
ABSTRACT
Coxsackievirus A16 (CA16), a main causative agent of hand, foot, and mouth disease (HFMD), has become a serious public health concern in the Asia-Pacific region. Here, we generated an anti-CA16 monoclonal antibody, DMA2017, derived from an epidemic strain CA16. Surprisingly, although DMA2017 could not neutralize the original and circulating CA16 strains in vitro, the passive transfer of DMA2017 (10 μg/g) could protect suckling mice from a lethal challenge with CA16 in vivo. Then, we confirmed the protective effect of DMA2017 relies on the Fc-dependent effector functions, such as antibody-dependent cellular cytotoxicity (ADCC). The linear epitope of DMA2017 was mapped by phage display technique to a conserved patch spanning residues 143–148 (NSHPPY) of the VP2 EF-loop of CA16. DMA2017 could inhibit the binding of the antibodies present in the sera of naturally infected children to CA16, indicating that the epitope of DMA2017 is immunodominant for CA16. Our results confirm, for the first time, that a potential preventive and therapeutic effect could be mediated by a non-neutralizing antibody elicited against CA16. These findings bring a hitherto understudied protective role of non-neutralizing antibodies during viral infections into the spotlight and provide a new perspective on the design and evaluation of CA16 vaccines.
Introduction
Coxsackievirus A16 (CA16) belongs to the Enterovirus genus of the Picornaviridae family and is a major causative agent of hand, foot, and mouth disease (HFMD) [Citation1]. Each year seasonal outbreaks of HFMD are reported globally [Citation2]. The disease is prevalent in young children, particularly in those under five years of age [Citation3]. CA16 is a single, positive-stranded RNA virus with a capsid of icosahedral symmetry. CA16 has been classified into three genogroups and several subgroups (A, B1a, B1b, B2a, B2b and C) based on phylogenetic analyses of the VP4 or VP1 gene [Citation1,Citation4]. B1 and B2 constituted the major epidemic strains in recent years [Citation5,Citation6].
CA16 was first isolated in 1951 [Citation7]. Large outbreaks associated with CA16 have been reported worldwide, including in Australia, England and Wales, Singapore, India and China [Citation8]. Most CA16-associated HFMD infections present mild symptoms, such as skin rash, mucosal blister and fever [Citation9]. However, recent clinical data have shown that CA16 infections can lead to severe neurological complications and even lethal myocarditis [Citation10]. Enterovirus 71 (EV71) is another major pathogen of HFMD. The coinfection of CA16 and EV71 causes more severe complications of the nervous system, worse conditions, longer duration and even higher critical illness transfer rates [Citation11]. Concerningly, there are no effective therapeutics or vaccines available for protection against CA16 infections.
Neutralizing antibody titer is a recognized key indicator of the efficacy of vaccines against enteroviruses. In the evaluation of enterovirus vaccines, more attention is often paid to neutralizing antibodies, and the role of non-neutralizing antibodies is ignored. However, in the development of CA16 vaccines by some manufacturers, antibodies induced by the CA16 vaccine with low neutralizing titer showed protective effects in animals. Moreover, previous research in our laboratory has found that poorly neutralizing polyclonal antibody elicited by circulating strains of CA16 could prevent a lethal challenge [Citation12]. Similar instances of non-neutralizing antibodies conferring protection have also been reported for West Nile Virus (WNV), flaviviruses, alphaviruses, rhabdoviruses, and coronaviruses [Citation13]. Therefore, whether the evaluation of neutralizing antibodies can adequately reflect the effectiveness of the CA16 vaccine has become one of the key issues, which hindering the evaluation and development of the CA16 vaccine. To address this issue, we first developed a monoclonal antibody DMA2017 against CA16, which could protect 100% suckling mice from the lethal challenge of CA16 in vivo. These findings highlight important roles for non-neutralizing antibodies during the course of viral infections and raise a new perspective on the design as well as evaluation of CA16 vaccines.
Materials and methods
Cells, virus, monoclonal antibody and human serum
Human muscular rhabdomyosarcoma (RD) cells were cultivated in Minimal Essential Medium (MEM, THERMO) supplemented with 10% fetal bovine serum (FBS) at 37°C in 5% CO2 atmosphere. All Coxsackievirus A16 (CA16), including G10, 190, 731, BJCA08 and 523/Enterovirus 71(EV71) were grown in RD cells (Supplementary Table 1). The anti-CA16 monoclonal antibody DMA2017 was produced and purified by Autobio company with the purified NIFDCK02/CA16. Seven human sera were collected from healthy children aged 12–35 months in Jiangsu in 2012. Among them, 5403, 5407, 7137 and 6807 were the convalescent serum samples after CA16 natural infection with neutralizing antibody (NT) titers of 1536, 1024, 1536 and 768, respectively. By contrast, 5406, 6776 and 6850 with NT titers <8 served as the negative serum samples. Written informed consent was received from all donors’ guardians involved in our study.
