
ABSTRACT
Congenital virus infections, for example cytomegalovirus and rubella virus infections, commonly affect the central nervous and hematological systems in fetuses and offspring. However, interactions between emerging congenital Zika virus and hematological system—bone marrow and blood—in fetuses and offspring are mainly unknown. Our overall goal was to determine whether silent in utero Zika virus infection can cause functional and molecular footprints in the bone marrow and blood of fetuses and offspring. We specifically focused on silent fetal infection because delayed health complications in initially asymptomatic offspring were previously demonstrated in animal and human studies. Using a well-established porcine model for Zika virus infection and a set of cellular and molecular experimental tools, we showed that silent in utero infection causes multi-organ inflammation in fetuses and local inflammation in the fetal bone marrow. In utero infection also caused footprints in the offspring bone marrow and PBMCs. These findings should be considered in a broader clinical context because of growing concerns about health sequelae in cohorts of children affected with congenital Zika virus infection in the Americas. Understanding virus-induced molecular mechanisms of immune activation and inflammation in fetuses may provide targets for early in utero interventions. Also, identifying early biomarkers of in utero-acquired immunopathology in offspring may help to alleviate long-term sequelae.
Introduction
Transplacental virus infections, for example cytomegalovirus and rubella virus infections, commonly affect the central nervous and hematological systems in fetuses and offspring [Citation1]. Zika virus (ZIKV) emerged in the Americas in 2015 posing ongoing public health concerns. Unlike other human flaviviruses, ZIKV infection showed unusual clinical phenotype in pregnant women with transplacental infection, fetal pathology, and birth defects in offspring. Zika virus has tropism to different placental cells, brain radial glia, astrocytes, neuronal progenitor cells, and mature neurons, causing placental and brain lesions [Citation2–6]. Given this phenotype, most studies are focused on pathogenesis in placental and nervous tissues. However, ZIKV-induced hematological effects in fetuses and immunopathology sequelae in offspring are poorly defined. Recently, systemic immune inflammation with disrupted cytokines in blood plasma and cerebrospinal fluid was described in human newborns affected with ZIKV infection during in utero life [Citation7, Citation8]. Our animal model studies also showed interferon-alpha (IFN-α) sequelae in offspring [Citation9] where in utero affected neonates showed altered IFN-α levels in blood plasma in the normal environment and during social stress. However, it is unknown whether in utero ZIKV infection affects the hematological system—bone marrow and blood—in fetuses and offspring.
Severe outcomes of ZIKV infection in pregnancy include fetal death, microencephaly, and developmental abnormalities in fetuses and offspring. However, the majority of fetal infections are silent, with difficult to diagnose birth defects [Citation10]. Alarmingly, delayed health complications of silent in utero ZIKV infection in initially asymptomatic offspring were demonstrated in our animal studies [Citation9], in mouse studies [Citation11], and in human studies [Citation12–14.] But the full complexity and duration of long-term sequelae of silent in utero ZIKV infection remain unknown. The definition of silent in utero ZIKV infection in our studies is based on our animal model [Citation9, Citation15–17], non-human primate [Citation18], and human research [Citation12–14]: In utero infection that does not cause severe lesions in the brain, in utero growth restriction, and other readily identifiable abnormalities in fetuses and newborns, however, imposes significant health risks in virus-negative offspring due to persistent molecular and cellular sequelae. We, and others, have developed porcine models that reproduce critical aspects of in utero ZIKV infection in humans with persistent infection in the fetal brain, fetal membranes, and placenta [Citation9, Citation15–17, Citation19]. Also, using the porcine model, we studied molecular pathology in the fetal brain and molecular sequelae in the brain of offspring after silent in utero ZIKV infection [Citation9, Citation17]. Systemic maternal inoculation with ZIKV does not cause maternal viremia and transplacental infection in pigs [Citation19]; but, similar to in vitro cell cultures and organoid systems, our model permits controlled in utero delivery of the virus for a focused study of pathogenesis in fetuses. The advantage of our pig model over in vitro cultures is that ZIKV infection can be analyzed in the context of pregnancy and intact fetal immune responses. Importantly, infection in mother and fetus may affect offspring differently; most infection sequelae studies showed the importance of maternal inflammation, while the specific role of fetal immunopathology is difficult to study in traditional models. In contrast, our fetal pig model allows us to dissect and to study specific fetal immunopathology during isolated in utero infection.
Here, we used the well-established porcine model to study whether isolated silent in utero ZIKV infection can affect the hematological system—bone marrow and blood—in fetuses and offspring. In fetuses, we studied systemic and local bone marrow immune activation, inflammation, and molecular footprints. In offspring, we studied functional and molecular footprints in whole bone marrow cells, bone marrow hematopoietic stem progenitor cells (HSPCs), and peripheral blood monocyte cells (PBMCs).
Materials and methods
Fetal experiment
Animal experiments were performed following the Canadian Council on Animal Care guidelines and university approved Animal Use Protocol #20180012. All efforts were made to minimize animal suffering. Pigs were euthanized with an anesthetic overdose followed by exsanguination. Conventional time-pregnant Landrace-cross pigs were purchased from the university high-health status herd free from porcine reproductive and respiratory syndrome virus (PRRSV), porcine parvovirus (PPV), porcine circovirus 2 (PCV2), and porcine circovirus 3 (PCV3), which can cause fetal infection in pigs. Accordingly, maternal, fetal, and piglet samples were negative for PRRSV, PPV, PCV2, and PCV3 in virus-specific PCR assays [Citation17].
Two pregnant pigs were housed at the Vaccine and Infectious Disease Organization, University of Saskatchewan biosafety level 2 facility. One of the pigs was in utero inoculated with the ZIKV H/PF/2013 strain at 50 gestation days (the total duration of porcine pregnancy is 114–115 days); and the second control pig was inoculated with virus-free media. For precise inoculation, we used an ultrasound-guided technique that verifies fetal viability before and after inoculation [Citation9, Citation15–17]. Four conceptuses (a fetus with fetal membranes) were inoculated in each pig with the ZIKV or control media. Each conceptus was inoculated with 2 × 105 TCID50 of ZIKV intraperitoneally + intra-amniotic (100 μl + 100 μl). Conceptuses were labelled with non-absorbable surgical sutures on the adjacent uterine walls to identify directly inoculated fetuses during sampling. Pigs were euthanized and sampled 28 days after ZIKV in utero inoculation as we previously described [Citation15–17]. For maternal tissues, we sampled blood plasma and uterine lymph nodes. Uteri with fetuses were removed, and all samplings were performed in the direction from a most distant non-manipulated conceptus toward inoculated fetuses with dedicated instruments for each fetus. In ZIKV and Control pigs, amniotic fluids, amniotic membranes, and uterine wall with the placenta (fetal placental compartment was subsequently dissected from the maternal endometrium) were collected from each conceptus and rapidly frozen. Umbilical cord blood was aspirated from each fetus with sterile syringes and needles before fetal tissue dissection. After blood centrifugation (2,000 g, 20 min, + 4 °C), plasma was aliquoted and frozen (−80 °C). Fetuses were visually examined, and gross pathology was recorded. Body and brain weights of viable fetuses were measured. Then, fetal organs were sampled and frozen in liquid nitrogen: Fetal brains, thymus, inguinal lymph nodes, spleen, and liver. Virology data from the fetal placenta, brain, and lymph nodes were partially reported in our previous study [Citation20]. The femoral and tibia bones of both hind legs were collected in clean plastic bags, placed on wet ice, and transferred into a biosafety cabinet for isolation of bone marrow cells as described in Supplementary Methods.
