
Abstract
Objective
In Norway, 89% of patients with Amyotrophic lateral sclerosis (ALS) lacks a genetic diagnose. ALS genes and genes that cause other neuromuscular or neurodegenerative disorders extensively overlap. This population-based study examined whether patients with ALS have a family history of neurological disorders and explored the occurrence of rare genetic variants associated with other neurodegenerative or neuromuscular disorders.
Methods
During a two-year period, blood samples and clinical data from patients with ALS were collected from all 17 neurological departments in Norway. Our genetic analysis involved exome sequencing and bioinformatics filtering of 510 genes associated with neurodegenerative and neuromuscular disorders. The variants were interpreted using genotype-phenotype correlations and bioinformatics tools.
Results
A total of 279 patients from a Norwegian population-based ALS cohort participated in this study. Thirty-one percent of the patients had first- or second-degree relatives with other neurodegenerative disorders, most commonly dementia and Parkinson’s disease. The genetic analysis identified 20 possible pathogenic variants, in ATL3, AFG3L2, ATP7A, BICD2, HARS1, KIF1A, LRRK2, MSTO1, NEK1, NEFH, and SORL1, in 25 patients. NEK1 risk variants were present in 2.5% of this ALS cohort. Only four of the 25 patients reported relatives with other neurodegenerative or neuromuscular disorders.
Conclusion
Gene variants known to cause other neurodegenerative or neuromuscular disorders, most frequently in NEK1, were identified in 9% of the patients with ALS. Most of these patients had no family history of other neurodegenerative or neuromuscular disorders. Our findings indicated that AFG3L2, ATP7A, BICD2, KIF1A, and MSTO1 should be further explored as potential ALS-causing genes.
Introduction
Amyotrophic lateral sclerosis (ALS) affects both the upper and lower motor neurons, leading to muscular weakening and paralysis. Approximately 10% of the diagnosed patients have a family history of ALS (fALS), while the remaining 90% are simplex cases (sALS) (Citation1). ALS is a heterogenic disease and more than 30 genes have been firmly linked to ALS (Citation2, Citation3). C9orf72, SOD1, TARDBP, and FUS are the most frequently mutated genes (Citation3, Citation4). In Europe, including Norway, a monogenic cause has been identified in approximately 50% of the fALS and 5–10% of the sALS cases (Citation5–8). The low diagnostic yield of sALS is consistent with recent studies demonstrating that ALS is multifaceted; a mix of genetics, environment, and age likely contributes to its development (Citation9). ALS heritability has been estimated at 50–60% (Citation10, Citation11).
Most identified ALS genes are pleiotropic, i.e., causing more than one disease phenotype, and are involved in other neuromuscular or neurodegenerative disorders (). For example, the most common genetic cause of ALS, the C9orf72 repeat expansion, may also cause frontotemporal dementia (Citation12), parkinsonism (Citation13), and psychiatric diseases including schizophrenia and mania (Citation14). Although ALS and frontotemporal dementia are distinct clinical entities, cognitive impairment occurs in both (Citation15). Furthermore, the ANG gene increases the risk of both ALS and Parkinson’s disease (PD) (Citation16), whereas KIF5A causes ALS, hereditary spastic paraplegia (HSP), and neuropathy (Citation17, Citation18).
Figure 1 Genetic pleiotropy of amyotrophic lateral sclerosis (ALS) genes. The figure is based on information from Online Mendelian Inheritance in Man (OMIM) (Citation19), GeneReviews (Citation20) and the study by Goutman et al. “Recent advances in the diagnosis and prognosis of amyotrophic lateral sclerosis” (Citation21). Abbreviations: SMA = spinal muscular atrophy; PLS = primary lateral sclerosis; HSP = hereditary spastic paraplegia.
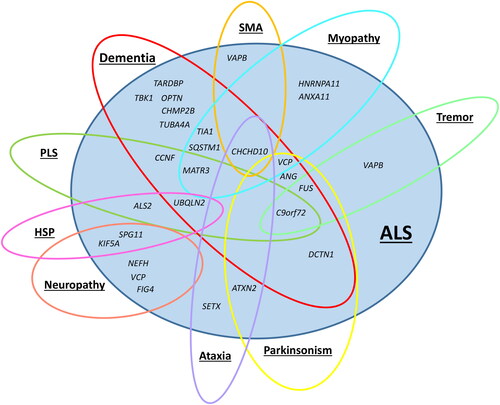
The variable expression of known ALS genes suggests that part of the missing heritability in ALS could be accounted for by genes related to other neuromuscular or neurodegenerative disorders (Citation22). To test this hypothesis, we first investigated the occurrence of neuromuscular or neurodegenerative disorders among the relatives of patients with ALS included in a Norwegian population-based cohort. Second, we explored whether the studied patients with ALS carried possible pathogenic gene variants known to cause other neuromuscular or neurodegenerative phenotypes.
