
ABSTRACT
Klebsiella pneumoniae is a notorious nosocomial pathogen causing a wide range of infections. The increasing trend of antimicrobial resistance obtained by the species immensely highly challenges the clinical treatment, representing a large threat to the global health care network. In particular, the recent convergence of multidrug resistance and hypervirulence in K. pneumoniae further worsens clinical outcomes, resulting in high mortality. Developments of new therapeutics become urgent, and immunotherapy based on antagonizing the anti-immune strategies of pathogens is a promising strategy, which requires the understanding of immune evasion mechanism in the context of the host–pathogen interactions. However, the underlying mechanisms employed by K. pneumoniae to counteract host immune responses, especially autophagy and cell death, have not been systematically reviewed and discussed yet. This review aims to summarize the tremendous progress that has been made to illuminate the landscape of cell signalling triggered by K. pneumoniae infection, especially in aspects of manipulating autophagy, cell death, and cytokine production.
Introduction
Over the past decades, K. pneumoniae has become one of the major causes of hospital- and community-acquired infections worldwide. In particular, the prevalence of multidrug-resistant (MDR) K. pneumoniae highly challenges antibiotic therapy, and infections caused by MDR K. pneumoniae are frequently associated with a high mortality [Citation1]. Of greater concern, a convergence of carbapenem resistance and hypervirulence has recently emerged in epidemic clones worldwide, e.g. sequence type (ST) 11 and 258 [Citation2,Citation3]. Such convergence immensely exacerbates clinical outcomes of infections caused by the “superbug” resulting in a higher mortality, thus representing an emerging threat to the public health network. Developments of novel therapeutic approaches therefore become urgent to tackle the problem.
Accumulating evidence shows that immunotherapy is one of the promising strategies for the treatment of “superbug” [Citation4]. Immunotherapy approaches based on passive immunization against Klebsiella spp. have been extensively studied. In particular, capsule polysaccharide (CPS)/lipopolysaccharide (LPS)-specific antibody and hyperimmune intravenous immunoglobulin (IVIG) from patients immunized with Klebsiella CPS/LPS have been tested in human clinical trials [Citation5,Citation6]. However, such strategies are largely limited by the highly diverse serotypes of Klebsiella spp.. Immunotherapy based on antagonizing the anti-immune strategies of pathogens could be an alternative solution, and a better understanding of the cross-regulation of the host and pathogen is a prerequisite for the development of host-directed therapeutics against pathogens. Current immunological concepts in bacteriology suggest that autophagy, cell death and cytokine production are critical host defense mechanism against invading bacteria [Citation4,Citation7,Citation8,Citation9,Citation10]. However, pathogens have correspondingly deployed devised immune evasion strategies, e.g. controlling cell death and limiting the emission of danger signals from dying cells to avoid elimination by the host. Despite K. pneumoniae is commonly considered as an extracellular pathogen, it can enter into macrophages through endocytosis dependent on host cytoskeleton, cell plasma membrane lipid rafts, and the activation of phosphoinositide 3-kinase (PI3K), and further persist intracellularly within a vacuolar compartment inside host cells in vivo [Citation11,Citation12]. Furthermore, K. pneumoniae can manipulate phagosome maturation, inhibit apoptosis, impair efferocytosis, and promote necrosis/necroptosis of host cells to survive in the host.
Understanding the molecular pathways responsible for manipulating cell signalling provides critical insights into the bacterial pathogenesis. In this study, we aim to systematically review the current knowledge to illuminate the landscape of host–pathogen interactions associated with K. pneumoniae, especially in aspects of autophagy, cell death, and cytokine production.
Virulence factors (VFs) of K. pneumoniae involved in the host–pathogen interactions
Currently, a number of VFs have been identified in K. pneumoniae, and a set of VFs associated with immune evasion have been well characterized, e.g. CPS, LPS, siderophores, and fimbriae. Besides, a few untypical VFs, including out membrane proteins (OMPs), porins, efflux pumps, iron transport systems, and genes involved in allantoin metabolism, also play an important role in the pathogenicity of K. pneumoniae [Citation13]. These VFs can influence the immune responses of host cells by various patterns, e.g. modulating cytokine secretions, and regulating autophagy and cell death, to escape the host killing. In this section, we will focus on those involved in circumventing early host responses for immune evasion during infection ().
Table 1. Key VFs of K. pneumoniae involved in the immune evasion.
Asymptomatic colonization is the first step for the infection development by K. pneumoniae [Citation13–15]. K. pneumoniae is a versatile colonizer which mainly colonizes in the upper airways and the intestine [Citation16,Citation17]. Colonization by K. pneumoniae to human carcinoma or intestinal cell lines at the early stage of infection is usually a prerequisite for the pathogenicity, which is mediated by adhesive structures, mainly including type 1 and 3 fimbriae, F-28 fimbriae, a nonfimbrial factor called CF29K, and a capsule-like material [Citation13,Citation18]. Type 1 and 3 fimbriae are the major adhesive structures of K. pneumoniae, and for CF29K and KPF-28, their structures and functions were unclear in animal model systems. It is found that genes encoding type 1 fimbriae of K. pneumoniae are expressed in the urinary tract but not in the gastrointestinal tract or lungs, indicating that type 1 fimbriae contributes to the urinary tract colonization of K. pneumoniae but without effect on the ability to colonize the intestine or to infect the lung [Citation19,Citation20]. Besides colonization, type 1 fimbriae triggers an opsonin-independent form of phagocytosis called lectinophagocytosis which leads to increased bacterial killing [Citation21]. Cooperative effect of type 1 fimbriae and LPS triggers oxygen-dependent apoptosis in human neutrophils. Type 3 fimbriae acts as a major contributor to the biofilm formation by K. pneumoniae, and mediates in vitro adhesion to epithelial cells and kidney and lung tissues [Citation22]. Additionally, type 3 fimbriae stimulate an oxidative response in neutrophils [Citation23] and mediate immunity to the infection in the murine model of respiratory diseases [Citation24].
CPS was also involved in K. pneumoniae colonization. In the upper airway, the adult microbiota primes upper airway defenses through interleukin (IL) 17A, however, encapsulation allows K. pneumoniae to overcome this barrier to establish colonization [Citation25]. In adult mice, the encapsulated K. pneumoniae colonizes the upper airways at higher levels than an isogenic, unencapsulated mutant [Citation26], and the ability to withstand microbiota-mediated defences is restored by complementation of the unencapsulated mutant [Citation25], indicating the importance of CPS in the successful colonization by K. pneumoniae. Additionally, CPS is the first component that contacts with the immune cells during infection [Citation27]. CPS can inhibit phagocytosis induced by immune cells [Citation18,Citation28], hinder the bactericidal action of antimicrobial peptides [Citation18,Citation29,Citation30], and prevent the early immune response [Citation31–34]. In an elegant study using a mouse model, it is found that in the early stages of infection, the levels of tumour necrosis factor (TNF)-α and IL 6 in broncho-alveolar lavage fluid (BALF) of mice infected with an non-capsulate mutant were significantly higher than those infected with a capsulate strain [Citation31]. The results suggest that capsule allows K. pneumonia to grow in vivo by suppressing the host immunological responses. Besides, CPS can prevent K. pneumoniae from the adherence to the membrane of host cells, and reduce bacterial internalization [Citation35]. Additionally, CPS plays an important role in the intracellular survival of K. pneumoniae [Citation12,Citation36,Citation37]. Compared with the wild-type strain, an isogenic wzy knockout strain was significantly more susceptible to the killing by human heparinized blood, serum, and neutrophils [Citation37]. Of note, some capsular serotypes are highly associated with hypervirulence, e.g. K1 and K2. Such hypervirulence variants are much more resistant than classic K. pneumoniae to in vitro killing by serum or to phagocytosis by neutrophils and macrophages, and induce high colonization levels. The enhanced virulence could be resulted from the specific structures of CPS. For instance, the K1/K2 capsules lack Man-α2-Man structure that can be recognized by macrophages and SP-A protein [Citation21], while they contain sialic acid involved in the resistance to phagocytosis [Citation38]. Additionally, K1/K2 capsules can induce less respiratory burst in polymorphonuclear leukocytes (PMNLs) [Citation39,Citation40].
LPS is an important VF for K. pneumoniae to resist eliminations by the host. It prevents complement C1q from binding with bacteria, thus inhibits the activation of complement pathway [Citation18,Citation27]. LPS also protects K. pneumoniae against C3 by binding to C3b, resulting in the prevention of interactions between C3b and cell membrane, and abrogating pore formation [Citation41,Citation42]. Of note, a recent study revealed that capsule and LPS together can promote K. pneumoniae infection by decreasing the levels of small ubiquitin-like modifier (SUMO)-conjugated proteins which leading to the limited activation of inflammatory responses and increased intracellular survival in macrophages during infection [Citation36].
A number of studies have shown that K. pneumoniae is able to persist in the host cells in vivo [Citation43–46]. During this process, K. pneumoniae utilizes host nutrients to replicate, and iron is a limited resource for K. pneumoniae to thrive in the host. To cope with the situation, K. pneumoniae secretes siderophores to “steal” iron from host iron-chelating proteins or to scavenge it from the environment [Citation47,Citation48]. Siderophores expressed in K. pneumoniae mainly include enterobactin, yersiniabactin, salmochelin, and aerobactin [Citation13]. The productions of more than one type of siderophore by K. pneumoniae may be as a means of optimizing successful colonization of different tissues and/or avoiding neutralization by the host [Citation47,Citation49]. And the diverse siderophores highly facilitate K. pneumoniae adaptations to the complex environment in vivo.
Notably, some membrane proteins (e.g. OMPs, porins and efflux pumps) as untypical VFs play a crucial role in modulating host cell immune responds. For example, OmpA is one of the major outer membrane proteins of Gram-negative bacteria, and it has been shown to be implicated in preventing the activation of toll-like receptors (TLRs) to inhibit inflammatory responses and responsible for resistance to antimicrobial peptides [Citation50–54]. Besides, OmpA was found to induce IL-1β and IL-18 production in host cells, resulting in caspase-1 activation, which simultaneously stimulated pyroptosis, thus leading to the death of the host cells [Citation55]. In K. pneumoniae, the porin OmpK36, a homology of OmpC, prevents phagocytosis of neutrophils and alveolar macrophages [Citation56,Citation57], but it is not required for the serum resistance [Citation54]. AcrAB is an efflux pump mainly contributing to the resistance against antibiotics [Citation58,Citation59]. Padilla et al. found that the AcrAB mutant is more susceptible than the wild type to cationic antimicrobial peptides (CAMPs) presenting in human bronchoalveolar lavage fluid (BALF) and to human antimicrobial peptides [Citation60]. Therefore, AcrAB is important for K. pneumoniae to cope with the killings of antimicrobial peptides produced by hosts. Additionally, the secreted proteins maybe involved in the pathogenicity of K. pneumoniae as well [Citation61–66]. A study shows that ten of fourteen (71%) sera collected from patients with K. pneumoniae pyogenic liver abscess (PLA) were immunoreactive with the recombinant secreted protein KP1_p307, and it is further confirmed that antibodies against KP1_p307 protein were elicited after infection. Hence this secreted protein could be used as an antigen for early serodiagnosis of K. pneumoniae PLA [Citation66]. However, more functional study of KP1_p307 should be performed to understand its contribution to the pathogenicity of K. pneumoniae.