Offspring experiment
In the offspring experiment, two pregnant pigs were in utero inoculated at 53 gestation days (the total duration of porcine pregnancy is 114–115 days) with the PRVABC59 ZIKV strain or control media as described above. Births were monitored closely, and individual amniotic membranes surrounding each newborn piglet were collected and snap-frozen on dry ice. At birth (day 0; 61 days after in utero inoculation), we tagged piglets, recorded gender, and measured cranium and body length. After delivery, piglets were monitored for 28–42 days for clinical signs. Shortly after birth, amniotic membranes from all piglets were tested for ZIKV as described in Supplementary Methods. Mothers were separated from piglets at 21 days after birth and sampled (blood plasma and uterine lymph nodes). Blood from piglets was collected at 26 days after birth for cytokine measurements in blood plasma and flow cytometry in PBMCs, and after euthanasia at 28–42 days for PBMC stimulation and next-generation sequencing (NGS) as described in Supplementary Methods. Blood was collected in sterile EDTA tubes with a vacutainer blood-sampling system (BD) by puncturing the vena cava.
To make sampling and painstaking work with bone marrow cells feasible, piglets were euthanized within 28–42 days after birth. To avoid the effects of circadian rhythms on gene expression and proteins in the bone marrow cells and blood cells, each pair of ZIKV-affected and Control piglets were sampled simultaneously within a short period of time. Licensed veterinarians euthanized animals with an anesthetic overdose followed by exsanguination. After injecting the anesthetic, complete unconsciousness was confirmed by loss of pedal and palpebral reflexes, and piglets were rapidly exsanguinated to ensure a quick death. This method minimizes animal distress and is consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association and approved by the University of Saskatchewan’s Animal Research Ethics Board. Immediately after exsanguination, liver, spleen, testis, mesenteric and bronchial lymph nodes were sampled and preserved in liquid nitrogen. The femurs and humeri were collected in clean plastic bags, placed on wet ice, and transferred into a biosafety cabinet to isolate whole bone marrow cells and CD117 + hematopoietic stem progenitor cells (HSPCs) as described in Supplementary Methods.
Cellular and molecular assays
Supplementary Methods contain detailed information on well-established protocols: ZIKV stock preparation, isolation and testing whole bone marrow cells in fetuses and offspring, bone marrow CD117+ HSPCs in offspring, PBMCs in offspring, quantification of virus loads in tissues, cytokine measurements in blood plasma from fetuses and offspring, immunohistochemistry, western blotting, kinome analysis, RNA-seq and bioinformatics.
Statistics
We used GraphPad PRISM 8 software. The difference with p < 0.05 was considered significant. Data were expressed as individual values and mean ± standard deviation. The number of dead fetuses and newborn piglets were compared with the Yates-corrected χ2-test. Brain–body weight ratio in fetuses and head-body size ratio in offspring were compared with Mann–Whitney U-test. Viral loads in tissues and cytokine levels in blood plasma were compared with Mann–Whitney U-test. The percentage of sialoadhesin stained area in the fetal organs and the expression of sialoadhesin in the fetal and offspring bone marrow were compared with Mann–Whitney U-test. We used the Spearman and Pearson correlations to evaluate relationships between ZIKV loads in placental tissues or amniotic fluids and the number of sialoadhesin-positive macrophages in fetal organs. To compare the number of granulocyte/macrophage colony-forming units and the total number of granulocyte/macrophage progenitor cells, CD3 + and CD14 + cells frequency in PBMCs, and the IL-1β levels in LPS-stimulated PBMCs, we used Mann–Whitney U-test. P values in graphs are shown only for statistically significant data.
Results
Zika virus infection causes systemic inflammation in fetuses
To identify whether silent ZIKV infection—an infection that does not cause fetal death and apparent clinical pathology—leads to systemic fetal inflammation, we used the established fetal pig model [Citation9, Citation15–17] (A). Among ZIKV-exposed fetuses, 13% were dead with other fetuses alive and with no visible pathology (B); brain/body ratio was also not affected in fetuses (not shown). This death rate is in line with reported rates of fetal mortality in apparently healthy pigs [Citation17]. Decomposition of tissues in dead fetuses prevented extraction of RNA suitable for testing and virus-specific PCR was negative. All four directly-injected fetuses were viable with no visible pathology.
Figure 1. Fetal data. (A) Experimental setup in the fetal study. (B) Fetal mortality (Yates-corrected χ2-test); % shows live fetuses. In the Control group, there were 8 viable fetuses and no dead fetuses. The ZIKV group had 13 viable fetuses and 2 dead decomposed fetuses. (C) Zika virus infection in fetuses (Mann-Whitney U-test). Samples from all live and dead fetuses were tested. (D) Cytokine responses in fetal blood plasma (Mann-Whitney U-test). Samples from live fetuses were tested. The dotted lines represent the limit of detection (LOD) or limit of quantification (LOQ). The solid line represents the mean. In all graphs, squares and circles indicate individual fetuses. Filled red circles represent fetuses directly inoculated with ZIKV.
