
Abstract
Objectives
We aimed to estimate the age-related risk of ALS in first-degree relatives of patients with ALS carrying the C9orf72 repeat expansion.
Methods
We included all patients with ALS carrying a C9orf72 repeat expansion in The Netherlands. Using structured questionnaires, we determined the number of first-degree relatives, their age at death due to ALS or another cause, or age at time of questionnaire. The cumulative incidence of ALS among first-degree relatives was estimated, while accounting for death from other causes. Variability in ALS risk between families was evaluated using a random effects hazards model. We used a second, distinct approach to estimate the risk of ALS and FTD in the general population, using previously published data.
Results
In total, 214 of the 2,486 (9.2%) patients with ALS carried the C9orf72 repeat expansion. The mean risk of ALS at age 80 for first-degree relatives carrying the repeat expansion was 24.1%, but ranged between individual families from 16.0 to 60.6%. Using the second approach, we found the risk of ALS and FTD combined was 28.7% (95% CI 17.8%–54.3%) for carriers in the general population.
Conclusions
On average, our estimated risk of ALS in the C9orf72 repeat expansion was lower compared to historical estimates. We showed, however, that the risk of ALS likely varies between families and one overall penetrance estimate may not be sufficient to describe ALS risk. This warrants a tailor-made, patient-specific approach in testing. Further studies are needed to assess the risk of FTD in the C9orf72 repeat expansion.
Keywords:
Introduction
Genetic discoveries play a crucial role in unraveling the complex nature of Amyotrophic Lateral Sclerosis (ALS) and have led to promising gene-based therapies now being assessed in clinical trials (Citation1, Citation2). The pathophysiological mechanism has not yet been elucidated, but is thought to involve an interaction between genetic and environmental factors over time (Citation3, Citation4). The GGGGCC hexanucleotide repeat expansion in C9orf72 (C9orf72 repeat expansion) has been identified as the most common genetic cause of ALS in Europe and North America (Citation5). The C9orf72 repeat expansion is genetically pleiotropic and has most frequently been associated with a phenotypic spectrum ranging from ALS to frontotemporal dementia (FTD) (Citation3). Testing for the C9orf72 repeat expansion as part of routine clinical care is becoming increasingly important (Citation6). However, considering the autosomal dominant inheritance pattern, identification of the C9orf72 repeat expansion in patients with ALS also has implications for their relatives (Citation6).
An important question that emerges when patients carry a C9orf72 repeat expansion is the risk of relatives developing an associated disorder (Citation6). For asymptomatic relatives, the risk of developing an associated disorder, especially in the absence of an effective therapy, is an important factor in the decision to undergo genetic testing (Citation7). Thus far, studies have reported near fully penetrant estimates of the C9orf72 repeat expansion in ALS at older ages (eTable 1) (Citation5, Citation8–11). These estimates were, however, based on populations in which the outcome of ALS was already known, leading to an overestimation of penetrance. Also, 11/7,579 (0.15%) subjects in the UK 1958 birth cohort appeared to carry a C9orf72 repeat expansion and in Project MinE, a large international collaboration, we have reported a C9orf72 repeat expansion in 2 out of 1,051 controls (0.19%) (Citation12, Citation13). Both estimates are in agreement, but much higher than expected when assuming high risks of developing ALS and FTD, given the lifetime risks of these conditions.
In this study, we used first-degree family history data of patients with ALS, drawn from a nationwide registry, and known to carry the C9orf72 repeat expansion, to estimate the age-related risk of ALS, thereby minimizing the risk of selection bias. We aimed to establish the risk of ALS in the C9orf72 repeat expansion in first-degree relatives and explore the variability in baseline hazards between families.
Methods
Design and participants
The Prospective ALS study the Netherlands (PAN) was initiated in 2006 and is an ongoing population-based registry (Citation14). For this study, we included patients carrying a C9orf72 repeat expansion, diagnosed with ALS between January 2006 and January 2020, hereafter referred to as index patients. To prevent including duplicate nuclear families, no more than one sibling from each nuclear family was included as an index patient. This was verified by creating a kinship matrix using data available from genome-wide association studies (data were available for 90.7% of the index patients) (Citation15, Citation16). Within one nuclear family the most recent data was used in the analysis, thus taking into account all known ALS cases in estimating ALS risk.