Production of DMA2017
CA16 was propagated and concentrated from virus-infected RD cells, emulsified in complete Freund’s adjuvant at a column ratio of 1:1. Healthy female BALB/C mice aged 6–8 weeks were immunized subcutaneously with a dose of 10 μg/mouse. The mice have boosted twice at 21 days intervals with CA16 in incomplete Freund’s adjuvant. After the final injection, splenocytes from the immunized mice were fused with Sp2/0-Ag-14. Antibodies against CA16 in hybridoma supernatant were screened by enzyme-linked immunosorbent assay (ELISA). Positive wells were cloned at least twice. Ascetic fluid produced from a single clone of positive cells was purified by protein A chromatography (GE Healthcare). The eluted antibodies were desalted by Sephadex G-25 to obtain purified DMA2017.
Neutralization assays
An in vitro neutralization assay was performed to test the ability of DMA2017 to neutralize CA16. Briefly, RD cells were seeded at 2 × 104 cells per well into 96-well plates (Corning). DMA2017 (1 mg/mL) was serially diluted in a 2-fold dilution from 1:8 to 1:16,384 and incubated with 100 cell culture infective dose 50% (CCID50) of CA16 at 37°C for 2 h. The virus/MAb mixtures were added into starvation cells and then incubated at 37°C for 7 days. The neutralization titer was defined as the highest dilution with over 50% cytopathic effect. Each assay was repeated three times under identical conditions.
Evaluation of in vivo protective effect of DMA2017 in vivo
All animal experiments were carried out in accordance with the guidelines of the National Institute for Food and Drug Control for the Care and Use of Laboratory Animals. Suckling mice were used to find out whether the anti-CA16 DMA2017 could confer protection against CA16 infections. All groups of newborn BALB/c mice (n = 5) were challenged with BJCA08/CA16 (54 CCID50). Then, five groups were injected intraperitoneally (i.p.) with serially 5-fold diluted CA16 DMA2017 (10–0.016 μg/g, respectively), and one control group was treated with the same volume of Minimal Essential Medium (MEM). To further explore the protective mechanism, the Fab and F(ab’)2 segment were acquired by papain(Thermo, 20341) and pepsin(Thermo, 20343). 10 μg/g Fab, F(ab’)2, DMA2017 and PBS were injected intraperitoneally (i.p.) to protect suckling mice from BJCA08/CA16 attack. Mice were monitored for survival and severity of clinical symptoms for 21 days. The grade of clinical disease was scored as follows: 0, healthy; 1, wasting/inactivity; 2, forelimb weakness; 3, hind legs paralysis; 4, quadriplegic; 5, moribund and death.
Histology and immunohistochemistry analysis
To further characterize the protective effect of DMA2017, two groups of newborn mice (n = 5) were challenged i.p with CA16 BJCA08(54 CCID50). At day 1 post-infection, one group was administered DMA2017 (10 μg/g per body weight), while the other group was given MEM medium. In the control group, the mice were challenged by MEM instead of CA16. After 7 days, the animals were euthanized, and tissues including brain, kidney, spinal cord, heart, liver, lung, intestine and limb muscle were collected and fixed in 4% paraformaldehyde for at least 2 days. Hematoxylin and eosin (HE) as well as immunohistochemistry (IHC) staining were performed as described in a previous study[Citation14]. Primary antibody against the VP1 region of CA16 was used at 1:32,000 dilution.
SDS-PAGE and Western blot analysis
BJCA08/CA16 was used to identify which capsid protein DMA2017 could bind. Proteins were resolved by SDS-PAGE and electro-blotted onto nitrocellulose membranes (Bio-Rad). The BJCA08/CA16 was subjected to 4-20% SDS-PAGE. The membrane was blocked in 5% blotting grade milk (diluted with PBS), and incubated in DMA2017 solution for 1 h, followed by incubation in HRP-conjugated goat anti-mouse IgG at 1:10,000 dilution (Dako Cytomation) for another hour. The membranes were washed three times for 5 min in 0.1% Tween-20 (diluted in TBS), and developed with ECL Western Blotting Substrate reagent (ThermoFisher), followed by color development with Amersham imager 600 (General Electric Company).
To detect the effect of enzyme digestion, the DMA2017 and digestion products Fab and F(ab’)2 were subjected to 12% non-reduced SDS-PAGE. The resulting gel was stained using Coomassie blue.
Viral RNA extraction and RT-PCR assay
Two groups of newborn mice were challenged i.p with BJCA08 (54 CCID50). At day 1 post-infection, one group was administered DMA2017 (10 μg/g per body weight), while the other group was given MEM. In the health control group, the mice were only challenged by MEM. After 7 days, three mice of all groups were euthanized, and tissues including blood, brain, kidney, spleen, heart, liver, lung, intestine and limb muscle were collected. Blood and homogenized tissues were harvested for RNA by a Mag Max viral isolation kit (Applied Biosystems, A52990). According to the instructions of the One-Step RT–PCR kit (Takara, RR096A), cDNA was synthesized from RNA by reverse transcription at 42°C for 5 min and 92°C for 10 s, followed by 40 cycles of 95°C for 5 s and 60°C for 30 s (Applied Biosystems, QuantStudio). Real-time quantitative PCR (RT–PCR) assays were performed using the following CA16-specific primes: forward primer, 5′-CACCTCCAAGCGAATGACCT-3′ and reverse primer 5′-ATCCATGCCCTGACGTGTTT-3′.