Materials and methods
Study population
Between August 2019 and August 2021, 279 fALS and sALS cases were recruited from all 17 neurological departments in Norway. The clinical characteristics of the study population, inclusion rates and results from the analysis of known ALS genes have previously been described (Citation8). At the time of enrollment, a questionnaire regarding clinical characteristics, and family history of ALS and other neuromuscular or neurodegenerative disorders among first- and second-degree relatives was answered. Patients who reported a family history of ALS among first-degree, second-degree, or other relatives were categorized as fALS. The study was approved by the Regional Committees for Medical and Health Research Ethics (REK) #2018/1916, the Norwegian Center for Research Data, #426990, and the data protection officers at the different hospitals involved in the study. All participants provided written informed consent for their involvement in the study.
Genetic analysis
Next-generation sequencing was performed as previously described (Citation8). Sequencing data from all patients were bioinformatically filtered to retrieve data on 510 genes known to cause neuromuscular or neurodegenerative disorders (Supplementary File 1). The genes were selected using Genomics England PanelApp (https://panelapp.genomicsengland.co.uk/). Genes, in which only expansions contributed to disease development, and genes located in the mitochondrial genome, were excluded. Thirty previously investigated ALS genes were not included, but previous genetic findings (Citation8) were considered during the interpretation. ALS risk genes present in the adult-onset neurodegenerative gene panel from Genomics England were included in the analysis.
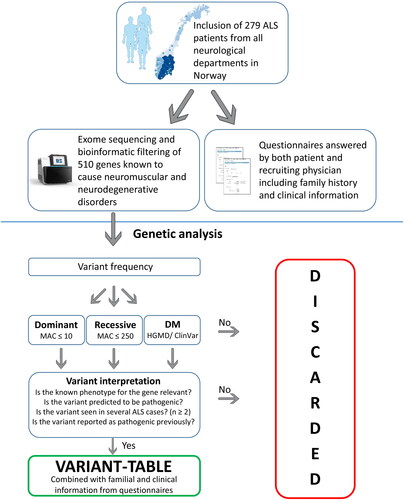
The identified variants were filtered based on dominant and recessive inheritance models using gnomAD (https://gnomad.broadinstitute.org/) (Citation23) with a minor allele count of ≤ 10 and ≤ 250, respectively. Known disease-causing variants registered in the Human Gene Mutation Database (HGMD®) (Citation24) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) were examined regardless of the gnomAD allele count. Intronic regions, untranslated regions, and synonymous variants were discarded before further interpretation unless they had been previously reported as pathogenic. The remaining variants were interpreted using gnomAD, frequencies from a Norwegian in-house hospital database (Department of Medical Genetics, Oslo University hospital) consisting of 12 874 individuals, the in silico prediction tool REVEL, Alamut Visual Interface (SOPHiA GENETICS), HGMD®, ClinVar, the Project MinE data browser (http://databrowser.projectmine.com/), and literature (Citation24–28). Special emphasis was given to variants identified in more than one participant, variants previously reported as pathogenic, loss-of-function variants where the loss-of-function is a known disease mechanism or where the gnomAD data predict non-tolerance for loss-of-function variants. An overview of patient inclusion, genetic analysis, and variant interpretation is shown in .
Figure 2 Overview of methods and variant interpretation. DMs = disease-causing mutations, HGMD = the Human Gene Mutation Database, MAC = minor allele count.
Results
Neurodegenerative and neuromuscular disorders among family members
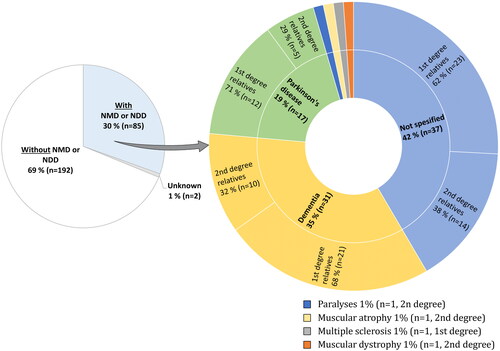
The questionnaires revealed that 30.5% (85/279) of the patients had first- or second-degree relatives with neuromuscular or neurodegenerative disorders, among which dementia and PD were the most common (). In our cohort, 32 patients (11.5%) reported a family history of ALS, as specified in our previous report (Citation8). Eight fALS patients (4 first-degree and 4 second-degree) reported relatives with other neurological disorders. Forty-two percent (37/85) did not specify the type of neurological disease reported among relatives. Nearly twice as many patients with ALS reported a family history of neurodegenerative or neuromuscular disorders among first-degree relatives than among second-degree relatives.
