
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Paroxetine is not suitable for oral administration due to its extensive first-pass metabolism, thus resulting in less bioavailability. This study aimed to prepare novel paroxetine-loaded solid lipid nanoparticles (SLNs) based sustained-release transdermal patches to overcome these problems by enhancing drug absorption and bioavailability. Nine formulations of paroxetine SLNs were prepared by the hot melt-homogenization method using different concentrations of glycerol monostearate (Kolliwax) and Tween 80. Then these prepared SLNs were incorporated in a matrix type transdermal patch having a matrix of ethyl cellulose and polyvinyl pyrrolidone in 3:2 with polyvinyl alcohol. The SLNs showed a particle size range of 113–230 nm and an entrapment efficiency of 85.14%. The SLNs showed sustained paroxetine release (77.86–95.63% release) up to 48 h. FTIR studies showed no interaction between drug and formulation components. Paroxetine is evenly distributed in an amorphous form in SLNs, as demonstrated by DSC as well as PXRD analysis. SLNs formulated patches showed higher drug permeation through the skin than drug-based transdermal patches., Draize patch test revealed no sign of erythema after applying paroxetine-loaded SLN patches (score 0) as observed with the marketed product. The developed SLNs based transdermal patches showed increased permeability and sustained release behaviour.
Introduction
Paroxetine is one of the most potent clinically available selective serotonin reuptake inhibitors (SSRI) antidepressants [Citation1]. It acts by blocking serotonin reuptake [Citation2], but its action is less selective at the serotonin site than other SSRIs, i.e. fluvoxamine or sertraline [Citation3]. Paroxetine is, clinically available in oral dosage forms (such as tablets, capsules and in oral suspension) [Citation4], readily absorbed from the gastrointestinal tract (GIT) and extensively metabolized in the liver [Citation5]; thus, a much lesser amount of the drug reaches the systemic circulation, resulting in poor bioavailability [Citation6]. The t1/2 varies according to the dosage and administration duration. The half-life of up to 21 h following oral paroxetine dosing of 30 mg per day [Citation2]. The pharmacodynamics of paroxetine is not suitable for its oral administration due to its extensive first-pass metabolism, thus resulting in less bioavailability [Citation7].
To overcome these problems, solid lipid nanoparticle (SLN)-based transdermal patches can be developed to ease the patient; furthermore, by developing patches, the first-pass effect can also be bypassed [Citation8]. For improving drug bioavailability, lipid-based, nanoparticulate systems proved to be very useful. Solid lipid nanoparticles (SLNs) are sub-micron colloidal carriers composed of lipids, fatty acids, waxes and triglycerides dispersed in an aqueous surfactant solution or water. They are generally safe for oral, topical and parenteral administration [Citation9,Citation10]. SLNs are novel colloidal drug carriers having the advantages of enhancing drug stability, improving drug release profile and thus bioavailability and biocompatibility [Citation9,Citation11].
SLNs system provides safe and effective drug delivery to the site of action with minimum side effects observed in other nanoparticle systems. They are used as alternatives to the existing nanocarriers like liposomes, polymeric nanoparticles and nano-emulsions, etc [Citation10]. have been selected as a substitute for liposomes and polymeric nanoparticles since 1990 because they have overcome several problems encountered in previous nanocarrier systems such as SLNs are safer than polymeric nanoparticles because organic solvents are not used in their production as well as they have better drug stability and prolonged release compared with liposomes [Citation12]. Preparation and evaluation methods of SLNs are well-documented, and scale-up production is also possible [Citation13].
It is recognized that drug delivery through the skin is considered a suitable tool for drug transport in many local and systemic diseases [Citation14]. In the last few years, transdermal delivery of drugs earned great popularity due to bypassing the first-pass effect, avoiding fluctuations in plasma level and improving patient compliance [Citation15]. Transdermal drug delivery systems, commonly referred to as ‘patches’, have many advantages, including controlled release, fewer systemic adverse effects, user-friendliness, painlessness and better patient compliance via a single dosage for multiple days. Transdermal patches transport drugs to the blood through the skin. The main advantage of the transdermal route is the possibility of attaining the steady-state plasma concentration and sustained action of the drug [Citation16].
The objective of this study was to explore the SLN-based tool to sustain drug release from transdermal patches, as reported by Mangesh et al. [Citation17]. Various characterizations, including particle size and charge, interactions studies, drug release behaviour and permeation studies, were performed to optimize the SLN formulations. The Draize patch test was used to evaluate the skin irritation potential of paroxetine-loaded SLN transdermal patches. The release behaviour of the drug was investigated by kinetic modelling of the drug release data. To the best of our knowledge, no study previously reports the fabrication of paroxetine-based SLN transdermal patches for the sustained anti-depressant effects.
Materials and methods
Chemicals
Surge Laboratories, Sheikhupura, Pakistan gifted paroxetine. Glycerol monostearate (Kolliwax GMS II) was obtained as a gift sample from BASF Chemicals Asia Pacific region Karachi Pakistan, and Polysorbate 80 (Tween 80) was purchased from Sigma-Aldrich, Germany. All other chemicals and reagents used were analytical grade.
Methodology
Formulation of SLNs
Three different levels of lipid amount (75, 100, 125 mg) and surfactant amount (350, 400, 450 mg) were taken as independent variables, and nine formulations were prepared as shown in . The effect of independent variables was checked on particle size, polydispersity index and zeta potential, entrapment efficiency, drug release and drug permeation of paroxetine-loaded SLNs. The influence of the concentration of Kolliwax GMS and Tween-80 on the entrapment efficiency and drug release of paroxetine in SLN was assessed.
Table 1. Composition of paroxetine-loaded solid lipid nanoparticles containing different lipid and surfactant concentrations.