Epitope mapping
The Ph.D.-12 Phage Display Peptide Library Kit E8110S used for epitope mapping was purchased from New England Biolabs. Panning was carried out by incubating 2 × 1011 phage-displayed peptides on a plate coated with 20 μg DMA2017 followed by blocking with casein at 37°C for 1 h. Next, 4 × 1010 phage library was added and incubation was carried out for 1 h at room temperature. Unbound phage was washed away using TBST (0. 5% Tween 20 in PBS), and the specifically bound phage were eluted by adding BJCA08/CA16. Then, the eluted phage were amplified and taken through addition amplification cycles to enrich the pool in favor of binding sequences. After three rounds, individual clones were characterized by DNA sequencing.
ELISA
Plates were individually coated with 50 μg/mL of each synthetic peptide in coating buffer (0.1M NaHCO3, PH8.6) and incubated overnight at 4°C. After the end of the incubation period, the plates were blocked with casein at 37°C for 1 h. Then 1 μg/mL DMA2017 was added. After 1 h incubation at 37°C, the binding of the DMA2017 to the immobilized peptides was detected by anti-mouse IgG-HRP conjugate, followed by color development with TMB substrate. The binding efficiency was estimated by reading the absorbance at 450/630 nm on a spectrophotometer (Bio-Rad). The procedure adopted for performing binding ELISA and competitive ELISA was the same as described above for peptides ELISA. For binding ELISA, an excess of CA16 polyclonal antibody was coated on plates and incubated with different viruses at 5 × 104 CCID50/mL at 37°C for 1 h. Then DMA2017 was added (0.625 μg/well) and the plates were incubated for 30 min at 37°C. For competitive ELISA, the 96-well plates were coated at 4°C overnight with CA16. Human serum was 2-fold serially diluted (1:10–1:20480) and added to the plates. Plates were incubated at 37°C for 60 min. After this DMA2017 was added (0.0625 μg/well) and the plates were incubated further for 60 min at 37°C. Binding of DMA2017 was estimated in the assays by anti-mouse IgG-HRP conjugate, followed by color development with TMB substrate. The absorbance value (A450nm/630nm) was converted to percentage inhibition (PI) using the formula: PI (%) = (1-ODsample/ODDMA2017) *100%. The ODDMA2017 represents the well-containing DMA2017 alone.
ADCC reporter bioassay
The ADCC effect of DMA2017 was measured according to the protocol of Murine FcγRIII ADCC Bioassay (CS1779B06) from Promega. In briefly, 293 T expressing CA16 P1 was taken as the target cell and Jurkat expressing murine FcγRIII receptor as effector cell. DMA2017 or a none CA16 antibody as control was diluted in two-fold series and added into 96 well plate in triplicate. 75,000 cells per well of the effector cell and 12,500 cells per well of target cell were added to the plate, then the plate was cultured in 37°C, 5% CO2 cell culture incubator for 24 h. In the next day, add 100μL of Bright-Glo™ reagent (E2620, Promega) to each well and incubate 2 min, then the luminescence was measured.
Homology analysis
To investigate the conservation of the DMA2017 epitope among CA16 strains, sequence alignment of the amino acids spanning residues 143–148 of the VP2 from different CA16 strains was performed. A similar primary sequence analysis was also performed between CA16 and EV71 strains.
Structural analysis
Based on the published crystal structure of the capsid of CA16, the location of the epitope was analyzed using Chimera.
Statistical analysis
The data statistical analyses were performed using Graphpad prism version 9. P < 0.05 was considered as statistical significance. The health scores were shown as means. The significant differences of survival rate were analyzed by Log-rank (Mantel–Cox) tests. The significant differences of health score and viral load were analyzed by pair T-tests.
Result
Generation and characterization of monoclonal antibody DMA2017
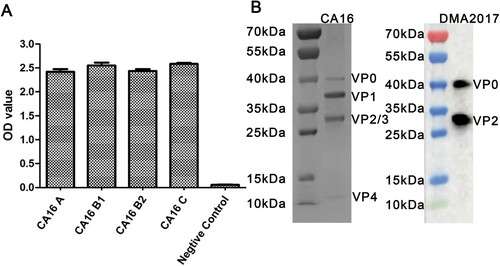
To produce antibodies against CA16, hybridomas were generated by fusing spleen cells collected from mice inoculated with the CA16 KX02 strain with myeloma cells. Antibodies against CA16 present in the supernatant of the hybridomas were screened by ELISA. Amongst 23 antibodies identified, DMA2017 was selected for further characterization, with best protection efficiency (Data not shown). The ability of DMA2017 to bind A, B1, B2 and C subgenotypes of CA16 was tested. The results showed that DMA2017 could bind different subgenotypes of CA16 ((A)). The natural CA16 empty particle contains proteins VP0, VP1, and VP3. In mature virus, VP0 undergo autocleavage to produce VP2 and VP4. Results of the Western blot analysis revealed that DMA2017 recognized VP2 as indicated by a band at 28 kDa. The antibody also recognized the precursor protein VP0, as indicated by a band at around 40 kDa ((B)). Taken together, the results of SDS-PAGE and Western Blot suggested that the linear epitope of DMA2017 was located on VP2.
Figure 1. Characterization of DMA2017. (A) Characterization of the binding ability of DMA2017 with A, B1, B2 and C subgenotypes of CA16. (B) SDS-PAGE analysis of CA16 and Western blotting of CA16 bound with DMA2017.