Figure 3 Neuromuscular or neurodegenerative disorders among the relatives of patients with amyotrophic lateral sclerosis (ALS). Left circle: patients with ALS (n = 279) reporting relatives with or without other neuromuscular or neurodegenerative disorders. Right circle: neuromuscular and neurodegenerative disorders reported among relatives of the 85 patients with ALS and a positive family history. Four cases had relatives with both PD and dementia; these individuals are shown twice in the right circle. Abbreviation: NMD or NDD = neuromuscular or neurodegenerative disorders.
Genetic analysis
Our genetic analysis of 510 genes associated with neuromuscular and neurodegenerative disorders revealed 20 potentially pathogenic variants in 25 patients with ALS. All variants had low frequencies both in gnomAD an in the Norwegian hospital database. One patient reported a family history of ALS. Variants were identified in genes causing ataxia, HSP, PD, mitochondrial disorders, sensory neuropathy, spinal muscular atrophy (SMA), and Charcot–Marie–Tooth disease (CMT), and in genes known to increase ALS risk (). None of the 25 patients had pathogenic variants of the 30 previously investigated ALS genes (Citation8). Four of the 25 patients reported having relatives with neuromuscular or neurodegenerative disorders, one patient reported PD in a first-degree relative, and three patients did not specify the type of disorder or kinship. The clinical characteristics of the patients are shown in .
Table 1 Variants identified from the genetic analysis of 510 genes known to cause neuromuscular and neurodegenerative disorders.
Table 2 Patient phenotypes at inclusion.
Neurodegenerative disorder genes
Case 1 carried a heterozygous nonsense AFG3L2 variant, which likely has a loss-of-function effect. Pathogenic AFG3L2 variants cause autosomal recessive spastic ataxia, autosomal-dominant spinocerebellar ataxia, and optic atrophy (Citation19). Loss-of-function AFG3L2 variants have been reported to cause AFG3L2-related disorders (Citation29, Citation30). Biallelic loss-of-function variants tend to have an earlier onset than heterozygous nonsense variants (Citation29–33). This variant was previously reported as likely pathogenic in ClinVar but without a phenotype.
Case 2, with early disease onset, carried a heterozygous missense KIF1A variant. KIF1A variants cause both autosomal dominant and recessive HSP as well as recessive hereditary sensory and autonomic neuropathy type 2 (Citation19). Recently, heterozygous KIF1A variants were reported in patients with ALS (Citation34). The variant identified in Case 2 is located in the kinesin motor domain close to other pathogenic variants and is predicted to be pathogenic (Citation35, Citation36). This variant has been reported as a variant of uncertain significance (VUS) for HSP in ClinVar and has been identified in a patient with ALS in Project MinE. Based on the recent study showing a potential link between KIF1A and ALS, as well as on the identification of variants located in the C-terminal cargo-binding domain instead of the commonly affected kinesin motor domain (Citation34), we also included variants located in other regions. Two potentially pathogenic heterozygous variants were identified (Cases 3 and 4). The variant identified in Case 4 was a VUS in ClinVar.
In Cases 5 and 6, a pathogenic LRRK2 variant frequently reported to cause PD (Citation37) was identified. This variant has a high carrier frequency and reduced penetrance in Norway (Citation38).
In Cases 7 and 8, an identical heterozygous splice variant in MSTO1 was identified. MSTO1 variants cause autosomal recessive mitochondrial and ataxic myopathy with childhood onset (Citation19). Case 7 had bulbar onset in her thirties and reported a first-degree relative with PD, whereas Case 8 reported a negative family history. The identified variant has been reported as pathogenic (biallelic form) in both the literature (Citation39) and ClinVar. Two other MSTO1 splice variants predicted to be likely deleterious were reported among patients with ALS in Project MinE.
In Cases 9–15, constituting 7/279 (2.5%) of our cohort, one splice variant and two heterozygous loss-of-function NEK1 variants were identified. Loss-of-function NEK1 variants increase the risk of ALS (Citation40, Citation41), but were not included in our previous study (Citation8). Although NEK1 has not been reported to cause neuromuscular or neurodegenerative disorders other than ALS, it was included in our study because it is a part of the adult-onset neurodegenerative gene panel from Genomics England. NEK1 variants have been suggested to display reduced penetrance (Citation42) which explains the relatively high carrier frequency of the p.(Ser1036*) variant identified in four of our cases.
Cases 16 and 17 carried frameshift and splice SORL1 variants, respectively, both of which are predicted to have a loss-of-function effect. Loss-of-function SORL1 variants increase the risk of early-onset Alzheimer’s disease (Citation43, Citation44), in which the identified splice variant has been previously reported in early-onset Alzheimer’s (Citation45). Neither Case 16 nor Case 17 showed cognitive impairment and did not report relatives with dementia.