Preparation of paroxetine-loaded SLNs
The hot melt encapsulation technique was employed to fabricate paroxetine-loaded SLN with slight modification as per requirement [Citation1]. In this method, the lipid (Kolliwax GMS) was heated at 75 °C, 5 to 10 °C above the actual melting point of solid lipid. The weighed amount of the drug was added to the melted lipid, and the drug was completely dissolved in the lipid phase. Tween 80 was dissolved in distilled water and heated at 75 °C to prepare the aqueous phase. Subsequently, the lipid and aqueous phases were mixed under magnetic stirring at 600 rpm, maintaining the temperature at 75 °C. The stirring was continued at 600 rpm till the room temperature was reached [Citation1]. The un-entrapped drug was separated from SLNs in the suspension by centrifugation at 14,000 rpm at room temperature. The supernatant was removed and separated SLNs were washed with distilled water twice to remove any other un-entrapped drug. SLNs obtained after centrifugation was then lyophilized to obtain dried SLNs, which were further incorporated in the transdermal patch.
Characterization of SLNs
Paroxetine-loaded SLNs were assessed for entrapment efficiency, particle size, PDI, zeta potential, morphology, drug–polymer interaction, release kinetics, thermal stability and amorphous nature.
Particle size, PDI and zeta potential
Mean particle size, zeta potential and polydispersity index are essential parameters for the stability of any formulation [Citation2]. The particle size, PDI and zeta potential of prepared dispersion of SLNs were investigated using a dynamic light scattering method using zeta sizer ZS-90 (Malvern instruments England). The sample was prepared by diluting 1 ml of SLN dispersion with 1 ml of deionized water and added into a zeta sizer cuvette. The experiment was conducted in triplicate for zeta potential, particle size and polydispersity index [Citation5].
Morphology
The surface morphology of SLNs was investigated by scanning electron microscopy (JSM-IT-100, JEOL, Japan) by adopting the following procedure. SLN sample was taken in the grid and dried under vacuum on an aluminium stub. The sample was then coated with a thin gold layer under an argon environment. The grid was inserted in the microscope, and scanning was done between 15 and 30 kV. The best morphology result was obtained at 50,000× magnification, and a 100 nm screen area was taken [Citation6].
Fourier transform infra-red (FTIR) spectroscopy
Fourier transforms infra-red (FTIR) spectra were acquired for chemical interaction study. Spectra of pure lipid, drug, surfactant and drug-loaded SLNs were taken. A small amount (100 mg) of each sample was taken on the slit, contacted with the piston, and spectra were measured between 500 nm to 4000 nm using FTIR spectrometer (Bruker, alpha-P, Germany).
Entrapment efficiency and drug loading
The centrifugation method was employed to calculate the entrapment efficiency of SLN dispersion [Citation18]. SLN dispersion containing equivalent to 2 mg of drug was centrifuged in an Eppendorf tube at 14,000 rpm at room temperature for 20 min. The supernatant was collected and again centrifuged at the same speed for 20 min. The supernatant liquid was collected and diluted with a phosphate buffer of pH 7.4. Entrapment efficiency was measured using an indirect method, i.e. by calculating the amount of free drug in the supernatant. The absorbance was taken at 293 nm using a UV spectrophotometer (Halo SB-10, Dynamica, USA), and entrapment efficiency was calculated using the following formula [Citation19].
(1)
(1)
The following formula was used to calculate drug loading in SLNs:
(2)
(2)
In vitro dissolution studies
In vitro dissolution studies of drug-loaded SLNs were carried out by performing a dissolution test. The experiment was performed by using semi-automated USP type II apparatus (Pharma test, Germany). Dissolution was performed for all formulations at 37 °C, 50 rpm paddle speed, and 500 ml volume of Phosphate buffer of pH 7.4 [Citation9]. 5 ml of paroxetine-loaded SLN dispersion was added in dialysis bag of molecular weight cut off 12,000 Da and fastened from both ends. The dialysis bag was immersed in 500 ml of phosphate buffer (pH 7.4) and was used as dissolution media at 37 °C with the speed of 50 rpm. Aliquots of 5 ml were taken at the specified time interval and replaced with 5 ml of fresh phosphate buffer. The absorbance of all samples was taken by UV spectrophotometer at the wavelength of 293 nm. Each experiment was carried out in triplicate, and the mean was calculated [Citation9].
Drug release kinetics
The release data of paroxetine-loaded SLNs were kinetically analysed using various models such as zero order, first order, Higuchi model and Korsmeyer-Peppas model. The value of n obtained from the Korsmeyer-Peppas model was used to interpret the results as follows: n = 0.5 indicates the mechanism of Fickian diffusion; n > 0.5 reflects non-Fickian diffusion; n = 1 indicates case-II transport mechanism; n > 1 suggested super case-II transport [Citation20].
Differential scanning calorimetry (DSC)
TA unit from the Q5000 series (West Sussex, UK) was utilized to carry out differential scanning calorimetry (DSC) studies of dried SLN A small amount of the sample (1 and 5 mg) was placed in a DSC cell and stored in a sealed crucible for analysis. For reference, a second crucible was utilized but without the sample. The sample was then heated for 15 min to 500 °C while nitrogen was constantly purged [Citation21].
Powder X-ray diffractometry (PXRD)
Diffraction studies were carried out using Bruker D8 powder diffractometer (Bruker Kahlsruhl, Germany). The samples were used for PXRD experiments after being exposed to CuKa source with a 1.542 wavelength as well as 1 mm slits, materials were analysed (5°–50° 2θ) at a rate of 1° 2θ/min to note XRD peak positions [Citation22].
