
Abstract
ALS is a neurodegenerative disease characterized by loss of motor neurons, resulting in progressive weakness and wasting of muscles. The average survival time is 2–5 years, mostly due to respiratory failure. Since current therapies can prolong survival time by only a few months, multidisciplinary care remains the cornerstone of the management of ALS. At the ALS Expert Centre of University Hospitals Leuven, a large proportion of Belgian ALS patients are seen for diagnosis and a significant number is also in follow-up with the multidisciplinary team. In this retrospective study, we compared the outcome of incident patients who were in follow-up at our site with patients who were not in follow-up. We included 659 patients of which 557 (84.5%) received specialized care at the ALS Expert Centre. After adjusting for clinically relevant prognostic parameters, multidisciplinary follow-up significantly prolonged survival (p = 0.004; HR = 0.683; CI 95% [0.528 − 0.884]). This increase in survival is mainly driven by patients with spinal onset (p = 0.035; HR = 0.746; CI 95% [0.568 − 0.980]), since no significant increased survival time was observed in patients with bulbar onset (p = 0.28; HR = 0.778; CI 95% [0.495 − 1.223]). These data confirm that multidisciplinary follow-up contributes to a better outcome of patients, emphasizing the importance of multidisciplinary specialized care in ALS.
Keywords:
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common motor neuron disease, characterized by loss of upper and lower motor neurons and has an incidence of 2–3/100.000/year in Europe (Citation1). Due to respiratory muscle weakness and respiratory failure, the mean survival after symptom onset is limited to 3 years (Citation1). Although multiple risk factors have been associated with the development of ALS, the etiology of sporadic ALS remains largely unknown (Citation2). In 10% of cases, ALS is a heritable disorder, most commonly caused by mutations in C9orf72, SOD1, FUS, TDP43 or TBK1 (Citation3). However, over 60 other genetic risk factors have been described that contribute to neuromuscular degeneration in both the familial and sporadic form of ALS (Citation4). As there is no cure for ALS, multidisciplinary care including nutritional and respiratory support remains the cornerstone of disease management in ALS (Citation5). The only EMA-approved drug for ALS so far is the sodium-channel inhibitor Riluzole, which counters neuronal glutamatergic excitotoxicity, prolonging survival by 36 months on average (Citation6–8). Currently, the antisense oligonucleotide drug Tofersen for the treatment of SOD1 patients has been approved by the FDA, yet it is still under consideration by EMA. Tofersen demonstrated effects on slowing down disease progression in patients with SOD1 mutations, even though this accounts for only 2% of all cases (Citation9).
Due to heterogeneity in the clinical phenotype, the progression rate and the degree of extra-motor manifestations, the disease trajectory of each patient remains variable and unpredictable (Citation10). Therefore, follow-up by a multidisciplinary team that provides tailored advice to cover the different aspects of the disease is needed (Citation11). In Europe, follow-up by a multidisciplinary specialized Centre has already been proven to have beneficial effects on survival in several populations (Citation12–14). However, both the composition and experience of multidisciplinary teams are different between Centres, as well as the fact that patients are not always immediately forwarded to a multidisciplinary clinic. When consulting general neurologists outside a multidisciplinary clinic, patients usually discuss their initial complaints, which are most often muscle weakness or problems with swallowing/speech. Consequently, a general neurologist will either evaluate these first complaints in the light of the broad spectrum of (neuro)muscular diseases or forward the patient to a multidisciplinary clinic. However, this is not a harmonized workflow, which contributes to the average diagnostic delay in ALS patients of 1 year.