Neuromuscular disorder genes
In Case 18, a heterozygous ATL3 missense variant was identified. ATL3 variants cause autosomal dominant sensory neuropathy (Citation19). The identified ATL3 variant was predicted to be pathogenic by in silico prediction tools. It is located in the guanylate-binding protein domain and has been reported in ClinVar as a VUS in type 1F hereditary sensory neuropathy. Interestingly, Case 18 showed sensory findings.
Cases 19 and 20 carried hemizygous ATP7A variants. ATP7A variants are associated with X-linked recessive distal SMA, Menkes disease, and optical horn syndrome. The onset of X-linked SMA usually occurs in the first decade of life; however, adult-onset X-linked SMA can occur as well (Citation19). Case 19 had an early onset in his thirties. A missense ATP7A variant was recently reported in an individual with ALS (Citation46). The two variants identified here are highly conserved, predicted to be pathogenic, and located close to other pathogenic variants associated with SMA and Menkes disease, and have been listed as VUS in ClinVar. Case 19 also carried a potentially pathogenic NEFH variant (), which will be discussed later.
Cases 21–23 carried different heterozygous BICD2 variants. This gene is associated with autosomal dominant SMA, with symptom onset usually in the first decade of life (Citation47). Cases 21 and 22 carried different BICD2 variants altering the same amino acid at position 265 (). The p.(Met265Val) substitution has a relatively high carrier frequency, whereas the p.(Met265Leu) variant is absent in gnomAD. The former was previously reported in a patient with HSP (Citation48), but has also been reported as likely benign in ClinVar. Case 23 carried a nonsense variant predicted to have a loss-of-function effect. According to gnomAD, BICD2 cannot tolerate loss-of-function variants. To the best of our knowledge, this is the first patient report of a loss-of-function variant in BICD2.
Case 24 harbored a heterozygous missense HARS1 variant. This gene causes autosomal dominant axonal CMT type 2W, which can resemble lower motor neuron ALS (Citation19). The identified variant affects a highly conserved amino acid and is predicted to be pathogenic. Case 24 had only lower motor neuron symptoms.
Heterozygous missense NEFH variants were identified in Cases 25 and 19. NEFH variants cause autosomal dominant CMT type 2CC, and this gene is believed to be a susceptibility gene for ALS (Citation19). The variant identified in Case 25 was located close to other variants implicated in ALS (Citation24), whereas that identified in Case 19 changed a highly conserved proline residue in the NEFH tail domain. This domain contains distinctive regions of lysine-serine-proline repeats (Citation49), and deletion, insertion, and missense variants in this region have been suggested to be causative factors of ALS (Citation50–52).
Discussion
The extensive clinical overlap between ALS and other neurodegenerative or neuromuscular disorders suggests that some of the missing heredity in ALS is caused by genetic variants underlying related disorders (Citation22). Accordingly, a recent genome-wide association study found that the genetic overlap between different neurodegenerative disorders, including ALS, is more extensive than expected (Citation59). To explore this hypothesis further, we examined rare variants in 510 genes implicated in neurodegenerative and neuromuscular phenotypes in 279 patients with ALS and investigated the possible coexistence of a neurodegenerative or neuromuscular family history and rare variants in genes causing similar disorders.
Nearly one-third of the patients with ALS reported relatives with related neuromuscular or neurodegenerative disorders. Unfortunately, nearly half of them did not specify the disorder type, making it difficult to compare our results with those of similar studies. Among the patients that specified the phenotype, neurodegenerative disorders, particularly dementia and PD, were much more common than neuromuscular disorders. Some (Citation53, Citation54), but not all previous studies (Citation55, Citation56) have found an increased prevalence of PD and Alzheimer’s disease among relatives of patients with ALS. Furthermore, twice as many patients with ALS in our study reported other neurological disorders among their first-degree relatives than among their second-degree relatives. This may reflect a lack of knowledge about second-degree relatives.
Among the 510 neurodegenerative and neuromuscular genes, we identified 20 possible pathogenic variants. Fifteen patients had variants in genes causing other neurological phenotypes, whereas nine patients had variants in genes increasing ALS risk. One patient had a variant in both ATP7A and NEFH, the latter being an ALS risk gene. Intriguingly, only three of the 15 patients with possible pathogenic variants in other neurodegenerative or neuromuscular genes reported relatives with such disorders, and one patient with a NEK1 variant reported a family history of ALS. Whether this was because of the absence of a causal relationship, reduced penetrance, or missing information regarding disorders among family members is unknown.
