
ABSTRACT
Coxiella burnetii (C. burnetii) is the causative agent of Q fever, a zoonotic disease. Intracellular replication of C. burnetii requires the maturation of a phagolysosome-like compartment known as the replication permissive Coxiella-containing vacuole (CCV). Effector proteins secreted by the Dot/Icm secretion system are indispensable for maturation of a single large CCV by facilitating the fusion of promiscuous vesicles. However, the mechanisms of CCV maintenance and evasion of host cell clearance remain to be defined. Here, we show that C. burnetii secreted Coxiella vacuolar protein E (CvpE) contributes to CCV biogenesis by inducing lysosome-like vacuole (LLV) enlargement. LLV fission by tubulation and autolysosome degradation is impaired in CvpE-expressing cells. Subsequently, we found that CvpE suppresses lysosomal Ca2+ channel transient receptor potential channel mucolipin 1 (TRPML1) activity in an indirect manner, in which CvpE binds phosphatidylinositol 3-phosphate [PI(3)P] and perturbs PIKfyve activity in lysosomes. Finally, the agonist of TRPML1, ML-SA5, inhibits CCV biogenesis and C. burnetii replication. These results provide insight into the mechanisms of CCV maintenance by CvpE and suggest that the agonist of TRPML1 can be a novel potential treatment that does not rely on antibiotics for Q fever by enhancing Coxiella-containing vacuoles (CCVs) fission.
Introduction
Coxiella burnetii is a gram-negative, obligate intracellular bacterium responsible for the worldwide zoonotic disease Q fever. Many species of mammals, birds, and ticks are natural reservoirs of C. burnetii [Citation1]. Human infections often occur through contact with infected animals and their biological products, and the clinical presentation may manifest as an acute febrile illness with pneumonia or as a chronic infection with accompanying endocarditis [Citation2].
Upon internalization by phagocytic cells, C. burnetii traffics through the endolysosomal pathway and replicates inside the lysosome-derived Coxiella-containing vacuoles (CCVs). CCVs are highly fusogenic and merge with endocytic vesicles, autophagosomes, and other CCVs to create a large organelle that rapidly occupies much of the cytoplasmic space of the host cell [Citation3]. Mechanistically, CCVs is the acidic environment (pH ~ 5.0), which is required to activate C. burnetii metabolism and the Dot/Icm Type 4 B Secretion System (T4BSS), a major virulence factor required for intracellularreplication. The lower pH (~5.0) of CCVs is an important factor for C. burnetii growth and virulence [Citation4]. Lysosomal hydrolases are also needed for normal CCVs development, but are not required for C. burnetii survival and growth [Citation5].
C. burnetii effector proteins, which are secreted into the host cell cytoplasm through the T4BSS, manipulate host cell processes to support CCV development [Citation6]. For instance, the effector protein CvpB facilitates CCV fusogenicity by stabilizing PI(3)P in early endosomes and CCV membranes [Citation7]. The effector proteins Cig57 and CvpA support CCV expansion by interacting with host factors involved in clathrin-mediated endocytosis [Citation8]. The CCV containing wild type C. burnetii displayed robust accumulation of the autophagosome protein LC3; however, the vacuoles formed by the Cig2 mutant did not contain detectible amounts of LC3. The Cig2 is important for interactions between the CCVs and host autophagosomes [Citation9]. The effector protein CvpF is also required for CCV formation and C. burnetii-induced LC3 lipidation via interactions with the autophagy-related RAB GTPase Rab26 [Citation10]. In addition to the effectors mentioned above, proteins within the C. burnetii effector repertoire are also involved in CCV development through distinct or uncharacterized mechanisms.
In mammalian cells, lysosomes receive and degrade cargo via endocytosis, phagocytosis, and autophagy [Citation11]. Intracellular lysosomal membrane trafficking, including fusion and fission, is crucial for cellular homoeostasis and normal cellular functions [Citation12]. The replacement of Rab7 for Rab5 occurs during maturation from early endosomes to late endosomes [Citation13]. Efficient membrane fusion requires Rabs labelling of fusion site, interaction of Rab-recruited tethers to soluble N-ethylmaleimide-sensitive fusion protein (NSF) attachment protein receptor (SNARE) [Citation14], and chaperoning of Sec1/Munc18 (SM) proteins [Citation15]. Broadly, lysosome fission may occur via vesiculation (formation of a vesicle), tubulation (generation of a tubular intermediate), and splitting (equatorial fission) which were regulated by several factors including autophagic lysosome reformation (ALR) [Citation16], and lipopolysaccharide (LPS) [Citation17]. Overall, fusion and fission events must be coordinated to achieve steady-state lysosome number and size [Citation18].
Treatment of cells with inhibitors targeting lysosomal modulators leads to lysosomal enlargement, a phenotype that strongly resembles the histological features of lysosomal storage disorders (LSDs) [Citation19]. Although CCV itself is an abnormally enlarged lysosome that contains many lysosomal markers on the membrane and displays robust hydrolytic activity, it is unknown whether C. burnetii disrupts lysosome fusion-fission dynamics to facilitate the development of an extensive CCV.
In this study, we identified Coxiella vacuolar protein E (CvpE), a T4BSS effector protein of C. burnetii that is involved in the disruption of lysosome fusion-fission dynamics in mammalian cells. CvpE is located in the CCV and binds to lysosome-derived PI(3)P, which perturbs the binding of PI3 phosphate 5-kinase PIKfyve to lysosomes. Inhibition of PI(3,5)P2 generation causes lysosomal fission defects and enlargement by blocking the PI(3,5)P2-TRPML1-Ca2+ signalling pathway, which is necessary for lysosomal tubulation and reformation [Citation20]. The treatment of C. burnetii-infected cells with ML-SA5, a transient receptor potential channel mucolipin 1 (TRPML1) agonist, impaired CCV expansion and bacterial replication, highlighting the importance of preventing lysosomal fission for CCV maintenance.
Material and methods
Bacterial strains, cell lines, and mice
The Coxiella burnetii Nine Mile Phase II (NMII) strain was maintained in our laboratory. NMIIpJB-FLAG-CvpE was generated by our lab and cultured axenically in liquid ACCM-2 supplemented with chloramphenicol, as appropriate. NMIIpdCas9 and NMIIpdCas9-sgCvpE were generated by the electroporation of pJB-mCherry-P1169-dCas9 or pJB-mCherry-P1169-dCas9-sgCvpE. For CvpE, the chosen small guide RNA sequence was 5’-ctaaaaaaaaaattaacgat-3.’ These strains were generated using previously described methods [Citation16]. To complement the CvpE knockdown strain, CvpE was inserted into pJB-mCherry-P1169-dCas9-sgCvpE as a BspEI/BsrGI fragment to generate NMIIpdCas9-sgCvpE-comp. These strains were cultured in ACCM-2 medium supplemented with chloramphenicol for 7 days and infected Vero cells at an appropriate MOI. The expression levels of CvpE in these strains were determined using qRT-PCR and Western Blotting. HeLa, Vero and COS-7 cell lines were purchased from ATCC and cultured in DMEM containing 10% foetal bovine serum (FBS). HeLa cells stably expressing FLAG or FLAG-CvpE which were induced by 0.5 μg/mL doxycycline and Vero cells stably expressing Lamp1-GFP were generated by Cyagen (Suzhou, China) and cultured in the same medium as Vero cells with the addition of 0.75 μg/mL puromycin. All the cell lines were cultured at 37°C and 5% CO2. SCID mice (6 weeks old) were purchased from the Vital River Laboratory (Beijing, China).