Fabrication of SLN loaded transdermal patch
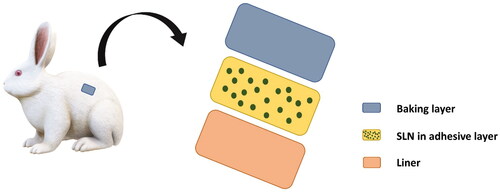
The solvent evaporation method was used to prepare matrix–type SLN loaded transdermal patches with the backing membrane of polyvinyl alcohol (PVA) illustrated in .
Figure 1. Illustration of the SLN patches with different parts and the components.
Preparation of backing membrane
The backing membrane was prepared by dissolving 4 g of PVA in distilled water (4% solution). Then this solution was stirred at 800 rpm for 2 h at 80 °C. Solution temperature was decreased for 5 min by cooling, and then sonication was done for 3–10 min to get rid of bubbles. Then 15 ml of this solution was measured and added to the petri dish with a surface area of at least 61 cm2 and dried in a dust-free environment at room temperature for 24 h [Citation23].
Development of SLN loaded matrix patch
Accurately measured 10 ml of cold ethanol (15 °C) was added in the conical flask, and measured quantities of ethyl cellulose (EC), polyvinyl pyrrolidone (PVP) in 3:2 was added in it, and di-butyl-phthalate (DBP) was used as plasticizer (). All the chemicals were dissolved, and then 5.12% of dried drug-loaded SLNs were added to them, and a matrix solution was formed. Then this matrix solution was poured on the already dried backing membrane present in the petri dish and dried at room temperature by using an inverted funnel to avoid rapid drying [Citation24]. When the matrix was completely dried, the film was taken out of the petri dish. A diameter of 1.54 cm2 was cut with the cutter and wrapped in aluminium foil and stored in the cool, dry container until use. Almost 10–12 patches were prepared from one petri dish [Citation23]. Three different transdermal patches of the same composition but containing different formulations of SLNs were prepared. F2, F6, and F9 formulations of SLNs were incorporated individually in three different patches with the polymeric matrix’s exact composition.
Table 3. Composition of transdermal patch.
Characterization of SLN loaded matrix patch
Thickness variation, weight variation and folding endurance of all formulations were done with the help of suitable gadgets [Citation25].
Thickness variation
Digital Vernier calliper was used to evaluate the patches’ thickness in three distinct places, and thus the mean value was subsequently calculated.
Weight variation
Three randomly chosen patches were separately weighed to determine weight variation. Each formulation performed such a determination, and the mean value was subsequently determined.
Folding endurance
The test for folding endurance involved folding individual patches repeatedly until they broke. The values of the folding endurance test measured how many times the fabricated film could be twisted in the same direction without breaking.
Mechanical test
Using a microprocessor-based modern force gauge (Ultra Test, Mecmesin, UK) with such a load cell of 25 kg, the mechanical characteristics of the fabricated films were assessed. The film strip that was 60 × 10 mm in size and free of air bubbles or other physical flaws was placed between two clamps that were placed 3 cm apart. The strips were pulled during measurement by a top clamp at such a rate of 2 mm/s until the film cracked or broke. When the film broke, the force, as well as elongation, were recorded. For each fabricated patch, measurements were carried out four times [Citation26].
Ex vivo skin permeation studies by using Franz diffusion cell
This study was approved (Approval No.: 34/Pharm/2018/IUB) by the Ethical Committee of the Pharmacy Department, The Islamia University of Bahawalpur, Pakistan, and conducted according to international guidelines for animal use in lab studies [Citation13]. For ex vivo permeation studies, 4 mice with weights ranging from 230–250 g and 6–8 weeks old were selected. The abdominal skin of mice was excised after removing hair with an electrical clipper. Extra fats were carefully removed, and the skin was washed with normal saline. Ex-vivo skin permeation study for the transdermal patch was performed in the Franz diffusion cell apparatus (Perme Gear Inc 4 G-01-00-15-12, India) using rat skin as a permeation membrane [Citation27,Citation28]. The excised abdominal skin of the rat was mounted on Franz cell using phosphate buffer pH 7.4 as permeation medium, which was filled in the receptor compartment, and the temperature was maintained at 32 ± 0.5 °C [Citation28]. The rat skin was carefully mounted on the receptor compartment, and a patch of area 3.14 cm2 was applied to the rat skin. Then the glass disc was placed over it as a donor compartment, and both compartments were fixed with the help of stainless steel clamp. This Franz diffusion cell was mounted on the magnetic stirrer and agitated at 600 rpm during the whole test. Ex-vivo permeation studies were performed for one control and three different SLNs formulation loaded transdermal patches. The control patch contains a pure drug quantity equivalent to the dried SLN-based patches. An liquot of 1 ml was taken at the predetermined time points. The amount of paroxetine permeated through rat skin was determined after 24 h of applying the transdermal test patches and control transdermal patches. A fresh buffer replaced the sample taken at each sampling time. The samples were analysed for absorbance by a UV spectrophotometer at the wavelength of 293 nm [Citation29] The permeability coefficient was determined by dividing the drug flux at steady state by the initial drug load and the drug flux was computed by dividing the slope of the linear segment of the curve through the exposed area of the skin surface [Citation30].
Draize patch test
Skin irritation study of paroxetine-loaded SLN patches was conducted on albino rabbits of 2.5–3.0 kg, divided into three groups with three animals in each. Group one was treated with a control formulation, group two with the marketed paroxetine-loaded transdermal patch, and group 3 with a paroxetine-loaded SLN patch for 72 h.