Albeit the biological function of most VFs discussed here have been well characterized, the role of VFs in the manipulation of cell death and autophagy is largely unknown. In the next sections, we will discuss in detail how K. pneumoniae counters with the host immune system in aspects of impairing autophagy, triggerring apoptosis, promoting necroptosis, interfering efferocytosis, and inducing pyroptosis.
K. pneumoniae manipulates autophagy-related cell signalling to survive in host cells
Autophagy is a process that controls the quality and quantity of intracellular biomass in eukaryotic cells, whereby cytoplasmic components are enveloped and sequestered by membranous structures that subsequently fuse to lysosomes for degradation. In the aspect of pathogen cleaning, autophagy can directly eliminate the intracellular microbes and/or their products via a process termed xenophagy, in which the selective capture and degradation of intracellular microbes are completed by lysosomes [Citation67]. Upon autophagy activation, serine/threonine kinases ULK1 and Beclin-1 combine with autophagy-related protein (Atg) 14 and type III phosphatidylinositol 3-kinase Vps34 together with other Atgs to promote the formation of a cup-shaped isolation membrane to engulf the cargo [Citation68]. Subsequently, the cup-shaped isolation membrane elongates into a double membrane vesicle, named autophagosome. The autophagosome further fuses with lysosomes to form an autolysosome in which the engulfed cargo will be degraded. Additionally, other cellular processes, e.g. neutrophil extracellular traps (NETs) formation, phagocytosis and inflammation activation, are also involved in the autophagy-mediated elimination of K. pneumoniae. The underlying mechanism is discussed here.
K. pneumoniae can activate autophagy during infection, but the underlying mechanism remains poorly understood
Autophagy serving as an innate immune response to defend against pathogens invading inside the host cells has been extensively studied on a set of intracellular microorganisms [Citation69]. Despite K. pneumoniae is an extracellular pathogen, it can induce autophagy of alveolar type II epithelial (A549) cells as shown in a recent study [Citation70], and autophagy constitutes an important mechanism to control intracellular growth of K. pneumoniae in macrophages. Enhancement of K. pneumoniae intracellular growth was observed when the autophagy process of macrophages was inhibited by bafilomycin A1, suggesting that autophagy is important for the physiological control of intracellular K. pneumoniae [Citation71]. In a model of K. pneumoniae-induced sepsis, autophagy was enhanced by the activation of Beclin-1 through forced expression of active mutant Becn1F121A or treatment with TB-peptide, and adverse outcomes of pneumonia-induced sepsis were significantly alleviated [Citation72]. These data underpin the role of autophagy in host defense against K. pneumoniae infection. It has been known that mTOR is an important inhibitor of autophagy [Citation73], and PI3K-AKT signalling is a major pathway through mTOR to regulate cell survival and proliferation [Citation74]. A recent study identifies that autophagy induced by K. pneumoniae infection is dependent on PI3K-AKT-mTOR pathway, and inhibition of PI3K-AKT-mTOR signalling pathway increased the susceptibility of the host to K. pneumoniae infection [Citation75].
Although K. pneumoniae is able to initiate autophagy, the underlying mechanism has been poorly defined. Generally, two pathways are individually involved in the activation of autophagy, i.e. the cell surface recognition and cytosolic sensing of PAMPs and DAMPs [Citation76]. In the pathway of cell surface recognition, pathogens bind to surface receptors (e.g. TLRs), and trigger LC3 conjugation directly onto the phagosomal membrane upon phagocytosis, followed by the initiation of autophagy [Citation76]. For the cytosolic sensing pathway, the representative is M. tuberculosis-induced autophagy. M. tuberculosis through its ESX-1 secretion system damages the phagosomal membrane, and cytosolic sensor c-GAS recognizes its DNA, resulting in the ubiquitination of the bacterium to initiate autophagy [Citation77]. It is unclear whether K. pneumoniae could damage the phagosomal membrane. Limited evidence shows that TLR4 can recognize LPS of K. pneumoniae, and is involved in autophagy activation during K. pneumoniae infection [Citation78]. OmpA is also associated with TLR2 signalling activation [Citation4]. We thus assume that K. pneumoniae may initiate autophagy through cell surface recognition of PAMPs and DAMPs. More studies are needed to further elucidate the pathway of K. pneumoniae-induced autophagy.
K. pneumoniae elimination by autophagy is associated with NETs formation
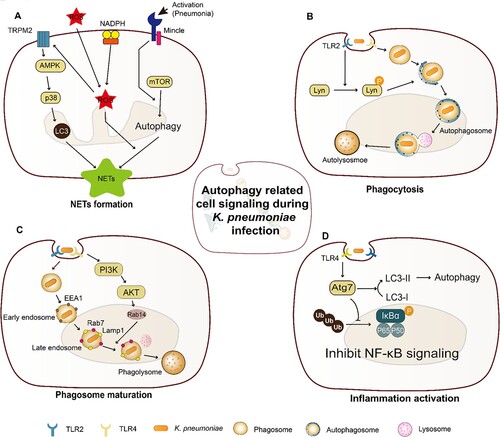
Neutrophils have been regarded as first line of defense to capture and destroy extracellular pathogens, and their activity is highly dependent on the formation of NETs. Recent studies suggest that TRPM2- and Mincle-mediated NETs formation is associated with autophagy and involved in the defence against K. pneumoniae infection ((A)). During K. pneumoniae infection, autophagy is required for transient receptor potential melastatin 2 (TRPM2)-AMPK-p38- and Mincle-mediated NETs formation in a reactive oxygen species (ROS)-dependent and -independent manner, respectively [Citation79–81]. Trpm2−/− neutrophils exhibited defective NET formation, impaired autophagy activation, and significantly reduced phosphorylation of AMPKα1 and p38 MAPK, and inhibition of AMPKα1 and p38 MAPK signalling additionally impaired H2O2-induced autophagy and NETs formation. Consistenly, Trpm2−/− mice displayed more susceptible to pneumoseptic infection with K. pneumoniae [Citation80]. These results indicate that the involvement of autophagy in TRPM2-mediated NETs formation via AMPK-p38 signalling pathway is an important antimicrobial mechanism for the protection against K. pneumoniae. Likewise, significantly reduced phagocytosis and NETs formation were observed in Mincle−/− neutrophils, resulting in impaired autophagy activation, and attenuated neutrophil-mediated bacterial uptake and clearance. The deficiency of NETs formation can be rescued by the activation of autophagy in Mincle-/- neutrophils, indicating that NETs formation is regulated by Mincle via the modulation of autophagy [Citation79,Citation82]. Further studies are needed to understand the molecular signalling pathway connecting autophagy and NETs formation.
Figure 1. Cell signalling associated with K. pneumoniae induced autophagy. (A) Autophagy is required for TRPM2-AMPK-p38- and Mincle-mediated NETs formation in a ROS-dependent and an independent manner, respectively. In the ROS-dependent pathway, TRPM2 can sense ROS through AMPK-p38-autophagy to promote NETs formation during K. pneumoniae infection; In a ROS-independent manner, Mincle-mediated activation of autophagy, occurring downstream of mTOR, is required for NETs formation in response to pneumonic infection, and this autophagy activation precedes ROS effect on NETs formation; (B) The activated TLR2 receptor through Lyn promotes the fusion of autophagosome and lysosome, which enhances autophagy; (C) K. pneumoniae enters macrophage through endocytosis, which needs PI3K activation. The activated PI3K phosphorylate AKT regulates the recruitment of Rab14 to the phagosome, and manipulates phagosome maturation; (D) Atg7 is responsible for the elongation of autophagosome membrane. During K. pneumoniae infection, Atg7 can interact with phosphorylated IκBα to inhibit IκBα ubiquitination and reduce the activation of NF-κB signalling pathway and inflammation activation.
K. pneumoniae manipulates phagosome maturation to survive in the host cells
As an important immune response, the role and machinery of bacteria phagocytosis-induced autophagy have been identified in multiple studies ((B)). Autophagy initiated by bacteria infection can regulate phagocytosis to facilitate the host to eliminate pathogens, and this process is co-modulated by Lyn, TLR2, and Rab [Citation76,Citation83]. Once autophagy is blocked, phagocytosis and subsequent bacterial clearance by alveolar macrophages are reduced [Citation76,Citation83]. For some intracellular pathogens, e.g. M. bovis and M. tuberculosis, autophagy can regulate phagocytosis indirectly by altering the surface expression of phagocytic receptors [Citation84]. Although it is unclear whether K. pneumoniae possesses a similar mechanism, a previous study reported that OmpA of K. pneumoniae can engage TLR2 to activate NF-κB signalling [Citation50]. Further work should be conducted to understand whether K. pneumoniae could through OmpA regulate TLR2 activation and modulate Lyn dependent autophagy.
In general, the host can through autophagosome or phagosome to eliminate intracellular pathogens after phagocytosis being induced. A recent study has shown that K. pneumoniae can avoid killing by regulating phagosome maturation to persist within phagosome compartment inside host cells in vivo [Citation12]. Although K. pneumoniae-containing phagosomes are acidic, they are negative for cathepsin D, a marker for the lysosomal soluble content, indicating that the fusion of the phagosome with lysosomes was prevented [Citation12]. The perturbed maturation of the K. pneumoniae-containing phagosomes is further found to be associated with the recruitment of Rab14 through PI3K-AKT signalling pathway [Citation12]. These results demonstrate that K. pneumoniae perturbs the fusion of the K. pneumoniae-containing phagosomes with the hydrolases-rich lysosomal compartment by targeting a PI3K-AKT-Rab14 pathway, and the efficient clearance of bacteria is subsequently disturbed in the intracellular niche of macrophages ((C)).
K. pneumoniae manipulates autophagy-related Atg7 to decrease inflammation activation
The initiation of autophagy can further activate inflammation to cope with bacterial infection. As a critical E1-like ubiquitin, Atg7 is responsible for the elongation of autophagosomes membrane during autophagy [Citation85]. The expression level of Atg7 significantly increased in murine alveolar macrophages upon K. pneumoniae infection, and Atg7-knockout mice exhibited decreased survival rates, attenuated bacterial clearance, and intensified lung injury [Citation86,Citation87]. Simultaneously, excessive proinflammatory cytokines and superoxide were further induced in the lung of Atg7-knockout mice [Citation87]. Loss of Atg7 in alveolar macrophages resulted in the upregulation of IL-1β and pyroptosis, and Atg7−/− mice manifested more intense proinflammatory responses and exhibited activated NF-κB and p38 MAPK following K. pneumoniae infection, suggesting that NF-κB activity is suppressed by Atg7 [Citation78,Citation86,Citation87]. The suppression mechanism has been further studied, in which Atg7 binding to p-IκBα negatively regulates the ubiquitylation of p-IκBα and the activation of NF-κB signalling pathway [Citation78] ((D)).
In summary, autophagy plays a significant role in host defense against K. pneumoniae infection () (). However, in the context of bacterial pathogenesis, key virulence factors (VFs) and receptors involved in K. pneumoniae-induced autophagy and the inhibition of phagosome maturation are largely unknown.