Reagents, proteins, and plasmids construction
The antibodies used in this study were rabbit anti-Rab5 (Abcam, ab218624), rabbit anti-Rab7 (Abcam, ab137029), rabbit anti-Lamp1 (CST, 9091S), rabbit anti-Rab11 (Abcam, ab180504), rabbit anti-Calreticulin (Proteintech 27,298–1-AP), rabbit anti-GM130 (Abcam, ab52649), mouse anti-strep tag (MBL Beijing Biotech, M211–3), mouse anti-GST tag(Proteintech 66,001–2-Ig), mouse anti-FLAG tag (Sigma, F1804), mouse anti-His tag (Proteintech 66,005–1-Ig), mouse anti-GFP tag (Proteintech 66,002–1-Ig), mouse anti-cas9 (CST, 14697T), rabbit anti-cathepsin D (Proteintech 21,327–1-AP), mouse anti-p62 (Abcam, ab280086), mouse anti-LC3B (CST, 3868), rabbit anti-AP2α1 (Abcam, ab151720), rabbit anti-AP2β1(Proteintech 15,690–1-AP), rabbit anti-AP2γ1(Abcam, ab167153), rabbit anti-AP2σ1 (Abcam, ab128950), mouse anti-PIP5KIII/PIKfyve (SantaCruz, sc -100,408), mouse anti-Fig4/Sac3 (NeuroMab, 75–201), rabbit anti-Vac14 (Proteintech 15,771–1-AP), and mouse anti-GAPDH (Proteintech 60,004–1-Ig). Mouse anti-C. burnetii polyclonal, mouse anti-CvpE, and mouse anti-com1 mAbs were generated in our laboratory. The following chemicals were used in this study: chloramphenicol (Millipore 220,551), Vacuolin-1 (Selleck, S6912), YM201636 (MC E, HY-13228), ML-SA5 (MCE, HY-152182), and PI(3)P diC16 (Echelon Biosciences, p-3016).All recombinant His or GST tagged fusion proteins involved in this article were purchased from CUSABIO (Wuhan, China). Optimized cbu1863 sequence was constructed into pEGFP-c1 as a BspEI/BamHI fragment, pStrep-mCherry as a double EcoRI fragment, and pRK5-flag as a EcoRI/BamHI fragment by using homologous recombination. The lc3 sequence was constructed into pcDNA3.1-mcherry as a BamHI/XbaI fragment. The trpml1 and pikfyve sequences were constructed into pEGFP-c1 as a BspEI/BamHI fragment. The 2×fyve probe sequence was constructed into pcDNA3.1 as a HindIII/XbaI fragment with a 3’-terminal HA tag by using homologous recombination.
Cell transfection
For ectopic expression of plasmids in mammalian cells, cells were grown to 60% confluence and transfected with Lipofectamine™ 3000 transfection reagent (Invitrogen) according to the manufacturer’s recommendations. At the indicated time points post transfection, the cells were fixed and processed for immunofluorescence or harvested for western blotting.
Confocal laser scanning microscopy assays
HeLa, Vero, or COS-7 cells were seeded in 24-well plates (Cellvis, P24–0-N), cultured overnight, and transfected with indicated eukaryotic recombinant plasmids. Twenty-four hours after transfection, cells were fixed with 4% paraformaldehyde (Beyotime, P0099), permeabilized with 0.2% Triton X-100 (Beyotime, P0096) for 15 min, blocked with 1% bovine serum albumin (BSA), and then probed with indicated antibodies, followed by goat anti-mouse or rabbit IgG Alexa Fluor 488/594 antibody (Abcam, ab150077/ab150116). After three washes with phosphate-buffered saline (PBS), the cell nuclei were stained with DAPI. The samples were observed using a spinning disk confocal microscope (Nikon Ti-Eclipse). Colocalization was quantified for fluorescent signals using the colocalization plugin in ImageJ, and Pearson’s Correlation Coefficient was used to analyse colocalization.
Tubulation of lysosomes in Vero cells infected with C. burnetii was observed using ImageXpress Micro Confocal (Molecular Devices).
Co-immunoprecipitation and glutathione S-transferase (GST) pulldown
Recombinant mCherry-strep-tagged CvpE was transfected into HEK293T cells seeded in T25 flasks. Twenty-four hours post transfection, cells were harvested and treated with 500 μL lysis buffer containing protease inhibitor for 20 min. Supernatants of cell lysates were incubated with 50 μL slurry of Protein A/G plus-agarose conjugating with 1 μg anti-strep antibody overnight at 4°C. Agarose was washed five times with lysis buffer and then boiled in loading buffer for 10 min. Samples were analysed by western blotting using 1 μg of anti-strep antibodies and AP2 subunit antibodies. For GST-pulldown, purified 1 μg GST-CvpE protein and purified 1 μg His-TRPML1 protein (generated by CUSABIO, Wuhan, China) were mixed in reaction buffer (mM Tris-HCl, mM glycine, and mM NaCl, pH7.5) for 8 h at 4°C. The mixture was then incubated with 50 μL slurry of Protein A/G plus-agarose conjugating with 1 μg anti-GST antibody overnight at 4°C. Agarose was washed five times with lysis buffer and then boiled in loading buffer for 10 min. Samples were analysed by western blotting using 1 μg anti-GST tag or anti-His tag antibodies.
Lipid–protein interaction assay
PIP strip membranes (Echelon Biosciences, p-6001) were blocked with PBS containing 0.1% Tween-80 and 3% BSA (TBS-T/BSA) for 1 h followed by incubation with purified GST or GST-CvpE for 1 h at room temperature. The membranes were washed three times with TBS-T/BSA and immunoblotted with anti-GST HRP-conjugated antibodies for 1 h at room temperature. After three washes with TBS-T/BSA, the membranes were probed with ECL for detection.
Bio-layer interferometry (BLI) assays
To detect the affinity between proteins and lipids, BLI assays were conducted on Sartorius Octet R8. Phosphate buffer solution (PBS) with 0.1% Tween-20 (PBST) was used as the kinetic buffer and diluent for all components. To prepare test probes, anti-GST biosensors were run at baseline in PBST for 60 s, loaded in 200 μL of GST-tagged full-length CvpE or truncations at 50 μg/mL for 360 s, and run at baseline again in PBST for 60 s. For binding kinetics assays, a serial dilution of five concentrations of phosphatidylinositol 3-phosphate [PI(3)P] dissolved in PBST was added to a black polypropylene 96-well microplate with PBST. One row was used as the PBS-only negative control. The assay cycle consisted of 180 s of association in the compound solution, followed by 180 s of dissociation in PBST. BLI results were analysed using Octet BLI Analysis software version 12.2. A 1:1 binding model was used for binding kinetics analysis. The KD, Kon, Koff, and R2 values have been reported.