Data collection
At time of inclusion in the registry, the index patients completed a structured family history questionnaire including information on the number of first-degree relatives (parents and siblings) and their history of ALS. The questionnaire was self-administered and, if necessary, patients were contacted by telephone to complete missing data. From September 2011 onwards, questions about relatives’ age at death, cause of death, and age at time of questionnaire were added. The questionnaire data were verified and updated using more recent information collected from several sources. First, we used data from the familial ALS study, in which patients with (suspected) familial ALS and their relatives were invited to participate (Citation17). Second, we used familial history questionnaires completed by first-degree relatives of index patients included in the registry, and last, we used patients’ medical records. Disease characteristics were collected from index patients’ medical records on the day of diagnosis, and follow-up of survival was done until June 30, 2022. Additionally, data on carriership of the C9orf72 repeat expansion was collected for individuals included in the familial ALS study, of whom all siblings were tested for the repeat expansion, and with at least one known carrier in their nuclear family (Citation17).
Genetic analysis
DNA was extracted from venous blood samples of the index patient according to standard protocols and procedures. Presence of the C9orf72 repeat expansion was determined using one of two previously validated methods: either by a repeat-primed PCR for the C9orf72 hexanucleotide repeat defining alleles with 30 or more repeats as mutated, or by using ExpansionHunter to detect the presence of the C9orf72 repeat expansion in whole-genome sequencing (WGS) data (Citation12, Citation18). Although repeat-primed PCR is considered the gold standard, the ExpansionHunter showed an expansion detection accuracy of more than 99% (Citation12).
Statistical analysis
The primary aim of the analysis was to estimate the age-related risk of ALS in the C9orf72 repeat expansion. Information available for each index patient comprised the number of first-degree relatives, the relative’s event, defined as death from ALS, death from other causes, or censor when alive at time of questionnaire, and the relative’s age at event. A sample of the data is presented in for a single index patient. We assumed that the C9orf72 repeat expansion was transmitted with Mendelian probability of 0.5 (Citation19). In addition, we assumed that de novo mutations do not occur. As such, 50% of first-degree relatives were assumed to carry the C9orf72 repeat expansion on population level (Citation19). To support this assumption, we calculated the percentage of carriers of the C9orf72 repeat expansion amongst all siblings included in the familial ALS study, excluding index patients (Citation17). To prevent giving an underestimation of ALS risk, we assumed that all ALS cases in our population were attributed to carriership, we thus aimed to estimate the “worst-case” scenario (upper bound) of the age-related risk of ALS in C9orf72. We estimated the risk by multiplying the cumulative incidence of ALS among all first-degree relatives by two. Hereby answering the question: what is my risk of ALS if I carry the C9orf72 repeat expansion and I have a family member with ALS who carries the repeat expansion?
Figure 1 Sample of available data of first-degree relatives of index patients.
Age indicates age at onset for index patient, age at time of questionnaire (censor) for living relatives, or age at death for other relatives.
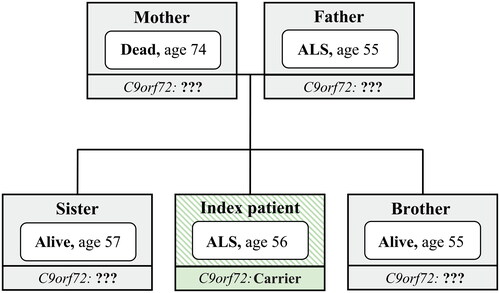
We used a cumulative incidence curve to model the occurrence of ALS among first-degree relatives while accounting for the competing event of death due to other causes (Citation20). Missing data on age at death, age at censoring, or age at ALS death (missing in 59 of the 933 cases (6.3%)), were imputed by creating multiple imputed datasets (n = 100), disregarding the first 100 iterations (burn-in). The imputation model contained the following variables: age at event, event (censor, death, ALS), sex of the relative, sibling or parent, age and sex of the index patient, year of survey and family size (since family size could serve as an important confounder for disease risk) (Citation21). Imputed values were generated using a curtailed non-linear regression model using bootstrapping. Cumulative incidence curves across imputations were pooled using Rubin’s rules based on a complementary log-log transformation (Citation22). Due to the clustering of observations within families, the variance around the cumulative incidences was estimated by means of bootstrapping (n = 10000) and averaged across imputations (“MI Boot”) (Citation23).