Table 2. Strategies and mechanism involved in counteracting autophagy and cell death trigged by K. pneumoniae.
K. pneumoniae induces apoptosis and further modulates apoptosis and efferocytosis of apoptotic cells to survive and emit in the host
Apoptosis is vital in counteracting microbe infection by restricting intracellular bacteria replication and delivering antigens to immune cells. Apoptosis triggered by intracellular pathogens has been intensively studied, while that by extracellular pathogens, like K. pneumoniae, is poorly characterized. Limited studies showed that K. pneumoniae enables to promote apoptosis of macrophages, platelet, epithelial cells, and liver cancer cells [Citation12,Citation88–90]. Cano and co-workers found that K. pneumoniae triggered programmed cell death in macrophages, and apoptosis features appeared at 6 h post infection [Citation12]. To our knowledge, this is the orphan study of K. pneumoniae-induced macrophage apoptosis during the preparation of this work. Since only phenotypical characterizations were reported in that study, the underlying mechanism remains unclear. Compared with macrophage apoptosis, the molecular mechanism of apoptosis induced by K. pneumoniae in liver hepatocellular cells HepG2 has been studied relatively clearly. Apoptosis of HepG2 cells is induced at the early stage of K. pneumoniae infection, and the expression of Bax is increased and that of Bcl-xL is decreased, resulting in mitochondrial release of Cytochrome c [Citation88]. The released Cytochrome c interacts with Apaf-1 to activate caspase 9, leading to apoptosis in the early hours of infection [Citation88]. These results indicate that K. pneumoniae-triggered apoptosis of HepG2 cells is dependent on the intrinsic pathway, also known as BCL-2 family-mediated apoptosis pathway.
The factors of K. pneumoniae invovled in triggering apoptosis are largely unknown. A recent study identified a relationship between hypermucoviscosity and apoptosis. Although both hypermucoviscous K. pneumoniae (hmKP) and non-hmKP clinical strains can induce apoptosis of platelets and cause decreased level of platelet in vivo, the highest apoptosis proportions (45%) were observed in the infection caused by the hmKP strain [Citation89]. It suggests that hypermucoviscosity could perhaps enhance apoptosis during infection. However, a discrepancy was found in another study that no significant differences were detected for apoptosis of bovine mammary epithelial cells triggered by hmKP and non-hmKP isolates at 9 h post infection, and the proportion of apoptosis cells was very low (ca. 6%) [Citation90]. Of note, K. pneumoniae strains used in this study were isolated from bovine clinical mastitis, and the hypermucoviscous phenotype was negatively correlated to the pathogenicity. We thus suppose that the discrepancy might be due to the diverse pathogenicity of strains used in the two studies.
K. pneumoniae has evolved capacities to inhibit host cell apoptosis signalling to survive intracelluarly. Delayed apoptosis and impaired efferocytosis are supposed to be two major strategies for this bacteria to counteract apoptosis, and the details are discussed below.
K. pneumoniae delays apoptosis to survive in the host
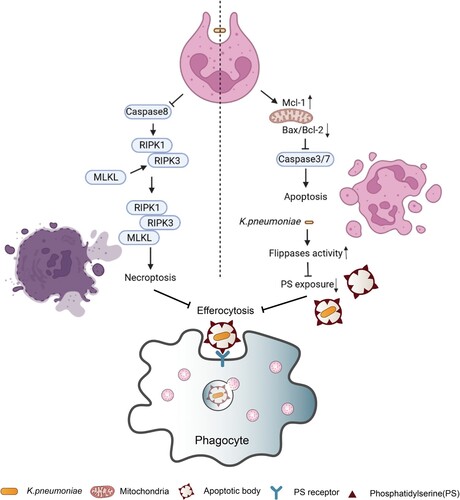
A study has shown that the interaction of K. pneumoniae with neutrophils inhibits the surface exposure of PS induced by anti-Fas antibody, an agonist known to cause neutrophil apoptosis, suggesting that K. pneumoniae can delay neutrophil apoptosis to escape the killing of neutrophils [Citation91]. Capsule, an important virulence factor of K. pneumoniae, was identified as a factor involved in delaying neutrophil apoptosis. Compared with acapsular mutants, the strain with K1-type capsule significantly delayed the apoptosis of infected neutrophils [Citation92]. Additionally, the K1-type capsule can modulate the anti-apoptotic effects by downregulating the ratio of Bax to Bcl-2 and by upregulating the expression of Mcl-1 (). This leads to the delay of caspase-3 activation in the neutrophils, accompanied by the induction of anti-apoptotic cytokine IL-8 [Citation92].
Figure 2. Cell death triggered by K. pneumoniae in neutrophils. K. pneumoniae modulates the anti-apoptotic effects by downregulating the ratio of Bax to Bcl-2 and upregulating the expression of Mcl-1, resulting in the delay of Caspase 3/7 activation and the inhibition of apoptosis. K. pneumoniae also can increase the activity of flippase to reduce PS exposure, which is the signal of “eat me” for efferocytosis, followed by decreasing the efferocytic uptake of neutrophils by macrophage. In parallel, K. pneumoniae inhibits caspase 8, which promotes the formation of necroptosome complex RIPK1/RIPK3/MLKL to induce necroptosis. Necroptosis emits K. pneumoniae to the external environment, and decreases efferocytosis. (Created with BioRender. com).
K. pneumoniae impairs efferocytosis to emit in the host
Apoptosis is not the last step to eliminate intracellular pathogens, and efferocytosis is responsible for the control of bacterial transmissions after apoptosis. If apoptotic cells are not swiftly eliminated by efferocytosis, secondary necrosis will be initiated mediated by gasdermin E- or DFNA5 (deafness, autosomal dominant 5), causing bacterium spread, autoimmunity and persistent inflammation [Citation93]. Therefore, the capacity of inhibiting efferocytosis is crucial for pathogens to develop severe infection. A recent study indicated that K. pneumoniae can impair the efferocytic uptake of neutrophils in vitro and in vivo with the assistance of macrophages [Citation94]. Impaired efferocytosis is linked to reduced externalization of PS via the modulation of flippase activity, and obstructs the recognition of apoptotic cells by macrophages to promote K. pneumoniae dissemination (). Efferocytosis thereby is inhibited resulting in the promotion of K. pneumoniae dissemination. Together, these results evidence that K. pneumoniae is able to impair efferocytosis by inhibiting apoptosis of neutrophils to establish infection () ().
K. pneumoniae promotes necrosis/necroptosis to escape the killing of hosts
Necrosis is a form of lytic cell death employed by some bacteria to disseminate in vivo and to evade host immune responses. Compared with necrosis, necroptosis is a programmed cell death, which also promotes pathogens’ transition. Necrosis occupies an important role in the pathogenicity of K. pneumoniae. Apoptotic HepG2 cells induced by K. pneumoniae infection further progress to secondary necrosis and primary necrosis, and PARP is inactivated by caspase 7 at this stage [Citation88]. As the infection persists over time, Endo G, AIF, and DFF40 are translocated from mitochondria to the nucleus, causing DNA fragmentation. The DNA damages lead to PARP activation and ATP depletion, and calpain 2 is subsequently upregulated resulting in the digestion of cellular components, followed by the induction of necrosis at the final, which promotes K. pneumoniae dissemination [Citation88] (). Alternatively, K. pneumoniae activates cell necroptosis through the RIPK1/RIPK3/MLKL cascade to escape the host killings [Citation95], and can further interfere apoptosis activation to inhibit efferocytosis. Inhibition of necroptosis by RIPK1 and RIPK3 restores the efferocytic uptake of K. pneumoniae infected neutrophils by macrophages in vitro, suggesting that K. pneumoniae induces necroptosis of neutrophils [Citation94]. Therefore, inhibition of necroptosis is able to rescue the efferocytic uptake for K. pneumoniae-infected neutrophils (). Compared with WT, Ripk3–/– mice promoted bacterial clearance from the BALF and the lung at 96 h post infection [Citation95]. These data suggest that necroptosis facilitates K. pneumoniae survival in the host by reducing the efferocytic uptake of infected neutrophils via macrophages (). In the context of therapy, inhibitions of necrosis/necroptosis could be an assistant treatment for K. pneumoniae infection.
K. pneumoniae is counteracted by pyroptosis/pyronecrosis during infection
Pyroptosis is a programmed cell death that depends on the inflammatory caspases, including caspase-1, murine caspase-11, and human caspase-4/5 [Citation96]. The most important character of pyroptosis is the maturation and secretion of pro-inflammatory cytokines (IL-1β, IL-18, and IL-33) promoted by active caspase 1 [Citation97]. Recently, pyronecrosis is proposed as a new form of pathogen-induced cell death similar to pyroptosis [Citation98]. The common feature of pyroptosis and pyronecrosis is the release of pro-inflammatory cytokines, and both can be stimulated by similar factors and produce similar outcomes. However, compared with pyroptosis, pyronecrosis needs cathepsin B instead of caspases’ activities [Citation99]. Abundant evidence supports the significance of pyroptosis/pyronecrosis in the anti-infection by hosts that pyroptosis/pyronecrosis of infected cells removes the pathogens’ protective intracellular niche and promotes neutrophil-mediated killing.
K. pneumoniae induced macrophage pyronecrosis is NLRP3 and ASC dependent
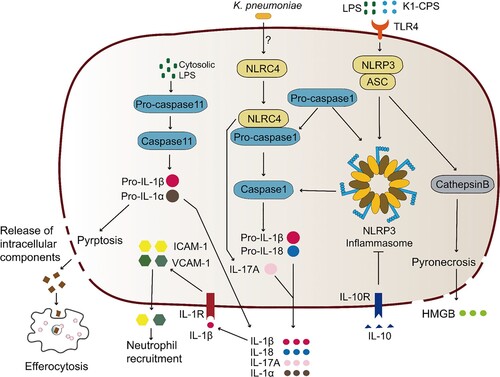
A few studies have suggested that K. pneumoniae can induce pyroptosis/pyronecrosis, and the underlying mechanism has been explored. In a K. pneumoniae-infected human macrophage cell line, NLRP3 or ASC knockdown abrogated IL-1β release and cell death, and further attenuated the release of high-mobility group box 1 protein (HMGB1), suggesting that K. pneumoniae induced macrophages pyronecrosis is dependent on NLRP3 and ASC characterized by IL-1β release, cell death, and HMGB1 release [Citation44]. As a result, deficiency of Nlrp3 and Asc significantly increased the mortality of mice infected by K. pneumoniae [Citation44], validating the protective function of NLRP3 and ASC in the host against K. pneumoniae ().
The factors of K. pneumoniae responsible for triggering pyroptosis/pyronecrosis are largely unknown. Recently, K1-CPS was identified as one of such factors in an orphan study. Caspase-1 activation and IL-1β secretion in the presence of ATP were induced by K1-CPS using mouse J774A.1 macrophages, and the reduced expression of NLRP3 and ASC by shRNA plasmids attenuated caspase-1 activation and IL-1β secretion [Citation100], which is consistent with the previous study aforementioned [Citation44]. K1-CPS-induced NLRP3 expression was significantly lower in TLR4 knockdown cells than in control, indicating that TLR4 is involved in the NLRP3 inflammasome activation induced by K1-CPS [Citation100]. K1-CPS induced NLRP3 inflammasome activation is also associated with ROS generation, mitogen-activated protein kinase phosphorylation, and NF-κB activation [Citation100]. These findings indicate that K1-CPS is a factor that induces IL-1β secretion through the NLRP3 inflammasome ().