Briefly, the back hairs of the rabbits were removed with the help of an electric clipper 24 h before the patch test. Precisely weighed amounts of formulations (containing 5 mg of drug) were applied on a 2.54 cm square area and covered using adhesive tape. The evaporation of volatile entities was prevented by wrapping the rabbit trunk in the rubberized cloth. The rabbits bearing patches were retained in restrainers. The patch applied area was examined for any visible change and graded for erythema/edoema at 0 (initial), 24, 48 and 72 h after the application of transdermal patches [Citation31]. The mean erythema score was assigned based on Draize dermal scoring criteria for skin irritation [Citation32] and the erythema index was also determined by adding the average erythema scores of skin before and after the application of patch at 0 24, 48 and 72 h and dividing by 4 [Citation33].
Statistical analysis
Statistical analysis was performed using one-way ANOVA. All the experiments were conducted in triplicate, and results were presented as mean ± SD. The level of significance was taken at p < .05.
Results and discussions
Formulation of SLNs
Paroxetine-based SLN formulations were optimized based on entrapment efficiency and in vitro drug release. The influence of formulation variables, including the concentration and lipid and tween-80, was evaluated with a single fact test. It was confirmed from results that all independent variables influenced the entrapment efficiency and in vitro drug release properties of paroxetine-loaded SLNs. A higher amount of lipid (100 mg) will likely increase drug entrapment efficiency in SLNs. For the surfactant concentration, a low level (350 mg) will be suitable for increasing drug entrapment and sustained drug release from the paroxetine SLNs. Formulation F4 was optimized on the selected criteria of high entrapment efficiency and sustained drug release. The results are presented in .
Table 2. Particle size, zeta potential, PDI and entrapment efficiency of formulations.
Preparation and optimization of paroxetine-loaded SLNs
This study was based on the formulation of SLNs of paroxetine for topical drug delivery by applying transdermal patches. Kolliwax GMS II (glycerol monostearate) was used as solid lipid and Tween 80 as a surfactant in different ratios. SLNs were prepared by the hot-melt encapsulation method. The fabricated nanoparticles were free of any toxic solvent. SLNs prepared by Reithmeier et al. using the hot melt encapsulation method predicted that relatively small size nanoparticles could be prepared by this method without any further dilution of the formulation [Citation34].
Characterization of paroxetine-loaded SLNs
Determination of particle size, PDI and zeta potential
The particle size of all formulations ranged from 113 nm to 230 nm, while F3 showed the smallest particle size (113 nm). It was observed that with the increase in lipid concentration, the size of nanoparticles was increased. This phenomenon could be explained as the lipid concentration increases, and the degree of homogenization decreases [Citation2]. Kumar et al. also studied the effect of lipid concentration on particle size. They concluded that the particle size of nanoparticles reduced from 103 nm to 61 nm when lipid content was decreased from 300 mg to 250 mg with constant atorvastatin concentration [Citation35]. Furthermore, the effect of surfactant concentration on particle size revealed that particle size reduced with an increase in the amount of surfactant because a higher amount of surfactant reduces the surface tension of lipid drops, which helped lipid drops break down into smaller droplets. Generally, at a low concentration of surfactant, particle aggregate to form clusters due to insufficient repulsion, resulting in SLNs of larger size [Citation3].
Zeta potential represents the charge on the surface of the particles, which indicates the extent of repulsion between the similarly charged and closely located particles [Citation3]. The values of zeta potential for all formulations range from −3.10 to −16 mV (). Nanoparticle dispersions are thermodynamically unstable systems; thus, they show good stability at sufficient zeta potential [Citation36]. Particle aggregation is most likely to occur when the zeta potential of the formulation is significantly less because of less repulsion between the particles. Enough repulsion is required to prevent particle aggregation, which leads to the instability of the particle [Citation37]. As we know, surfactant imparts repulsion between the particles to prevent aggregation, improving the stability of dispersion; thus, it also enhances the zeta potential of the dispersion. So higher the surfactant concentration, the higher the zeta potential; thus, the higher will be the stability of the dispersion [Citation38].
PDI is defined as a ratio that gives information about the homogeneity of the particle size distribution of the colloidal dispersion. The appropriate value of the polydispersity index is below 0.3 which represents a monodisperse and homogenous system [Citation2]. Values of PDI of all formulations are below 0.3 except for F1 which shows 0.64 PDI are shown in . These results showed that all the formulations are homogenous and mono disperse systems except one formulation. F1 shows high PDI because of low lipid concentration. This occurs due to the high drug to lipid ratio as lipid concentration is less but the amount of drug remains the same [Citation3]. Higher the poly dispersity index lower will be the homogeneity of particles in the dispersion [Citation39]. Surfactant also helps to maintain low values of PDI and better homogeneity. Higher the concentration of surfactant, lower will be the value of PDI [Citation3]. When F1, F2 and F3 were compared to check the effect of surfactant concentration on PDI, results showed that with the increase in the concentration of surfactant from 350 mg to 450 mg, the value of PDI lowers from 0.64 to 0.20.
Entrapment efficiency and drug loading
The entrapment efficiency of formulation varied from 73.60% to 85.14% (). F7 shows the highest value of entrapment efficiency, i.e. 85.14%. When F1, F2 and F3 were compared to check the effect of surfactant on the entrapment efficiency of nanoparticles, the results showed that there was a decrease in entrapment efficiency with the increase in surfactant concentration. This phenomenon is attributed to the increased solubility of drugs in the aqueous phase as the percentage of surfactants increases [Citation40]. When the effect of lipid concentration was checked on the entrapment efficiency of SLNs, it was observed that the entrapment efficiency of nanoparticles increased from 81.16% to 85.14% on increasing lipid concentration from 75 to 125 mg. This situation can be explained as an increase in lipid concentration providing more space for drug encapsulation. Similar trends were observed in other studies [Citation22].