The multidisciplinary team at the ALS Expert Centre of University Hospitals Leuven (UZ Leuven) consists of neurologists, rehabilitation physicians, specialized nurses, (neuro)psychologists, dietitians, occupational therapists, speech therapists, social workers and physiotherapists. In UZ Leuven, multidisciplinary follow-up visits are based on the needs of each individual patient. Depending on the progression rate of the disease and the manifestation of symptoms, the duration between visits and the appointments with the different disciplines can vary, respectively, after discussing with the patient. Contrary to other multidisciplinary Centres, not all disciplines are seen by the patient every visit in UZ Leuven because this would be too time-consuming, since each discipline has an appointment with the patient of roughly 1 hour. Attained specialized nurses are the main point of contact for each patient. In accordance with the multidisciplinary team and the ALS neurologist, these specialized nurses discuss their patients in terms of disease progression, medication, timing of symptomatic interventions (i.e., NIV, PEG) and which disciplines would be useful for them to see during the following visit. These specialized nurses also provide personalized advise concerning the use of symptomatic aids/appliances, psychological counseling and palliative care. Additionally, UZ Leuven provides genetic testing and genetic consulting for all patients and family members.
This team closely collaborates with gastroenterologists and pulmonologists to assure timely gastrostomy placement and initiation of noninvasive ventilation and adheres to European guidelines on the management of ALS (Citation11). In this study, we aimed to evaluate the outcome of patients in follow-up in an ALS Expert Centre.
Materials & methods
Study Population
Subjects used for these analyses were patients diagnosed with ALS who were either in follow-up at the ALS Expert Centre of UZ Leuven or patients who were in follow-up externally. We categorized patients as ‘Follow-up in multidisciplinary Centre of UZ Leuven: “yes” or “no”’. All subjects had given consent to use clinical data for research through data collection approved by the Ethics Committee of UZ Leuven. Patients who were not in follow-up at the ALS Expert Centre were consulted by a neurologist at UZ Leuven at least once (e.g. for a second opinion), which is why we did have clinical data of these patients. Subjects that are in follow-up externally could either be in follow-up at a general neurology clinic, in follow-up at another multidisciplinary Centre or not in follow-up at all, but we did not have full access to this information. Patient data was collected from patients that were diagnosed and started multidisciplinary follow-up in the ALS Expert Centre between 1/jan/2012 and 31/dec/2022. We included subjects of which extensive clinical data was available (for more details see ) and consent was given to use their clinical data for research, which resulted in the exclusion of 348 patients due to incomplete clinical data. If only one clinical parameter was missing, patients were still included for the analysis, whereas patients were excluded if more than one clinical parameter was missing. Survival of the included subjects was calculated as time from date of diagnosis to date of death, with date of death occurring between 1/jan/2012 and 31/dec/2022. Date of death of patients not in follow-up at the ALS Expert Centre could be obtained using regular updates from the national registry through National Registration Numbers.
Table 1. Demographic and clinical characteristics of study cohorts.
Clinical parameters
In the multivariate analysis, we included clinically relevant variables that reflect both progression rate and severity of the disease at diagnosis. We compared patients that demonstrated bulbar onset of the disease with patients that demonstrated onset in other regions (e.g. spinal, respiratory, generalized). Furthermore, patients that met the revised El Escorial criteria for the “definite ALS” diagnostic category were compared to all other revised El Escorial diagnostic categories (i.e., Suspected ALS, Possible ALS, Probable ALS lab supported and Probable ALS). Site of onset and revised El Escorial diagnostic category were determined by a neurologist of UZ Leuven at the time of diagnosis. Percentage of weight loss at diagnosis was measured as the difference in percentage between weight at diagnosis and the reference weight before disease onset. Forced Vital Capacity (FVC) was measured either the day of diagnosis or shortly thereafter. In the analyses, we used the first FVC recorded in the ALS Expert Centre. Most patients underwent gene testing. C9orf72 repeat status was labeled as expanded when the cutoff of >29 repeats is exceeded.