Among the neurodegenerative genes, AFG3L2, KIF1A, and MSTO1 have previously been directly or indirectly linked to ALS, making their variants particularly interesting. AFG3L2 is an important SPG7 paralogue. Together, they encode an m-AAA protease that degrades damaged proteins and regulates mitochondrial ribosome synthesis (Citation57). Heterozygous SPG7 variants have recently been associated with increased ALS risk (Citation58). KIF1A is part of the anterograde transport machinery (Citation59), and was recently proposed as an ALS gene (Citation34). KIF5A, another kinesin motor gene involved in anterograde transport, is known to increase ALS risk (Citation60). MSTO1, a gene important for mitochondrial dynamics, has been implicated in pathways perturbed in ALS (Citation61). MSTO1 has not yet been linked to monoallelic disease; however, it is not uncommon for neurodegenerative genes to cause both a severe early-onset recessive phenotype and a late-onset dominant phenotype, as observed for the ALS genes FIG4 and GLE1 (Citation19). To our knowledge, neither SORL1 nor LRRK2 have been implicated in ALS pathology; thus, it is uncertain whether the variants identified in our study are relevant to ALS or merely an incidental finding.
Among the neuromuscular genes, variants were identified in two genes, namely ATP7A and BICD2, known to cause SMA or distal motor neuropathy. These disorders may resemble spinal ALS, but usually have a much earlier onset (Citation19, Citation62). Notably, one of our patients carrying an ATP7A-variant had ALS onset in his thirties. BICD2 variants usually cause childhood-onset SMA; however, its onset in adulthood has recently been reported (Citation47, Citation63). In our study, two patients with BICD2 had spinal-onset ALS. Furthermore, BICD2 is involved in retrograde transport along axon microtubules (Citation19, Citation59) together with two other motor neuron associated genes, namely DYNC1H1 (Citation64–66) and DCTN1 (Citation19, Citation67), suggesting a plausible role in ALS pathogenesis. Two patients carried variants in genes involved in peripheral neuropathy, namely ATL3 and HARS1 (Citation19). To our knowledge, these genes have not been associated with ALS. Notably, ATL3 is a sensory neuropathy gene, and Case 18 had spinal ALS with sensory neuropathy; in contrast, HARS1 is an axonal neuropathy gene, and Case 24 reported spinal ALS with pure lower motor signs at the time of diagnosis.
We identified variants in two genes that increase the ALS risk: NEFH and NEK1 (Citation19). NEFH was first proposed as an ALS risk gene because it encodes the heavy neurofilament protein that is a biomarker of neuronal damage, as the neurofilament accumulation may play a causal pathogenic role in ALS development (Citation68, Citation69). However, reports confirming this relationship have been lacking until recently, when especially variants in the NEFH tail domain that disturb distinctive lysine-serine-proline repeats, have been reported to be disease-causing (Citation70, Citation71). Case 19 carried an NEFH variant that changed a highly conserved proline residue in the tail domain, making this variant particularly interesting. This patient had ALS onset in his thirties and he also carried a potentially pathogenic ATP7A variant; therefore, it could be speculated in a possible digenic effect.
Seven (2.5%) patients with ALS in our cohort carried NEK1 variants predicted to cause loss-of-function. Loss-of-function NEK1 variants have been reported to increase ALS risk (Citation40, Citation41). In a meta-analysis involving 8603 cases from five European and three Asian studies, loss-of-function variants were reported in 1% of the patients from Europe and 0.7% from Asia (Citation72). This indicates a high prevalence of NEK1 loss-of-function variants in our Norwegian cohort compared with other populations.
The main limitation of the present study is that family history was based on self-reported information; thus, it is possible that the numbers are falsely low, either caused by early death, unknown family history, misunderstandings, forgetfulness, denial, or, in some cases misdiagnosis because of unclear symptoms. The lack of a matched control group makes it difficult to interpret whether there is an enrichment of these disorders among relatives of our ALS patients. Furthermore, it cannot be ruled out that some variants have not been called or that our strict bioinformatics filtering discarded some relevant variants with higher frequency and reduced penetrance. The relationship between the genes identified in this study and ALS pathogenesis is unclear, and a causal relationship needs to be further confirmed. It should also be stressed that although genetic variants are rare, they are mostly not disease-causing. Several studies have shown that rare genetic variants are much more common than previously thought (Citation73, Citation74), and the minor allele frequencies of 99% of the variants in the ExAC frequency database are lower than 1% (Citation74). Nevertheless, we believe that several variants and genes identified in the present study are good candidates for further studies. The low frequencies of these variants in the Norwegian hospital database indicates that they are not part of the normal genetic background in Norway.
In conclusion, a substantial proportion (9%) of Norwegian ALS patients carry rare genetic variants associated with other neurodegenerative or neuromuscular disorders, regardless of whether they report a family history of such disorder or not. ALS risk variants in NEK1 seems to be relatively frequent in the Norwegian population.
Supplemental Material
Download MS Word (15.8 KB)Acknowledgments
We thank the participating individuals and their families for their cooperation as well as the nurses and technical personnel for assisting with the inclusion of individuals and genetic analysis.
Disclosure statement
The authors report there are no competing interests to declare.
Data availability statement
Data supporting the findings of this study are available from the corresponding author upon request.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23.