Surface morphology of nanoparticles
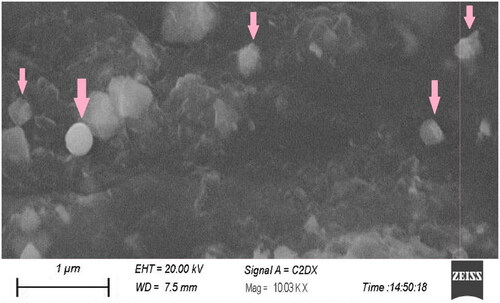
Surface morphology of the SLN formulation embedded in a patch prepared with optimized parameters was observed sby scanning electron microscope as shown in . The study showed that nanoparticles were primarily spherical with a smooth outer surface. It was also found that the particle size of the optimized formulation was in the nanometre range [Citation41].
Figure 2. Scanning electron microscope image of paroxetine-loaded SLNs embedded in patch formulation.
FTIR spectra of paroxetine-loaded nanoparticles formulation
FTIR spectra of drug, lipid, surfactant and paroxetine-loaded SLN formulation are shown in . Paroxetine FTIR spectrum showed an intense peak at 3556.99 cm−1 and 3499.56 cm−1 this is mainly due to the stretching of N–H group whereas –CH symmetrical stretching appeared at 2940.22 cm−1. Another characteristic peak of paroxetine appeared at 1519.22 cm−1 which demonstrated the C-C stretching. 1220.32 cm−1 indicated the CO– stretching [Citation42].
Figure 3. FT-IR of pure drug (a), lipid (b), surfactant (c) and drug loaded SLN formulation (d).
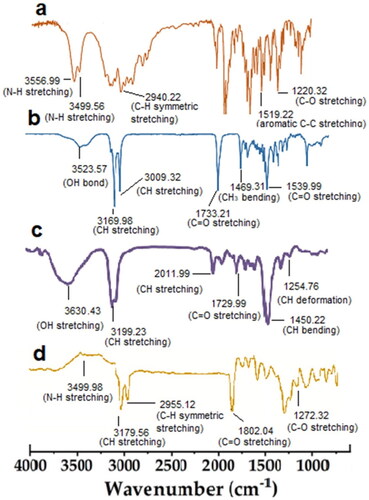
FTIR spectrum of GMS (Kolliwax) showed bands due to characteristic stretching of C–H bond at 3169.98 cm−1 as well as at 3009.32 cm−1 whereas C=O stretching showed at 1539.22 cm−1. OH bond vibration appeared at 3523.57 cm−1 [Citation43]. Sharp and broader characteristic peak of tween 80 showed at 3630.43 cm−1 mainly due to OH stretching whereas CH stretching appeared at 3199.23 cm−1 and 2011.99 cm−1. The peak at 1729.99 cm−1 illustrated the C=O stretching. CH bending appeared at 1450.22 cm−1 [Citation44]. FTIR spectra of formulated SLNs showed that the slightly shifting of characteristic peaks of the drug which confirmed the entrapment of the drug in the lipid matrix. Therefore it is confirmed that there is no chemical reaction between drug and polymers during the SLNs formulation [Citation45].
In vitro release studies
In vitro drug release from all SLN formulations was in the range of 77.86–95.63%. The varying the percentage of the drug to SLN matrix ratios in the various formulations may possibly be the cause of the variations in the release profile of drug. F2 showed the highest drug release and F4 showed minimum drug release in 48 h. When the effect of lipid concentration was observed by comparing the formulations F2, F5 and F8, it was seen that with increasing lipid concentration from 75 mg to 125 mg, the initial burst release of drug from nanoparticles occurred. This happened because as the lipid concentration increases, more amount of drug is encapsulated in the core due to decreased solubility in the lipid phase [Citation46]. In addition, with the increase in surfactant concentration, there was an increase in the drug release from SLNs. It was also observed the same effect of surfactant concentration on tolnaftate release from SLNs. This phenomenon could be explained as the higher the amount of surfactant used results in an increase in drug solubility in the aqueous phase; thus higher is the release of drug from nanoparticles [Citation2]. Another study suggested the same drug release pattern from ramipril-based SLNs [Citation18]. The best fit kinetic model for optimized formulation F4 having maximum entrapment and sustained release effect was the Higuchi model (R2 = 0.9863) (), showing that the drug release from the paroxetine based SLNs is dependent on the solubility in the vehicle and diffusion of the drug from the matrix system [Citation47]. The in vitro release of pure drug and optimized drug-loaded SLNs formulation and described as cumulative drug release versus time as shown in . The figure showed that the release of drug from control was more significant than the drug released from SLNs; thus, SLN formulation effectively provides the sustained release of paroxetine.
Figure 4. Comparison of in-vitro drug release profile with lipid concentration (a). Comparison of in-vitro drug release profile with surfactant concentration (b). Comparison of in-vitro drug release profile of all formulation with control (c).
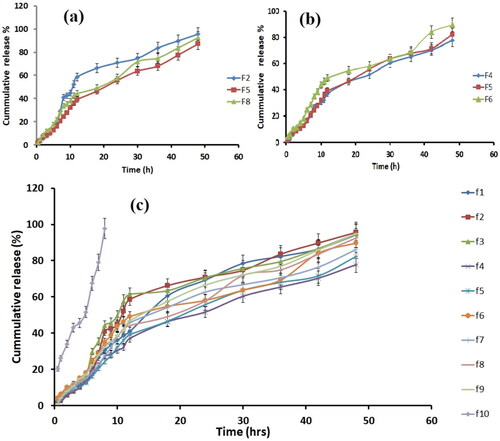
Table 4. Kinetic constants and regression coefficients of kinetic models for drug release from F4.