ADP glo kinase assay
The kinase activity of CvpE was determined using the ADP Glo Kinase assay following the manufacturer’s instructions. Briefly, 500 ng of purified GST-CvpE, 100 μM PIs [PI(3)P, PI(4)P, or PI(5)P] and 100 μM ATP were mixed in a 96-well plate and incubated for 2 h. ADP-GloTM Reagent was then added to the reaction, 1 h later, the mixture was incubated with the kinase detection reagent for 1 h. Finally, chemiluminescence of the reaction was detected using SpectraMax i3X.
Acetate ringer’s treatment
HeLa cells stably expressing FLAG or FLAG-CvpE treated with 0.5 μg/mL doxycycline were incubated in Acetate Ringer’s medium (80 mM NaCl, 70 mM Sodium acetate, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 2 mM NaH2PO4, 10 mM Hepes, 10 mM glucose and 0.5 mg/mL bovine serum albumin) for 1 h at 37°C to allow lysosomes to fragment and then placed back in normal growth media for the indicated times to allow lysosomes to refuse.
Activity determination of TRPML1 channel
HeLa cells stably expressing FLAG or FLAG-CvpE were transfected with pcDNA3.1-TRPML1 containing a C-terminal GECO1 tag. Changes in cytosolic Ca2+ levels were monitored by following changes in TRPML1-GECO1 fluorescence for 5 min upon addition of 30 μM ML-SA5 in Ca2+-free Hanks’ balanced salt solution (HBSS) using the video mode of OLYMPUS IX72P1F microscopy. TRPML1-GECO1 fluorescence was measured and analysed using ImageJ, and TRPML1 channel activity was represented as ΔF/F0.
Quantitative analysis of C. burnetii
Total DNA was extracted from ACCM-2 cultured C.burnetii or C.burnetii infected cells using a DNeasy Tissue kit, following the manufacturer’s instructions. Taqman Universal Master Mix was used to perform real-time PCR on Applied Biosystems QuantStudio 3. Diluted pQGK-com1 plasmid was used as the standard. The primers and probes used for quantification were as follows: com1-F,5’-aaaacctccgcgttgtcttca-3;’ com1-R,5’-gctaatgatactttggcagcgtattg-3;’ com1-probe,5’-6-FAM-agaactgcccatttttggcggcca-BHQ-X-3.’
Quantitative real time-PCR (qRT-PCR)
Total RNA was extracted from Vero cells infected with NMII, NMII-dCas9, and NMII-dCas9-sgCvpE using the PureLinkTM RNA Mini Kit following the manufacturer’s instructions. Quantitative real-time PCR was performed using the One Step TB Green PrimeScript™ PLUS RT-PCR Kit and Applied Biosystems QuantStudio 3. The specific primers used for cbu1861 (sense primer:5’-atgacagggatttcagacgc-3,’ and antisense primer:5’-ataacgtacgttatgggcg-3’), cbu1862 (sense primer:5’-agcacaatggtttctcgc-3,’ and antisense primer:5’-tgcttaaaggcgtcttgga-3’), cbu1863 (sense primer:5’-atgatactcattcaacgtt-3,’ and antisense primer:5’-agcagaatcagtcacgtct-3’), cbu1864 (sense primer:5’-tgagtttgtggattggtttg-3,’ and antisense primer:5’-agtttttattgacaaggag-3’) and cbu1865 (sense primer:5’-gaacttcggaatacggcgtc-3,’ and antisense primer:5’-tgacgtcaacgccttctaa-3’) were synthesized by Sangon Company. All samples were examined in triplicate, and the Rpob gene (sense primer:5’-cgtcatttccaccatcgtccctatc-3’ and antisense primer:5’-gcccaacccagatgagtttccag-3’) was used as the reference gene.
Animal experiment
SCID mice (n = 7) were challenged with 1 × 106 NMIIpdCas9, NMIIpdCas9-sgCvpE or NMIIpdCas9-CvpE-comp strains, respectively. Two weeks post infection, the mice were sacrificed, and spleens were weighed to determine the degree of splenomegaly (spleen weight/body weight). The genome equivalents of C. burnetii in the spleen were determined using qPCR.
Ethics statement
All animal experiments were approved by the Institute of Animal Care and Use Committee (IACUC) of the Academy of Military Medical Sciences (approval no. IACUC-DWZX-2023-P020), and all experiments were performed in accordance with the regulations and guidelines of the committee.
Statistics analysis
Statistical significance of differences in C. burnetii genome equivalents (GE), mRNA level of C. burnetii, fluorescence intensity, size of CvpE-positive particles or CCV, and ratio of spleen weight to body weight were determined using one-way or two-way variance (ANOVA). The data were analysed using GraphPad Prism 8.0 software (GraphPad). For all analyses, a p value < 0.05 was deemed to be significant.
Results
CvpE is a virulence factor of C. burnetii and contribute to CCV development
Several T4BSS effector proteins of C. burnetii localize to the CCVs membrane and promote the development of CCVs through distinct mechanisms [Citation7,Citation10,Citation21–24]. Among these effectors, the role of CvpE in the biogenesis of CCVs remains unclear. To fill this gap, we first generated a recombinant NMII strain that stably overexpressed 3×FLAG-CvpE (NMIIpJB-CvpE) and examined its subcellular localization using immunofluorescence microscopy. The translocated CvpE was localized to the membrane of Lamp1-positive CCVs in NMIIpJB-CvpE infected cells, as detected by anti-FLAG monoclonal antibody staining (). Consistent with previous observations, ectopically expressed GFP-CvpE also colocalized with LAMP1 on the membranes of CCVs in NMII-infected cells (). Collectively, these results demonstrate that CvpE traffics to the membranes of CCVs during infection.
Figure 1. CvpE is a virulence factor of C. burnetii and contribute to CCV development.
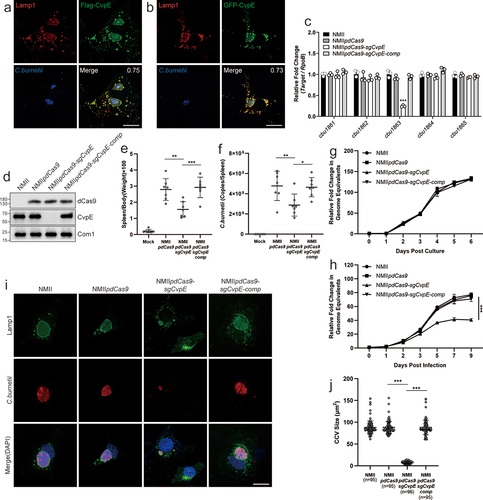
To explore the role of CvpE in CCV development and virulence in C. burnetii, we repressed the expression of CvpE in C. burnetii (NMIIpdCas9-sgCvpE) using a constitutive CRISPRi system [Citation25] and complemented the CvpE-knockdown strain (NMIIpdCas9-sgCvpE-comp) by electroporation of recombinant plasmids, according to Zhang [Citation26]. The mRNA levels of CBU1861, CBU1862, CBU1864 and CBU1865 which are adjacent to the cbu1863 in NMIIpdCas9-sgCvpE were almost equal to those in NMII or NMIIpdCas9 by qRT-PCR (), confirming the specificity of the small guide RNA targeting CvpE. CvpE expression in Vero cells infected with NMIIpdCas9-sgCvpE was almost completely suppressed compared to NMIIpdCas9 and NMII as measured by western blotting, and the CvpE of NMIIpdCas9-sgCvpE-comp was almost equal to that of NMIIpdCas9 and NMII ().