Statistics
Significance of survival time in the Kaplan-Meier curves was calculated by Log Rank test (Mantel-Cox). Multivariate analyses were performed with Cox Regression analysis and significance of each variate in the equation was calculated by Chi-Square testing. Missing cases were automatically dropped from the Cox analyses (N = 8 (1.2%) for all patients, N = 2 (1.1%) for bulbar patients). To examine the effect of eliminating patients with missing data, we performed a sensitivity analysis including all patients. Hazard Ratios for survival were analyzed in SPSS (CI 95%). Between groups, significance of numeric datasets (FVC, age at diagnosis, weight loss at diagnosis, diagnostic delay) was calculated with non-parametric Mann-Whitney U testing and significance of categorical datasets (Sex, revised El Escorial category, site of onset, riluzole use, C9orf72 status, ENCALS risk profile) was calculated with Fisher’s exact tests. Normality of parameters was tested with Shapiro-Wilk testing. Statistical data and figures were analyzed and created using IBM SPSS Statistics and Graphpad Prism v9. In all analyses, statistical significance was attained at a p-value < 0.05.
Results
Demographics and clinical data
At the time of diagnosis, ALS patients are informed about the possibility to receive multidisciplinary monitoring at the ALS Expert Centre or a multidisciplinary Centre closer to home. Between January 2012 and December 2022, 1007 patients were seen at the ALS Expert Centre. Of those, 659 fulfilled the inclusion criteria. The most important reason for exclusion was incomplete data concerning the clinical prognostic predictors, used for the multivariate analysis. The baseline characteristics are shown in . Most of the parameters did not differ significantly between the patients in follow-up at the ALS Expert Centre and the patients in follow-up externally (), except for age at diagnosis (p < 0.001). Out of 659 patients, 557 (84.5%) received specialized care in the ALS Expert Centre. We observed an average time of 57.6 days (1.9 months) between receiving the diagnosis and the first contact at the ALS Expert Centre.
Progression rate at diagnosis is equal across groups
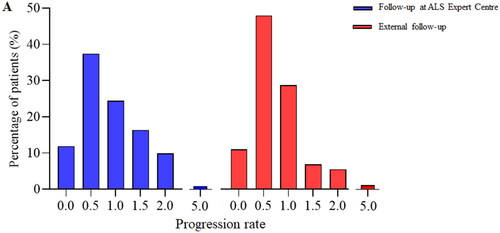
To ensure comparability in disease progression between the two groups, we assessed the progression rate (defined as points lost on ALS-FRS-R at diagnosis divided by the diagnostic delay in months) of each subject at the time of diagnosis (Citation15, Citation16). For some patients, there was no ALS-FRS-R score at diagnosis available, resulting in a small decrease in sample size for both groups (N = 515 for patients in follow-up at the ALS Expert Centre, N = 83 for patients in follow-up externally). Progression rate of subjects was normally distributed and did not differ significantly between the groups (, p = 0.091). Additionally, we calculated the risk profile from the ENCALS-survival model proposed in (Citation17), which attains a personalized prognosis to each ALS patient at time of diagnosis. With the data available, it was possible to calculate this predictor for 599 patients from our study cohort (N = 511 for patients in follow-up at the ALS Expert Centre, N = 88 for patients in follow-up externally). Interestingly, after comparing this predictor between groups, we observed a higher risk profile in patients in follow-up at the ALS Expert Centre (p = 0.024). The median ENCALS prediction score was −3.87 for patients in follow-up at the ALS Expert Centre and −4.53 for patients in follow-up externally.
Figure 1. Clinical profile at diagnosis is similar between groups. A. Progression rate is defined by the difference between the maximal ALS-FRS-R score and the ALS-FRS-R score at diagnosis divided by the diagnostic delay in months. B. Significance of progression rate between groups was tested with non-parametric Mann-Whitney U testing. Progression rate does not differ between groups (p = 0.091).
