, et al. Novel clinical and pathologic findings in a heartland virus-associated death. Clin Infect Dis. 2017;64:510–512. doi: 10.1093/cid/ciw766
- Carlson AL, Pastula DM, Lambert AJ, et al. Heartland virus and hemophagocytic lymphohistiocytosis in immunocompromised patient, Missouri, USA. Emerg Infect Dis. 2018;24(5):893–897. doi: 10.3201/eid2405.171802
- Ahlers CG, Matthews H, Perez R, et al. Secondary hemophagocytic lymphohistiocytosis due to heartland virus. BMJ Case Rep. 2022;15(12):e253082. doi: 10.1136/bcr-2022-253082
- Liu S, Kannan S, Meeks M, et al. Fatal case of heartland virus disease acquired in the Mid-Atlantic Region, United States. Emerg Infect Dis. 2023;29(5):992–996. doi: 10.3201/eid2905.221488
- Bosco-Lauth AM, Calvert AE, Root JJ, et al. Vertebrate host susceptibility to heartland virus. Emerg Infect Dis. 2016;22(12):2070–2077. doi: 10.3201/eid2212.160472
- Clarke LL, Ruder MG, Mead D, et al. Experimental infection of white-tailed deer (odocoileus virginanus) with heartland virus. Am J Trop Med Hyg. 2018;98(4):1194–1196. doi: 10.4269/ajtmh.17-0963
- Fujii H, Tani H, Egawa K, et al. Susceptibility of type I interferon receptor knock-out mice to heartland bandavirus (HRTV) infection and efficacy of favipiravir and ribavirin in the treatment of the mice infected with HRTV. Viruses. 2022;14(8):1668. doi: 10.3390/v14081668
- Fujii H, Fukushi S, Yoshikawa T, et al. Pathological and virological findings of type I interferon receptor knockout mice upon experimental infection with heartland virus. Virus Res. 2023;340:199301. doi: 10.1016/j.virusres.2023.199301
- Reynolds ES, Wooldridge JT, Stevenson HL, et al. The lone star tick, amblyomma americanum, salivary factors exacerbate the clinical outcome of heartland virus disease in a small animal model. Sci Rep. 2023;13(1):13304. doi: 10.1038/s41598-023-40397-x
- Taniguchi S, Inagaki T, Tajima S, et al. Reverse genetics system for Heartland bandavirus: NSs protein contributes to Heartland bandavirus virulence. J Virol. 2022;96(7):e0004922. doi: 10.1128/jvi.00049-22
- Westover JB, Rigas JD, Van Wettere AJ, et al. Heartland virus infection in hamsters deficient in type I interferon signaling: protracted disease course ameliorated by favipiravir. Virology. 2017;511:175–183. doi: 10.1016/j.virol.2017.08.004
- Carty M, Guy C, Bowie AG. Detection of viral infections by innate immunity. Biochem Pharmacol. 2021;183:114316. doi: 10.1016/j.bcp.2020.114316
- Min YQ, Ning YJ, Wang H, et al. A RIG-I–like receptor directs antiviral responses to a bunyavirus and is antagonized by virus-induced blockade of TRIM25-mediated ubiquitination. J Biol Chem. 2020;295(28):9691–9711. doi: 10.1074/jbc.RA120.013973
- Yamada S, Shimojima M, Narita R, et al. RIG-I-Like receptor and toll-like receptor signaling pathways cause aberrant production of inflammatory cytokines/chemokines in a severe fever with thrombocytopenia syndrome virus infection mouse model. J Virol. 2018;92(13). doi: 10.1128/JVI.02246-17
- Song P, Zheng N, Zhang L, et al. Downregulation of interferon-β and inhibition of TLR3 expression are associated with fatal outcome of severe fever with thrombocytopenia syndrome. Sci Rep. 2017;7(1):6532. doi: 10.1038/s41598-017-06921-6