To evaluate the effects of CvpE on the pathogenicity of C. burnetii, SCID mice were infected with NMIIpdCas9, NMIIpdCas9-sgCvpE or NMIIpdCas9-sgCvpE-comp. Two weeks after infection, the spleens were removed for weighing and quantification of bacterial load. As shown in , the spleen weight and bacterial burden of NMIIpdCas9-sgCvpE-infected mice were significantly lower than those of NMIIpdCas9-infected mice. As expected, the growth of NMIIpdCas9-sgcvpE was significantly decreased in Vero cells but not in axenic medium (), and the CCV area was remarkably reduced in Vero cells infected with NMIIpdCas9-sgcvpE (). More importantly, the reduction in intracellular replication and CCV size could be complemented to wild-type levels by expressing another version of CvpE driven by the P1169 promoter on pdCas9-sgCvpE (). Taken together, these results demonstrate that CvpE is an essential virulence factor for SCID mouse infection, and that CvpE contributes to CCV development and intracellular replication of C. burnetii.
CvpE induces lysosome-like vacuole (LLV) enlargement
To gain insight into the biological function of CvpE, the C. burnetii effector was ectopically expressed as a GFP-tagged fusion protein in HeLa, Vero, and COS-7 cells. Accumulation of cytoplasmic vacuoles was observed in eukaryotic cells, and GFP-CvpE was localized on the surface of the vacuoles (). Transfected cells were then stained with antibodies against Rab5 (marker of early endosome), Rab7 (marker of late endosome/lysosome), Rab11 (marker of recycling endosome), Lamp1 (marker of lysosome), GM130 (marker of trans-Golgi network) and Calreticulin (marker of endoplasmic reticulum). As shown in , CvpE colocalized with Rab7 and Lamp1 on the membrane of vacuoles. indicating that the cytoplasmic vacuoles triggered by CvpE are enlarged lysosomes. The Rab7 or Lamp1 cannot distinguish a certain single organelle adequately, because the markers localized to lysosomes, late endosomes, or hybrid lysosomal organelles fused with phagosomes, autophagosomes, endosomes [Citation27–30]. Therefore, the enlarged Rab7 and Lamp1-positive vacuoles induced by CvpE are described as lysosome-like vacuole (LLV) in this study.
Figure 2. CvpE induces lysosomal enlargement.
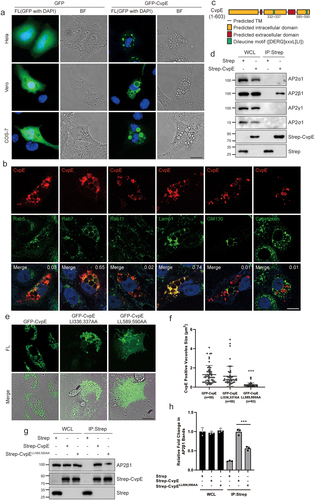
CvpE contains two endocytic sorting motifs ([DERQ]XXXL[LI]) () similar to those recognized by the tetrameric clathrin adaptor complex AP2 [Citation31]. Co-immunoprecipitation assays revealed that only AP2 subunit β1 strongly interacted with CvpE (). To evaluate the importance of endocytic sorting motifs on the function of CvpE, we constructed mutant constructs CvpE-LI336,337AA and CvpE-LL589,590AA and examined their subcellular localization in transfected Vero cells. In contrast to the total vacuole localization of wild-type CvpE and CvpE-LI336,337AA, CvpE-LL589,590AA were mainly localized on the plasma membrane and formed less vacuoles (). Subsequently, AP2β1 weakly interacted with CvpE-LL589,590AA (). Taken together, these data suggest that the C-terminal endocytic sorting motif participates in CvpE LLV localization and vacuoles formation via interaction with AP2.
CvpE-induced LLV enlargement resulted from inhibition of lysosome fission
Lysosome size is regulated by a balance between vesicle fusion and fission and can be reversibly altered by acidifying the cytoplasm using Acetate Ringer’s solution or by incubating with Vacuolin-1 [Citation32]. To determine whether the enlarged LLV induced by CvpE were due to increased lysosome fusion, HeLa cells stably expressing FLAG-CvpE (HeLaCvpE) were incubated with Acetate Ringer’s solution, which was used to fragment enlarged lysosomes, and removal of Acetate Ringer’s solution led to lysosomal refusion. Concurrently, HeLa cells stably expressing a FLAG tag (HeLaflag) treated with Vacuolin-1, a drug that induces lysosome enlargement by preventing fission but not by promoting fusion [Citation32], were used as controls. The fragmented lysosomes of HeLaCvpE and Vacuolin-1 treated HeLaflag cells following Acetate Ringer’s solution treatment were almost the same size (). After removal of Acetate Ringer’s solution, lysosomes of both cells gradually regained their original size over time, and the t1/2 to maximum size of lysosomes in HeLaCvpE cells was not significantly different from that in HeLaflag treated with Vacuolin-1 (). These data suggest that the abnormally enlarged lysosomes triggered by CvpE do not result from increased lysosome fusion.
Figure 3. CvpE inhibited lysosome fission.
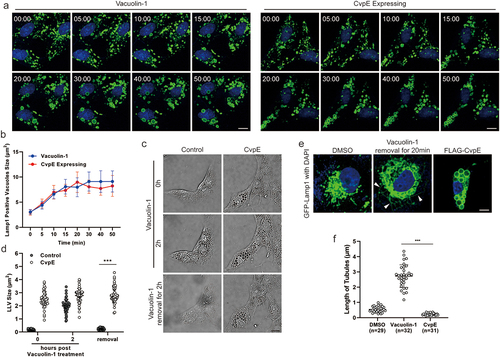
Besides the possibility of regulating lysosome fusion, CvpE is probably involved in suppressing the lysosome fission. To test this, CvpE-transfected and WT cells were treated with Vacuolin-1 for 2 h, which revealed multiple enlarged vacuoles. The enlarged lysosomes in WT cells recovered to normal size after 3 h of Vacuolin-1 removal, whereas CvpE-expressing Vero cells showed constant enlarged vacuoles after Vacuolin-1 removal (). Subsequently, the fission of lysosomes by tubulation was visualized in Vero cells stably expressing GFP-Lamp1. Vero cells treated with DMSO exhibited a small number of shorter tubules on lysosomal surface. Cells with removed Vacuolin-1 showed many longer tubules that were barely found in CvpE-expressing Vero cells (), confirming that CvpE inhibited lysosome fission. Taken together, these results demonstrate that CvpE triggers LLV enlargement by inhibiting lysosomal fission.