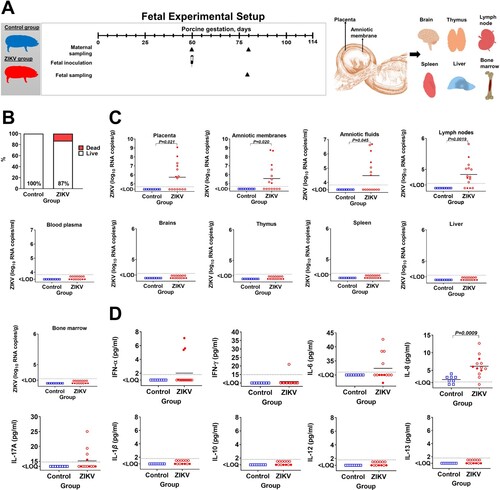
As expected [Citation9, Citation15–17], in utero ZIKV inoculation resulted in trans-fetal virus spread from directly injected to non-manipulated siblings and silent infection. High virus loads were detected in directly injected and trans-infected fetuses (C). Zika virus caused persistent infection with high virus loads in the placenta (53% of fetuses) and fetal lymph nodes (73%). The virus was also identified in amniotic fluids and amniotic membranes (C). Altogether, all 13 liveborn fetuses had ZIKV in one or several tissues and were exposed to the virus for around four weeks. Fetal brain, thymus, spleen, liver, blood plasma (C), and maternal samples were negative for the virus. In accordance with our animal studies [Citation9, Citation16, Citation17, Citation21] in ZIKV-affected fetuses and studies in human neonates [Citation7, Citation22], fetuses exposed to the virus showed an increase of cytokines. Three directly injected virus-positive fetuses out of four had immune activation with elevated IFN-α in blood plasma (D). Fetuses also showed systemic inflammation with elevated interleukin (IL)−6, IL-8, and IL-17a in blood plasma (D). While the difference in cytokine levels did not reach statistical significance (except for IL-8), the trend for IFN-α, IL-6, and IL-17a was clear. Interleukin-γ was detected in one fetus from the ZIKV group (21 pg/ml). Interleukin-1β, IL-10, IL-12 and IL-13 were below the quantification limits in both groups.
As an inflammatory marker in fetal tissues, we used sialoadhesin. Sialoadhesin is the macrophage inflammatory marker; ZIKV-induced sialoadhesin upregulation was reported in cells from human adults [Citation23] and primate placenta [Citation24]. The number of sialoadhesin-positive macrophages was significantly higher in virus-positive tissues—fetal lymph nodes, and in virus-negative tissues—liver and thymus (A and B). In other virus-positive (placenta and amniotic membranes) and virus-negative (brain and spleen) tissues the increase of sialoadhesin-positive macrophages did not reach statistical significance, but had the trend to increase. Zika virus loads in the placenta significantly correlated with the number of sialoadhesin-positive macrophages (C). Also, ZIKV loads in placental tissues and virus loads in amniotic fluids significantly correlated with the number of sialoadhesin-positive macrophages in virus-positive tissues—amniotic membranes, lymph nodes, and in virus-negative tissues—brain and spleen (Suppl. Figure S1).
Figure 2. Sialoadhesin expression in fetal organs. (A) Sialoadhesin-specific immunohistochemistry in fetal organs. The red staining represents sialoadhesin-positive cells. In fetal organs (except the brain) the staining pattern was diffused. In the brain, positive cells were localized in the meninges. (B) Digital quantification of the sialoadhesin-positive area in tissues. The solid line represents the mean. The percentage of sialoadhesin stained area in the fetal organs was compared with Mann-Whitney U-test. (C) Spearman correlation between sialoadhesin and ZIKV loads in the placenta. The X-axis represents ZIKV log10 RNA copies per g. In all graphs, squares and circles indicate individual fetuses. Filled red circles represent fetuses directly inoculated with ZIKV.
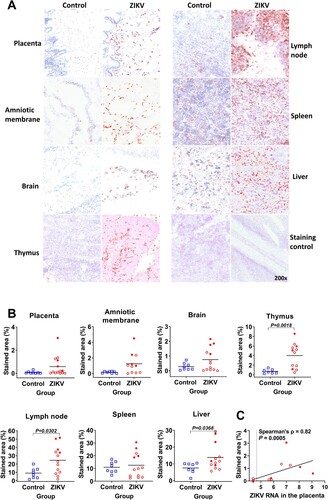
Altogether, silent ZIKV infection can cause systemic inflammation in fetuses represented by increased proinflammatory cytokines in the blood and sialoadhesin overexpression in virus-positive and virus-negative fetal organs.
Zika virus infection causes molecular footprints in the fetal bone marrow
We isolated the whole bone marrow cells from ZIKV and Control fetuses and quantified sialoadhesin with the western blot. In accordance with inflammation in organs of infected fetuses (), sialoadhesin was overexpressed in fetal bone marrow cells (A, Suppl. Figure S2). Sialoadhesin loads in the bone marrow correlated with ZIKV loads in amniotic fluids (Pearson correlation, ρ=0.68, P=0.03)
Figure 3. Molecular pathology in the bone marrow of Zika-affected fetuses and offspring. (A) Western blot semi-quantification of sialoadhesin (Sn) expression in the whole bone marrow cells of fetuses (Mann-Whitney U-test). Corresponding western blot images are in Suppl. Figure S2. Squares and circles indicate individual fetuses. Filled red circles represent fetuses directly inoculated with ZIKV. (B and C) Differentially phosphorylated proteins (p < 0.05) in whole bone marrow cells of Zika-affected fetuses identified with kinome analysis. Raw data are in Table S1-A. Some proteins have more than one differently phosphorylated site; all differently phosphorylated sites for each protein are shown. (D) Western blot semi-quantification of sialoadhesin expression in the whole bone marrow cells of offspring (Mann-Whitney U-test). Squares and circles indicate individual piglets. (E and F) Differentially phosphorylated proteins (p < 0.05) in whole bone marrow cells of Zika-affected offspring identified by kinome analysis. Raw data are in Table S1-C. Some proteins have more than one differently phosphorylated site; all differently phosphorylated sites for each protein are shown.
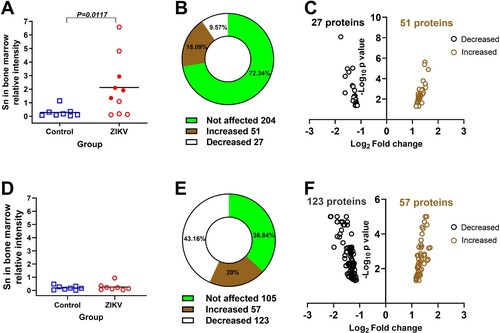
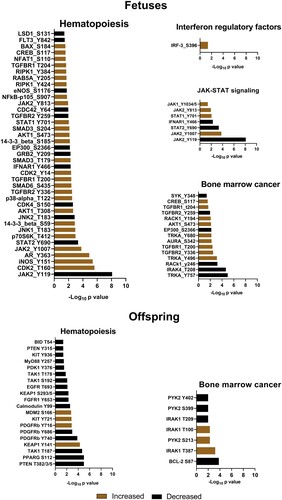
Next, to better understand molecular footprints associated with bone marrow inflammation in fetuses, we compared kinase activities with high-throughput kinome analysis. In total, ZIKV fetuses had 78 differentially phosphorylated sites in 67 proteins (B and C; Table S1-A). Among affected proteins, there were IFNAR1, JAK1, JAK2, STAT1, and STAT2 ()—components of the canonical JAK-STAT signaling pathway that plays a role in bone marrow hematopoietic function [Citation25]. The bone marrow provides a niche for hematopoietic stem cells in fetuses at the late term of the development and afterbirth in offspring. Kinome profiling in fetal bone marrow cells showed that silent in utero ZIKV infection may affect hematopoiesis. Specifically, the number of proteins previously linked to the biology of the HSC (hematopoietic stem cell) niche and homeostatic and stress hematopoiesis were affected (; references are in File S1). In addition to proteins involved in interferon responses and hematopoiesis, a set of differently phosphorylated proteins in fetuses was related to bone marrow cancer (; references are in File S1).