The neutralizing ability of DMA2017
Different CA16 viral strains covering A, B1, B2 and C subgenotypes were used for detecting the neutralizing ability of DMA2017. Anti-CA16 neutralizing MAb NA11F12 was used as a positive control [Citation15]. The titer of DMA2017 for neutralizing strains belonging to the CA16 A, B1, B2 and C subgenotypes and EV71 were below 1:8, suggesting that DMA2017 could not neutralize CA16 and EV71 ().
Table 1. Neutralization titers of DMA2017 against four CA16 subgenotypes and EV71. Note: NA11F12 is a known CA16-neutralizing antibody.
Non-neutralizing DMA2017 could protect mice from CA16 lethal attack
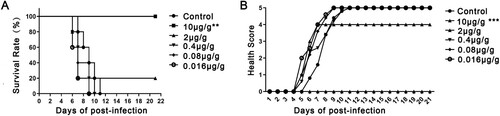
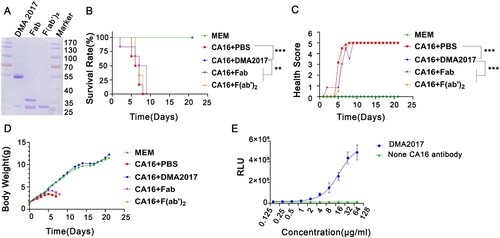
Groups of newborn BALB/c mice were challenged i.p. with BJCA08/CA16 (GenBank no. JXe81738) 54 CCID50/mouse. Test groups were administered serially diluted DMA2017 (10–0.016 μg/g), while the control group was given MEM at day 1 post-infection. Inactivity occurred in the control group treated with Minimal Essential Medium (MEM) at 5 days post-infection, and the symptoms worsened gradually to forelimb weakness, followed by paralysis of hind legs and quadriplegic, eventually leading to death at 7–11 days post-infection. In the high dose group, all the mice that received 10 μg/g DMA2017 survived and were healthy, showing no clinical symptoms of infection. The results of survival rate and health score showed statistically significant difference (P<0.01) between 10 μg/g DMA2017 treatment group and control. In contrast to the highest dose group, the survival rate of mice treated with 2 μg/g of DMA2017 reduced to 20%, and all the mice of other groups (0.4, 0.08 and 0.016 μg/g) showed inactivity and quadriplegia as the disease progressed; eventually leading to their death (). There was no significant difference between the treatment dose lower than 10 μg/g group and the control group (P > 0.05). The median effective dose (ED50) was calculated by Reed and Munch method to be 3.65 μg/g.
Figure 2. Protection efficacy of DMA2017 against CA16 challenge in suckling mice. (A) Groups of mice (n = 5) were infected with 54 CCID50 BJCA08/CA16. One day post-challenge, the mice were treated with different doses of DMA2017 (10, 2, 0.4, 0.08 and 0.016 μg/g) via intraperitoneal route. The asterisk indicates significant differences at **P < 0.01 and ***P < 0.001. (B) Clinical symptoms of mice were monitored and recorded for 21 days.
Viral load changes in DMA2017-treated mice infected with CA16
As shown in Supplementary Figure 1, CA16 viruses could be detected in all tissues and blood of MEM-treated group. The virus was ubiquitous in all tissues, such as the heart (107.97 copies/mg), liver (109.53 copies/mg), spleen (107.97 copies/mg), lung (108.84 copies/mg), kidney (108.37 copies/mg), brain (107.54 copies/mg), intestine (106.92 copies/mg), leg muscle (1011.24 copies/mg) and blood (106.61 copies/mg). However, CA16 viruses were undetected in the mice of DMA2017-treated group and control group. The viral load of DMA2017-treated group and MEM-treated group showed significant differences (P < 0.0001).
Pathological changes in DMA2017-treated mice infected with CA16
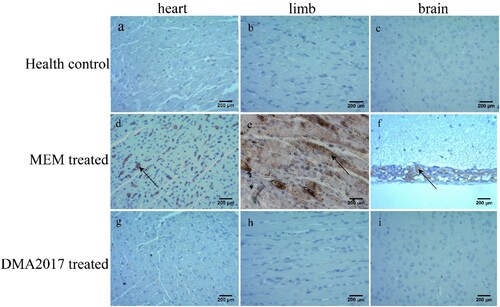
To monitor the effectiveness of the treatment of DMA2017, HE staining and IHC assays were performed mice from control (MEM-treated) and DMA2017-treated groups. Inflammatory cell infiltration was observed in the brain ((f)) and severe necrosis was found in the heart ((d)) and limb muscles ((e)) in the MEM-treated group. Positive CA16 antigen was found in the heart, limb muscle and brain in MEM-treated mice ((d–f)). However, no pathological change and CA16 antigen were detected in DMA2017-treated mice ( and (g–i)). These results show that the treatment of 10 μg/g DMA2017 was able to protect mice from CA16-induced histopathology, paralysis and death.
Figure 3. Histological examination results. In the group of health control and DMA2017 treated, no histological change was observed in the heart, brain and limb muscle. (d) In the MEM-treated group of mice, myocardial cell necrosis occurred in the heart. (e) MEM-treated mice exhibited severe muscle fiber necrosis in the limb muscle (arrow). (f) In the brain of mice, eosinophilic neuronal necrosis was observed occasionally. All the pathological changes are indicated by arrow. Representative images are shown at a magnification of 200×. Scale bar: 200 μm.