Figure 3. Pyrotosis triggered by K. pneumoniae infection. TLR4 receptor can recognize K. pneumoniae LPS and K1-CPS, and activate NLRP3 inflammasome and caspase-11. Inflammasome formation allows caspase-1 activation, resulting in mature IL-1β and IL-18 release by cleavage of pro-IL-1β and pro-IL-18, respectively. Subsequently, pore formation and release of intracellular soluble components, such as LDH and HMGB, is culminated by the induction of pyroptosis/pyronecrosis. IL-1β together with other mediators recruit new cells to the infection site, promoting efferocytosis of pyroptotic-infected cells and bacterial clearance. But this progress can be inhibited by IL-10. Simultaneously, K. pneumoniae can activate NLRC4 to promote caspase1 activation, and further increase the release of IL-1β and IL-18. NLRC4 is also associated with IL-17 releasing, which was independent with caspase1. The released IL-1β through the interaction with its receptor IL-1R promotes the secretion of ICAM-1 and VCAM-1, and further promotes neutrophil recruitment.
K. pneumoniae infection is inhibited by neutrophil recruitment through NLRC4and caspase11 activation
A set of pyroptosis-related proteins is additionally involved in the protection against K. pneumoniae infection, such as NLRC4 and caspase11. Cleavage of caspase-1, IL-1b, and IL-18 were reduced in NLRC4-deficient mice, suggesting that the activation of caspase-1 and cleavage of IL-1b and IL-18 are NLRC4 dependent [Citation101] (). NLRC4-/- mice further exhibited higher lung and spleen bacterium burden, suggestive of a defect of bacterial clearance and dissemination in the NLRC4-/- mice. This well explains the decreased survival of NLRC4-/- mice caused by K. pneumoniae infection [Citation101]. Notably, NLRC4, did not induce pyroptosis in pulmonary macrophages and neutrophils though it promoted IL-1b and IL-18 production, and the main function of NLRC4 signalling coping with K. pneumoniae infection is neutrophil recruitment and innate immunity [Citation101]. Additionally, Caspase11 was activated by K. pneumoniae in BMDMs and mice, and caspase-11 deficiency blocked the secretion of IL-1α and IL-1β in BMDMs, indicating that caspase-11 mediates K. pneumoniae induced BMDM pyroptosis [Citation102] (). Caspase-11 deficiency also impaired neutrophil recruitment and bacterial clearance in the early stage of pulmonary infection caused by K. pneumoniae [Citation102], suggesting that caspase-11 is involved in defending against K. pneumoniae infection by recruiting neutrophils in the early stage of infection [Citation102].
K. pneumoniae clearance by pyroptosis needs efferocytic uptake of the pyroptotic cells
Similar to apoptotic cells, pyroptotic cells also launch “eat me” signals to promote efferocytosis by phagocytes. However, if the infected pyroptotic cells cannot be cleared in time by hosts, it can promote the spread of pathogens. Indeed, Codo et al. showed that the survival of K. pneumoniae in macrophages increased when the infected pyroptotic cells had not been engulfed through efferocytosis [Citation103]. Although pyroptotic cells were engulfed efficiently after being co-cultured with freshly collected macrophages, this process could be inhibited by IL-10, followed by the further inhibition of inflammasome activation, pyroptosis, and efferocytosis of infected cells [Citation103] (). This study demonstrates that the assistance from efferocytic uptake of the pyroptotic cells is needed for the clearance of K. pneumoniae by pyroptosis (). Additionally, K. pneumoniae might employ pyroptosis to harness the host immune response to promote its infection [Citation103]. We therefore suppose that promoting pyroptosis induction and/or enhancing the efferocytosis of pyroptotic cells by phagocytes may be feasible to improve the treatment efficacy for K. pneumoniae infection.
K. pneumoniae induced cytokine network during infection
Cytokines, secreted by immune and non-immune cells, are cell-to-cell signalling small proteins (∼6–70 kDa). Cytokines make up an exceptionally large and diverse group of pro- and anti-inflammatory factors, acting as regulators of host responses to infection, immune responses, inflammation, and trauma [Citation104]. The release of pro-inflammatory cytokines leads to the activation of immune cells and to the further release of other cytokines. Inflammation may bring beneficial effects to hosts in terms of pathogen clearance and phagocytosis of apoptotic cells, however, uncontrolled inflammation could cause adverse outcomes resulting in a variety of chronic inflammatory diseases [Citation104]. To maintain the balance, anti-inflammatory factors can neutralize excessive inflammation induced by pro-inflammatory cytokines. Simultaneous release of pro- and anti-inflammatory cytokines is suggested to be mandatory in any immune responses, and inflammatory cytokines are crucial for hosts to defend against infections. Currently, a set of cytokines has been reported associated with K. pneumoniae infection ().
Table 3. The function of cytokines coping with K. pneumoniae infection.
K. pneumoniae promotes the production of chemokines responsible for neutrophil migration and subverts the activation of CXCL8 secretion to survive in the host
Chemokines are an extended family of small-size chemoattractant cytokines that facilitate leukocyte migration and positioning via binding to receptors located at the target cell surface. Multiple chemokines have been identified involved in counteracting K. pneumoniae infection, like CXCL15, MIP-1α/CCL3, CXCL1 (keratinocyte cell-derived chemokine, KC), CXCL8 (IL-8), and CXCL5. CXCL15, a novel CXC chemokine, mediates neutrophil migration from the lung parenchyma into the airspace [Citation105], and MIP-1α/CCL3, a member of the CC chemokine family, is required for the phagocytic activity of alveolar macrophages during K. pneumoniae infection [Citation106]. Mice lacking CXCL15 (lungkine) or MIP-1α/CCL3 was susceptible to K. pneumoniae infection [Citation105,Citation106]. Transient intrapulmonary expression of MIP-1α/CCL3 or CXCL1 showed a striking improvement in survivals associated with recruitment of neutrophils [Citation107–109]. Hence, CXCL15, MIP-1α/CCL3 and CXCL1 are necessary for K. pneumoniae elimination by hosts.
CXCL8 is mainly secreted by leukocytes and endothelial cells dependent on the activation of NF-κB signalling pathway under special conditions, such as exposure to IL-1 or TNF-α, bacterial or viral products and cellular stress[Citation110]. Similar to the function of CXCL15 and MIP-1α/CCL3, CXCL8 recruits neutrophils and other immune cells to the sites of infection to cope with K. pneumoniae [Citation111]. Correspondingly, the pathogen has developed multiple strategies to subvert the activation of CXCL8 secretion (). K. pneumoniae attenuates pro-inflammatory mediators-induced CXCL8 secretion by upregulating the expression of CYLD and MKP-1 in alveolar epithelial cells [Citation34]. Alternatively, through Rho GTPase (RacI)–NOD1–CYLD and MKP-1 pathways, K. pneumoniae regulates NF-κB and mitogen-activated protein kinases (MAPKs) signalling activation to attenuate IL-1β-induced CXCL8 secretion [Citation34]. A mechanistic study further showed that the activation of NF-κB is attenuated by K.pneumoniae via activating an EGF receptor (EGFR)-phosphatidylinositol 3-OH kinase (PI3K)–AKT–PAK4–ERK–GSK3β signalling pathway, and CYLD is identified as the downstream effector of the signalling pathway [Citation112]. The activation of EGFR is stimulated by CPS dependent on a TLR4–MyD88–c-SRC pathway [Citation112]. However, the factors responsible for RacI activation remain unknown.
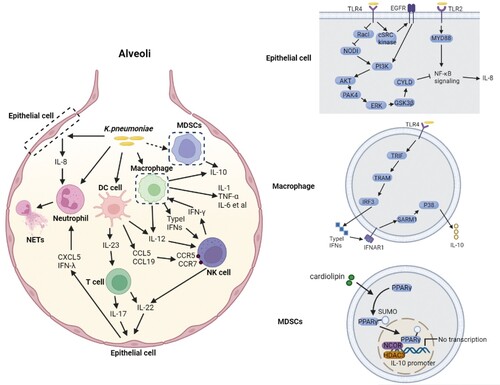
Figure 4. Complex network of cytokines coping with K. pneumoniae infection. K. pneumoniae can directly interact with macrophages, neutrophils, DC cells and epithelial cells in alveoli (left panel). The macrophage is the major immune cell in alveoli, and can produce type I IFNs upon infection with K. pneumoniae, resulting in NK cell migration and IFN-γ production. IFN-γ further activates the macrophage anti-microbial armament; both macrophages and DC cells can produce IL-12, which is required for IFN-γ production. IFN-γ production can be additionally promoted by K. pneumoniae matured DC cells through the interaction of CCL5-CCR5 receptor and CCL19-CCR7 receptor on NK cells. K. pneumoniae-infected DC cells secrete IL-23 which further induces IL-17 production by T cells to promote bacterial clearance. IL-17 promotes CXCL5 production through IL-17R on the epithelial cells to induce the recruitment of neutrophils. The recruitment of neutrophils can also be promoted by IL-8 and IFN-λ secreted by K. pneumoniae infected epithelial cells. Neutrophils are involved in the elimination of K. pneumoniae through NETs formation. IL-22 interacts with IFN-λ to help maintain the integrity of epithelial barrier to inhibit neutrophil transmigration and microbial invasion. The MDSCs produced IL-10 also has a crucial role in K. pneumoniae clearance. The synergy of these cytokines constructs the complex network of host defense against K. pneumoniae. The detailed signalling pathways of IL-8 and IL-10 production upon K. pneumoniae infection are shown on the right panel. (Created with BioRender. com).
K. pneumoniae induces the pro-inflammatory cytokines production which mediates K. pneumoniae elimination
The synergy of multiple pro-inflammatory cytokines constructs the key network of host resistance to K. pneumoniae, mainly including IL-1β, IL-6, TNF-α, IFN-γ, IL-12, IL-17, and IL-23. As the most well-known pro-inflammatory cytokines, IL-1β, IL-6, and TNF-α are critical for hosts to cope with infection caused by various pathogens. Indeed, the expression of IL-1β, IL-6, and TNF-α is upregulated during K. pneumoniae infection [Citation44,Citation100,Citation113–115]. TNF-α receptors (TNFR1) deficiency and neutralization of TNF-α consistently caused an increased mortality resulting from impairing lung antibacterial host defense and/or dysregulating production of cytokines detected in the mouse model [Citation113,Citation114]. CPS is an essential factor to stimulate the production of TNF-α and IL-6 in macrophages, dependent on TLR4/ROS/PKC-δ/NF-κB, TLR4/PI3-kinase/AKT/NF-κB, and TLR4/MAPK signalling pathways [Citation116].