We conducted the following sensitivity analyses: (Citation1) complete case analysis, excluding all first-degree relatives with missing age or event, (Citation2) fully imputed analysis, including all first-degree relatives, (Citation3) excluding data from parents to minimize under-ascertainment of cases in older generations, (Citation4) excluding questionnaires completed prior to 2011 to exclude data prior to the change in questionnaire, (Citation5) excluding all data originating from outside the registry to exclude a potential effect of nonrandom missingness, (Citation6) assuming Mendelian inheritance probabilities ranging from 0.4 to 0.6, excluding that results were driven by random fluctuations of mutant allele transmission, and (Citation7) excluding subsets of relatives based on the age of the index patient and family size (e.g. excluding relatives of index patients with small families), to exclude potential under-ascertainment of cases.
Furthermore, to test the robustness of our primary analysis, we used a second distinct approach to estimate the disease risk using estimates regarding the prevalence of the C9orf72 repeat expansion in the general population, based on the 1958 UK birth cohort (n = 7,579), and lifetime risks of dying from C9orf72-related ALS or FTD. We combined the uncertainty around published estimates of C9orf72 prevalence in the general population, number of ALS and FTD patients with a C9orf72 repeat expansion and the lifetime risk of dying from ALS or FTD using the Delta method to estimate a confidence interval.
In addition, we used a mixed effects Cox proportional hazards model with a random intercept term for the baseline hazard to evaluate the variability in ALS risk between families and extracted the relative risk for each family. A relative risk of 2.0, for example, corresponds to a 100% higher hazard of ALS for members in that family compared to the average risk in families. The absolute family-specific risk of ALS could be calculated as: 1 – (1 – overall risk of ALS)Family-specific HR. We used a likelihood ratio test to evaluate the significance of the random effects and the variability in baseline hazard between families. In addition, we used a permutation test, randomly allocating family identifiers, to understand the variability in results if family was not associated with the baseline hazard.
Standard protocol approvals, registrations, and patient consents
The medical ethics committee and institutional review board of the University Medical Center Utrecht (METC NedMec) approved this study (Study Registration Number: METC 19–190). Written consent was obtained from all study participants before this study.
Data availability
All protocol, analyses, and anonymized data will be shared on request. We take full responsibility for data, analyses and interpretation, and conduct of the research.
Results
Between 1 January 2006 and 1 January 2020, 2,734 patients diagnosed with ALS provided informed consent, and 2,486 (90.9%) participated in the study. Of these, 2,316 were screened for the presence of the C9orf72 repeat expansion and 214 (9.2%) patients carried the pathogenic mutation, in line with previous reports (eTable 2) (Citation5, Citation18, Citation24, Citation25). For ALS patients carrying the repeat expansion, there was no relationship between having familial ALS and participation in our nationwide population-based ALS study (eTable 3). Including one sibling from each nuclear family resulted in 207 index patients with a total of 1,160 first-degree relatives (). For the primary analysis, relatives with an unknown event (censor, death, ALS) were excluded. Of the remaining 933 relatives from 172 index patients (), 345 were deceased, 506 were alive at the time of the questionnaire, and there were 82 ALS cases.
Table 1 Clinical characteristics of index patients.
Table 2 Characteristics of included first-degree relatives.