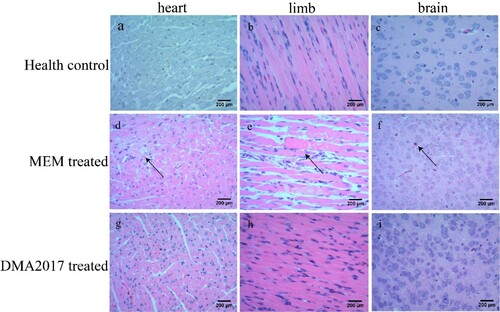
Figure 4. Immunohistochemical results. An anti-CA16 VP1 antibody was used to detect CA16 antigen. Numerous viral antigen positive reactions were observed in the heart, limb muscle and brain (arrows) in the MEM-treated mice. In contrast, no viral antigen was observed in the heart, limb muscle and brain of the health control and DMA2017-treated mice. Representative images are shown at a magnification of 200×. Scale bar: 200 μm.
Fc is essential for the protection of DMA2017 against CA16 infection
Antibody-dependent protection against viruses is facilitated by two main functions, as follows: Fab-mediated virus neutralization and Fc-dependent effector functions. Given the non-neutralization characteristic of DMA2017, we speculated that the protective effect is related to Fc fragment. The F(ab’)2 and Fab fragments of DMA2017 which lack Fc were generated by enzyme digestion ((A)) and subjected to CA16 challenge experiment. The mice treated with intact DMA2017 survived, but all the mice received F(ab’)2 and Fab of DMA2017 developed waste, limb paralysis and died ((B)). As shown in (B,C), there are statistical difference in the survival rate and health score of the DMA2017 group compared to the PBS group, Fab group and F(ab’)2 group (all P<0.01). These results indicate DMA2017 without Fc lost its protection against CA16 infection, indicating the Fc fragment is essential for DMA2017 protection. Fc fragment could mediate verities of mechanism, among which antibody-dependent cellular (ADCC) effect plays an important role in the protection of antibodies against viral infection. To figure out if the ADCC effect accounts for the protection of DMA2017 against CA16, we first measured the ADCC activity of DMA2017. As shown in (E), with the increase of DMA2017 concentration, the RLU value also increased. The ED50 of DMA2017 was 26.39 μg/mL. Consistent with our conjecture, Fc fragment is essential for DMA2017 protection, which could be through the ADCC effect.
Figure 5. The Fc fragment is essential for DMA2017 against CA16 infection. (A) SDS-PAGE of Fab and F(ab’)2. Groups of mice (n = 6) were infected with 54 CCID50 BJCA08/CA16. The mice were treated with 10 μg/g of DMA2017, Fab, F(ab’)2 or PBS via intraperitoneal route. (B)The survivorship curve of each group. (C) Clinical symptoms of mice were monitored and recorded for 21 days. (D) Weight change of each group. (E) DMA2017 mediated the ADCC between Murine FcγRIII Jurkat cell and CA16 P1 expressing 293 T. The asterisk indicates significant differences at **P < 0.01 and ***P < 0.001.
Epitope mapping of DMA2017
The positive phage clones binding to the DMA2017 were selected from the PhD.12 phage library. In the panning process, the enrichment of phage was monitored by determining the yield of phage (number of phages eluted/number of phages applied). After three rounds of selection, an approximate 370,000-fold increase in the number of eluted phages was observed (Supplementary Table 2). We randomly picked 12 individual clones after the third round of panning to test the specific interaction of the peptides with DMA2017. Sequencing of the peptides helped us deduce the amino acid sequences of the selected polypeptides that bound DMA2017. As shown in Supplementary Table 3, the alignment of the sequences revealed a number of conserved residues in the selected peptides. Nine out of twelve clones have a conserved PPY sequence. In addition, two clones share a very similar PPW sequence. There is only one amino acid residue difference; however, Tyr and Trp are both aromatic amino acids with similar chemical characteristics. Notably, we found the conserved PPY signature sequence recognized by DMA2017 in VP2, encompassing residues 146-148.
To further characterize the epitope of DMA2017, we tested the ability of TGNENSHPPYATT (residues 139–151 of VP2) to bind DMA2017 using ELISA, with YEPATFPPYYVR (the most frequent phage clone) and GSGSGSGSGSGS as positive control and negative control, respectively. The results showed that TGNENSHPPYATT could bind DMA2017 with a high affinity ((A)). Based on these results, the epitope of DMA2017 is probably located on VP2 spanning the region 139TGNENSHPPYATT151.
Figure 6. NSHPPY is the minimal epitope recognized by DMA2017. (A) DMA2017 could bind the peptide 139TGNENSHPPYATT151 located on the capsid protein VP2. (B) Assessing the interaction of individual peptides with DMA2017 by ELISA. (C)Alignment of VP2 amino acid sequence (143-148) of the different CA16 and EV71 subgenotypes. (D)An enlarged protomer of the mature CVA16 particle. Different parts of the capsid are colored as follows - VP1, blue; VP2, green; VP3, red; VP4, yellow. The epitope is shown in hot pink. (E) VP2 EF-loop, BC-loop and HI loop are signed (black arrows) [Citation16]. The epitope of DMA2017 is located in EF-loop (red arrow).