Figure 4. Differentially phosphorylated proteins related to immune activation, hematopoiesis, and cancer in the bone marrow of Zika-affected fetuses and offspring. Some proteins have more than one differently phosphorylated site; all differently phosphorylated sites for each protein are shown. All proteins affected in fetuses (except MSK2, SOC3, AR, JNK1, SMAD6, BAX, TGFBR2, IRAK4) were also affected in offspring (upper panel). In addition to proteins commonly affected in fetuses and offspring, offspring also had individually affected proteins (lower panel). Raw data are in Tables S1-A, C.
Based on differentially phosphorylated protein targets, ZIKV fetuses had 515 (FDR <0.01) altered biological pathways including pathways related to interferon responses, immunity, and cancer (Table S1-B). Among the top ten affected pathways, there were pathways involved in bone marrow function—osteoclast differentiation [Citation26] and IL-7 [Citation27].
To identify whether bone marrow inflammation resulted from local virus replication, we tested cells with the sensitive virus-specific PCR and did not find ZIKV RNA in bone marrow cells from directly inoculated and trans-infected fetuses (C).
To summarize, silent ZIKV infection causes the molecular footprint in the fetal bone marrow with altered sialoadhesin expression and altered protein phosphorylation.
In utero Zika virus infection causes molecular footprints in the offspring bone marrow
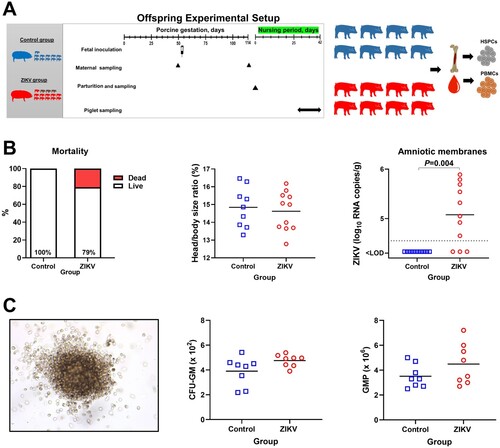
To induce subclinical in utero infection, we inoculated four conceptuses from a pregnant pig with ZIKV at 53 gestation days that resulted in persistent in utero infection for 61 days till the birth (A). As expected [Citation9, Citation15–17], in utero inoculation caused silent infection in many fetuses with no apparent clinical signs in live-born offspring. Although 21% of ZIKV-exposed newborn piglets were dead (B). In contrast, in the control group, all offspring were alive and healthy at birth. Individual amniotic membranes collected at birth and tissues collected postmortem from Control offspring were negative for ZIKV. In ZIKV offspring, individual amniotic membranes collected from three live fetuses immediately after birth were also negative (B). High virus loads—4.6-5.9 log10 ZIKV RNA copies/g—were detected in individual amniotic membranes from 8 live newborn piglets, showing virus persistence in fetal membranes for around two months (in utero inoculation at 53 gestation days and sampling 61 days later, at birth). Testing of other tissues (liver, spleen, testis, mesenteric and bronchial lymph nodes, whole bone marrow cells, CD117 + progenitor cells, blood plasma and PBMCs tested with RT-qPCR) collected from offspring postmortem did not show ZIKV; maternal samples were also virus-negative. In three dead fetuses, amniotic membranes were decomposed; internal organs from dead fetuses were also negative for ZIKV. Samples from 8 fetuses with ZIKV in amniotic membranes were used for all experimental analyses, except testing cytokines in the blood plasma where samples from all 11 liveborn piglets were tested.
Figure 5. Offspring data. (A) Experimental setup in the offspring study. (B) Mortality (Yates-corrected χ2-test; % shows live piglets), head/body size ratio (Mann-Whitney U-test), and Zika loads in individual amniotic membranes from newborn piglets (Mann-Whitney U-test). There were 11 viable piglets in the Control group with no dead piglets. In the ZIKV group, there were 11 viable and 3 dead piglets. The dotted line represents the limit of detection (LOD). The solid line represents the mean. (C) The colony-forming unit assay. The image represents a granulocyte/macrophage colony-forming unit. The first graph shows the number of CFU-GM (granulocyte/macrophage colony-forming units) in Control and Zika offspring. For each animal, the total number of colonies was counted in 6 technical well replicates and the average is shown. The second graph shows the absolute number of GMP in Control and Zika offspring. GMP: granulocyte/macrophage progenitor cells. CFU-GM were disrupted to single-cell suspension, and the total number of GMP cells was calculated from 6 technical well replicates. Solid lines represent the mean. Mann–Whitney U-test was used to compare the number of CFU-GM and GMP. In all graphs, squares and circles indicate individual piglets.
Quantitative IHC did not show changes in sialoadhesin in the liver, spleen, and lymph nodes of affected offspring; tissues from both ZIKV and Control offspring had a similar pattern of staining (data not shown). Also, there was no difference in sialoadhesin expression by the western blot in the whole bone marrow cells from ZIKV offspring (D). However, more sensitive kinome analysis showed bone marrow molecular footprints: In total, ZIKV offspring had 180 differently phosphorylated protein sites (E and F; Table S1-C). Interestingly, most proteins that were differently phosphorylated in fetuses (except MSK2, SOC3, AR, JNK1, SMAD6, BAX, TGFBR2, IRAK4; Table S1-A), including proteins related to type I IFN responses, biology of HSC niche, hematopoiesis, and bone marrow cancer, were also affected in offspring (Table S1-C). Accordingly, among the top twelve affected biological pathways in fetal (Table S1-B) and offspring (Table S1-D) bone marrow cells, there were nine commonly affected pathways related to bone marrow function, HSC niche function, immune responses, type I IFN responses, and cancer (Table S1-E). One of the top three affected pathways in fetal and offspring bone marrow cells was “osteoclast differentiation” (Table S1-B and D). Interestingly, it has been recently shown that ZIKV infection can affect osteoclast and osteoblast functions [Citation28, Citation29]. In addition to commonly affected proteins in fetuses and offspring, we also identified proteins uniquely affected in ZIKV offspring that were related to hematopoiesis (13 proteins) and bone marrow cancer (3 proteins) (; references are in File S1).