The estimated risk of ALS at age 80 for first-degree relatives carrying the repeat expansion ranged from 18.3% to 30.3%, mean 24.1% (). We found that the risk of ALS was low prior to the age of 30 and reached a plateau at age 80 years or older, similar to known ALS incidence rates in the Netherlands (Citation26). The results of the sensitivity analyses are shown in . All sensitivity analyses showed a risk of ALS in line with the primary analysis. Using data from the familial ALS study, we found 76 of the 145 (52.4%; 95% CI 44.3% to 60.5%) siblings carried the repeat expansion (eTable 4).
Figure 2 Age-related risk of ALS in the C9orf72 repeat expansion.
Cumulative incidence curve of ALS in 933 first-degree relatives, adjusted for death from other causes. Censored indicates relative was alive at time of questionnaire. Shaded area represents bootstrapped 95% percentile interval.
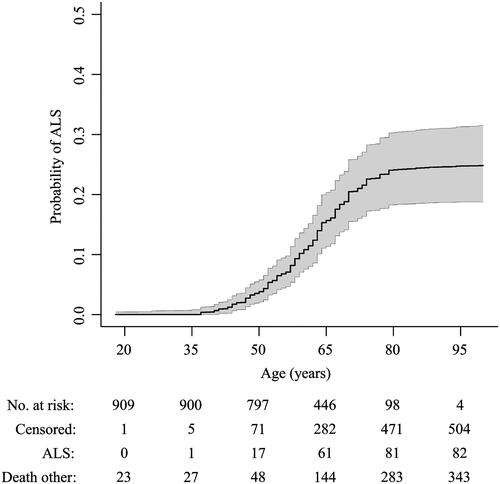
Figure 3 Sensitivity analyses of risk of ALS in the C9orf72 repeat expansion.
Primary analysis included relatives with a known event, imputing missing age, by creating multiple imputed datasets (n = 100) disregarding the first 100 iterations (burn-in), and assuming 50% of relatives are mutation carriers. Complete cases only included relatives with known age and event. Fully imputed included all relatives, imputing both age and event. Data since 2011 indicates questionnaires completed prior to 2011 were excluded. Registry data indicates data collected through the registry, excluding data originating from other sources. 40% carrier and 60% carrier indicate the assumed number of relatives carrying the repeat expansion was changed to 40% and 60%, respectively, reflecting the uncertainty in the Mendelian inheritance probability. Age at onset ≥ 60 year included only index patients with an age at disease onset ≥ 60 years. Family size ≥ 6 only included index patients with a family size ≥ 6.
As a second distinct approach, we used independent, published data to estimate risk of ALS or FTD in the general population: in 2017, 1 in 322 deaths in the Netherlands was due to ALS, and of the individuals who died from ALS, 8.5% carried the repeat expansion (Citation26). Hence, 1 in 3,803 individuals is expected to die of C9orf72 related ALS. A study conducted in the United Kingdom showed the lifetime risk of FTD is 1 in 742 and a European study showed mutation frequency in FTD is 10.0% (Citation27, Citation28). Based on a study conducted in the United Kingdom, 1 in 689 of the general population is a carrier of the C9orf72 repeat expansion (Citation13). This resulted in an estimated risk of ALS for C9orf72 repeat expansion carriers of 18.4% (95% CI 9.9% to 41.7%) and risk of FTD of 9.4% (95% CI 5.1% to 21.2%), and to a combined estimate of 28.7% (95% CI 17.8% to 54.3%)
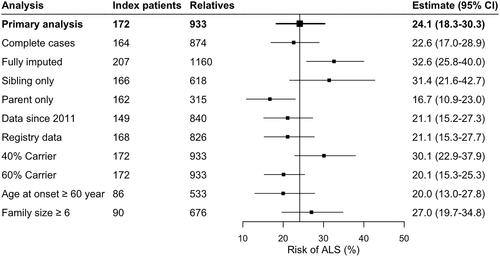
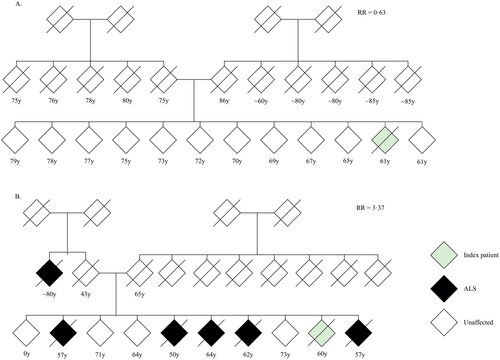
Finally, we determined interfamilial differences in risk of ALS by allowing the baseline risk to vary between families (). The baseline hazard of ALS differed between families (p = 0.023), where relative familial risks ranged from a minimum of 0.63 to a maximum of 3.37 times the average familial risk. This translates to risk of ALS ranging from 16.0% to 60.6% at age 80 in single families. We found familial ALS risk was independent from family size (Pearson 0.08, p > 0.29), and the variation in family-specific ALS risk was larger than what would be expected if the risk was identical for each family (). and eFigure 1 illustrate pedigrees of similar sized families with a family-specific ALS risk ranging from 0.63 to 3.37.