![Figure 6. NSHPPY is the minimal epitope recognized by DMA2017. (A) DMA2017 could bind the peptide 139TGNENSHPPYATT151 located on the capsid protein VP2. (B) Assessing the interaction of individual peptides with DMA2017 by ELISA. (C)Alignment of VP2 amino acid sequence (143-148) of the different CA16 and EV71 subgenotypes. (D)An enlarged protomer of the mature CVA16 particle. Different parts of the capsid are colored as follows - VP1, blue; VP2, green; VP3, red; VP4, yellow. The epitope is shown in hot pink. (E) VP2 EF-loop, BC-loop and HI loop are signed (black arrows) [Citation16]. The epitope of DMA2017 is located in EF-loop (red arrow).](/cms/asset/ce98dba3-3b67-46d3-aed6-6f3275e1da9e/temi_a_2149352_f0006_oc.jpg)
In order to identify the minimum effective epitope of DMA2017, we proceeded to screen synthetic peptides that could interact with DMA2017 by ELISA. A total of 20 overlapping peptides corresponding to the sequence of VP2 126-151aa were synthesized (GL Biochem, Shanghai). Each peptide contained 15 amino acids with 14 residues overlapping with the adjacent peptides. As shown in (B), DMA2017 interacted with peptides P7-P16, but not with P1-P6, P17-P20. There is a common stretch of sequence SHPPY between P7 and P16. While deleting the Asn143 of P15, the binding affinity declined significantly from 1.543 to 0.339. Taken together, the motif NSHPPY is the defined minimal epitope for DMA2017.
We evaluated the conservation of the NSHPPY epitope among CA16 and EV71 viruses. Analysis of 17 sequences from different CA16 and EV71 isolates covering all subgenotypes demonstrates that the epitope recognized by DMA2017 is conserved among CA16 strains. Interestingly, as shown in (C), there is a two amino acid difference between the putative epitopes of CA16 and EV71: D/N (143) and S/T (144). N (143) was found in all CA16 strains and EV71-A isolate BrCr, while the D was limited to most EV71 strains. S (144) was conserved in CA16 strains and most EV71 strains. However, the substitution of T (144) was reported in EV71-C4 China strains.
The mature CVA16 virus particle contains VP1∼4. The structure of monomer structure of the mature CA16 marked with color is shown in (D). The location of the epitope is marked in hot pink. By comparing with (E), the epitope was located at the EF loop of VP2, which is known to participate in virus expansion and genome release, together with the GH loop of VP1.
Competitive ELISA of DMA2017 against human sera
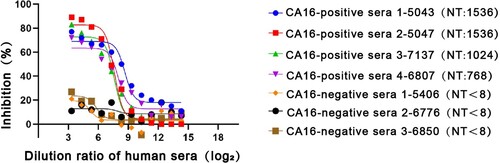
Protection in mice may not reflect a protective effect in humans. To further investigate the relative affinities of DMA2017 and antibodies present in human sera, we performed competitive ELISA assays. Four CA16-positive and three CA16-negative human serum samples from CA16-infected patients confirmed by neutralization assay were used in the study. As shown in , DMA2017 could block almost 80% of the interaction of CA16-positive human sera with CA16. The inhibition obviously decreased with increasing serial dilution of the four CA16-positive sera. In contrast, the control groups using CA16-negative human sera showed the highest inhibition of about 25%. The difference in inhibition between CA16-positive and CA16-negative human sera indicated that DMA2017 recognizes immunodominant epitopes, and the epitope bound by DMA2017 is abundant in CA16-infected humans.
Figure 7. Competitive ELISA of DMA2017 against human sera. The DMA2017 competed with four sera from convalescent children after CA16 natural infection with NT titers 1536, 1024, 1536 and 768, respectively. Three CA16-negative human sera were used as control with NT titer <8. NT represents the neutralization titer of human sera.
Discussion
CA16 and EV71 are the main causative agents of HFMD. Prophylactic EV71 vaccines have been successfully developed by three companies (Beijing Vigoo Biological, Sinovac Biotech Co. Ltd, Institute of Medical Biology) in China [Citation3]. However, the alternating as well as co-circulation of CA16 and EV71 makes it difficult to control the epidemic of HMFD. Therefore, many companies have carried out CA16 monovalent or multiple vaccines research and development. Most of them are still at the stage of clinical trials [Citation17]. The poorly neutralizing polyclonal antibodies against CA16 circulating strains and their uncanny ability to confer protection against a lethal challenge were unearthed during this stage, giving rise to questions regarding the regulatory guidelines promulgated on the design and evaluation of the performance of vaccines that put emphasis on the elicitation of neutralizing antibodies by vaccines for approval [Citation18–20].
Neutralizing antibody titer is a major indicator to evaluate the protective efficacy of many vaccines. In the middle of the last century, studies have discovered and confirmed the existence of neutralizing antibodies against enteroviruses [Citation21–23]. These antibodies were primarily used for identifying and classifying enteroviruses. Then, with the development and application of enterovirus vaccine, large number of clinical and animal experiments have demonstrated the correlation between the neutralizing antibodies and protective effect of vaccines, and consequently a protective threshold has been established [Citation24,Citation25]. In recent years, many studies have shown that CA16 could induce neutralizing antibodies which could protect mice from lethal CA16 attack. For example, anti-CA16 MAb 8C4 and NA11F12 could protect mice from CA16-induced diseases, with differing protective efficacies [Citation15,Citation26]. Some studies believed that although the antiviral activity of these anti-stem antibodies in vivo and in vitro is generally less potent than that of anti-head antibodies, they can exert better protective efficacy in vivo by cooperating with non-neutralizing antibodies [Citation27]. However, there are very few studies suggesting that non-neutralizing antibodies against enteroviruses could play a role in protection against the viruses. As a result, the role of non-neutralizing antibody is ignored in the surveillance of epidemic and CA16 vaccine development and evaluation.