IFN-γ is one of the most well-studied pro-inflammatory cytokines involved in defence against K. pneumoniae infection. Different from those common pro-inflammatory cytokines aforementioned, IFN-γ signalling directly activates numerous antimicrobial programmes in host defense, such as the production of reactive oxygen species (ROS), reactive nitrogen species (RNS), and generation of antimicrobial peptides and cytokines [Citation117]. Intratracheal inoculation of IFN-γ knockout mice infected by K. pneumoniae caused a significantly increased mortality, associated with a 100-fold increase of pulmonary bacteria within 2 days post infection and the upregulation of lung-associated IL-10 mRNA [Citation118,Citation119]. At the late stage of infection, the level of serum IL-6 in IFN-γ-/- mice was significantly higher than that of controls, suggestive of the defective immunological host response [Citation119]. These results support the critical role of IFN-γ in the host defense against K. pneumoniae.
The mechanism of IFN-γ stimulation by K. pneumoniae has also been characterized recently (). IFN-γ is mainly secreted by natural killer (NK) cells during infection, highly dependent on the DC signalling pathway or macrophage type I IFN signalling pathway [Citation120,Citation121]. In the DC signalling pathway, NK cells are actively recruited after physical contact with membrane fractions of K. pneumoniae-matured DC, either in a CCR5-dependent manner or by the induction of CCR7 expression in CD56 (dim) CD16 (+) NK cells [Citation120]. Both pathways stimulate enhanced migratory responsiveness of NK cells to the lymph node, and increase the production of IFN-γ [Citation120]. It is accordingly hypothesized that in vivo K. pneumoniae infection is responsible for the induction of innate immune responses characterized by DC-dependent NK-cell migration accompanied by IFN-γ production, and NK–DC interactions are facilitated both in the periphery and in the lymphoid tissue [Citation120]. However, in vivo validations are needed for this conclusion. Alternatively, the IFN-γ production can be promoted by the NK-macrophage communication. Type I IFN is mainly secreted by alveolar macrophages, and IFN-γ can significantly enhance the killing of K. pneumoniae by macrophages. Type I IFN signalling-deficient mice (Ifnar1-/-) were susceptible to K. pneumoniae infection, exhibiting a markedly decreased survival, higher bacterial lung burden, increased dissemination to spleen and liver and severe bronchopneumonia, which were due to the defection of both NK cell accumulation and NK cell-derived IFN-γ production in the lung [Citation121]. Bacterial clearance and NK cell IFN-γ production were rescued by the supplementation of wild-type NK cells in Ifnar1-deficient hosts, and the crosstalk between alveolar macrophages and NK cells were restored by the administration of exogenous IFN-γ [Citation121]. Therefore, K. pneumoniae-induced type I IFNs, as the signal factor between alveolar macrophages and NK cells, promotes NK cell IFN-γ production which in turn activates the anti-microbial armament of macrophages [Citation121]. Taken together, the DC-NK and macrophage-NK signalling pathways are responsible for NK accumulation and NK cell-derived IFN-γ production to counteract K. pneumoniae infection.
IL-12 is regarded as another important inducer for the production of IFN-γ during K. pneumoniae infection. Significantly attenuated lung IFN-γ levels were found in IL-12 deficient K. pneumoniae-infected mice [Citation122]. Consistently, overexpression of IL-12 in K. pneumoniae-infected mice promoted host survival, and the treatment using IFN-γ antibody partially attenuated this survival benefits [Citation123], suggesting that the synergy of IL-12 and IFN-γ can improve host defense against K. pneumoniae. The production of IFN-γ may be regulated by IL-12 signalling through STAT4, however, this conclusion needs to be further validated [Citation124,Citation125].
The other well-studied pro-inflammatory cytokine involved in K. pneumoniae elimination is IL-17. IL-17 is produced in a compartmentalized fashion in the lung after being challenged by K. pneumoniae. IL-17R-deficient mice displayed a significant delay in neutrophil recruitment into the alveolar space resulting in a greater dissemination of K. pneumoniae, and were exquisitely sensitive to intranasal K. pneumoniae with 100% mortality after 48 h [Citation126]. This defect in response to the K. pneumoniae challenge was associated with a significant reduction in lung chemokine gene expression, e.g. granulocyte colony stimulating factor (G-CSF) and macrophage inflammatory protein (MIP)-2 [Citation126]. Consistently, overexpression of IL-17 in the pulmonary compartment can improve K. pneumoniae clearance and host survival [Citation127]. These data demonstrate that IL-17R signalling is critical for the lung chemokine gene expression and early lung neutrophil recruitment against K. pneumoniae [Citation126]. IL-17 induced neutrophil recruitment is associated with the expression level of the chemokine CXCL5. Reduced expression of CXCL5 was found in the IL-17R deficient epithelium, and recombinant CXCL5 administration restored neutrophil recruitment and K. pneumoniae clearance [Citation128]. The results suggest that IL-17 through epithelial IL-17R signalling regulates Cxcl5 expression followed by the recruitment of neutrophils to clean K. pneumoniae in lungs.
The regulation mechanism of IL-17 production during K. pneumoniae infection has been further revealed. IL-17 was induced in CD4+ and CD8+ T cells response to conditioned medium from K. pneumoniae-pulsed DC, and the signalling requires biologically active IL-23 [Citation129]. Blocking IL-23 signalling can significantly reduce the amount of IL-17 in CD4+ and CD8+ T cells [Citation129], and a decreased survival of IL-23 deficient mice with pulmonary K. pneumoniae challenge was observed [Citation122,Citation130]. TLR4 signalling in the lung is additionally required for the induction of the IL-23 p19 subunit transcript and IL-17 protein elaboration [Citation129]. Defective bacterial clearance in IL-23 p19−/− mice was abolished by the administration of exogenous IL-17, and the treatment further restored the reduced production of lung G-CSF and LPS-induced C-X-C chemokine (LIX) [Citation122]. These findings indicate that IL-23 is dependent on IL-17 to protect against K. pneumoniae. IL-17 can also be produced by γδ17 cells (a subset of γδ T cells) in vivo in the presence of IL-23 and IL-1β. However, IL-17 production is negatively regulated by STAT6 signalling, since IL-4 through IL-4R of γδ17 cells increases STAT6 phosphorylation, which further inhibits the production of IL-17A in γδ17 cells in vitro [Citation131]. Notably, K. pneumoniae has no effect on IL-4 production in vivo [Citation131], thus the signalling of IL-17 production in γδ17 cells during K. pneumoniae infection needs to be further studied.
K. pneumoniae manipulates the production of anti-inflammatory cytokines
Together with pro-inflammatory cytokines, a few anti-inflammatory cytokines are involved in protection against K. pneumoniae to balance the immunity reaction of the host. One of the most well-studied anti-inflammatory cytokines is IL-10, a core member of IL-10 family. In early studies, IL-10 was considered as the assistance of pathogens to counteract the host immune system. Neutralization of IL-10 leads to a significant decrease of K. pneumoniae loads in both lung homogenates and plasma harvested at 48 h post infection, resulting in a significant increase in survival. The expression of pro-inflammatory cytokines, such as TNF, MIP-2, and MIP-1α, was simultaneously increased [Citation132]. In agreement, a lower 5-day survival rate of IL-10 overexpression mice was observed when being infected by K. pneumoniae. Alveolar macrophages from the IL-10 overexpression mice displayed increased alternative activation (M2 macrophages), whereas those from control exhibited classical activation (M1 macrophages) with a much higher intracellular bacterial killing potential [Citation133]. These results suggest that K. pneumoniae exploits IL-10 to attenuate host immune response.
In contrast, a recent study showed that IL-10 produced by myeloid-derived suppressor cells (MDSCs) protected the host against K. pneumoniae infection, albeit it functioned divergently in the defense against carbapenem-resistant clinical isolates (CR-Kp) and the hypervirulent strain (KPPR1) [Citation134,Citation135]. During CR-Kp infection, MDSCs-produced IL-10 (MDSCs-IL-10) was highly responsible for the pathogen elimination. Overexpression of IL-10 in MDSCs not only improved CR-Kp clearance but also slightly reduced the lung injury. Using IL-10-/- mice, the protective effect of MDSCs-IL-10 was found through the prevention of lung damage and improvement of host survival to enhance CR-Kp clearance in the airways [Citation134]. The immune response is an early recruitment of neutrophils and MDSCs that are able to produce IL-10 as well as other suppressive effectors to promote the pathogen elimination, resulting in tempering the inflammatory response and minimizing lung injury at early time points [Citation134]. Nevertheless, MDSCs-IL-10 was thought to be essential for the resolution of lung inflammation but not for bacterial clearance upon KPPR1 infection [Citation135]. Although the bacteria was eliminated in IL-10−/− mice, an increased morbidity with progressive weight loss and persistent lung inflammation was found in the later phase [Citation135]. In assistant with IL-10, the MDSCs efficiently efferocytosed apoptotic neutrophils, which was critical to promote lung recovery [Citation135]. In comparison to CR-Kp, the immune response against KPPR1 is a much slower infiltration of MDSCs but companied with a rapid bacterial clearance and a rapid infiltration of neutrophils [Citation134], suggesting that the time of MDSCs recruitment and MDSCs-IL-10 production are divergently triggered by CR-Kp and KPPR1, which determines the outcome. However, the underlying mechanism remains unclear. Considered that KPPR1 is a hypervirulent strain, it is reasonable to assume that different virulence genes carried by CR-Kp and KPPR1 may be responsible for the functional divergence of MDSCs-IL-10 observed.
Two pathways have been identified as responsible for the production of IL-10 during K. pneumoniae infection, namely the cardiolipin pathway and SARM1 pathway (). In the cardiolipin pathway, PPARγ SUMOylation is induced by cardiolipin at the onset of infection, which causes the recruitment of a repressive NCOR/HDAC3 complex to the IL-10 promoter, resulting in the attenuated transcription level of the IL-10 [Citation136]. Alternatively, SARM1 is required for IL-10 induction by fine-tuning the p38-type I IFN axis in the SARM1 pathway. The level of IL-10 transcription and p38 activation was reduced in the absence of SARM1 during K. pneumoniae infection. In turn, the activation of p38 and IL-10 production was increased in Sarm1-/- macrophages treated with IFNAR1 receptor blocking antibody. These results suggest that K. pneumoniae induced p38 activation is modulated by SARM1 through negatively regulating the expression levels of type I IFN, which further induces the production of IL-10 [Citation137].
Another two members of IL-10 family, IFN-λ and IL-22, are also involved in synergistically regulating the host immune response against K. pneumoniae. Both IFN-λ and IL-22 are associated with mucosal defenses to regulate the barrier functions, but with differing kinetics. In the early infection, the elevated IFN-λ amplifies epithelial permeability, which enables bacterial transmigration from the airway lumen into the lung and bloodstream, and also promotes the recruitment of neutrophils across a polarized monolayer of airway epithelial cells. The production of IL-22 is subsequently increased, which helps in maintaining the integrity of epithelial barrier to block dextran permeability and to inhibit neutrophil transmigration and microbial invasion, resulting in enhanced bacterial clearance and bacteraemia prevention. The divergent and opposing expression of these two related cytokines suggests a regulated interaction in the host response to K. pneumoniae infection [Citation138].