Differential scanning calorimetry (DSC)
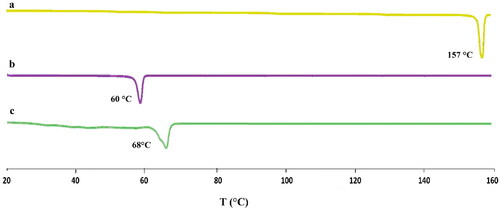
The DSC curves for paroxetine, Kolliwax GMS, and loaded SLNs are shown in . The sharp melting peak of paroxetine is indicated by thermograms to be about 157 °C. the Kolliwax GMS reached its endothermic peak at 60 °C. In lyophilized loaded SLNs, there wasn’t a melting peak for paroxetine at 157 °C. This shows that paroxetine is encapsulated in the SLNs lipid matrix in the amorphous state rather [Citation1]. Slightly shifting of loaded SLNs endothermic peak towards lower temperature at 68 °C in comparison to the pure drug is justified by the incorporation of paroxetine and the melting point depression was mainly due to the Thomson effect brought by the reduction in size to the nanodimension [Citation48].
Figure 5. DSC of pure drug (a), lipid (b) and drug loaded SLN formulation (c).
Powder X-ray diffractometry (PXRD)
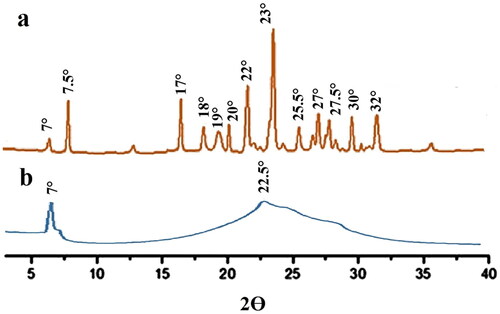
The PXRD graph for paroxetine and loaded SLNs are shown in . PXRD graph of paroxetine showed intense peaks at 2Ɵ of 7°,7.5°, 17°, 18°, 19°, 20°, 22°, 23°, 25.5°, 27.5°, 30° and 32°, which indicates its crystallinity [Citation49] whereas loaded SLNs represented the single sharp peak at 2Ɵ of 7° and broader peak at 2Ɵ from 18° to 32° which demonstrated the amorphous nature of SLNs as well as dispersion of pure drug at the molecular level in the SLNs lipid matrix [Citation50].
Figure 6. XRD of pure drug (a) and drug loaded SLN formulation (b).
Fabrication of transdermal patch
SLN-based matrix-type transdermal patches with polyvinyl alcohol backing were fabricated by solvent evaporation technique.
Physicochemical characterization of transdermal patches
Weight variation and thickness of all transdermal patches were found to be uniform. Transdermal patches exhibited good resistance to cleavage and have good plasticity whereas mechanical testing demonstrated that elongation at the break having lower values and tensile strength were shown higher values. These findings show that patches weren’t brittle but instead were strong as well as flexible ().
Table 5. Physiochemical properties of transdermal patches.
Ex vivo permeation studies
Ex vivo permeation studies were performed using Franz diffusion cell coupled with rat skin as a permeation membrane. Phosphate buffer (pH 7.4) was used as a permeation medium. Pure drug-based transdermal patches showed less permeation through the skin thenP1,P2 and P3 SLN formulated patch at 24 h () () and the steady state flux along with permeability coefficient represented in . It has been reported that PVP increases the amount of drug released from the patch as its concentration increases because it decreases the crystallinity of the drug [Citation24].
Figure 7. Comparison of drug permeation of P1 with control (a). Comparison of drug permeation of P2 with control (b). Comparison of drug permeation of P3 with control. (c) Comparison of drug permeation of all formulations with control (d).
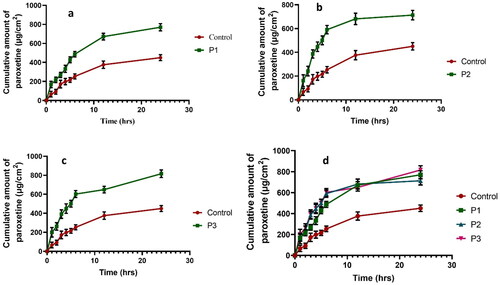
Table 6. Cumulative drug permeation and skin irritation studies..
Table 7. The steady state flux and permeability coefficient.
Dibutyl-phthalate was used as a plasticizer, as it helps to soften the polymer matrix by diffusing it. It reduces the interactions between the polymer molecules like hydrogen bonding and also forms a bond with polymer molecules that helps in the film formation [Citation23]. The effect of particle size on permeation was also analysed. The results showed that permeation increases with decreased particle size, i.e. the smaller the particle size, the higher the surface of particles, and more intimate their contact with corneocytes, thus a greater the penetration of the drug through stratum corneum [Citation36]. When the effect of surfactant on permeation was analysed, it was observed that there was an increase in permeation of drug through the skin with an increase in surfactant to lipid ratio. Likely, the surfactant helps to loosen the lipid bilayers of the stratum corneum, thus enhancing drug penetration. Similar observations were explained in other studies [Citation51].
Draize patch test
The results of the Draize patch test revealed no sign of erythema after applying paroxetine-loaded SLN patches (score 0) as observed with the marketed product. Moreover, no skin irritation was observed at 48 h (score 0–1) and 72 h (score 0–1) for marketed product and test formulation of paroxetine-loaded SLNs as well as erythema index confirmed that there was no difference in erythema values before and after the application of control, P1, P2 and P3 formulated patch (). This was the indication of the suitability of paroxetine-loaded SLNs transdermal patches for skin application.
Table 8. Skin irritation test.