Figure 4 Family-specific risk of developing ALS.
(A) Variability in baseline risk across families. Relative risks were calculated using a mixed effects Cox proportional hazards model with a random intercept term for the baseline hazard. A value of 2.0 indicates that the hazard in that family is twice the hazard of the entire study. (B) Overall, there was no clear correlation between family-specific hazard ratios and family size (Pearson 0.08, p = 0.29). The shaded grey area indicates the expected random variability in baseline hazards for each family size if the baseline hazard would be identical across families, expressed as 95% confidence interval. The majority of the family-specific hazard ratios fall out-side the 95% confidence interval, indicating that the variation in family-specific hazard ratios is larger than expected under random chance.
Figure 5 Pedigree charts of two index patients.
(A) Pedigree of index patient classified as sporadic ALS. (B) Pedigree of index patient classified as familial ALS. Given age indicates age at onset for index patient, age at time of family history questionnaire for living relatives and age at death for other relatives. RR = family-specific relative ALS risk; y = years.
Discussion
We estimated the risk of ALS in the C9orf72 repeat expansion among first-degree relatives using family history data of patients known to carry the expansion. We found the mean risk of ALS at age 80 was 24.1%. We showed however, that the risk of ALS ranged between individual families from 16.0 to 60.6%, and one overall penetrance estimate may not be sufficient to describe ALS risk. In addition, using a second distinct approach based on previously published data, we found an estimate consistent with the results of our primary analysis (mean 18.4%; 95% CI 9.9% to 41.7%). Combined with published data on FTD, we found an overall risk of ALS or FTD in the general population of 28.7% (95% CI 17.8% to 54.3%). Our findings have important implications when counseling patients who carry the C9orf72 repeat expansion and their relatives, and for decisions related to investigating preventive gene therapy in asymptomatic carriers of the repeat expansion.
Although previous studies reported the penetrance of the C9orf72 repeat expansion to be age-related and incomplete, their estimated ALS risks were considerably higher, showing nearly full penetrance in older ages (Citation5, Citation8–11). This discrepancy could be due to previous studies including solely study populations in which ALS as outcome was already known, leading to an overestimation of overall penetrance. The range in risk of ALS found in this study is in line with the expected risk based on the frequency of the C9orf72 repeat expansion in the general population (Citation12, Citation13, Citation26–28). These findings were recently confirmed by another study, with penetrance estimates falling within the range found in our study (Citation29).
Another important finding of our study is the interfamilial differences in relative risk of developing ALS. We found the variation in family-specific ALS risk was larger than what would be expected if ALS risk was identical for each family. This indicates that the risk may be family-specific, resulting in families with a higher or lower prevalence of ALS. The variability in ALS risk aligns with the multistep hypothesis of ALS, suggesting the C9orf72 repeat expansion, in isolation, is insufficient to cause ALS, but other genetic, epigenetic, or environmental factors are needed for the disease to manifest (Citation30). The exact estimate of family-specific risk of ALS is, however, limited by family size (Citation21). In this study, only first-degree relatives were included due to limited data. Larger familial studies, including second- and third-degree relatives, may provide more accurate family-specific estimates and help to refine counseling.