In this study, based on cell culture neutralization assays, we found that a DMA2017 could not neutralize different CA16 strains covering A, B and C subgenotypes. However, 10 μg/g DMA2017 could fully protect suckling mice from lethal CA16 challenge in vivo. Beyond neutralization, Abs mediate an array of additional antiviral functions via their ability to interact with Fc-domain receptors (FcR) found on all innate immune cells [Citation28]. When the Fc fragment was removed, the DMA2017 could not protect mice from CA16 attack. It indicated that the non-neutralizing antibodies work in conjunction with other immune processes via Fc-mediated, like antibody-dependent cellular cytotoxicity (ADCC) or phagocytosis (ADCP) to clear CA16. Antibodies bound to antigen interact with Fcγ-receptor-bearing immune effector cells, such as macrophages or NK cells, through Fc region cross-linking that triggers clearance of the antigen-expressing cells [Citation29]. In our study, DMA2017 could mediate ADCC, which measured Jurkat effector cells. These findings indicated the Fc of DMA2017 play an important role in the clearance of CA16 infections in vivo. Indeed, there are numerous instances where even the most potent neutralizing MAbs are significantly compromised in their ability to confer antiviral protection in vivo when Fc-FcγR interactions are abrogated; conversely, MAbs with poor neutralizing activity in vitro assays can provide robust antiviral protection in vivo, suggesting that antiviral protection of these MAbs are dependent on activating FcγR engagement [Citation30]. Broadly neutralizing anti-H1 MAbs targeting the HA stalk required Fc-FcγR interactions to protect from a lethal HINI infection by FcRγ−/− mice and FcγR knock-out mice [Citation31]. When Fc function is ablated by the introduction of the LALA mutations, the therapeutic protection of MR228 is lost in mice, which is a non-neutralizing anti-Marburg virus MAb [Citation32]. Therefore, the role of poorly neutralizing antibodies and Fc-mediated effectors should be emphasized in the development and evaluation of vaccines.
VP1 is believed to contain important epitopes among VP1∼4 capsid proteins [Citation33]. Researchers have successfully identified six CA16 neutralizing liner epitopes within the VP1 protein. Anti-epitope antisera could neutralize CA16 strains, with titers ranging from 8∼64 [Citation34]. The epitope of DMA2017 has been identified by phage display and peptide ELISA. Sequential deletions of the VP2 capsid protein were used to exactly map the epitope to amino acids 143–148 of VP2 (NSHPPY), which is located on VP2 EF loop and is the “puff” region that forms the “southern rim” of the canyon according to the high-resolution crystal structure of CA16[Citation16]. In the atomic structure, the VP2 epitope appeared to lie close to VP1 GH loop, and it was exposed on the surface of CA16 EF loop of VP2, which has been shown to participate in the release of RNA in conjunction with the GH loop of VP1 and VP3 during CA16 infections [Citation35–37].
Because EV71 and CA16 have the closest genetic relationship among enteroviruses, we conducted a literature search of EV71 studies on similar epitopes [Citation38]. Several studies have shown that similar epitopes in VP2 of EV71 containing residues 136–150 or 141–155 were identified as neutralizing epitopes [Citation39, Citation40]. And the good protection effect of aa141-155 has been proved, but a MAb (7C7) with shorter epitope mapped to amino acids 142–146 (EDSHP) had neither neutralizing activity nor protective ability [Citation41]. It is indicated that this region of VP2 is a key epitope-related region for both CA16 and EV71. Moreover, a similar phenomenon has been observed for influenza virus where a non-neutralizing MAb 65C3 epitope is close to the epitope recognized by a neutralizing MAb 65E5 [Citation42]. The reasons underpinning the ability of an epitope to influence neutralizing need to be further researched.
To understand the conservation of the exact VP2 epitope (NSHPPY), blast analysis was carried out among different genotypes of CA16 and EV71. The results showed that it was conserved among all genotypes of CA16. However, two single amino acid variations between CA16 and EV71 family: D (143) was found in most EV71 strains, while the N (143) was limited to CA16 and EV71-A isolate BrCr. T (144) substitution was only reported in EV71-C4 China strains. In other studies, anti-EV71-B5 serum could not detect VP2 protein in the EV71-C4 strain, and anti-EV71-C4 serum contained no antibodies against VP2 [Citation40]. The mutation of serine (144) to threonine (144) in EV71 epitope confers a loss of VP2 antigenicity on EV71-C4 strains. In conclusion, this epitope is conserved in CA16 subgenotypes, and there are two amino acids variation in EV71.