Conclusion
Transdermal patches based on SLNs of the anti-depressant drug paroxetine have been successfully prepared and characterized. The present study demonstrated SLN loaded patches of paroxetine (20 mg) as a possible alternative to the conventional drug delivery system (i.e. tablets). This could be a novel drug delivery system to reduce all the side effects associated with initial high plasma concentration after oral administration and fluctuations in plasma levels and also to reduce the first pass effect to improve the bioavailability of paroxetine as well as provide a more patient-friendly dosage form. Further studies are suggested.
Author contributions
Fahad Pervaiz, Haya Yasin, Manal Buabeid and Ghulam Murtaza had substantial contributions to the conception or design of the work and interpretation of data for the work; Fahad Pervaiz and Haya Yasin managed the resources; Ayesha Saba and Sobia Noreen were involved in the acquisition, analysis, or interpretation of data for the work; Fahad Pervaiz, Haya Yasin, Manal Buabeid, Abida Kalsoom Khan and Ghulam Murtaza drafted the work and revised it critically for important intellectual content and all the authors finally approved the version to be published; Fahad Pervaiz agrees to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available from the corresponding author, [GM], upon reasonable request.
Additional information
Funding
References
- Rehman M, Asadullah Madni AI, Khan WS, et al. Solid and liquid lipid-based binary solid lipid nanoparticles of diacerein: in vitro evaluation of sustained release, simultaneous loading of gold nanoparticles, and potential thermoresponsive behavior. Int J Nanomed. 2015;10:2805.
- Abousamra MM, Mohsen MA. Solid lipid nanoparticles and nanostructureed lipid carriers of tolnaftate: design, optimization and in-vitro evaluation measurement. Int J Pharm Pharm Sci. 2016;10:100.
- Das S, Ng WK, Tan RB. Are nanostructured lipid carriers (NLCs) better than solid lipid nanoparticles (SLNs): development, characterizations and comparative evaluations of clotrimazole-loaded SLNs and NLCs? Eur J Pharm Sci. 2012;47(1):139–151.
- Paroxetine Medscape. 2022. Available from: https://reference.medscape.com/drug/paxil-brisdelle-paroxetine-342959
- Shah R, Eldridge D, Palombo E, et al. Optimisation and stability assessment of solid lipid nanoparticles using particle size and zeta potential. J Phys Sci. 2014;25(1):59–75.
- Vijayanand P, Jyothi V, Aditya N, et al. A. Development and characterization of solid lipid nanoparticles containing herbal extract: in vivo antidepressant activity. J Drug Delivery. 2018;2018:1–7.
- El-Nabarawi MA, Bendas ER, El Rehem RTA, et al. Transdermal drug delivery of paroxetine through lipid-vesicular formulation to augment its bioavailability. Int J Pharm. 2013;443(1-2):307–317.
- Prajapati P, Oza M, Shah HM, et al. Formulation and evaluation of sublingual tablet of paroxetine Hcl. Int J Pharma Bio Sci. 2014;3(2):1019–1036.
- Patel K, Padhye S, Nagarsenker M. Duloxetine HCl lipid nanoparticles: preparation, characterization, and dosage form design. AAPS PharmSciTech. 2012;13(1):125–133.
- Mukherjee S, Ray S, Thakur R. Solid lipid nanoparticles: a modern formulation approach in drug delivery system. Indian J Pharm Sci. 2009;71(4):349.
- Suresh G, Manjunath K, Venkateswarlu V, et al. Preparation, characterization, and in vitro and in vivo evaluation of lovastatin solid lipid nanoparticles. AAPS Pharmscitech. 2007;8(1):E162–E170.
- Naseri N, Valizadeh H, Zakeri-Milani P. Solid lipid nanoparticles and nanostructured lipid carriers: structure, preparation and application. Adv Pharm Bull. 2015;5(3):305–313.
- Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery–a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177.
- Valenta C, Auner BG. The use of polymers for dermal and transdermal delivery. Eur J Pharm Biopharm. 2004;58(2):279–289.
- Kurz A, Farlow M, Lefevre G. Pharmacokinetics of a novel transdermal rivastigmine patch for the treatment of Alzheimer’s disease: a review. Int J Clin Pract. 2009;63(5):799–805.
- Wokovich AM, Prodduturi S, Doub WH, et al. Transdermal drug delivery system (TDDS) adhesion as a critical safety, efficacy and quality attribute. Eur J Pharm Biopharm. 2006;64(1):1–8.
- Bhalekar MR. Solid lipid nanoparticles incorporated transdermal patch for improving the permeation of piroxicam. Asian J Pharm. 2016;10(1):45–51.
- Ekambaram P, Sathali AAH. Formulation and evaluation of solid lipid nanoparticles of ramipril. J Young Pharm. 2011;3(3):216–220.
- Onal A, Kepekçi SE, Oztunç A. Spectrophotometric methods for the determination of the antidepressant drug paroxetine hydrochloride in tablets. J AOAC Int. 2005;88(2):490–495.
- Abdellatif AA, El-Telbany DFA, Zayed G, et al. Hydrogel containing PEG-Coated fluconazole nanoparticles with enhanced solubility and antifungal activity. J Pharm Innov. 2018;14:1–11.
- Ding Y, Nielsen KA, Nielsen BP, et al. Lipid-drug-conjugate (LDC) solid lipid nanoparticles (SLN) for the delivery of nicotine to the oral cavity–optimization of nicotine loading efficiency. Eur J Pharm Biopharm. 2018;128:10–17.
- Mohamed RA, Abass HA, Attia MA, et al. Formulation and evaluation of metoclopramide solid lipid nanoparticles for rectal suppository. J Pharm Pharmacol. 2013;65(11):1607–1621.
- Idrees A, Rahman NU, Javaid Z, et al. In vitro evaluation of transdermal patches of flurbiprofen with ethyl cellulose. Acta Pol Pharm. 2014;71(2):287–295.