- Goutman SA, Chen KS, Paez-Colasante X, Feldman EL. Emerging understanding of the genotype-phenotype relationship in amyotrophic lateral sclerosis. Handb Clin Neurol 2018;148:603–23.
- Mathis S, Goizet C, Soulages A, Vallat JM, Masson GL. Genetics of amyotrophic lateral sclerosis: A review. J Neurol Sci. 2019;399:217–26.
- Al-Chalabi A, Van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol. 2017;13:96–104.
- Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88:540–9.
- Van Daele SH, Moisse M, Van Vugt JJFA, Zwamborn RAJ, Van der Spek R, Van Rheenen W, et al. Genetic variability in sporadic amyotrophic lateral sclerosis. Brain. 2023;146:3760–9.
- Van Blitterswijk M, Van Es MA, Hennekam EA, Dooijes D, Van Rheenen W, Medic J, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21:3776–84.
- Olsen CG, Busk ØL, Aanjesen TN, Alstadhaug KB, Bjørnå IK, Braathen GJ, et al. Genetic Epidemiology of Amyotrophic Lateral Sclerosis in Norway: A 2-Year Population-Based Study. Neuroepidemiology 2022;56:271–82.
- Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9:617–28.
- Al-Chalabi A, Fang F, Hanby MF, Leigh PN, Shaw CE, Ye W, et al. An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry. 2010;81:1324–6.
- Ryan M, Heverin M, McLaughlin RL, Hardiman O. Lifetime Risk and Heritability of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2019;76:1367–74.
- Sha SJ, Takada LT, Rankin KP, Yokoyama JS, Rutherford NJ, Fong JC, et al. Frontotemporal dementia due to C9ORF72 mutations: clinical and imaging features. Neurology 2012;79:1002–11.
- Bourinaris T, Houlden H. C9orf72 and its Relevance in Parkinsonism and Movement Disorders: A Comprehensive Review of the Literature. Mov Disord Clin Pract. 2018;5:575–85.
- Ducharme S, Bajestan S, Dickerson BC, Voon V. Psychiatric Presentations of C9orf72 Mutation: What Are the Diagnostic Implications for Clinicians? J Neuropsychiatry Clin Neurosci. 2017;29:195–205.
- Kirola L, Mukherjee A, Mutsuddi M. Recent Updates on the Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Mol Neurobiol. 2022;59:5673–94.
- Van Es MA, Schelhaas HJ, Van Vught PWJ, Ticozzi N, Andersen PM, Groen EJN, et al. Angiogenin variants in Parkinson disease and amyotrophic lateral sclerosis. Ann Neurol. 2011;70:964–73.
- Morais S, Raymond L, Mairey M, Coutinho P, Brandão E, Ribeiro P, et al. Massive sequencing of 70 genes reveals a myriad of missing genes or mechanisms to be uncovered in hereditary spastic paraplegias. Eur J Hum Genet. 2017;25:1217–28.
- Crimella C, Baschirotto C, Arnoldi A, Tonelli A, Tenderini E, Airoldi G, et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin Genet. 2012;82:157–64.
- Online Mendelian Inheritance in Man. OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD): 1995 [cited 2023 Jan 10]. Available from: https://omim.org/.
- Siddique N, Siddique T, Amyotrophic Lateral Sclerosis Overview. In: Adam M, Everman D, Mirzaa G, Pagon R, Wallace S, Bean L, et al. editors. 2023. GeneReviews [Internet]: Seattle (WA): University of Washington, Seattle; 1993–2023.; 2001 Mar 23 [Updated 2021 Sep 30].
- Goutman SA, Hardiman O, Al-Chalabi A, Chió A, Savelieff MG, Kiernan MC, et al. Recent advances in the diagnosis and prognosis of amyotrophic lateral sclerosis. Lancet Neurol. 2022;21:480–93.
- Goutman SA, Hardiman O, Al-Chalabi A, Chió A, Savelieff MG, Kiernan MC, et al. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022;21:465–79.
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43.
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, et al. The Human Gene Mutation Database (HGMD(®)): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207.
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet. 2016;99:877–85.
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067.
- Van Rheenen W, Pulit SL, Dekker AM, Al Khleifat A, Brands WJ, Iacoangeli A, et al. Project MinE: study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur J Hum Genet 2018;26:1537–46.
- Stenson PD, Ball E, Howells K, Phillips A, Mort M, Cooper DN. Human Gene Mutation Database: towards a comprehensive central mutation database. J Med Genet. 2008;45:124–6.
- Coutelier M, Hammer MB, Stevanin G, Monin ML, Davoine CS, Mochel F, et al. Efficacy of Exome-Targeted Capture Sequencing to Detect Mutations in Known Cerebellar Ataxia Genes. JAMA Neurol. 2018;75:591–9.