Our study has several limitations that need to be considered. First, we assumed a 50% carrier rate based on an autosomal dominant inheritance pattern, which could be incorrect (Citation31, Citation32). However, in the familial ALS study, we found a similar number of individuals carried the repeat expansion (52.4%; 95% CI 44.3% to 60.5%). In addition, we chose to estimate the “worst-case” scenario, attributing all ALS cases to carrying the repeat expansion, which might be incorrect, since even in highly penetrant conditions, an independent polygenic or environmental component might be at play (phenocopies). Nevertheless, if that is the cases, true penetrance estimates would even be lower than presented here. Similarly, we found a higher risk of ALS when including only siblings of the index patients, suggesting potential under-ascertainment of ALS amongst parents. The use of questionnaires could lead to under-ascertainment of patients with cognitive or behavioral symptoms, or patients of older age, leading to an underestimation of the risk of ALS. Nevertheless, completing a variety of sensitivity analyses based on these limitations, we obtained estimates all falling within range of the primary analysis. Significantly, besides ALS, the repeat expansion has been associated with a range of other diseases, most importantly FTD (Citation3, Citation33), which was not included in our questionnaire. Further studies are needed to estimate the risk of developing any of the diseases associated with the C9orf72 repeat expansion.
In conclusion, we showed that the risk of ALS in the C9orf72 repeat expansion may not be one fixed number but varies between individual families and on average is lower than previously reported. Our findings have important clinical implications. The range in risk of ALS challenges the practice of routinely offering C9orf72 testing to all patients for genetic counseling purposes (Citation6). Alternatively, taking an extensive family history in patients prior to genetic testing, can help to distinguish familial ALS from truly sporadic ALS, and thus families with a potential high and low disease risk, should they carry the repeat expansion. The increased uncertainty in individual ALS risk should be considered in genetic counseling, as this could impact their decision to undergo genetic testing. Genetic testing could routinely be offered but then with the explicit intent to include patients in new clinical trials targeted at C9orf72 mutation carriers. Moreover, our findings also have consequences for gene-based clinical trials, and potentially future therapies, in asymptomatic carriers. The increased uncertainty in individual ALS risk highlights the need for preclinical markers, including neurofilaments, neurophysiology or imaging markers, to predict who is at increased short-term risk of developing ALS and needs to be treated in the pre-symptomatic phase (Citation34).
Supplemental Material
Download MS Word (228.5 KB)Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
References
- Smukowski SN, Maioli H, Latimer CS, Bird TD, Jayadev S, Valdmanis PN. Progress in amyotrophic lateral sclerosis gene discovery: reflecting on classic approaches and leveraging emerging technologies. Neurol Genet. 2022;8:e669.
- Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC, et al. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2022;387:1099–110.
- van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390:2084–98.
- Westeneng H-J, van Veenhuijzen K, van der Spek RA, Peters S, Visser AE, van Rheenen W, et al. Associations between lifestyle and amyotrophic lateral sclerosis stratified by C9orf72 genotype: a longitudinal, population-based, case-control study. Lancet Neurol. 2021;20:373–84.
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–30.
- Roggenbuck J. C9orf72 and the care of the patient with ALS or FTD: progress and recommendations after 10 years. Neurol Genet. 2021;7:e542.
- Benatar M, Stanislaw C, Reyes E, Hussain S, Cooley A, Fernandez MC, et al. Presymptomatic ALS genetic counseling and testing: experience and recommendations. Neurology 2016;86:2295–302.
- Murphy NA, Arthur KC, Tienari PJ, Houlden H, Chiò A, Traynor BJ. Age-related penetrance of the C9orf72 repeat expansion. Sci Rep. 2017;7:2116.
- Le Ber I, Camuzat A, Guillot-Noel L, Hannequin D, Lacomblez L, Golfier V, et al. C9ORF72 repeat expansions in the frontotemporal dementias spectrum of diseases: a flow-chart for genetic testing. J Alzheimers Dis. 2013;34:485–99.
- McCann EP, Williams KL, Fifita JA, Tarr IS, O’Connor J, Rowe DB, et al. The genotype-phenotype landscape of familial amyotrophic lateral sclerosis in Australia. Clin Genet. 2017;92:259–66.