- Peng C, Wang H, Zhang W, et al. Decreased monocyte subsets and TLR4-mediated functions in patients with acute severe fever with thrombocytopenia syndrome (SFTS). Int J Infect Dis. 2016;43:37–42. doi: 10.1016/j.ijid.2015.12.009
- Li S, Li H, Zhang YL, et al. SFTSV infection induces BAK/BAX-dependent mitochondrial DNA release to trigger NLRP3 inflammasome activation. Cell Rep. 2020;30(13):4370–4385.e7. doi: 10.1016/j.celrep.2020.02.105
- Dalskov L, Gad HH, Hartmann R. Viral recognition and the antiviral interferon response. Embo J. 2023;42(14):e112907. doi: 10.15252/embj.2022112907
- Mo Q, Xu Z, Deng F, et al. Host restriction of emerging high-pathogenic bunyaviruses via MOV10 by targeting viral nucleoprotein and blocking ribonucleoprotein assembly. PLOS Pathog. 2020;16(12):e1009129. doi: 10.1371/journal.ppat.1009129
- Miura TA, Carlson JO, Beaty BJ, et al. Expression of human MxA protein in mosquito cells interferes with LaCrosse virus replication. J Virol. 2001;75(6):3001–3003. doi: 10.1128/JVI.75.6.3001-3003.2001
- Kochs G, Janzen C, Hohenberg H, et al. Antivirally active MxA protein sequesters La Crosse virus nucleocapsid protein into perinuclear complexes. Proc Natl Acad Sci U S A. 2002;99(5):3153–3158. doi: 10.1073/pnas.052430399
- Reichelt M, Stertz S, Krijnse-Locker J, et al. Missorting of LaCrosse virus nucleocapsid protein by the interferon-induced MxA GTPase involves smooth ER membranes. Traffic. 2004;5(10):772–784. doi: 10.1111/j.1600-0854.2004.00219.x
- Andersson I, Bladh L, Mousavi-Jazi M, et al. Human MxA protein inhibits the replication of crimean-congo hemorrhagic fever virus. J Virol. 2004;78(8):4323–4329. doi: 10.1128/JVI.78.8.4323-4329.2004
- Bridgen A, Dalrymple DA, Weber F, et al. Inhibition of dugbe nairovirus replication by human MxA protein. Virus Res. 2004;99(1):47–50. doi: 10.1016/j.virusres.2003.10.002
- Habjan M, Penski N, Wagner V, et al. Efficient production of rift Valley fever virus-like particles: the antiviral protein MxA can inhibit primary transcription of bunyaviruses. Virology. 2009;385(2):400–408. doi: 10.1016/j.virol.2008.12.011
- Chang M, Min YQ, Xu Z, et al. Host factor MxA restricts dabie bandavirus infection by targeting the viral NP protein to inhibit NP-RdRp interaction and ribonucleoprotein activity. J Virol. 2024;98(1):e0156823. doi: 10.1128/jvi.01568-23
- Mudhasani R, Tran JP, Retterer C, et al. IFITM-2 and IFITM-3 but not IFITM-1 restrict rift Valley fever virus. J Virol. 2013;87(15):8451–8464. doi: 10.1128/JVI.03382-12
- Xu-Yang Z, Pei-Yu B, Chuan-Tao Y, et al. Interferon-induced transmembrane protein 3 inhibits hantaan virus infection, and its single nucleotide polymorphism rs12252 influences the severity of hemorrhagic fever with renal syndrome. Front Immunol. 2016;7:535. doi: 10.3389/fimmu.2016.00535
- Du S, Wang Y, Wang J, et al. IFITM3 inhibits severe fever with thrombocytopenia syndrome virus entry and interacts with viral gc protein. J Med Virol. 2024;96(3):e29491. doi: 10.1002/jmv.29491
- Hevey MA, JA O, Jagger BW, et al. Heartland virus infection in a heart transplant recipient from the Heartland. Transplant Infectious Dis. 2019;21(4):e13098. doi: 10.1111/tid.13098