Multidisciplinary follow-up prolongs survival of ALS patients
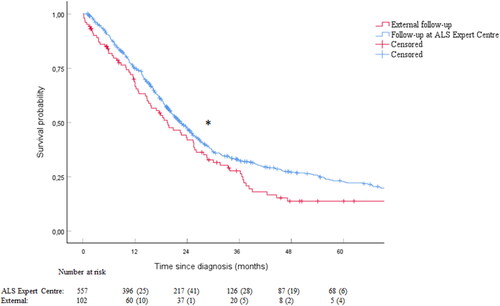
We performed a Cox proportional hazards regression for differences in survival between both groups with extensive clinical data and plotted a five-year survival rate from diagnosis of patients who were in follow-up in the ALS Expert Centre (N = 557) compared to patients who were in follow-up externally (N = 102) (). Survival rate was higher for the patients in follow-up at the ALS Expert Centre, as indicated by the difference between curves (p = 0.046; HR = 0.683; CI 95% [0.528 − 0.884]). Median survival time is 685 days (IQR: 621.6;748.4) (22.5 months) for the patients in follow-up at the ALS Expert Centre and 591 days (IQR: 432.7;749.3) (19.4 months) for the patients in follow-up externally. The excluded patients did not bias the results as also in the sensitivity analysis including all 1007 patients, multidisciplinary care significantly prolonged survival (p < 0.001; HR = 0.685; CI 95% [0.569 − 0.825].
Figure 2. Follow-up in the ALS Expert Centre prolongs survival of ALS patients. A. Kaplan-Meier analysis displays an increased survival rate in the ALS Expert Centre cohort (N = 557) versus the external cohort (N = 102) (p = 0.046). After 5 years, death had occurred in 391 patients (70.2%) in follow-up at the ALS Expert Centre and in 76 patients (74.5%) in follow-up externally by the time of this analysis. Numbers at risk and the respective censored values at each year are listed below the graph. * p < 0.05. B. Clinical parameters added in the multivariate analysis. All parameters except age at diagnosis contribute significantly to survival outcome.
Since we saw an overall beneficial effect of follow-up at the ALS Expert Centre on survival in our univariate analysis, we wanted to validate these results in a multivariate analysis. The following clinically relevant parameters were added in this analysis: site of onset, revised El Escorial score, diagnostic delay, FVC, age at diagnosis and percentage of weight loss at diagnosis (). This multivariate analysis confirmed our findings, demonstrating that follow-up in the ALS Expert Centre significantly increases survival (p = 0.004; HR = 0.683; 95% CI = 0.528 − 0.884). These results indicate that follow-up at the ALS Expert Centre is an independent contributor to prolonged survival of ALS patients.
Different survival effects are observed in patients with bulbar versus spinal onset
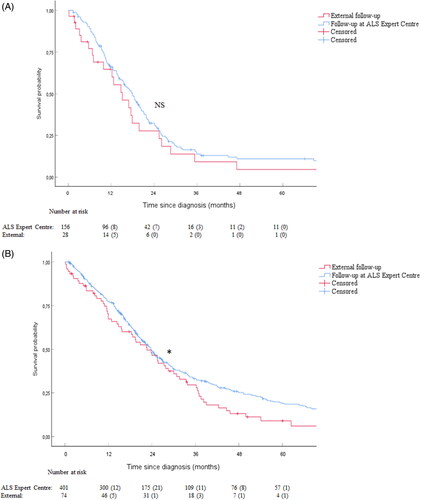
Overall, patients with bulbar onset tend to have a worse prognosis, indicated by a faster progression rate and a shorter survival time as compared to spinal onset (Citation18). Therefore, we wanted to evaluate whether the beneficial effect of multidisciplinary follow-up was also observed in bulbar patients. No significant difference is observed in the survival time of bulbar ALS patients in follow-up at the ALS Expert Centre as compared to the external patients (, p = 0.28; HR = 0.778; CI 95% [0.495 − 1.223]). Median survival time of bulbar patients in follow-up at the ALS Expert Centre was 547.0 days (IQR: 467.4;626.6) (18.0 months) and 460.0 days (IQR: 272.3;647.7) (15.1 months) in the external patients. On the contrary, we did observe a prolonged survival for patients with spinal onset ALS in follow-up at the ALS Expert Centre as compared to the external patients (, p = 0.035; HR = 0.746; CI 95% [0.568 − 0.980]). Median survival time of spinal patients in follow-up at the ALS Expert Centre was 722.0 days (IQR: 648.4;795.6) (23.7 months) and 689.0 days (IQR: 505.5;872.5) (22.6 months) in the external patients.