- Williams KL, Fifita JA, Vucic S, Durnall JC, Kiernan MC, Blair IP, et al. Pathophysiological insights into ALS with C9ORF72 expansions. J Neurol Neurosurg Psychiatry. 2013;84:931–5.
- Dolzhenko E, van Vugt J, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 2017;27:1895–903.
- Beck J, Poulter M, Hensman D, Rohrer JD, Mahoney CJ, Adamson G, et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am J Hum Genet. 2013;92:345–53.
- Huisman MHB, de Jong SW, van Doormaal PTC, Weinreich SS, Schelhaas HJ, van der Kooi AJ, et al. Population based epidemiology of amyotrophic lateral sclerosis using capture–recapture methodology. J Neurol Neurosurg Psychiatry. 2011;82:1165–70.
- van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet. 2016;48:1043–8.
- van Rheenen W, van der Spek RAA, Bakker MK, van Vugt J, Hop PJ, Zwamborn RAJ, et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet. 2021;53:1636–48.
- van Blitterswijk M, van Es MA, Hennekam EA, Dooijes D, van Rheenen W, Medic J, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21:3776–84.
- van Rheenen W, van Blitterswijk M, Huisman MH, Vlam L, van Doormaal PT, Seelen M, et al. Hexanucleotide repeat expansions in C9ORF72 in the spectrum of motor neuron diseases. Neurology 2012;79:878–82.
- Goutman SA, Hardiman O, Al-Chalabi A, Chió A, Savelieff MG, Kiernan MC, et al. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022;21:465–79.
- Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94:496–509.
- Al-Chalabi A, Lewis CM. Modelling the effects of penetrance and family size on rates of sporadic and familial disease. Hum Hered. 2011;71:281–8.
- Morisot A, Bessaoud F, Landais P, Rébillard X, Trétarre B, Daurès JP. Prostate cancer: net survival and cause-specific survival rates after multiple imputation. BMC Med Res Methodol. 2015;15:54.
- Schomaker M, Heumann C. Bootstrap inference when using multiple imputation. Stat Med. 2018;37:2252–66.
- Brown CA, Lally C, Kupelian V, Flanders WD. Estimated prevalence and incidence of amyotrophic lateral sclerosis and SOD1 and C9orf72 genetic variants. Neuroepidemiology 2021;55:342–53.
- Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88:540–9.
- de Jongh AD, van Eijk RPA, Peters SM, van Es MA, Horemans AMC, van der Kooi AJ, et al. Incidence, prevalence, and geographical clustering of motor neuron disease in the Netherlands. Neurology 2021;96:e1227–e36.
- van der Zee J, Dillen L, Van Langenhove T, Engelborghs S, Philtjens S, Vandenbulcke M, et al. A Pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat. 2013;34:363–73.
- Coyle-Gilchrist IT, Dick KM, Patterson K, Vázquez Rodríquez P, Wehmann E, Wilcox A, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology 2016;86:1736–43.
- Spargo TP, Opie-Martin S, Bowles H, Lewis CM, Iacoangeli A, Al-Chalabi A. Calculating variant penetrance from family history of disease and average family size in population-scale data. Genome Med. 2022;14:141.
- Chiò A, Mazzini L, D Alfonso S, Corrado L, Canosa A, Moglia C, et al. The multistep hypothesis of ALS revisited. Neurology 2018;91:e635-42–e642.
- Ryan M, Heverin M, McLaughlin RL, Hardiman O. Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol. 2019;76:1367–74.
- Greeff JM, Erasmus JC. Three hundred years of low non-paternity in a human population. Heredity (Edinb). 2015;115:396–404.
- Silverman HE, Goldman JS, Huey ED. Links between the C9orf72 repeat expansion and psychiatric symptoms. Curr Neurol Neurosci Rep. 2019;19:93.
- Benatar M, Wuu J, Lombardi V, Jeromin A, Bowser R, Andersen PM, et al. Neurofilaments in pre-symptomatic ALS and the impact of genotype. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:538–48.