CvpE induced accumulation of autophagosomal markers and impairing cathepsin D maturation
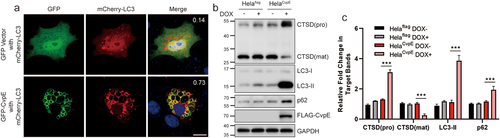
During C. burnetii infection, autophagosomes are recruited to the CCV, and the host lipoprotein LC3-II continuously resides on the CCV membrane. Several effector proteins have been reported to be involved in autophagy manipulation during C. burnetii infection [Citation9]. To determine whether CvpE-induced enlarged lysosomes fuse with autophagosomes, intracellular autophagosomes were visualized by mCherry-LC3 transfection. The LC3 signal was observed on the membrane of CvpE-positive vacuoles (). The data indicated that LC3-labelled CvpE vacuoles were autolysosomes. Next, autophagosomal biomarkers in CvpE-expressing cells were detected using western blotting. As shown in , doxycycline (DOX)-induced expression of CvpE increased the levels of LC3-II and p62, indicating that autolysosomes degradation was blocked and the autophagy flux was impaired in cells stably expressing CvpE. Moreover, the cellular levels of mature Cathepsin D (CTSD) in DOX induced HeLaCvpE cells were lower than those in untreated HeLaCvpE cells, and the levels of pro-CTSD in CvpE-expressing cells were significantly higher than those in controls (), suggesting that CvpE impaired enzymatic activity in lysosomes. In addition, the acidity of endosomes and lysosomes in cells expressing CvpE was not significantly changed, as detected by acid-sensitive Oregon Green 488 staining (data not shown). Taken together, these results indicated that CvpE induces the accumulation of autophagosomal markers by impairing autolysosome degradation.
Figure 4. CvpE induced accumulation of autophagosomal markers through impairing autolysosome degradation.
CvpE suppressed TRPML1 activity
It has been reported that transient receptor potential mucolipin 1 (TRPML1) Ca2+ channels promote lysosome fission through releasing intralysosomal Ca2+ and activating downstream Calmodulin and ALG2 [Citation33]. To investigate whether lysosomal Ca2+ channel activity was involved in CvpE-induced LLV enlargement, we treated CvpE-expressing Vero cells with ML-SA5, an agonist of TRPML1. ML-SA5 treatment completely prevented CvpE-induced cytoplasmic vacuolization (), suggesting that the activity of TRPML1 might be inhibited in CvpE-induced LLV enlargement. Because co-expression of TRPML1 with CvpE also precluded cytoplasmic vacuolization and made CvpE punctate distributions, we co-expressed TRPML1PtdMut, a mutant that cannot bind to PtdIns(3,5)P2 [Citation34], with CvpE and found that TRPML1PtdMut colocalized with CvpE on the membrane of enlarged LLV ().
Figure 5. CvpE suppressed TRPML1 activity.
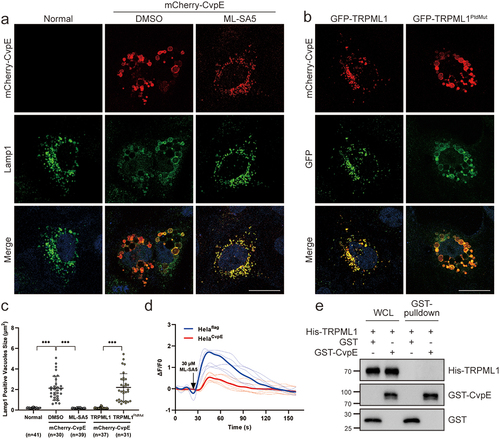
To determine whether CvpE perturbs TRPML1 activity, we constructed a plasmid containing the entire TRPML1 ORF with an N-terminal GECO1 tag, which revealed green fluorescence after Ca2+ outflow from the lysosomal lumen via the TRPML1 channel [Citation35]. The ΔF/F0 value indicating the activity of TRPML1 after ML-SA5 treatment was measured using ImageXpress MicroConfocal, and the fluorescence intensity was analysed and calculated using Image J software. The results showed that ΔF/F0 value in CvpE-expressing cells was significantly lower than that in the controls (), suggesti ng that CvpE inhibited the activity of TRPML1. However, the GST pull-down experiment revealed that purified His-TRPML1 did not bind to GST-CvpE directly (), indicating that CvpE may perturb the TRPML1 activity indirectly via suppressing upstream or downstream factors of TRPML1.
CvpE binds PI(3)P and perturbs PIKfyve activity on lysosomes
It has been reported that PtdIns(3,5)P2 binds and opens the TRPML1 Ca2+ channel to locally release intralysosomal Ca2+ to mediate lysosomal fission, suggesting that CvpE might perturb PI(3,5)P2 generation on the lysosomal membrane. To test this hypothesis, we first co-transfected CvpE with several phosphoinositide-binding probes and examined their subcellular localization. Among these probes, GFP-2×FYVE [PI(3)P binding probe] and GFP-pHlact C2 (phosphatidylserine-binding probe) colocalized with ectopically expressed CvpE (). Subsequently, the PIP strip assay showed that the purified recombinant GST-CvpE preferentially bound to PI(3)P (). PI(3)P is a major determinant of early endosome membrane identity [Citation36] and immediately undergoes phosphorylation to PI(3,5)P2 by PIKfyve kinase on the membrane of lysosomes [Citation37–39]. The accumulation of PI(3)P on CvpE triggered enlarged LLVs, consistent with the phenotype of YM201636 treatment, a specific PIKfyve inhibitor that suppresses the generation of PI(3,5)P2 from PI(3)P (), suggesting that CvpE perturbs the generation of PI(3,5)P2. Using an ADP-Glo kinase assay, we excluded the possibility that CvpE itself functions as a PI kinase (). Additionally, CvpE had no effect on the assembly of the PIKfyve-vac14-/Sac3 complex (). These results indicate that CvpE might compete with PIKfyve for PI(3)P binding, similar to the mechanism employed by another well-characterized effector, CvpB [Citation7].
Figure 6. CvpE binds PI(3)P and perturbs PIKfyve activity on lysosomes.
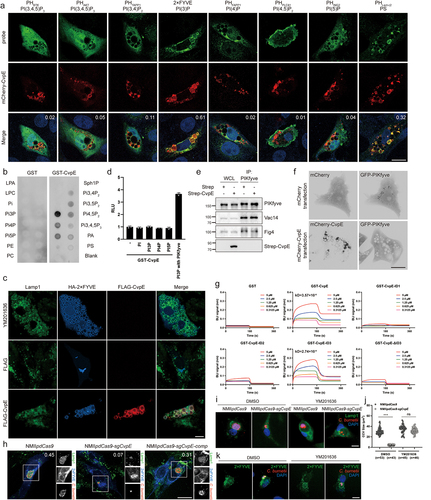
We co-transfected GFP-PIKfyve with mCherry-CvpE to estimate the distribution of GFP-PIKfyve. In control cells, co-expressing GFP-PIKfyve and mCherry-CvpE, GFP-PIKfyve showed diffuse localization instead of punctate distribution (). Collectively, these results indicate that CvpE perturbs PIKfyve activity by binding PI(3)P to the LLVs.