Next, we studied pathology in CD117+ HSPCs isolated from the offspring bone marrow. The colony-forming unit assay—the commonly used assay to measure proliferation and differentiation of HSPCs in humans, mice, and pigs [Citation30, Citation31]—showed that ZIKV offspring have a trend to the higher number of granulocyte/macrophage colony-forming units and individual granulocyte/macrophage progenitor cells (C); however, the difference was not statistically significant. Next, we identified whether silent fetal infection causes enduring molecular footprints in HSPCs of offspring. We used RNA-seq to compare global gene expression in bone marrow HSPCs from ZIKV and Control groups. With the individual gene analysis, two genes had significantly altered expression: thioredoxin-interacting protein (TXNIP) and methenyltetrahydrofolate synthetase domain containing (MTHFSD) protein (Table S2-A). Interestingly, TXNIP is essential for maintaining HSC quiescence and the interaction between HSCs and the bone marrow niche; it is induced by oxidative stress and maintains the hematopoietic cells by regulating intracellular ROS during oxidative stress [Citation32–34]. Functional set enrichment of Gene Ontology (GO) biological processes, which provides a more sensitive analysis than single-gene analysis, showed more significant molecular footprints in HSPCs. In total, ZIKV offspring had 93 affected GO biological processes (Table S2-B). Many positively enriched GO processes were related to immune responses and inflammation. Pathways involved in HSPC biology were also positively enriched, specifically “stem cell division,” “response to interleukin 3,” and “negative regulation of interleukin 12 production” [Citation35, Citation36] (Table S2-B).
To summarize, silent in utero ZIKV infection causes enduring molecular footprints in the offspring bone marrow.
In utero Zika virus infection causes functional and molecular pathology in PBMCs of offspring
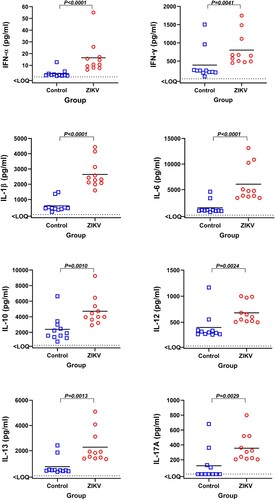
First, we quantified and compared cytokine responses in the blood plasma of offspring collected 26 days after birth. ZIKV offspring showed different pattern of cytokines compared to Control offspring (). Specifically, ZIKV offspring had increased levels of immune activation and inflammatory cytokines: IFN-α, IFN-γ, IL-1β, IL-6, and IL-17a. Anti-inflammatory cytokine IL-10 and a mediator of allergic inflammation IL-13 were also increased in ZIKV offspring.
Figure 6. Cytokines in the blood plasma of offspring affected by silent Zika virus infection during in utero life. Blood plasma samples from Control and Zika offspring were collected 26 days after birth. Dotted lines represent the limit of quantification (LOQ); solid lines represent mean values. Cytokine levels in offspring blood plasma were compared with Mann-Whitney U-test. Squares and circles indicate individual piglets.
Second, we quantified and compared CD3 + and CD14 + cells in PBMCs from both offspring groups using flow cytometry. Zika offspring showed statistically significant expansion of the CD3+ T-cells population and reduction of CD14+ T-cell population (A).
Figure 7. Functional and molecular pathology in PBMCs of offspring affected by silent Zika virus infection during in utero life. (A) Quantification of CD3 + and CD14 + cells in offspring PBMCs (Mann–Whitney U-test). Squares and circles indicate individual piglets. (B) Lipopolysaccharide (LPS)-induced IL-1β response in offspring PBMCs (Mann–Whitney U-test). Solid lines represent the mean. The dotted line represents the limit of quantification (LOQ). (C) Molecular pathology network in PBMCs of offspring affected with silent in utero Zika virus infection. The enrichment map of significantly altered GO biological processes. Red are pathways with positive and blue are with negative enrichment. All subnetworks with FDR-adjusted p < 0.25 and at least three connected nodes are shown. See raw data in Table S2-D for individual GO biological processes.
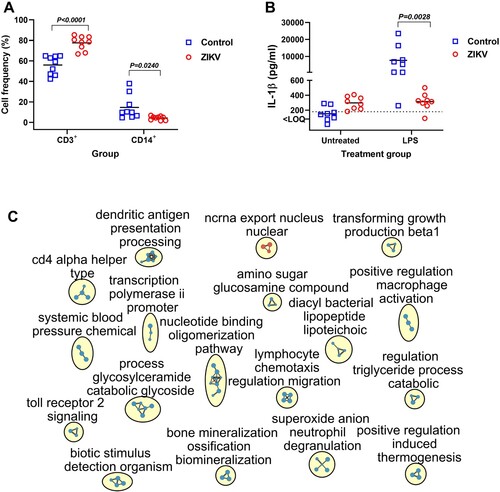
Third, we quantified and compared IL-1β responses in PBMCs from both offspring groups after stimulation with LPS. Interestingly, PBMCs from ZIKV offspring produced considerably higher levels of IL-1β before stimulation with LPS (B). After stimulation with LPS, as expected, PBMCs from Control offspring produced high levels of IL-1β, while PBMCs from ZIKV offspring showed unexpected shutdown of IL-1β response (B).
Finally, we compared global gene expression in PBMCs from Control and ZIKV offspring. Along with profound gene transcriptional changes (1,294 affected genes, p < 0.05, log2 fold change > 1; Table S2-C), ZIKV offspring had 197 affected GO biological processes (Table S2-D; C). Genes with altered expression in PBMCs were enriched for 37 GO processes related to immune responses (Table S2-D; C). Interestingly, in accordance with abnormal PBMC responses to in vitro LPS stimulation (B), eight GO processes related to Toll-like receptor 2, 4, and 9 pathways and LPS-mediated signaling were negatively enriched (Table S2-D; C).
Collectively, silent in utero ZIKV infection causes functional and molecular footprints in offspring PBMCs with different phenotypes, abnormal response to LPS stimulation, and altered global gene expression.