Figure 3. The effect of follow-up in the ALS Expert Centre differs between patients with bulbar onset and spinal onset ALS. A. No prolonged survival is observed in the subgroup of bulbar patients (p = 0.28) (N = 156 for patients in follow-up at the ALS Expert Centre, N = 28 for patients in follow-up externally). After 5 years, death had occurred in 128 patients (82.1%) in follow-up at the ALS Expert Centre and in 22 patients (75.9%) in follow-up externally by the time of this analysis. B. Survival is significantly prolonged in the subgroup of spinal patients (p = 0.035) (N = 401 for patients in follow-up at the ALS Expert Centre, N = 74 for patients in follow-up externally). After 5 years, death had occurred in 321 patients (80.0%) in follow-up at the ALS Expert Centre and in 63 patients (85.1%) in follow-up externally by the time of this analysis. Numbers at risk and the respective censored values at each year are listed below each graph. NS = Not significant, * p < 0.05.
Discussion
In this study, we demonstrated that multidisciplinary follow-up prolongs survival time, by comparing the survival time in ALS patients that received specialized care in the ALS Expert Centre at UZ Leuven versus patients that did not receive this care. We observed that this increased survival time is independent of clinically relevant variates which describe the disease phenotype of the patient. Of all these parameters, only age at diagnosis differed significantly between groups, with a higher mean age at diagnosis in the patients in follow-up at the ALS Expert Centre. One hypothetical explanation for this could be that somewhat older people tend to be more prone to seeking advice or help from a multidisciplinary team, whereas a younger population wants to be less dependent on help from others. Similarly, it can be hypothesized that patients with a higher progression rate would be more prone to specialized care. However, we demonstrated that ALS progression rate at diagnosis does not differ between groups, as defined by (48-the first recorded ALS-FRS-R score)/diagnostic delay. If anything, progression rate was slightly higher in the patients in follow-up at the ALS Expert Centre, thereby rejecting the idea that changes in survival would be influenced by a higher progression rate in the cohort in follow-up externally. Remarkably, this finding was confirmed by the risk profile based on the ENCALS-survival prediction model created in (Citation17), which demonstrated a higher risk profile score in the group of patients followed in the ALS Expert Centre. This indicates that patients in the ALS Expert Centre have a worse predicted prognosis at diagnosis, which gives even more value to the observed effect of multidisciplinary care on survival in this study.
In our cohort, we observed an increased survival time which appears to be mainly driven by patients with spinal onset ALS, as we did not obtain a significant increase in survival time in patients with bulbar onset. This is opposite to the findings from Papai et al, which demonstrated a greater increase in survival in bulbar patients (Citation13). However, the use of riluzole was higher in the patients receiving multidisciplinary care in this study, which might influence their survival in bulbar patients. In our study the number of bulbar patients in follow-up externally was small, which could have rendered our results underpowered.
Conflicting findings have been reported on the effect of multidisciplinary care on survival in ALS patients (Citation12, Citation14, Citation19, Citation20). For example, alleviating ALS-related complaints and pain through symptomatic strategies has shown to be beneficial for overall survival in both patients with spinal and bulbar onset, as demonstrated by a 7.5 months and 9.5 months increased survival time, respectively, even after correcting for riluzole use (Citation12). However, these studies have been criticized, as they are not controlled randomized studies and thus prone to bias and a low methodological quality (Citation21). In some studies, the cohorts that received specialized care had a higher proportion of riluzole use and were younger (Citation21). In our study, we did not observe a significant difference in progression rate at diagnosis or riluzole use between the two groups, but it has the limitation of being a single Centre retrospective study. Another limitation of this study is the fact that we have no information concerning the use of therapeutic interventions of external patients, which might influence their survival. Lastly, even though there is a certain risk of bias in our results because we excluded 348 patients of whom too many clinical parameters were missing to attain a clear clinical profile, we did perform a sensitivity analysis, in which we obtained similar results.