CvpE possesses four predicted transmembrane domains, two extracellular domains, and three intracellular domains (ID) (). To distinguish the PI(3)P binding area in CvpE, Octet Anti-Glutathione-S-Transferase (GST) biosensors were used to detect binding kinetics between purified GST-tagged CvpE IDs (ID1:1-199aa, ID2:255-414aa, ID3:498-603aa) and PI(3)P. As shown in , only GST-CvpE and GST-CvpE-ID3 bind to PI(3)P at high affinity in Bio-Layer Interferometry (BLI). Moreover, GST-CvpE-ΔID3 could not bind with PI(3)P, suggesting that the PI(3)P-binding domain of CvpE is located in ID3.
To further verify the biological function of CvpE during C. burnetii infection, we compared the colocalization of 2×FYVE with Lamp1-positive CCVs in NMIIpdCas9, NMIIpdCas9-sgCvpE and NMIIpdCas9-sgCvpE-comp infected Cells. As shown in , the colocalization of 2×FYVE with Lamp1 in NMIIpdCas9-sgcvpE-infected cells was significantly reduced. Furthermore, decreased CCVs in NMIIpdCas9-sgCvpE-infected Vero cells were restored to almost normal size (CCVs formed by NMIIpdCas9 infection) when treated with YM201636, even though only a small amount of C. burnetii was in the CCVs of NMIIpdCas9-sgCvpE-infected Vero cells (). To determine whether this YM201636-induced CCV expansion is a result of PI(3)P on the CCV, we compared PI(3)P localization between YM201636 and DMSO treated Vero cells that infected with NMIIpdCas9-sgCvpE. As shown in , expanded CCVs were positive for PI(3)P in YM201636 treatment, however, the smaller CCVs in controls were negative for PI(3)P. Taken together, these data demonstrate that CvpE directly binds to PI(3)P and perturbs PIKfyve activity in lysosomes during infection.
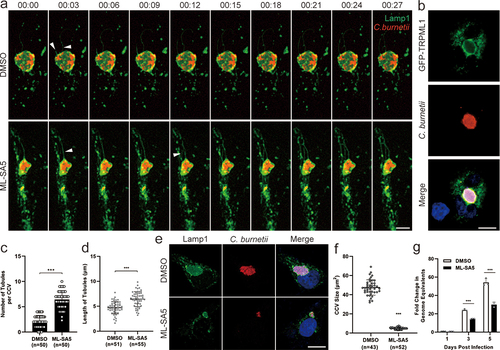
ML-SA5 treatment inhibited CCV biogenesis by promoting lysosomal tubulation and C. burnetii replication
To study the effects of lysosomal dynamics on CCV development and intracellular replication of C. burnetii, we treated NMII-infected Vero cells with ML-SA5. The number of Lamp1-positive tubules per CCV and the tubules length in ML-SA5 treated cells was significantly higher than that in DMSO treated cells, and the transiently expressed GFP-TRPML1 was localized on CCVs (), indicating that ML-SA5 facilitated CCV tubulation. In addition, long-term treatment with ML-SA5 significantly suppressed CCV development and bacterial replication in Vero cells (). Concurrently, the results indicated that lysosome fission is important for the biogenesis of CCVs.
Figure 7. ML-SA5 treatment inhibited CCV biogenesis by promoting lysosomal tubulation and suppressed C. burnetii replication.
Discussion
Living inside the host cell, C. burnetii directs the maturation of a phagolysosome-like compartment known as the replication-permissive CCV. Many effector proteins secreted by T4BSS are required for intracellular replication of C. burnetii and CCV biogenesis [Citation9,Citation40,Citation41]. Among these effector proteins, the mechanisms by which CvpE is involved in C. burnetii replication and CCVs development remain to be defined. Our data showed that CvpE maintained CCVs biogenesis by suppressing lysosomal fission rather than facilitating homologous fusion. These findings provide a novel perspective for revealing the causes of intracellular CCVs maintenance and host clearance evasion.
Different to the localization of CvpA and CvpB, ectopically, CvpE colocalized with PI(3)P and the late endosome/lysosome marker Rab7 and Lamp1. Usually, lysosomes locating PI(3)P are immediately phosphorylated by PIKfyve; therefore, PI(3)P colocalizing with Lamp1 is difficult to detect. In our data, since CvpE interacted with PI(3)P, interfering with its recruitment of PIKfyve, PI(3)P obviously colocalized with Lamp1 in CvpE-expressing cells, indicating that CvpE facilitates CCVs biogenesis via a mechanism distinct from that of CvpB. The inability of contact between PIKfyve and lysosomal PI(3)P results in a decrease in lysosomal PI(3,5)P2 which is a crucial endogenous agonist of the Ca2+ channel TRPML1 [Citation42–45], whose activation plays a critical role in lysosomal fission [Citation20,Citation44]. Consistent with the above results, ID3 of CvpE interacted with PI(3)P directly (), and the level of PI(3)P on CCV in NMIIpdCas9 infected cells was higher than that in NMIIpdCas9-sgCvpE (), suggesting that CvpE inhibits lysosomal fission by blocking the PI(3,5)P2–TRPML1 signalling pathway. However, the massive lysosomal enlargement observed with CvpE ectopic overexpression is not revealed in infected cells () suggesting that other effectors of C. burnetii may interfere with the capacity of CvpE during infection. In addition, CvpE triggered enlarged LLV did not fuse further to form a single vacuole regardless of long-term ectopic expression of CvpE, indicating that CvpE is not involved in the fusion of CCVs and inhibits fission of a single fused CCV.
Several studies have confirmed that autophagosome fusion contributes to the size of the CCVs. The absence of CLTC (clathrin heavy chain) disturbed the role of autophagosomes in the expansion of CCVs [Citation46]. CpeB promoted C. burnetii virulence by inducing LC3-II accumulation [Citation23]. CvpF manipulates endosomal trafficking and autophagy induction for optimal C. burnetii vacuole biogenesis [Citation10]. Our results show that ectopically expressed CvpE did not affect the fusion of lysosomes and autophagosomes. but disrupts lysosomal hydrolase activity and autolysosome degradation. It has also been reported that C. burnetii inhibits mTORC1 but does not accelerate autophagy or block autophagic flux triggered by cell starvation [Citation47], suggesting that other effector proteins might perform different roles from CvpE in the modulation of autophagy. During C. burnetii intracellular replication and CCVs maturation, autophagy levels show intricate results, indicating that the relationship between CCVs development and autophagy requires further investigation.