Discussion
Our overall goal was to find out whether silent in utero ZIKV infection can cause functional and molecular footprints in the bone marrow and blood of fetuses and offspring. There are two key findings from this study. First, silent ZIKV infection caused multi-organ inflammation in fetuses and local inflammation in the fetal bone marrow. Second, silent in utero ZIKV infection caused molecular footprints in the offspring bone marrow and PBMCs. Our data also suggest the persistent nature of these footprints. Moreover, we dissected and studied specific fetal inflammation and immunopathology during isolated in utero virus infection with no infection in maternal tissues. Infection in mother and fetus may affect offspring differently, and many previous sequelae studies in offspring showed the importance of maternal inflammation. In contrast, the specific role of fetal inflammation during viral infections is largely unknown. Here we showed that fetuses may develop inflammation independently from maternal infectious status, and the fetal bone marrow is not protected from inflammation.
Inflammation in different fetal organs was most probably caused directly by ZIKV replication and indirectly by increased levels of proinflammatory cytokines in fetal blood because sialoadhesin overexpression was detected both in virus-positive and virus-negative organs. While we found the positive correlation between sialoadhesin and virus loads (C, Suppl. Figure S1), it is difficult to identify a direct relationship between viral loads in specific tissues, concentrations of systemic cytokines, and sialoadhesin overexpression in tissues because in the present model fetuses were sampled at a single time after inoculation, and we cannot exclude earlier direct virus replication in tissues that follows by virus clearance before sampling.
Systemic inflammation may affect the composition and functions of bone marrow cells in adult animals [Citation37–39] Only one clinical study described bone marrow thrombocytopenic purpura in two adult humans infected with ZIKV [Citation40]. Here, we found inflammation and molecular footprints in the fetal bone marrow. Molecular footprints in the bone marrow were probably caused indirectly by increased levels of proinflammatory cytokines in blood because whole bone marrow cells were ZIKV-negative in the sensitive PCR test. In support, systemic inflammation and systemic cytokines can affect JAK2/STAT3 signaling (which was affected in the bone marrow of fetuses and offspring, ) in tissues of mice [Citation41]. However, like in other virus-negative organs, we cannot exclude earlier direct virus replication followed by virus clearance before sampling.
Next, we were interested to find out whether ZIKV-induced bone marrow footprints in fetuses persist after birth in offspring. While newborns only had ZIKV in their individual amniotic membranes and the virus was cleared in lymph nodes, exposure to silent in utero ZIKV infection caused the molecular footprints in the bone marrow of offspring after birth. Whole bone marrow cells from affected offspring retained altered kinase activity profiles like in bone marrow cells from affected fetuses suggesting the persistent nature of molecular footprints. The similar ZIKV-induced molecular footprint in the bone marrow of fetuses and offspring is remarkable because fetal and offspring studies were conducted as two independent experiments.
The higher number of proteins with altered phosphorylation in offspring than in fetuses suggests evolving molecular changes in the bone marrow during fetal and subsequently during offspring development. Accordingly, in utero-acquired progressively developing pathology is known; for example, immune activation during mouse pregnancy causes transiently elevated brain cytokines in offspring at birth, decreased levels postnatally, and then elevated levels during adulthood [Citation42].
The primary function of bone marrow is to maintain and replenish the constellation of mature short-living blood cells continuously for the entire life. In accordance with molecular changes in the bone marrow, ZIKV offspring had phenotypical, functional, and molecular changes in PBMCs. Phenotypical, functional, and molecular changes in PBMCs were identified in offspring after birth suggesting the persistent nature of footprints. Phenotypically, ZIKV offspring had significantly increased and decreased CD3 + and CD14 + cells, which is in agreement with studies in adult human patients and macaques [Citation43, Citation44]. Functionally, PBMCs from ZIKV offspring showed IL-1β shutdown after LPS stimulation; RNA-seq data also confirmed many affected genes related to LPS-mediated signaling. Interestingly, while we cannot directly compare our data from virus-negative offspring affected with ZIKV in utero and data from in vitro infected mouse bone marrow-derived macrophages, ZIKV infection in these mouse cells inhibited inflammasome activation and failed to trigger the secretion of IL-1β even in the presence of LPS priming [Citation45]. Another rhesus monkey study which is more suitable for direct comparison, showed that repeated episodes of maternal stress during pregnancy lead to significantly lower LPS-induced cytokine responses in PBMCs of juvenile offspring [Citation46]. Stimulation and release of IL-1β upon recognition of bacterial LPS activate protective mechanisms of monocytes, macrophages, and neutrophils and induce Th1 and Th17 adaptive cellular responses [Citation47]. In addition to altered IL-1β responses, other immunological and molecular footprints identified in the present study may affect the outcomes of infections. Thus, it will be essential to determine whether offspring affected with silent in utero ZIKV infection have higher risks of bacterial and viral infections. Significantly altered levels of eight tested cytokines in offspring blood plasma () also support PBMC dysfunction. For example, ZIKV-affected offspring have higher levels of IL-1β in blood plasma. In accordance with this in vivo finding, PBMCs from ZIKV-affected offspring produced considerably higher levels of IL-1β ex vivo before stimulation with LPS (B). Increased levels of IL-1β in both in vivo and ex vivo experimental systems suggest that increased cytokines in the blood plasma at least partially originate from PBMCs.
Our data suggest that altered cytokine profiles in offspring blood plasma, changes in the bone marrow and PBMCs are sequelae of previous exposure to in utero ZIKV infection. First, while we found high virus loads in individual amniotic membranes surrounding each newborn piglet at birth, the virus RNA was not identified in the liver, spleen, testis, mesenteric and bronchial lymph nodes, whole bone marrow cells, CD117 + progenitor cells, blood plasma, and PBMCs of piglets. Second, during in utero infection, we found immune activation and inflammation in fetuses. Immunological sequela in offspring after adversities that affect fetal immunity is known. For example, maternal immune activation (induced by polyI:C) during mouse pregnancy affects fetuses and causes altered brain cytokines in offspring at birth, postnatally, and during adulthood [Citation42]. While unlikely, offspring may still carry the persistent ZIKV in untested tissues, and this unnoticed persistent infection may at least partially contribute to altered cytokine profiles, molecular changes in the bone marrow, and PBMC pathology.