- Calandra CR, Buda G, Vishnopolska SA, Oliveri J, Olivieri FA, Pérez Millán MI, et al. Spastic ataxia with eye-of-the-tiger-like sign in 4 siblings due to novel compound heterozygous AFG3L2 mutation. Parkinsonism Relat Disord. 2020;73:52–4.
- Davis RL, Kumar KR, Puttick C, Liang C, Ahmad KE, Edema-Hildebrand F, et al. Use of Whole-Genome Sequencing for Mitochondrial Disease Diagnosis. Neurology 2022;99:e730–e742.
- Galatolo D, De Michele G, Silvestri G, Leuzzi V, Casali C, Musumeci O, et al. NGS in Hereditary Ataxia: When Rare Becomes Frequent. Int J Mol Sci 2021;22:8490.
- Musova Z, Kaiserova M, Kriegova E, Fillerova R, Vasovcak P, Santava A, et al. A novel frameshift mutation in the AFG3L2 gene in a patient with spinocerebellar ataxia. Cerebellum. 2014;13:331–7.
- Liao P, Yuan Y, Liu Z, Hou X, Li W, Wen J, et al. Association of variants in the KIF1A gene with amyotrophic lateral sclerosis. Transl Neurodegener. 2022;11:46.
- Pennings M, Schouten MI, Van Gaalen J, Meijer RPP, de Bot ST, Kriek M, et al. KIF1A variants are a frequent cause of autosomal dominant hereditary spastic paraplegia. Eur J Hum Genet. 2020;28:40–9.
- Nicita F, Ginevrino M, Travaglini L, D'Arrigo S, Zorzi G, Borgatti R, et al. Heterozygous KIF1A variants underlie a wide spectrum of neurodevelopmental and neurodegenerative disorders. J Med Genet. 2021;58:475–83.
- Tolosa E, Vila M, Klein C, Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol. 2020;16:97–107.
- Aasly JO, Toft M, Fernandez-Mata I, Kachergus J, Hulihan M, White LR, et al. Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann Neurol. 2005;57:762–5.
- Nasca A, Scotton C, Zaharieva I, Neri M, Selvatici R, Magnusson OT, et al. Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia. Hum Mutat. 2017;38:970–7.
- Kenna KP, Van Doormaal PT, Dekker AM, Ticozzi N, Kenna BJ, Diekstra FP, et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat Genet. 2016;48:1037–42.
- Naruse H, Ishiura H, Mitsui J, Takahashi Y, Matsukawa T, Yoshimura J, et al. Loss-of-function variants in NEK1 are associated with an increased risk of sporadic ALS in the Japanese population. J Hum Genet. 2021;66:237–41.
- Brenner D, Müller K, Wieland T, Weydt P, Böhm S, Lulé D, et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain. 2016;139:e28–e28.
- Verheijen J, Van den Bossche T, Van der Zee J, Engelborghs S, Sanchez-Valle R, Lladó A, et al. A comprehensive study of the genetic impact of rare variants in SORL1 in European early-onset Alzheimer’s disease. Acta Neuropathol. 2016;132:213–24.
- Nicolas G, Charbonnier C, Wallon D, Quenez O, Bellenguez C, Grenier-Boley B, et al. SORL1 rare variants: a major risk factor for familial early-onset Alzheimer’s disease. Mol Psychiatry. 2016;21:831–6.
- Bellenguez C, Charbonnier C, Grenier-Boley B, Quenez O, Le Guennec K, Nicolas G, et al. Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol Aging 2017;59:e1–e9.
- Bakkar N, Starr A, Rabichow BE, Lorenzini I, McEachin ZT, Kraft R, et al. The M1311V variant of ATP7A is associated with impaired trafficking and copper homeostasis in models of motor neuron disease. Neurobiol Dis. 2021;149:105228.
- Wan C, Wang Y, Zhou Q, Yu Y, Hong D, Zhu M. Adult-onset SMALED2 due to a novel BICD2 mutation presenting with asymmetrical lower limb involvement. Clin Neuropathol. 2019;38:109–17.
- D'Amore A, Tessa A, Casali C, Dotti MT, Filla A, Silvestri G, et al. Next Generation Molecular Diagnosis of Hereditary Spastic Paraplegias: An Italian Cross-Sectional Study. Front Neurol. 2018;9:981.
- Didonna A, Opal P. The role of neurofilament aggregation in neurodegeneration: lessons from rare inherited neurological disorders. Mol Neurodegener 2019;14:1–10.
- Figlewicz DA, Krizus A, Martinoli MG, Meininger V, Dib M, Rouleau GA, et al. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet. 1994;3:1757–61.
- Rooke K, Figlewicz DA, Han FY, Rouleau GA. Analysis of the KSP repeat of the neurofilament heavy subunit in familiar amyotrophic lateral sclerosis. Neurology 1996;46:789–90.