Furthermore, transmembrane helix prediction showed that CvpE contains at least four transmembrane domains, three intracellular domains, and two extracellular domains. Purified GST-tagged fragments of CvpE provided evidence that the third intracellular domain (493-603aa) is involved in binding PI(3)P to lysosomes. A literature search revealed that PI(3)P-binding motifs consist of DxHxxN and IDH, separated by 14 amino acids, and are conserved among eukaryotic PI kinases and host-associated bacteria, including species belonging to the genera Legionella, Vibrio, and Rickettsia, indicating that interaction with PI(3)P requires no more than 30 amino acids [Citation48]. However, we did not find any identified PI(3)P binding motif in ID3 of CvpE, suggesting that CvpE contains a unique PI(3)P binding motif, which requires further identification by constructing truncations or mutations. Considering the molecular weight and domains of CvpE, we suggested that CvpE has other biological functions that contribute to the intracellular replication of C. burnetii and CCVs biogenesis.
Recently, a synthetic agonist of TRPML1, ML-SA5, was reported to ameliorate Duchenne muscular dystrophy (DMD) [Citation49], a devastating disease caused by mutations in dystrophin that compromise sarcolemma integrity. ML-SA5 activated TRPML1 inhibited C. burnetii growth and CCVs development via lysosomal tubulation (). However, downstream signalling pathways of TRPML1 are also potential reasons. Calcium efflux from lysosomes facilitated by TRPML1 is essential for TFEB activation which regulates autophagy flux [Citation50]. Zinc release from lysosomes to cytosol triggered by TRPML1 affects autophagy as well [Citation51]. Acidic lysosomal pH influenced by TRPML1 [Citation52] may also act as an essential role in C. burnetii infection [Citation6]. Therefore, extensive research is necessary for revealing the effects of lysosomal iron channel on C.burnetii growth and CCVs development. Based on this study, the agonist of TRPML1 is expected to be a novel potential treatment that does not rely on antibiotics for Q fever by enhancing the fission of lysosomes.
Author contributions
M.Z. and X.X. designed and conceived the experiments, conducted the data analysis, and wrote and revised the manuscript. M.Z. performed experiments. S.Z., W.W., C.Z., N.L., and R.C. contributed to and shared the experimental materials, reagents, and instruments. Y.Y. and X.O. conducted the formal analyses. B.W., J.J. and X.X. revised and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Acknowledgements
We thank Lei Song for providing several phosphoinositide-binding probe plasmids.
Disclosure statement
We declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Data Availability statement
The authors confirmed that all data supporting the findings of this study are available within the article.
Additional information
Funding
References
- Eldin C, Mélenotte C, Mediannikov O, et al. From Q fever to coxiella burnetii infection: a paradigm change. Clin Microbiol Rev. 2017 Jan;30(1):115–17. doi: 10.1128/CMR.00045-16
- Maurin M, Raoult D. Q fever. Clin Microbiol Rev. 1999 Oct;12(4):518–553. doi: 10.1128/CMR.12.4.518
- Padmanabhan B, Fielden LF, Hachani A, et al. Biogenesis of the spacious coxiella-Containing vacuole depends on host transcription factors TFEB and TFE3. Infect Immun. 2020 Feb 20;88(3). doi: 10.1128/IAI.00534-19
- Heinzen RA, Scidmore MA, Rockey DD, et al. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of coxiella burnetii and chlamydia trachomatis. Infect Immun. 1996 Mar;64(3):796–809. doi: 10.1128/iai.64.3.796-809.1996
- Miller HE, Hoyt FH, Heinzen RA, et al. Replication of coxiella burnetii in a lysosome-like vacuole does not require lysosomal hydrolases. Infect Immun. 2019 Nov;87(11). doi: 10.1128/IAI.00493-19
- Samanta D, Clemente TM, Schuler BE, et al. Coxiella burnetii Type 4B secretion system-dependent manipulation of endolysosomal maturation is required for bacterial growth. PLOS Pathog. 2019 Dec;15(12):e1007855. doi: 10.1371/journal.ppat.1007855
- Martinez E, Allombert J, Cantet F, et al. Coxiella burnetii effector CvpB modulates phosphoinositide metabolism for optimal vacuole development. Proc Natl Acad Sci USA. 2016 Jun 7;113(23):E3260–9. doi: 10.1073/pnas.1522811113
- Thomas DR, Newton P, Lau N, et al. Interfering with autophagy: the opposing strategies deployed by legionella pneumophila and coxiella burnetii effector proteins. Front Cell Infect Microbiol. 2020;10:599762. doi: 10.3389/fcimb.2020.599762
- Newton HJ, Kohler LJ, McDonough JA, et al. A screen of coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLOS Pathog. 2014 Jul;10(7):e1004286. doi: 10.1371/journal.ppat.1004286
- Siadous FA, Cantet F, Van Schaik E, et al. Coxiella effector protein CvpF subverts RAB26-dependent autophagy to promote vacuole biogenesis and virulence. Autophagy. 2021 Mar;17(3):706–722. doi: 10.1080/15548627.2020.1728098
- Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. 2020 Feb;21(2):101–118. doi: 10.1038/s41580-019-0185-4
- de Araujo, MEG, Liebscher G, Hess MW, et al. Lysosomal size matters. Traffic. 2020 Jan;21(1):60–75. doi: 10.1111/tra.12714
- Huotari J, Helenius A. Endosome maturation. Embo J. 2011 Aug 31;30(17):3481–3500. doi: 10.1038/emboj.2011.286
- Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012 Oct 11;490(7419):201–207. doi: 10.1038/nature11320
- Baker RW, Hughson FM. Chaperoning SNARE assembly and disassembly. Nat Rev Mol Cell Biol. 2016 Aug;17(8):465–479. doi: 10.1038/nrm.2016.65
- Chen Y, Yu L. Recent progress in autophagic lysosome reformation. Traffic. 2017 Jun;18(6):358–361. doi: 10.1111/tra.12484
- Saric A, Hipolito VE, Kay JG, et al. mTOR controls lysosome tubulation and antigen presentation in macrophages and dendritic cells. Mol Biol Cell. [2016 Jan 15];27(2):321–333. doi: 10.1091/mbc.e15-05-0272
- Saffi GT, Botelho RJ. Lysosome fission: planning for an exit. Trends Cell Biol. 2019 Aug;29(8):635–646. doi: 10.1016/j.tcb.2019.05.003
- Cerny J, Feng Y, Yu A, et al. The small chemical vacuolin-1 inhibits Ca(2+)-dependent lysosomal exocytosis but not cell resealing. EMBO Rep. 2004 Sep;5(9):883–888. doi: 10.1038/sj.embor.7400243