Since functional decline affects all ALS patients at some point, strategies to minimize these complaints are important for the wellbeing of the patients (Citation22). The survival of patients receiving multidisciplinary care may improve because of improved well-being, but also because interventions such as noninvasive ventilation and gastrostomy placement are offered in a timely manner. In our dataset, we had no information about the use of noninvasive ventilation and gastrostomy in the patients in follow-up externally, so we were unable to investigate further which specific factors would drive a possible survival benefit. Furthermore, we did not investigate quality of life nor advanced care planning strategies in patients both in follow-up at the ALS Expert Centre or externally. It is also possible that the combination of all measures influences survival, rather than one single intervention, but we did not have the data to explore this further.
In conclusion, our data provides further evidence for a survival benefit of multidisciplinary care in an ALS Expert Centre. Further research is needed to better understand the interventions that drive this effect.
Declaration of interest
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17071.
- Ingre C, Roos PM, Piehl F, Kamel F, Fang F. Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol. 2015;7:181–93.
- Goutman SA, Hardiman O, Al-Chalabi A, Chió A, Savelieff MG, Kiernan MC, et al. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022;21:465–79.
- Rich KA, Roggenbuck J, Kolb SJ. Searching far and genome-wide: the relevance of association studies in amyotrophic lateral sclerosis. Front Neurosci. 2020;14:603023.
- Wales S, Kiernan C, DSc Cheah AM, Burrell M, Biostat BC, MBBS Zoing J, Kiernan B, MC, MC, et al. Seminar Amyotrophic lateral sclerosis. Lancet 2011;377:942–55. [Internet]. Available from: www.thelancet.com
- Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis/riluzole study group II. Lancet. 1996; 347:1425–31.
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med. 1994; 330:585–91.
- Miller RG, Mitchell JD, Moore DH,. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012;3:CD001447.
- Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC, et al. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2022; 387:1099–110.
- Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. 2014;10:661–70. Vol. Nature Publishing Group;
- Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A, Van Damme P, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS) - revised report of an EFNS task force. Eur J Neurol. 2012; 19:360–75.
- Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O. Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996-2000 [Internet]. J Neurol Neurosurg Psychiatry. 2003;74:1258–61. Available from: www.jnnp.com
- Paipa AJ, Povedano M, Barcelo A, Domínguez R, Saez M, Turon J, et al. Survival benefit of multidisciplinary care in amyotrophic lateral sclerosis in spain: Association with noninvasive mechanical ventilation. J Multidiscip Healthc. 2019;12:465–70.
- Zoccolella S, Beghi E, Palagano G, Fraddosio A, Guerra V, Lepore V, et al. ALS multidisciplinary clinic and survival: Results from a population-based study in Southern Italy. J Neurol. 2007; 254:1107–12.
- Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. J Neurol Sci. 1999;169:13–21. [Internet]. Vol. Available from: www.elsevier.com/locate/jns
- Bakker LA, Schröder CD, van Es MA, Westers P, Visser-Meily JMA, van den Berg LH. Assessment of the factorial validity and reliability of the ALSFRS-R: a revision of its measurement model. J Neurol. 2017; 264:1413–20.
- Westeneng HJ, Debray TPA, Visser AE, van Eijk RPA, Rooney JPK, Calvo A, et al. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol. 2018; 17:423–33.
- Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. 2009;10:310–23. Vol.
- Martin S, Trevor-Jones E, Khan S, Shaw K, Marchment D, Kulka A, et al. The benefit of evolving multidisciplinary care in ALS: a diagnostic cohort survival comparison. Amyotroph Lateral Scler Frontotemporal Degener. 2017; 8:569–75.
- Oliveira De Almeida FE, Kelly A, Santana C, Oliveira De Carvalho F. Multidisciplinary care in amyotrophic lateral sclerosis: a systematic review and meta-analysis. Neurol Sci. 2021;42(3):911–23.
- Ng L, Khan F, Mathers S,. Multidisciplinary care for adults with amyotrophic lateral sclerosis or motor neuron disease. Cochrane Database Syste Rev. 2009;(4):CD007425.
- Van Den Berg JP, Kalmijn S, Lindeman E, Veldink JH, De Visser M, Van Der Graaff MM, et al. Multidisciplinary ALS care improves quality of life in patients with ALS. 2005.