- Marriott H, Spargo TP, Khleifat AA, Fogh I, Consortium PMAS, Andersen PM, et al. Mutations in the tail domain of the neurofilament heavy chain gene increase the risk of amyotrophic lateral sclerosis. medRxiv 2022;2022:11.
- Campos CF, Gromicho M, Uysal H, Grosskreutz J, Kuzma-Kozakiewicz M, Pinto S, et al. Family history of neurodegenerative disorders in patients with amyotrophic lateral sclerosis: population-based case–control study. J Neurol Neurosurg Psychiatry. 2020;91:671–2.
- Cruz DC, Nelson LM, McGuire V, Longstreth WT. Physical Trauma and Family History of Neurodegenerative Diseases in Amyotrophic Lateral Sclerosis: A Population-Based Case-Control Study. Neuroepidemiology 1999;18:101–10.
- Huisman MHB, de Jong SW, Verwijs MC, Schelhaas HJ, van der Kooi AJ, de Visser M, et al. Family history of neurodegenerative and vascular diseases in ALS: a population-based study. Neurology 2011;77:1363–9.
- Fallis BA, Hardiman O. Aggregation of neurodegenerative disease in ALS kindreds. Amyotroph Lateral Scler. 2009;10:95–8.
- Almontashiri NA, Chen HH, Mailloux RJ, Tatsuta T, Teng AC, Mahmoud AB, et al. SPG7 variant escapes phosphorylation-regulated processing by AFG3L2, elevates mitochondrial ROS, and is associated with multiple clinical phenotypes. Cell Rep. 2014;7:834–47.
- Osmanovic A, Widjaja M, Förster A, Weder J, Wattjes MP, Lange I, et al. SPG7 mutations in amyotrophic lateral sclerosis: a genetic link to hereditary spastic paraplegia. J Neurol. 2020;267:2732–43.
- Sleigh JN, Rossor AM, Fellows AD, Tosolini AP, Schiavo G. Axonal transport and neurological disease. Nat Rev Neurol. 2019;15:691–703.
- Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018;97:1268–83.e6.
- Lissouba A, Liao M, Kabashi E, Drapeau P. Transcriptomic Analysis of Zebrafish TDP-43 Transgenic Lines. Front Mol Neurosci. 2018;11:463.
- Kaler SG, DiStasio AT, ATP7A-Related Copper Transport Disorders. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al. editors. GeneReviews. Seattle (WA): University of Washington, Seattle, 1993–2023; 1993.
- Aziz I, Davis M, Liang C. Late adult-onset spinal muscular atrophy with lower extremity predominance (SMALED). BMJ Case Rep 2022;15:e248297.
- Mentis AA, Vlachakis D, Papakonstantinou E, Zaganas I, Patrinos GP, Chrousos GP, et al. A novel variant in DYNC1H1 could contribute to human amyotrophic lateral sclerosis-frontotemporal dementia spectrum. Cold Spring Harb Mol Case Stud 2022;8:a006096.
- Scarlino S, Domi T, Pozzi L, Romano A, Pipitone GB, Falzone YM, et al. Burden of Rare Variants in ALS and Axonal Hereditary Neuropathy Genes Influence Survival in ALS: Insights from a Next Generation Sequencing Study of an Italian ALS Cohort. Int J Mol Sci 2020;21:3346.
- Tripolszki K, Gampawar P, Schmidt H, Nagy ZF, Nagy D, Klivényi P, et al. Comprehensive Genetic Analysis of a Hungarian Amyotrophic Lateral Sclerosis Cohort. Front Genet. 2019;10:732.
- Borg R, Farrugia Wismayer M, Bonavia K, Farrugia Wismayer A, Vella M, Van Vugt J, et al. Genetic analysis of ALS cases in the isolated island population of Malta. Eur J Hum Genet. 2021;29:604–14.
- Collard JF, Côté F, Julien JP. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature 1995;375:61–4.
- Al-Chalabi A, Andersen PM, Nilsson P, Chioza B, Andersson JL, Russ C, et al. Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum Mol Genet. 1999;8:157–64.
- Grassano M, Calvo A, Moglia C, Sbaiz L, Brunetti M, Barberis M, et al. Systematic evaluation of genetic mutations in ALS: a population-based study. J Neurol Neurosurg Psychiatry. 2022;93:1190–3.
- Shepheard SR, Parker MD, Cooper-Knock J, Verber NS, Tuddenham L, Heath P, et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2021;92:510–8.
- Yao L, He X, Cui B, Zhao F, Zhou C. NEK1 mutations and the risk of amyotrophic lateral sclerosis (ALS): a meta-analysis. Neurol Sci. 2021;42:1277–85.
- Check Hayden E. Humans riddled with rare genetic variants. Nature (London). 2012. https://doi.org/10.1038/nature.2012.10655
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91.