- Zhang SF, Yang ZD, Huang ML, et al. Preexisting chronic conditions for fatal outcome among SFTS patients: an observational cohort study. PLOS Negl Trop Dis. 2019;13(5):e0007434. doi: 10.1371/journal.pntd.0007434
- Wang M, Huang P, Liu W, et al. Risk factors of severe fever with thrombocytopenia syndrome combined with central neurological complications: a five-year retrospective case-control study. Front Microbiol. 2022;13:1033946. doi: 10.3389/fmicb.2022.1033946
- Ge HH, Wang G, Guo PJ, et al. Coinfections in hospitalized patients with severe fever with thrombocytopenia syndrome: a retrospective study. J Med Virol. 2022;94(12):5933–5942. doi: 10.1002/jmv.28093
- Zhang Y, Huang Y, Xu Y. Associated microbiota and treatment of severe fever with thrombocytopenia syndrome complicated with infections. J Med Virol. 2022;94(12):5916–5921. doi: 10.1002/jmv.28059
- Sun J, Min YQ, Li Y, et al. Animal model of severe fever with thrombocytopenia syndrome virus infection. Front Microbiol. 2021;12:797189. doi: 10.3389/fmicb.2021.797189
- Feng K, Zhang H, Jiang Z, et al. SFTS bunyavirus NSs protein sequestrates mTOR into inclusion bodies and deregulates mTOR-ULK1 signaling, provoking pro-viral autophagy. J Med Virol. 2023;95(1):e28371. doi: 10.1002/jmv.28371
- Zhao J, Lu QB, Li H, et al. Sex differences in case fatality rate of patients with severe fever with thrombocytopenia syndrome. Front Microbiol. 2021;12:738808. doi: 10.3389/fmicb.2021.738808
- Qu B, Qi X, Wu X, et al. Suppression of the interferon and NF-κB responses by severe fever with thrombocytopenia syndrome virus. J Virol. 2012;86(16):8388–8401. doi: 10.1128/JVI.00612-12
- Wu X, Qi X, Qu B, et al. Evasion of antiviral immunity through sequestering of TBK1/IKKε/IRF3 into viral inclusion bodies. J Virol. 2014;88(6):3067–3076. doi: 10.1128/JVI.03510-13
- Ning YJ, Wang M, Deng M, et al. Viral suppression of innate immunity via spatial isolation of TBK1/IKKε from mitochondrial antiviral platform. J Mol Cell Biol. 2014;6(4):324–337. doi: 10.1093/jmcb/mju015
- Santiago FW, Covaleda LM, Sanchez-Aparicio MT, et al. Hijacking of RIG-I signaling proteins into virus-induced cytoplasmic structures correlates with the inhibition of type I interferon responses. J Virol. 2014;88(8):4572–4585. doi: 10.1128/JVI.03021-13
- Ning YJ, Feng K, Min YQ, et al. Disruption of type I interferon signaling by the nonstructural protein of severe fever with thrombocytopenia syndrome virus via the hijacking of STAT2 and STAT1 into inclusion bodies. J Virol. 2015;89(8):4227–4236. doi: 10.1128/JVI.00154-15
- Ning YJ, Mo Q, Feng K, et al. Interferon-γ-directed inhibition of a novel high-pathogenic phlebovirus and viral antagonism of the antiviral signaling by targeting STAT1. Front Immunol. 2019;10:1182. doi: 10.3389/fimmu.2019.01182
- Choi Y, Park SJ, Sun Y, et al. Severe fever with thrombocytopenia syndrome phlebovirus non-structural protein activates TPL2 signalling pathway for viral immunopathogenesis. Nat Microbiol. 2019;4(3):429–437. doi: 10.1038/s41564-018-0329-x
- Hong Y, Bai M, Qi X, et al. Suppression of the IFN-α and -β induction through sequestering IRF7 into viral inclusion bodies by nonstructural protein NSs in severe fever with thrombocytopenia syndrome bunyavirus infection. J Immunol. 2019;202(3):841–856. doi: 10.4049/jimmunol.1800576