- Mutalik S, Udupa N. Glibenclamide transdermal patches: physicochemical, pharmacodynamic, and pharmacokinetic evaluations. J Pharm Sci. 2004;93(6):1577–1594.
- Rajabalaya R, Khanam J, Nanda A. Design of a matrix patch formulation for long-acting permeation of diclofenac potassium. Asian J Pharm Sci. 2008;3(1):30–39.
- Ayesha N. Development of tizanidine HCl transdermal patches: in-vitro and ex-vivo characterization. J Drug Deliv Therapeut. 2019;9(1-s):295–300.
- Lin Y-K, Huang Z-R, Zhuo R-Z, et al. Combination of calcipotriol and methotrexate in nanostructured lipid carriers for topical delivery. Int J Nanomed. 2010;5:117.
- Ubaidulla U, Reddy MV, Ruckmani K, et al. Transdermal therapeutic system of carvedilol: effect of hydrophilic and hydrophobic matrix on in vitro and in vivo characteristics. AAPS PharmSciTech. 2007;8(1):E13–E20.
- Kurakula M, Ahmed OAA, Fahmy UA, et al. Solid lipid nanoparticles for transdermal delivery of avanafil: optimization, formulation, in-vitro and ex-vivo studies. J Liposome Res. 2016;26(4):288–296.
- Gannu R, Vishnu V, Kishan Y, et al. Y. Development of nitrendipine transdermal patches: in vitro and ex vivo characterization. CDD. 2007;4(1):69–76.
- Bagchi A, Dey BK. Formulation, in-vitro evaluations and skin irritation study of losartan potassium transdermal patches. Iran J Pharm Sci. 2010; 6: 163–170.
- Draize JH. Methods for the study of irritation and toxicity of substances applied topically to the skin and mucous membranes. J Pharmacol Exp Ther. 1944;82:377–390.
- Villanueva-Martínez A, Hernández-Rizo L, Ganem-Rondero A. Evaluating two nanocarrier systems for the transdermal delivery of sodium alendronate. Int J Pharm. 2020;582:119312.
- Reithmeier H, Herrmann J, Göpferich A. Lipid microparticles as a parenteral controlled release device for peptides. J Control Release. 2001;73(2-3):339–350.
- Kumar PP, Gayatri P, Sunil R, et al. Atorvastatin loaded solid lipid nanoparticles: formulation, optimization, and in vitro characterization. IOSR J Pharm. 2012;2(5):23–32.
- Müller R, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy: rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001;47(1):3–19.
- Nasiri F, Faghfouri L, Hamidi M. Preparation, optimization and in-vitro characterization of α-Tocopherol-loaded solid lipid nanoparticles (SLNs). Drug Dev Ind Pharm. 2020;1:42-46.
- Asasutjarit R, Lorenzen S-I, Sirivichayakul S, et al. Effect of solid lipid nanoparticles formulation compositions on their size, zeta potential and potential for in vitro pHIS-HIV-Hugag transfection. Pharm Res. 2007;24(6):1098–1107.
- Shakeel F, Baboota S, Ahuja A, et al. Nanoemulsions as vehicles for transdermal delivery of aceclofenac. AAPS Pharmscitech. 2007;8(4):191.
- Venkateswarlu V, Manjunath K. Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J Control Release. 2004;95(3):627–638.
- Jain S, Jain S, Khare P, et al. Design and development of solid lipid nanoparticles for topical delivery of an anti-fungal agent. Drug Deliv. 2010;17(6):443–451.
- Cozar I, Szabó L, Mare D, et al. IR, Raman, SERS and DFT study of paroxetine. J Mol Struct. 2011;993(1-3):243–248.
- Weiseng N, Lee C, Cheng SF, et al. Biocompatible polyurethane scaffolds prepared from glycerol monostearate-derived polyester polyol. J Polym Environ. 2018;26: 2881–2900.
- Fu X, Weibo K, Zhang Y, et al. Novel solid-solid phase change materials with biodegradable trihydroxy surfactant for thermal energy storage. RSC Adv. 2015;5: 68881–68889.
- Bhalekar MR, Pokharkar V, Madgulkar A, et al. Preparation and evaluation of miconazole nitrate-loaded solid lipid nanoparticles for topical delivery. AAPS PharmSciTech. 2009;10(1):289–296.
- Baig MS, Ahad A, Aslam M, et al. Application of Box–Behnken design for preparation of levofloxacin-loaded stearic acid solid lipid nanoparticles for ocular delivery: optimization, in vitro release, ocular tolerance, and antibacterial activity. Int J Biol Macromol. 2016;85:258–270.
- Peng L-H, Wei W, Shan Y-H, et al. Sustained release of piroxicam from solid lipid nanoparticle as an effective anti-inflammatory therapeutics in vivo. Drug Dev Ind Pharm. 2017;43(1):55–66.
- Lan Y, Minnich AJ, Chen G, et al. Enhancement of thermoelectric figure‐of‐merit by a bulk nanostructuring approach. Adv Funct Mater. 2010;20(3):357–376.
- Sugi K, Itaya N, Katsura T, et al. Improved synthesis of paroxetine hydrochloride propan-2-ol solvate through one of metabolites in humans, and characterization of the solvate crystals. Chem Pharm Bull. 2000;48(4):529–536.
- Joshi SA, Jalalpure SS, Kempwade AA, et al. Fabrication and in-vivo evaluation of lipid nanocarriers based transdermal patch of colchicine. J Drug Delivery Sci Technol. 2017;41:444–453.
- Elnaggar YS, El-Massik MA, Abdallah OY. Fabrication, appraisal, and transdermal permeation of sildenafil citrate-loaded nanostructured lipid carriers versus solid lipid nanoparticles. Int J Nanomed. 2011;6:3195.