Besides IFN-λ, IL-22 can additionally work in concert with other inflammatory cytokines like IL-17. IL-17 and IL-22 cytokines are often co-expressed and further co-regulate CXC chemokine and G-CSF production in the lung to eliminate K. pneumoniae [Citation138–140]. Similar to IL-17, IL-22 expression was markedly attenuated in Il23a−/− mice, suggesting that IL-23 is required for the induction of IL-22 [Citation140]. In the liver, IL-22 additionally shows potent bacteriostatic activity against K. pneumoniae by producing antimicrobial peptides (i.e. lipocalin 2 and serum amyloid A2) through IL-22Ra1 and STAT3 singling [Citation141], suggestive of pleiotropic effects of IL-22 on K. pneumoniae elimination. Therefore, development of agents targeting IL-22-associated signalling pathways may provide selective alternatives for the management of K. pneumoniae infection.
Concluding remarks and future directions
With the exhausted traditional antibiotic pipeline, the emergence of MDR K. pneumoniae promotes the growing clinical needs for novel therapeutic strategies. At present, the therapeutic targeting is mainly gathered in metabolic pathway and certain virulence factors (e.g. LPS and CPS) of K. pneumoniae [Citation142]. Phages, phage polysaccharide depolymerases and antimicrobial peptides represent additional promising tools for antimicrobial therapy [Citation143,Citation144]. However, most of the strategies are highly specific, even strain-dependent, thus largely comprising their cost efficiency and/or outcomes in clinical settings. Accordingly, immunotherapy based on antagonizing the anti-immune strategies of pathogens could be an alternative solution to satisfy these requires. According to the various functions of cytokines during K. pneumoniae infection, compartmentalized cytokine delivery as adjuvant therapy may be a promising strategy. For instance, treatment with growth factor granulocyte-macrophage colony-stimulating factor (GM-CSF) can protect alveolar epithelial cells from apoptosis in response to K. pneumoniae. Intratracheal administration of GM-CSF significantly decreased albumin leak, lung cell apoptosis, and bacteraemia in K. pneumoniae infected mice [Citation145]. Additionally, recombinant IL-22 treatment efficiently ameliorated the ST258 pulmonary infection [Citation146]. Thus, GM-CSF and IL-22 treatment may represent a promising immunomodulatory method for the prevention or treatment of pneumonia. Development of agonists and inhibitors for cell signalling could also be an adjunct treatment. Toll-like receptor 4 (TLR4), which can recognize LPS of gram-negative bacteria, is a potential target for inhibiting undesired inflammatory responses. Some Traditional Chinese Medicine, e.g. Sodium houttuyfonate, LianQinJieDu decoction, and Ugonin M et al., which can modulate TLR4 signalling pathway, have been suggested to be the potential drugs for the treatment of gram-negative bacteria pneumonia [Citation147]. Use of complement inhibitors is another approach to downregulate the host's immune response to protect organs from damages caused by bacterial infection. For instance, blocking of complement C5a can protect lung and extrapulmonary organ failure in pneumonia-induced sepsis [Citation148].
To further promote the development of immunotherapy based on antagonizing the anti-immune strategies of pathogens, a better understanding of the interaction between K. pneumoniae and the host is indispensable. However, the underlying mechanisms have not yet been systematically studied, and numerous knowledge gaps are needed to be filled in. In particular, it is critical to identify the VFs involved in the host–pathogen interaction, especially those essential for the K. pneumoniae-induced autophagy, cell death and cytokines production. Furthermore, it is imperative to uncover the type(s) of cell death that is more efficient for hosts to cope with K. pneumoniae infection. It is largely unclear whether the triggers for each type of cell death are identical or strain/clone-dependent. Understanding these questions would provide us with novel insights into the pathogenicity of K. pneumoniae and the development of selective alternatives for the management of K. pneumoniae infection.
Acknowledgements
Sha Wei and Kai Zhou prepared the manuscript. Tingting Xu helped writing the manuscript. Kai Zhou designed the study. Kai Zhou and Yuxin Chen critically edited the manuscript. All authors had full access to the manuscript and results and accept responsibility of the submission for publication.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- De Oliveira DMP, Forde BM, Kidd TJ, et al. Antimicrobial resistance in ESKAPE pathogens. Clin Microbiol Rev. 2020 Jun 17;33(3):e00181-19.
- Zhou K, Xiao T, David S, et al. Novel subclone of carbapenem-resistant Klebsiella pneumoniae sequence type 11 with enhanced virulence and transmissibility, China. Emerg Infect Dis. 2020 Feb;26(2):289–297.
- Gu D, Dong N, Zheng Z, et al. A fatal outbreak of ST11 carbapenem-resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: a molecular epidemiological study. Lancet Infect Dis. 2018;18(1):37–46.
- Bengoechea JA, Sa Pessoa J. Klebsiella pneumoniae infection biology: living to counteract host defences. FEMS Microbiol Rev. 2019 Mar 1;43(2):123–144.
- Diago-Navarro E, Calatayud-Baselga I, Sun D, et al. Antibody-based immunotherapy to treat and prevent infection with hypervirulent Klebsiella pneumoniae. Clin Vaccine Immunol. 2017;24(1):e00456-16.
- Donta ST, Peduzzi P, Cross AS, et al. Immunoprophylaxis against Klebsiella and pseudomonas Aeruginosa infections. The federal hyperimmune immunoglobulin trial study group. J Infect Dis. 1996;174(3):537–543.
- Liu M, Li W, Xiang X, et al. Mycobacterium tuberculosis effectors interfering host apoptosis signaling. Apoptosis. 2015;20(7):883–891.
- Etna MP, Giacomini E, Severa M, et al. Pro- and anti-inflammatory cytokines in tuberculosis: a two-edged sword in TB pathogenesis. Semin Immunol. 2014;26(6):543–551.
- Periselneris J, Ercoli G, Pollard T, et al. Relative contributions of extracellular and internalized bacteria to early macrophage proinflammatory responses to Streptococcus pneumoniae. mBio. 2019;10(5):e02144-19.
- Zhang Y, Yao Y, Qiu X, et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat Immunol. 2019;20(4):433–446.
- Wanford JJ, Hames RG, Carreno D, et al. Interaction of Klebsiella pneumoniae with tissue macrophages in a mouse infection model and ex-vivo pig organ perfusions: an exploratory investigation. The Lancet Microbe. 2021;2(12):e695–e703.
- Cano V, March C, Insua JL, et al. Klebsiella pneumoniae survives within macrophages by avoiding delivery to lysosomes. Cell Microbiol. 2015;17(11):1537–1560.
- Paczosa MK, Mecsas J. Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol Mol Biol Rev: MMBR. 2016;80(3):629–661.
- Martin RM, Bachman MA. Colonization, infection, and the accessory genome of Klebsiella pneumoniae. Front Cell Infect Microbiol. 2018;8(4).
- Donskey CJ. The role of the intestinal tract as a reservoir and source for transmission of nosocomial pathogens. Clin Infect Dis. 2004;39(2):219–226.
- Martin RM, Cao J, Brisse S, et al. Molecular epidemiology of colonizing and infecting isolates of Klebsiella pneumoniae. mSphere. 2016;1(5):e00261-16.
- Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214.
- Li B, Zhao Y, Liu C, et al. Molecular pathogenesis of Klebsiella pneumoniae. Future Microbiol. 2014;9(9):1071–1081.
- Struve C, Bojer M, Krogfelt KA. Characterization of Klebsiella pneumoniae type 1 fimbriae by detection of phase variation during colonization and infection and impact on virulence. Infect Immun. 2008;76(9):4055–4065.
- Struve C, Bojer M, Krogfelt KA. Identification of a conserved chromosomal region encoding Klebsiella pneumoniae type 1 and type 3 fimbriae and assessment of the role of fimbriae in pathogenicity. Infect Immun. 2009;77(11):5016–5024.
- Sahly H, Keisari Y, Crouch E, et al. Recognition of bacterial surface polysaccharides by lectins of the innate immune system and its contribution to defense against infection: the case of pulmonary pathogens. Infect Immun. 2008;76(4):1322–1332.
- Stahlhut SG, Struve C, Krogfelt KA. Klebsiella pneumoniae type 3 fimbriae agglutinate yeast in a mannose-resistant manner. J Med Microbiol. 2012;61(Pt 3):317–322.
- Przondo-Mordarska A, Ko HL, Beuth J, et al. Chemiluminescence response of human polymorphonuclear leukocytes induced by purified, latex attached Klebsiella fimbriae. Z Bakteriologie: Int J Med Microbiol. 1991;275(4):521–529.
- Lavender H, Jagnow JJ, Clegg S. Klebsiella pneumoniae type 3 fimbria-mediated immunity to infection in the murine model of respiratory disease. Int J Med Microbiol: IJMM. 2005;295(3):153–159.
- Sequeira RP, McDonald JAK, Marchesi JR, et al. Commensal Bacteroidetes protect against Klebsiella pneumoniae colonization and transmission through IL-36 signalling. Nat Microbiol. 2020;5(2):304–313.
- Clements A, Tull D, Jenney AW, et al. Secondary acylation of Klebsiella pneumoniae lipopolysaccharide contributes to sensitivity to antibacterial peptides. J Biol Chem. 2007;282(21):15569–15577.
- Doorduijn DJ, Rooijakkers SH, van Schaik W, et al. Complement resistance mechanisms of Klebsiella pneumoniae. Immunobiology. 2016;221(10):1102–1109.
- Tan YH, Chen Y, Chu WHW, et al. Cell envelope defects of different capsule-null mutants in K1 hypervirulent Klebsiella pneumoniae can affect bacterial pathogenesis. Mol Microbiol. 2020;113(5):889–905.
- Campos MA, Vargas MA, Regueiro V, et al. Capsule polysaccharide mediates bacterial resistance to antimicrobial peptides. Infect Immun. 2004;72(12):7107–7114.
- Moranta D, Regueiro V, March C, et al. Klebsiella pneumoniae capsule polysaccharide impedes the expression of beta-defensins by airway epithelial cells. Infect Immun. 2010;78(3):1135–1146.
- Yoshida K, Matsumoto T, Tateda K, et al. Role of bacterial capsule in local and systemic inflammatory responses of mice during pulmonary infection with Klebsiella pneumoniae. J Med Microbiol. 2000;49(11):1003–1010.
- Evrard B, Balestrino D, Dosgilbert A, et al. Roles of capsule and lipopolysaccharide O antigen in interactions of human monocyte-derived dendritic cells and Klebsiella pneumoniae. Infect Immun. 2010;78(1):210–219.
- Yoshida K, Matsumoto T, Tateda K, et al. Induction of interleukin-10 and down-regulation of cytokine production by Klebsiella pneumoniae capsule in mice with pulmonary infection. J Med Microbiol. 2001;50(5):456–461.
- Regueiro V, Moranta D, Frank CG, et al. Klebsiella pneumoniae subverts the activation of inflammatory responses in a NOD1-dependent manner. Cell Microbiol. 2011;13(1):135–153.
- Walker KA, Treat LP, Sepulveda VE, et al. The small protein RmpD drives hypermucoviscosity in Klebsiella pneumoniae. mBio. 2020;11(5):e01750-20.
- Sa-Pessoa J, Przybyszewska K, Vasconcelos FN, et al. Klebsiella pneumoniae reduces SUMOylation to limit host defense responses. mBio. 2020;11(5):e01733-20.