- Zhang L, Fu Y, Zhang R, et al. Nonstructural protein NSs hampers cellular antiviral response through LSm14A during severe fever with thrombocytopenia syndrome virus infection. J Immunol. 2021;207(2):590–601. doi: 10.4049/jimmunol.2100148
- Ning YJ, Feng K, Min YQ, et al. Heartland virus NSs protein disrupts host defenses by blocking the TBK1 kinase–IRF3 transcription factor interaction and signaling required for interferon induction. J Biol Chem. 2017;292(40):16722–16733. doi: 10.1074/jbc.M117.805127
- Rezelj L VV, Chaudhary P, Elliott V, et al. Differential antagonism of human innate immune responses by tick-borne phlebovirus nonstructural proteins. mSphere. 2017;2. doi: 10.1128/mSphere.00234-17
- Moriyama M, Igarashi M, Koshiba T, et al. Two conserved amino acids within the NSs of severe fever with thrombocytopenia syndrome phlebovirus are essential for anti-interferon activity. J Virol. 2018;92(19): doi: 10.1128/JVI.00706-18
- Feng K, Deng F, Hu Z, et al. Heartland virus antagonizes type I and III interferon antiviral signaling by inhibiting phosphorylation and nuclear translocation of STAT2 and STAT1. J Biol Chem. 2019;294(24):9503–9517. doi: 10.1074/jbc.RA118.006563
- Liu S, Su Y, Lu Z, et al. The SFTSV nonstructural proteins induce autophagy to promote viral replication via interaction with Vimentin. J Virol. 2023;97(4):e0030223. doi: 10.1128/jvi.00302-23
- Yan JM, Zhang WK, Yan LN, et al. Bunyavirus SFTSV exploits autophagic flux for viral assembly and egress. Autophagy. 2022;18(7):1599–1612. doi: 10.1080/15548627.2021.1994296
- Hofmann H, Li X, Zhang X, et al. Severe fever with thrombocytopenia virus glycoproteins are targeted by neutralizing antibodies and can use DC-SIGN as a receptor for pH-dependent entry into human and animal cell lines. J Virol. 2013;87(8):4384–4394. doi: 10.1128/JVI.02628-12
- Tani H, Shimojima M, Fukushi S, et al. Characterization of glycoprotein-mediated entry of severe fever with thrombocytopenia syndrome virus. J Virol. 2016;90(11):5292–5301. doi: 10.1128/JVI.00110-16
- Sun Y, Qi Y, Liu C, et al. Nonmuscle myosin heavy chain IIA is a critical factor contributing to the efficiency of early infection of severe fever with thrombocytopenia syndrome virus. J Virol. 2014;88(1):237–248. doi: 10.1128/JVI.02141-13
- Zhang L, Peng X, Wang Q, et al. CCR2 is a host entry receptor for severe fever with thrombocytopenia syndrome virus. Sci Adv. 2023;9(31):eadg6856. doi: 10.1126/sciadv.adg6856
- Zhang LK, Wang B, Xin Q, et al. Quantitative proteomic analysis reveals unfolded-protein response involved in severe fever with thrombocytopenia syndrome virus infection. J Virol. 2019;93(10). doi: 10.1128/JVI.00308-19
- Park SJ, Kim YI, Park A, et al. Ferret animal model of severe fever with thrombocytopenia syndrome phlebovirus for human lethal infection and pathogenesis. Nat Microbiol. 2019;4(3):438–446. doi: 10.1038/s41564-018-0317-1
- Chen L, Chen T, Li R, et al. Recent advances in the study of the immune escape mechanism of SFTSV and its therapeutic agents. Viruses. 2023;15(4):940. doi: 10.3390/v15040940