Unique properties of bone marrow HSPCs are the long lifespan and multilineage differentiation that support renewal of blood cells. In the present study, ZIKV offspring showed only trends in HSPC alterations. Our findings in PBMCs however support these trends: The life span of peripheral blood monocytes is 1–7 days [Citation48], and monocytes should be continuously replenished by progenitors differentiated from bone marrow HSPCs. Thus, the observed PBMC pathology in ZIKV offspring sampled 4–6 weeks after birth may not directly represent residual or evolved changes in fetal PBMCs. In contrast, it suggests that pathology in offspring PBMCs—at least pathology determined by changes in short-living LPS-responsive monocytes—is acquired during differentiation from long-living self-renewing bone marrow HSPCs, via for example epigenetic memory and epigenetic inheritance [Citation49]. However, other peripheral blood cells, for example T cells, have a much longer life span [Citation50]; thus, specific T-cell pathology in offspring may directly represent residual or evolved changes in fetal PBMCs. Further studies are needed to better understand the complexity of in utero-acquired PBMC pathology in offspring, identify affected blood cell populations, and the role of bone marrow HSPCs.
The limitation of the present study is that we used different ZIKV strains for fetal and offspring inoculation; however, both strains are from the Asian lineage. Moreover, the similar pattern of molecular footprints in the bone marrow of fetuses and offspring induced by closely related but different virus strains suggests that the bone marrow sequelae are not strain-specific. Another limitation is that we used one pregnant pig per Control and ZIKV groups in fetal and offspring studies. However, each pregnant pig provides 8–15 fetuses or offspring. Each fetus and offspring is a replicate that provides sufficient data for statistical comparison. The similar ZIKV-induced molecular footprints in the bone marrow of fetuses and offspring in two independent experiments strongly support findings. To our knowledge, together with our previous studies [Citation9, Citation17], currently, this is the largest ZIKV offspring study with a non-rodent large animal model. Also, the model provides relevant data because pigs and humans have similar immune responses, and fetal and postnatal development [Citation51, Citation52].
Collectively, we identified that silent in utero ZIKV infection causes inflammation in fetal internal organs, including inflammation in the bone marrow. We also discovered that silent in utero ZIKV infection causes enduring sequelae in the offspring bone marrow and PBMCs. These findings should be considered in a broader clinical context because of growing concerns about health sequelae in cohorts of children affected with congenital ZIKV infection in the Americas, including increased blood cytokines and systemic inflammation [Citation7, Citation8, Citation12–14.] Understanding virus-induced molecular mechanisms of immune activation and inflammation in fetuses may provide targets for early in utero interventions. Also, the identification of early biomarkers of in utero-acquired immunopathology in offspring may help alleviate long-term sequelae.
Author contributions
Conceptualization: UK. Investigation: DU, IV, SL, SN, UK. Data analysis: DU, IV, SL, SN, UK. Funding: UK. Resources: UK and SN. Writing—original draft preparation: DU and UK. Writing—review and editing: DU, IV, SL, SN, UK.
Supplemental Material
Download Zip (5.1 MB)Acknowledgments
We thank VIDO animal care technicians and veterinarians for their help with animal experiments. We thank Dr. Hans Nauwynck for his generous gift of monoclonal antibodies against porcine sialoadhesin. Published as VIDO manuscript series number 984.
Data availability
An accession number for raw RNA-seq data is PRJNA814280 in NCBI BioProject.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Corrigan JJ. Hematologic manifestations of congenital infections. Clin Perinatol. 1981;8:499–507.
- Schwartz DA. Viral infection, proliferation, and hyperplasia of Hofbauer cells and absence of inflammation characterize the placental pathology of fetuses with congenital Zika virus infection. Arch Gynecol Obstet. 2017;295(6):1361–1368. doi:10.1007/s00404-017-4361-5.
- Wu K-Y, Zuo G-L, Li X-F, et al. Vertical transmission of Zika virus targeting the radial glial cells affects cortex development of offspring mice. Cell Res 2016. doi:10.1038/cr.2016.58.
- Schouest B, Peterson TA, Szeltner DM, et al. Transcriptional signatures of Zika virus infection in astrocytes. J Neurovirol. 2021;27:116–125.
- Li C, Xu D, Ye Q, et al. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell. 2016. doi:10.1016/j.stem.2016.04.017.
- Büttner C, Heer M, Traichel J, et al. Zika virus-mediated death of hippocampal neurons is independent from maturation state. Front Cell Neurosci. 2019;13; doi:10.3389/FNCEL.2019.00389.
- Vinhaes CL, Arriaga MB, de Almeida BL, et al. Newborns with Zika virus-associated microcephaly exhibit marked systemic inflammatory imbalance. J Infect Dis. 2020;222:670–680.
- Nascimento-Carvalho GC, Nascimento-Carvalho EC, Ramos CL, et al. Zika-exposed microcephalic neonates exhibit higher degree of inflammatory imbalance in cerebrospinal fluid. Sci Rep. 2021;11; doi:10.1038/S41598-021-87895-4.
- Trus I, Udenze D, Cox B, et al. Subclinical in utero Zika virus infection is associated with interferon alpha sequelae and sex-specific molecular brain pathology in asymptomatic porcine offspring. PLoS Pathog. 2019;15:e1008038.
- Nogueira ML, Nery Júnior NRR, Estofolete CF, et al. Adverse birth outcomes associated with Zika virus exposure during pregnancy in São José do Rio Preto, Brazil. Clin Microbiol Infect. 2018;24:646–652.
- Stanelle-Bertram S, Walendy-Gnirß K, Speiseder T, et al. Male offspring born to mildly ZIKV-infected mice are at risk of developing neurocognitive disorders in adulthood. Nat Microbiol. 2018;3:1161–1174.
- Nielsen-Saines K, Brasil P, Kerin T, et al. Delayed childhood neurodevelopment and neurosensory alterations in the second year of life in a prospective cohort of ZIKV-exposed children. Nat Med. 2019. doi:10.1038/s41591-019-0496-1.
- Mulkey SB, Arroyave-Wessel M, Peyton C, et al. Neurodevelopmental abnormalities in children with in utero Zika virus exposure without congenital Zika syndrome. JAMA Pediatr. 2020. doi:10.1001/jamapediatrics.2019.5204.
- Mulkey SB, DeBiasi RL. Do not judge a book by its cover: critical need for longitudinal neurodevelopmental assessment of in utero Zika-exposed children. Am J Trop Med Hyg. 2020;102:913–914.
- Trus I, Darbellay J, Huang Y, et al. Persistent Zika virus infection in porcine conceptuses is associated with elevated in utero cortisol levels. Virulence. 2018;9(1):1338–1343.
- Udenze D, Trus I, Berube N, et al. The African strain of Zika virus causes more severein uteroinfection than Asian strain in a porcine fetal transmission model. Emerg Microbes Infect. 2019;8:1098–1107. doi:10.1080/22221751.2019.1644967.