- Kobayashi SD, Porter AR, Freedman B, et al. Antibody-mediated killing of carbapenem-resistant ST258 Klebsiella pneumoniae by human neutrophils. mBio. 2018;9(2):e00297-18.
- Lee CH, Chang CC, Liu JW, et al. Sialic acid involved in hypermucoviscosity phenotype of Klebsiella pneumoniae and associated with resistance to neutrophil phagocytosis. Virulence. 2014;5(6):673–679.
- Follador R, Heinz E, Wyres KL, et al. The diversity of Klebsiella pneumoniae surface polysaccharides. Microbial Genomics. 2016;2(8):e000073.
- Sahly H, Aucken H, Benedi VJ, et al. Impairment of respiratory burst in polymorphonuclear leukocytes by extended-spectrum beta-lactamase-producing strains of Klebsiella pneumoniae. Eur J Clin Microbiol Infect Dis. 2004;23(1):20–26.
- Merle NS, Noe R, Halbwachs-Mecarelli L, et al. Complement system part II: role in immunity. Front Immunol. 2015;6:257.
- Shankar-Sinha S, Valencia GA, Janes BK, et al. The Klebsiella pneumoniae O antigen contributes to bacteremia and lethality during murine pneumonia. Infect Immun. 2004;72(3):1423–1430.
- Cortes G, Borrell N, de Astorza B, et al. Molecular analysis of the contribution of the capsular polysaccharide and the lipopolysaccharide O side chain to the virulence of Klebsiella pneumoniae in a murine model of pneumonia. Infect Immun. 2002;70(5):2583–2590.
- Willingham SB, Allen IC, Bergstralh DT, et al. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol. 2009;183(3):2008–2015.
- Greco E, Quintiliani G, Santucci MB, et al. Janus-faced liposomes enhance antimicrobial innate immune response in Mycobacterium tuberculosis infection. Proc Natl Acad Sci U S A. 2012 May 22;109(21):E1360–8.
- Fevre C, Almeida AS, Taront S, et al. A novel murine model of rhinoscleroma identifies Mikulicz cells, the disease signature, as IL-10 dependent derivatives of inflammatory monocytes. EMBO Mol Med. 2013;5(4):516–530.
- Miethke M, Marahiel MA. Siderophore-based iron acquisition and pathogen control. Microbiol Mol Biol Rev: MMBR. 2007;71(3):413–451.
- Shon AS, Bajwa RP, Russo TA. Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence. 2013;4(2):107–118.
- Bachman MA, Lenio S, Schmidt L, et al. Interaction of lipocalin 2, transferrin, and siderophores determines the replicative niche of Klebsiella pneumoniae during pneumonia. mBio. 2012;3(6):e00224-11.
- March C, Moranta D, Regueiro V, et al. Klebsiella pneumoniae outer membrane protein A is required to prevent the activation of airway epithelial cells. J Biol Chem. 2011;286(12):9956–9967.
- Jeannin P, Renno T, Goetsch L, et al. Ompa targets dendritic cells, induces their maturation and delivers antigen into the MHC class I presentation pathway. Nat Immunol. 2000;1(6):502–509.
- Soulas C, Baussant T, Aubry JP, et al. Outer membrane protein A (OmpA) binds to and activates human macrophages. J Immunol. 2000;165(5):2335–2340.
- Pichavant M, Delneste Y, Jeannin P, et al. Outer membrane protein A from Klebsiella pneumoniae activates bronchial epithelial cells: implication in neutrophil recruitment. J Immunol. 2003;171(12):6697–6705.
- Llobet E, March C, Gimenez P, et al. Klebsiella pneumoniae OmpA confers resistance to antimicrobial peptides. Antimicrob Agents Chemother. 2009;53(1):298–302.
- You HS, Lee SH, Kang SS, et al. Ompa of Klebsiella pneumoniae ATCC 13883 induces pyroptosis in HEp-2 cells, leading to cell-cycle arrest and apoptosis. Microbes Infect. 2020;22(9):432–440.
- Tsai YK, Fung CP, Lin JC, et al. Klebsiella pneumoniae outer membrane porins OmpK35 and OmpK36 play roles in both antimicrobial resistance and virulence. Antimicrob Agents Chemother. 2011;55(4):1485–1493.
- March C, Cano V, Moranta D, et al. Role of bacterial surface structures on the interaction of Klebsiella pneumoniae with phagocytes. PloS one. 2013;8(2):e56847.
- Bialek-Davenet S, Lavigne JP, Guyot K, et al. Differential contribution of AcrAB and OqxAB efflux pumps to multidrug resistance and virulence in Klebsiella pneumoniae. J Antimicrob Chemother. 2015;70(1):81–88.
- Wang X, Chen H, Zhang Y, et al. Genetic characterisation of clinical Klebsiella pneumoniae isolates with reduced susceptibility to tigecycline: role of the global regulator RamA and its local repressor RamR. Int J Antimicrob Agents. 2015;45(6):635–640.
- Padilla E, Llobet E, Domenech-Sanchez A, et al. Klebsiella pneumoniae AcrAB efflux pump contributes to antimicrobial resistance and virulence. Antimicrob Agents Chemother. 2010;54(1):177–183.
- Stamm CE, Pasko BL, Chaisavaneeyakorn S, et al. Screening Mycobacterium tuberculosis secreted proteins identifies Mpt64 as a eukaryotic membrane-binding bacterial effector. mSphere. 2019;4(3):e00354-19.
- Shirani I, Zhang H, Zhao G, et al. In silico identification of novel immunogenic secreted proteins of Mycoplasma bovis from secretome data and experimental verification. Pathogens. 2020;9(9):770.
- Satchidanandam V, Kumar N, Biswas S, et al. The secreted protein Rv1860 of Mycobacterium tuberculosis stimulates human polyfunctional CD8+ T cells. Clin Vacc Immunol. 2016;23(4):282–293.
- Trajkovic V. [The role of mycobacterial secretory proteins in immune response in tuberculosis]. Med Pregl. 2004;57(Suppl 1):25–28.
- Brinkworth AJ, Hammer CH, Olano LR, et al. Identification of outer membrane and exoproteins of carbapenem-resistant multilocus sequence type 258 Klebsiella pneumoniae. PloS one. 2015;10(4):e0123219.
- Lin TL, Chuang YP, Huang YT, et al. Identification of an immuno-dominant protein from Klebsiella pneumoniae strains causing pyogenic liver abscess: implication in serodiagnosis. BMC Microbiol. 2014;14:321.
- Levine B, Deretic V. unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7(10):767–777.
- Ktistakis NT, Tooze SA. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016;26(8):624–635.
- Hu W, Chan H, Lu L, et al. Autophagy in intracellular bacterial infection. Semin Cell Dev Biol. 2020;101:41–50.
- Shi Z, Li G, Zhang L, et al. Klebsiella pneumoniae infection inhibits autophagy by alveolar type II epithelial cells. Exp Ther Med. 2020 Oct;20(4):3703–3708.
- Rekha RS, Karadottir H, Ahmed S, et al. Innate effector systems in primary human macrophages sensitize multidrug-resistant Klebsiella pneumoniae to antibiotics. Infect Immun. 2020;88(8):e00186-20.
- Nikouee A, Kim M, Ding X, et al. Beclin-1-dependent autophagy improves outcomes of pneumonia-induced sepsis. Front Cell Infect Microbiol. 2021;11:706637.
- Wang Y, Zhang H. Regulation of autophagy by mTOR signaling pathway. Adv Exp Med Biol. 2019;1206:67–83.
- Heras-Sandoval D, Perez-Rojas JM, Hernandez-Damian J, et al. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014;26(12):2694–2701.
- Kamaladevi A, Balamurugan K. Global proteomics revealed Klebsiella pneumoniae induced autophagy and oxidative stress in Caenorhabditis elegans by inhibiting PI3K/AKT/mTOR pathway during infection. Front Cell Infect Microbiol. 2017;7:393.
- Bah A, Vergne I. Macrophage autophagy and bacterial infections. Front Immunol. 2017;8:1483.
- Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012;150(4):803–815.
- Ye Y, Tan S, Zhou X, et al. Inhibition of p-IkappaBalpha ubiquitylation by autophagy-related gene 7 to regulate inflammatory responses to bacterial infection. J Infect Dis. 2015;212(11):1816–1826.
- Sharma A, Simonson TJ, Jondle CN, et al. Mincle-Mediated neutrophil extracellular trap formation by regulation of autophagy. J Infect Dis. 2017;215(7):1040–1048.
- Tripathi JK, Sharma A, Sukumaran P, et al. Oxidant sensor cation channel TRPM2 regulates neutrophil extracellular trap formation and protects against pneumoseptic bacterial infection. FASEB J. 2018;32(12):6848–6859.
- Wang K, Chen Y, Zhang P, et al. Protective features of autophagy in pulmonary infection and inflammatory diseases. Cells. 2019 Feb 3;8(2):123.
- Sharma A, Steichen AL, Jondle CN, et al. Protective role of Mincle in bacterial pneumonia by regulation of neutrophil mediated phagocytosis and extracellular trap formation. J Infect Dis. 2014;209(11):1837–1846.
- Li X, He S, Zhou X, et al. Lyn delivers bacteria to lysosomes for eradication through TLR2-initiated autophagy related phagocytosis. PLoS Pathog. 2016;12(1):e1005363.
- Bonilla DL, Bhattacharya A, Sha Y, et al. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity. 2013;39(3):537–547.
- Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169(3):425–434.
- Pu Q, Gan C, Li R, et al. Atg7 deficiency intensifies inflammasome activation and pyroptosis in Pseudomonas sepsis. J Immunol. 2017;198(8):3205–3213.
- Ye Y, Li X, Wang W, et al. Atg7 deficiency impairs host defense against Klebsiella pneumoniae by impacting bacterial clearance, survival and inflammatory responses in mice. Am J Physiol Lung Cell Mol Physiol. 2014;307(5):L355–L363.
- Yang PY, Chen WX, Chang FY, et al. Hepg2 cells infected with Klebsiella pneumoniae show DNA laddering at apoptotic and necrotic stages. Apoptosis. 2012;17(2):154–163.
- Wang Z, Ren J, Liu Q, et al. Hypermucoviscous Klebsiella pneumoniae infections induce platelet aggregation and apoptosis and inhibit maturation of megakaryocytes. Thromb Res. 2018;171:45–54.
- Cheng J, Zhang J, Han B, et al. Klebsiella pneumoniae isolated from bovine mastitis is cytopathogenic for bovine mammary epithelial cells. J Dairy Sci. 2020;103(4):3493–3504.
- Kobayashi SD, Porter AR, Dorward DW, et al. Phagocytosis and killing of Carbapenem-resistant ST258 Klebsiella pneumoniae by human neutrophils. J Infect Dis. 2016;213(10):1615–1622.
- Lee CH, Chuah SK, Tai WC, et al. Delay in human neutrophil constitutive apoptosis after infection with Klebsiella pneumoniae serotype K1. Front Cell Infect Microbiol. 2017;7:87.
- Fernandez-Boyanapalli R, McPhillips KA, Frasch SC, et al. Impaired phagocytosis of apoptotic cells by macrophages in chronic granulomatous disease is reversed by IFN-gamma in a nitric oxide-dependent manner. J Immunol. 2010;185(7):4030–4041.