- Cao Q, Yang Y, Zhong XZ, et al. The lysosomal Ca(2+) release channel TRPML1 regulates lysosome size by activating calmodulin. J Biol Chem. 2017 May 19;292(20):8424–8435. doi: 10.1074/jbc.M116.772160
- Latomanski EA, Newton P, Khoo CA, et al. The effector Cig57 hijacks FCHO-mediated vesicular trafficking to facilitate intracellular replication of coxiella burnetii. PLOS Pathog. 2016 Dec;12(12):e1006101. doi: 10.1371/journal.ppat.1006101
- Larson CL, Beare PA, Howe D, et al. Coxiella burnetii effector protein subverts clathrin-mediated vesicular trafficking for pathogen vacuole biogenesis. Proc Natl Acad Sci USA. 2013 Dec 3;110(49):E4770–9. doi: 10.1073/pnas.1309195110
- Fu M, Zhang J, Zhao M, et al. Coxiella burnetii plasmid effector B promotes LC3-II accumulation and contributes to bacterial virulence in a SCID mouse Model. Infect Immun. 2022 Jun 16;90(6):e0001622. doi: 10.1128/iai.00016-22
- Larson CL, Beare PA, Voth DE, et al. Coxiella burnetii effector proteins that localize to the parasitophorous vacuole membrane promote intracellular replication. Infect Immun. 2015 Feb;83(2):661–670. doi: 10.1128/IAI.02763-14
- Fu M, Liu Y, Wang G, et al. A protein–protein interaction map reveals that the coxiella burnetii effector CirB inhibits host proteasome activity. PLOS Pathog. 2022 Jul;18(7):e1010660. doi: 10.1371/journal.ppat.1010660
- Zhang Y, Fu J, Liu S, et al. Coxiella burnetii inhibits host immunity by a protein phosphatase adapted from glycolysis. Proc Natl Acad Sci USA. 2022 Jan 4;119(1). doi: 10.1073/pnas.2110877119
- Harrison RE, Bucci C, Vieira OV, et al. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol. 2003 Sep;23(18):6494–6506. doi: 10.1128/MCB.23.18.6494-6506.2003
- Seto S, Tsujimura K, Koide Y. Rab GTPases regulating phagosome maturation are differentially recruited to mycobacterial phagosomes. Traffic. 2011 Apr;12(4):407–420. doi: 10.1111/j.1600-0854.2011.01165.x
- Johansson M, Lehto M, Tanhuanpää K, et al. The oxysterol-binding protein homologue ORP1L interacts with Rab7 and alters functional properties of late endocytic compartments. Mol Biol Cell. 2005 Dec;16(12):5480–5492. doi: 10.1091/mbc.e05-03-0189
- Schröder B, Wrocklage C, Pan C, et al. Integral and associated lysosomal membrane proteins. Traffic. 2007 Dec;8(12):1676–1686. doi: 10.1111/j.1600-0854.2007.00643.x
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72(1):395–447. doi: 10.1146/annurev.biochem.72.121801.161800
- Durchfort N, Verhoef S, Vaughn MB, et al. The enlarged lysosomes in beige j cells result from decreased lysosome fission and not increased lysosome fusion. Traffic. 2012 Jan;13(1):108–119. doi: 10.1111/j.1600-0854.2011.01300.x
- Li X, Rydzewski N, Hider A, et al. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat Cell Biol. 2016 Apr;18(4):404–417. doi: 10.1038/ncb3324
- Fine M, Schmiege P, Li X. Structural basis for PtdInsP2-mediated human TRPML1 regulation. Nat Commun. 2018 Oct 10;9(1):4192. doi: 10.1038/s41467-018-06493-7
- Zhao Y, Araki S, Wu J, et al. An expanded palette of genetically encoded Ca(2)(+) indicators. Science. 2011 Sep 30;333(6051):1888–1891. doi: 10.1126/science.1208592
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006 Oct 12;443(7112):651–657. doi: 10.1038/nature05185
- Ikonomov OC, Sbrissa D, Shisheva A. Mammalian cell morphology and endocytic membrane homeostasis require enzymatically active phosphoinositide 5-kinase PIKfyve. J Biol Chem. 2001 Jul 13;276(28):26141–26147. doi: 10.1074/jbc.M101722200
- McCartney AJ, Zolov SN, Kauffman EJ, et al. Activity-dependent PI(3,5)P2 synthesis controls AMPA receptor trafficking during synaptic depression. Proc Natl Acad Sci USA. 2014 Nov 11;111(45):E4896–905. doi: 10.1073/pnas.1411117111
- Ho CY, Alghamdi TA, Botelho RJ. Phosphatidylinositol-3,5-bisphosphate: no longer the poor PIP2. Traffic. 2012 Jan;13(1):1–8. doi: 10.1111/j.1600-0854.2011.01246.x
- Carey KL, Newton HJ, Luhrmann A, et al. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLOS Pathog. 2011 May;7(5):e1002056. doi: 10.1371/journal.ppat.1002056
- Lifshitz Z, Burstein D, Peeri M, et al. Computational modeling and experimental validation of the legionella and coxiella virulence-related type-IVB secretion signal. Proc Natl Acad Sci USA. 2013 Feb 19;110(8):E707–15. doi: 10.1073/pnas.1215278110
- McCartney AJ, Zhang Y, Weisman LS. Phosphatidylinositol 3,5-bisphosphate: low abundance, high significance. BioEssays. 2014 Jan;36(1):52–64. doi: 10.1002/bies.201300012
- Chen Q, She J, Zeng W, et al. Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nature. 2017 Oct 19;550(7676):415–418. doi: 10.1038/nature24035
- Dong XP, Shen D, Wang X, et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun. 2010 Jul 13;1(1):38. doi: 10.1038/ncomms1037
- Hirschi M, Herzik Jr MA Jr., Wie J, et al. Cryo-electron microscopy structure of the lysosomal calcium-permeable channel TRPML3. Nature. 2017 Oct 19];550(7676):411–414. doi: 10.1038/nature24055
- Latomanski EA, Newton HJ. Interaction between autophagic vesicles and the Coxiella-containing vacuole requires CLTC (clathrin heavy chain). Autophagy. 2018;14(10):1710–1725. doi: 10.1080/15548627.2018.1483806
- Larson CL, Sandoz KM, Cockrell DC, et al. Noncanonical inhibition of mTORC1 by coxiella burnetii promotes replication within a phagolysosome-like vacuole. MBio. 2019 Feb 5;10(1). doi: 10.1128/mBio.02816-18
- Ledvina HE, Kelly KA, Eshraghi A, et al. A phosphatidylinositol 3-kinase effector alters phagosomal maturation to promote intracellular growth of Francisella. Cell Host Microbe. 2018 Aug 8;24(2):285–295.e8. doi: 10.1016/j.chom.2018.07.003
- Yu L, Zhang X, Yang Y, et al. Small-molecule activation of lysosomal TRP channels ameliorates duchenne muscular dystrophy in mouse models. Sci Adv. 2020 Feb;6(6):eaaz2736. doi: 10.1126/sciadv.aaz2736
- Nakamura S, Shigeyama S, Minami S, et al. LC3 lipidation is essential for TFEB activation during the lysosomal damage response to kidney injury. Nat Cell Biol. 2020 Oct;22(10):1252–1263. doi: 10.1038/s41556-020-00583-9
- Qi J, Xing Y, Liu Y, et al. MCOLN1/TRPML1 finely controls oncogenic autophagy in cancer by mediating zinc influx. Autophagy. 2021 Dec;17(12):4401–4422. doi: 10.1080/15548627.2021.1917132
- Li M, Zhang WK, Benvin NM, et al. Structural basis of dual Ca(2+)/pH regulation of the endolysosomal TRPML1 channel. Nat Struct Mol Biol. 2017 Mar;24(3):205–213. doi: 10.1038/nsmb.3362