- Darbellay J, Cox B, Lai K, et al. Zika virus causes persistent infection in porcine conceptuses and may impair health in offspring. EBioMedicine. 2017;25:73–86. doi:10.1016/j.ebiom.2017.09.021.
- Adams Waldorf KM, Nelson BR, Stencel-Baerenwald JE, et al. Congenital Zika virus infection as a silent pathology with loss of neurogenic output in the fetal brain. Nat Med. 2018;24:368–374.
- Wichgers Schreur PJ, van Keulen L, Anjema D, et al. Microencephaly in fetal piglets following in utero inoculation of Zika virus. Emerg Microbes Infect. 2018;7:42.
- Daniel U, Ivan T, Henry M, Nathalie B, Uladzimir K. The Isolated in Utero Environment Is Conducive to the Emergence of RNA and DNA Virus Variants. Viruses. 2021. doi:10.3390/v13091827.
- Trus I, Walker S, Fuchs M, et al. A Porcine Model of Zika Virus Infection to Profile the In Utero Interferon Alpha Response. Methods Mol Biol. 2020;2142:181–195.
- Foo S-S, Chen W, Chan Y, et al. Biomarkers and immunoprofiles associated with fetal abnormalities of ZIKV-positive pregnancies. JCI Insight. 2018;3; doi:10.1172/jci.insight.124152.
- Michlmayr D, Eun-Young Kim EY, Rahman AH, et al Comprehensive immunoprofiling of pediatric Zika reveals key role for monocytes in the acute phase and no effect of prior dengue virus infection. Cell Rep. 2020;31:107569.
- Fenutria R, Maringer K, Potla U, et al. Zika virus infection in pregnant rhesus macaques causes placental dysfunction and immunopathology. Msphere. 2021;6; doi:10.1128/MSPHERE.00505-21.
- Dorritie KA, McCubrey JA, Johnson DE. STAT transcription factors in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leuk. 2014;28(2):248–257.
- Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342.
- Pillai M, Torok-Storb B, Iwata M. Expression and function of IL-7 receptors in marrow stromal cells. Leuk Lymphoma. 2004;45:2403–2408.
- Mumtaz N, Koedam M, van den Doel PB, et al. Zika virus infection perturbs osteoblast function. Sci Rep. 2018;8:16975.
- Mumtaz N, Koedam M, Van Leeuwen JPTM, et al. Zika virus infects human osteoclasts and blocks differentiation and bone resorption. Emerg Microbes Infect. 2022;11:1621–1634. doi:10.1080/2222175120222086069.
- Kondo M. Lymphoid and myeloid lineage commitment in multipotent hematopoietic progenitors. Immunol Rev. 2010;238:37.
- Ohshima S, Mori S, Shigenari A, et al. Differentiation ability of multipotent hematopoietic stem/progenitor cells detected by a porcine specific anti-CD117 monoclonal antibody. Biosci Trends. 2014;8:308–315.
- Jung H, Kim MJ, Kim DO, et al. TXNIP maintains the hematopoietic cell pool by switching the function of p53 under oxidative stress. Cell Metab. 2013;18:75–85.
- Jeong M, Piao Z-H, Kim MS, et al. Thioredoxin-interacting protein regulates hematopoietic stem cell quiescence and mobilization under stress conditions. J Immunol. 2009;183:2495–2505.
- Jung H, Choi I. Thioredoxin-interacting protein, hematopoietic stem cells, and hematopoiesis. Curr Opin Hematol. 2014;21:265–270.
- Ihle JN. Interleukin-3 and hematopoiesis. Chem Immunol. 1992;51:65–106.
- Jacobsen SE, Veiby OP, Smeland EB. Cytotoxic lymphocyte maturation factor (interleukin 12) is a synergistic growth factor for hematopoietic stem cells. J Exp Med. 1993;178:413–418.
- Leimkühler NB, Schneider RK. Inflammatory bone marrow microenvironment. Hematol Am Soc Hematol Educ Progr. 2019;2019:294.
- Pascutti MF, Erkelens MN, Nolte MA. Impact of viral infections on hematopoiesis: from beneficial to detrimental effects on bone marrow output. Front Immunol. 2016;7:364.
- Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood. 2017;130:1693.
- Chraïbi S, Najioullah F, Bourdin C, et al. Two cases of thrombocytopenic purpura at onset of Zika virus infection. J Clin Virol. 2016;83:61–62.
- Kim YK, Lee JY, Suh HN. Cytokine-induced JAK2-STAT3 activates tissue regeneration under systemic or local inflammation. Int J Mol Sci. 2022;23; doi:10.3390/IJMS23042262.
- Garay PA, Hsiao EY, Patterson PH, et al. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav Immun. 2013;31:54–68.
- Cimini E, Castilletti C, Sacchi A, et al. Human Zika infection induces a reduction of IFN-γ producing CD4 T-cells and a parallel expansion of effector Vδ2 T-cells. Sci Rep. 2017;7; doi:10.1038/S41598-017-06536-X.
- Silveira EL V, Rogers KA, Gumber S, et al. Immune cell dynamics in rhesus macaques infected with a Brazilian strain of Zika virus. J Immunol. 2017;199:1003–1011.
- Gim E, Shim DW, Hwang I, et al. Zika virus impairs host NLRP3-mediated inflammasome activation in an NS3-dependent manner. Immune Netw. 2019;19; doi:10.4110/IN.2019.19.E40.
- Coe CL, Kramer M, Kirschbaum C, et al. Prenatal stress diminishes the cytokine response of leukocytes to endotoxin stimulation in juvenile rhesus monkeys. J Clin Endocrinol Metab. 2002;87:675–681.
- Netea MG, Simon A, Van De Veerdonk F, et al. IL-1β processing in host defense: beyond the inflammasomes. PLoS Pathog. 2010;6:e1000661.
- Patel AA, Zhang Y, Fullerton JN, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med. 2017;214:1913–1923.
- Lacal I, Ventura R. Epigenetic inheritance: concepts, mechanisms and perspectives. Front Mol Neurosci. 2018;11:292.
- Vukmanovic-Stejic M, Zhang Y, Cook J, et al. Human CD4+ CD25hi Foxp3 + regulatory T cells are derived by rapid turnover of memory populations in vivo. J Clin Invest. 2006;116:2423–2433.
- Meurens F, Summerfield A, Nauwynck H, et al. The pig: a model for human infectious diseases. Trends Microbiol. 2012;20:50–57.
- Lind NM, Moustgaard A, Jelsing J, et al. The use of pigs in neuroscience: modeling brain disorders. Neurosci Biobehav Rev. 2007;31:728–751.