- Jondle CN, Gupta K, Mishra BB, et al. Klebsiella pneumoniae infection of murine neutrophils impairs their efferocytic clearance by modulating cell death machinery. PLoS Pathog. 2018;14(10):e1007338.
- Ahn D, Prince A. Participation of necroptosis in the host response to acute bacterial pneumonia. J Innate Immun. 2017;9(3):262–270.
- Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665.
- Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14(1):10–22.
- Fernandes-Alnemri T, Wu J, Yu JW, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14(9):1590–1604.
- Ting JP, Willingham SB, Bergstralh DT. NLRs at the intersection of cell death and immunity. Nat Rev Immunol. 2008;8(5):372–379.
- Hua KF, Yang FL, Chiu HW, et al. Capsular polysaccharide Is involved in NLRP3 inflammasome activation by Klebsiella pneumoniae serotype K1. Infect Immun. 2015;83(9):3396–3409.
- Cai S, Batra S, Wakamatsu N, et al. NLRC4 inflammasome-mediated production of IL-1beta modulates mucosal immunity in the lung against gram-negative bacterial infection. J Immunol. 2012;188(11):5623–5635.
- Wang J, Shao Y, Wang W, et al. Caspase-11 deficiency impairs neutrophil recruitment and bacterial clearance in the early stage of pulmonary Klebsiella pneumoniae infection. Int J Med Microbiol: IJMM. 2017;307(8):490–496.
- Codo AC, Saraiva AC, Dos Santos LL, et al. Inhibition of inflammasome activation by a clinical strain of Klebsiella pneumoniae impairs efferocytosis and leads to bacterial dissemination. Cell Death Dis. 2018;9(12):1182.
- Ramesh G, MacLean AG, Philipp MT. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediat Inflamm. 2013;2013:480739.
- Chen SC, Mehrad B, Deng JC, et al. Impaired pulmonary host defense in mice lacking expression of the CXC chemokine lungkine. J Immunol. 2001;166(5):3362–3368.
- Lindell DM, Standiford TJ, Mancuso P, et al. Macrophage inflammatory protein 1alpha/CCL3 is required for clearance of an acute Klebsiella pneumoniae pulmonary infection. Infect Immun. 2001;69(10):6364–6369.
- Zeng X, Moore TA, Newstead MW, et al. Intrapulmonary expression of macrophage inflammatory protein 1alpha (CCL3) induces neutrophil and NK cell accumulation and stimulates innate immunity in murine bacterial pneumonia. Infect Immun. 2003;71(3):1306–1315.
- Tsai WC, Strieter RM, Wilkowski JM, et al. Lung-specific transgenic expression of KC enhances resistance to Klebsiella pneumoniae in mice. J Immunol. 1998 Sep 1;161(5):2435–2440.
- Cai S, Batra S, Lira SA, et al. CXCL1 regulates pulmonary host defense to Klebsiella infection via CXCL2, CXCL5, NF-kappaB, and MAPKs. J Immunol. 2010;185(10):6214–6225.
- Hoffmann E, Dittrich-Breiholz O, Holtmann H, et al. Multiple control of interleukin-8 gene expression. J Leukocyte Biol. 2002;72(5):847–855.
- Gabellini C, Trisciuoglio D, Desideri M, et al. Functional activity of CXCL8 receptors, CXCR1 and CXCR2, on human malignant melanoma progression. Eur J Cancer. 2009;45(14):2618–2627.
- Frank CG, Reguerio V, Rother M, et al. Klebsiella pneumoniae targets an EGF receptor-dependent pathway to subvert inflammation. Cell Microbiol. 2013;15(7):1212–1233.
- Moore TA, Lau HY, Cogen AL, et al. Defective innate antibacterial host responses during murine Klebsiella pneumoniae bacteremia: tumor necrosis factor (TNF) receptor 1 deficiency versus therapy with anti-TNF-alpha. Clin Infect Dis. 2005;41(Suppl 3):S213–S217.
- Laichalk LL, Kunkel SL, Strieter RM, et al. Tumor necrosis factor mediates lung antibacterial host defense in murine Klebsiella pneumonia. Infect Immun. 1996;64(12):5211–5218.
- de Paula TP, Santos PC, Arifa R, et al. Treatment with atorvastatin provides additional benefits to imipenem in a model of gram-negative pneumonia induced by Klebsiella pneumoniae in mice. Antimicrob Agents Chemother. 2018;62(5):e00764-17.
- Yang FL, Yang YL, Liao PC, et al. Structure and immunological characterization of the capsular polysaccharide of a pyrogenic liver abscess caused by Klebsiella pneumoniae: activation of macrophages through Toll-like receptor 4. J Biol Chem. 2011;286(24):21041–21051.
- Jessop F, Buntyn R, Schwarz B, et al. Interferon gamma reprograms host mitochondrial metabolism through inhibition of complex II To control intracellular bacterial replication. Infect Immun. 2020;88(2):e00744-19.
- Moore TA, Perry ML, Getsoian AG, et al. Divergent role of gamma interferon in a murine model of pulmonary versus systemic Klebsiella pneumoniae infection. Infect Immun. 2002;70(11):6310–6318.
- Yoshida K, Matsumoto T, Tateda K, et al. Protection against pulmonary infection with Klebsiella pneumoniae in mice by interferon-gamma through activation of phagocytic cells and stimulation of production of other cytokines. J Med Microbiol. 2001;50(11):959–964.
- Van Elssen CH, Vanderlocht J, Frings PW, et al. Klebsiella pneumoniae-triggered DC recruit human NK cells in a CCR5-dependent manner leading to increased CCL19-responsiveness and activation of NK cells. Eur J Immunol. 2010;40(11):3138–3149.
- Ivin M, Dumigan A, de Vasconcelos FN, et al. Natural killer cell-intrinsic type I IFN signaling controls Klebsiella pneumoniae growth during lung infection. PLoS Pathog. 2017;13(11):e1006696.
- Happel KI, Dubin PJ, Zheng M, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med. 2005;202(6):761–769.
- Greenberger MJ, Kunkel SL, Strieter RM, et al. IL-12 gene therapy protects mice in lethal Klebsiella pneumonia. J Immunol. 1996 Oct 1;157(7):3006–3012.
- Deng JC, Zeng X, Newstead M, et al. STAT4 is a critical mediator of early innate immune responses against pulmonary Klebsiella infection. J Immunol. 2004;173(6):4075–4083.
- Bacon CM, Petricoin EF, Ortaldo JR, et al. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc Natl Acad Sci U S A. 1995;92(16):7307–7311.
- Ye P, Rodriguez FH, Kanaly S, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194(4):519–527.
- Ye P, Garvey PB, Zhang P, et al. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol. 2001;25(3):335–340.
- Chen K, Eddens T, Trevejo-Nunez G, et al. IL-17 Receptor signaling in the lung epithelium is required for mucosal chemokine gradients and pulmonary host defense against K. pneumoniae. Cell Host Microbe. 2016;20(5):596–605.
- Happel KI, Zheng M, Young E, et al. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J Immunol. 2003;170(9):4432–4436.
- Happel KI, Odden AR, Zhang P, et al. Acute alcohol intoxication suppresses the interleukin 23 response to Klebsiella pneumoniae infection. Alcohol Clin Exp Res. 2006;30(7):1200–1207.
- Bloodworth MH, Newcomb DC, Dulek DE, et al. STAT6 signaling attenuates interleukin-17-producing gammadelta T cells during acute Klebsiella pneumoniae infection. Infect Immun. 2016;84(5):1548–1555.
- Greenberger MJ, Strieter RM, Kunkel SL, et al. Neutralization of IL-10 increases survival in a murine model of Klebsiella pneumonia. J Immunol. 1995 Jul 15;155(2):722–729.
- Dolgachev VA, Yu B, Sun L, et al. Interleukin 10 overexpression alters survival in the setting of gram-negative pneumonia following lung contusion. Shock. 2014;41(4):301–310.
- Penaloza HF, Noguera LP, Ahn D, et al. Interleukin-10 produced by myeloid-derived suppressor cells provides protection to carbapenem-resistant Klebsiella pneumoniae sequence type 258 by enhancing Its clearance in the airways. Infect Immun. 2019;87(5):e00665-18.
- Poe SL, Arora M, Oriss TB, et al. STAT1-regulated lung MDSC-like cells produce IL-10 and efferocytose apoptotic neutrophils with relevance in resolution of bacterial pneumonia. Mucosal Immunol. 2013;6(1):189–199.
- Chakraborty K, Raundhal M, Chen BB, et al. The mito-DAMP cardiolipin blocks IL-10 production causing persistent inflammation during bacterial pneumonia. Nat Commun. 2017;8:13944.
- Feriotti C, Sa-Pessoa J, Calderon-Gonzalez R, et al. Klebsiella pneumoniae hijacks the Toll-IL-1R protein SARM1 in a type I IFN-dependent manner to antagonize host immunity. Cell Rep. 2022;40(6):111167.
- Ahn D, Wickersham M, Riquelme S, et al. The effects of IFN-lambda on epithelial barrier function contribute to Klebsiella pneumoniae ST258 Pneumonia. Am J Respir Cell Mol Biol. 2019;60(2):158–166.
- Ahn D, Prince A. Participation of the IL-10RB related cytokines, IL-22 and IFN-lambda in defense of the airway mucosal barrier. Front Cellular Infect Microbiol. 2020;10:300.
- Aujla SJ, Chan YR, Zheng M, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14(3):275–281.
- Zheng M, Horne W, McAleer JP, et al. Therapeutic role of interleukin 22 in experimental intra-abdominal Klebsiella pneumoniae infection in mice. Infect Immun. 2016;84(3):782–789.
- Ali S, Alam M, Hasan GM, et al. Potential therapeutic targets of Klebsiella pneumoniae: a multi-omics review perspective. Brief Funct Genomics. 2022;21(2):63–77.
- A MS NVV, et al AIB. Characterization and therapeutic potential of bacteriophage-encoded polysaccharide depolymerases with beta galactosidase activity against Klebsiella pneumoniae K57 Capsular Type. Antibiotics. 2020 Oct 25;9(11):732.
- de Souza CM, da Silva AP, Junior NGO, et al. Peptides as a therapeutic strategy against Klebsiella pneumoniae. Trends Pharmacol Sci. 2022;43(4):335–348.
- Standiford LR, Standiford TJ, Newstead MJ, et al. TLR4-dependent GM-CSF protects against lung injury in gram-negative bacterial pneumonia. Am J Physiol Lung Cellular Mol Physiol. 2012;302(5):L447–L454.
- Iwanaga N, Sandquist I, Wanek A, et al. Host immunology and rational immunotherapy for carbapenem-resistant Klebsiella pneumoniae infection. JCI Insight. 2020;5(8):e135591.
- Ding J, Liu Q. Toll-like receptor 4: A promising therapeutic target for pneumonia caused by Gram-negative bacteria. J Cell Mol Med. 2019;23(9):5868–5875.
- Muller-Redetzky H, Lienau J, Suttorp N, et al. Therapeutic strategies in pneumonia: going beyond antibiotics. Eur Resp Rev. 2015;24(137):516–524.