In recent years, several CA16 inactivated and recombinant vaccines have been researched and developed. A vaccine effective against CA16 and other HFMD pathogens is necessary for controlling HMFD in the future. In this study, we first found out that a non-neutralizing monoclonal antibody of enterovirus CA16, DMA2017, could protect mice from lethal attack in vivo via Fc-dependent ADCC. The epitope of CA16 was located on aa143-148 of VP2 capsid, which was conserved among different genotypes of CA16. More importantly, DMA2017 could inhibit the binding of the serum of naturally infected healthy children to CA16, which suggested that the epitope of DMA2017 is immunodominant for CA16 infection. This study suggests that routine single neutralizing antibody assessment may lead to incomplete assessment of humoral immunogenicity and highlights the need to bring non-neutralizing MAbs into evaluation of CA16 vaccines, and proposes the potential of VP2 EF loop as a target site for the development of a vaccine against both CA16 and EV71.
Supplemental Material
Download MS Word (92 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Perera D, Yusof MA, Podin Y, et al. Molecular phylogeny of modern coxsackievirus A16. Arch Virol. 2007;152(6):1201–1208.
- Cox B, Hand LF. Foot, and mouth disease. JAMA. 2018 Dec 18;320(23):2492.
- Mao QY, Wang Y, Bian L, et al. EV71 vaccine, a new tool to control outbreaks of hand, foot and mouth disease (HFMD). Expert Rev Vaccines. 2016 May;15(5):599–606.
- Hosoya M, Kawasaki Y, Sato M, et al. Genetic diversity of coxsackievirus A16 associated with hand, foot, and mouth disease epidemics in Japan from 1983 to 2003. J Clin Microbiol. 2007 Jan;45(1):112–120.
- Iwai M, Masaki A, Hasegawa S, et al. Genetic changes of coxsackievirus A16 and enterovirus 71 isolated from hand, foot, and mouth disease patients in Toyama, Japan between 1981 and 2007. Jpn J Infect Dis. 2009 Jul;62(4):254–259.
- Li L, He Y, Yang H, et al. Genetic characteristics of human enterovirus 71 and coxsackievirus A16 circulating from 1999 to 2004 in Shenzhen, People's Republic of China. J Clin Microbiol. 2005 Aug;43(8):3835–3839.
- Mutterer M Sicklesgm, Plager H Feorinop. Recently classified types of Coxsackie virus, group A; behavior in tissue culture. Proc Soc Exp Biol Med. 1955 Nov;90(2):529–531.
- Mao Q, Wang Y, Yao X, et al. Coxsackievirus A16: epidemiology, diagnosis, and vaccine. Hum Vaccin Immunother. 2014;10(2):360–367.
- Chang LY, Lin TY, Huang YC, et al. Comparison of enterovirus 71 and coxsackie-virus A16 clinical illnesses during the Taiwan enterovirus epidemic, 1998. Pediatr Infect Dis J. 1999 Dec;18(12):1092–1096.
- Goto K, Sanefuji M, Kusuhara K, et al. Rhombencephalitis and coxsackievirus A16. Emerg Infect Dis. 2009 Oct;15(10):1689–1691.
- Ma S, Zhang Y, Du C, et al. Dynamic constitution of the pathogens inducing encephalitis in hand, foot and mouth disease in Kunming, 2009-2011. Jpn J Infect Dis. 2015;68(6):504–510.
- Yao X, Mao Q, Li Y, et al. Poorly neutralizing polyclonal antibody in vitro against coxsackievirus A16 circulating strains can prevent a lethal challenge in vivo. Hum Vaccin Immunother. 2018 May 4;14(5):1275–1282.
- Vogt MR, Dowd KA, Engle M, et al. Poorly neutralizing cross-reactive antibodies against the fusion loop of West Nile virus envelope protein protect in vivo via Fcgamma receptor and complement-dependent effector mechanisms. J Virol. 2011 Nov;85(22):11567–11580.
- Mao Q, Wang Y, Gao R, et al. A neonatal mouse model of coxsackievirus A16 for vaccine evaluation. J Virol. 2012 Nov;86(22):11967–11976.
- Du R, Mao Q, Hu Y, et al. A potential therapeutic neutralization monoclonal antibody specifically against multi-coxsackievirus A16 strains challenge. Hum Vaccin Immunother. 2019;15(10):2343–2350.
- Ren J, Wang X, Zhu L, et al. Structures of Coxsackievirus A16 capsids with native antigenicity: implications for particle expansion, receptor binding, and immunogenicity. J Virol. 2015 Oct;89(20):10500–10511.
- Liu Q, Yan K, Feng Y, et al. A virus-like particle vaccine for coxsackievirus A16 potently elicits neutralizing antibodies that protect mice against lethal challenge. Vaccine. 2012 Oct 19;30(47):6642–6648.
- Liang Z, Mao Q, Gao Q, et al. Establishing China's national standards of antigen content and neutralizing antibody responses for evaluation of enterovirus 71 (EV71) vaccines. Vaccine. 2011 Dec 6;29(52):9668–9674.
- A guide to clinical management and public health response for hand, foot and mouth disease (HFMD). Available from: http://www.wpro.who.int/publications/docs/Guidanceforthec linicalmanagementofHFMD.pdf.
- Japanese Encephalitis: Vaccine Preventable Diseases Surveillance Standards. Available from:https://www.who.int/publications/m/item/vaccine-preventable-diseases-surveillance-standards-je.
- Takada A, Ebihara H, Jones S, et al. Protective efficacy of neutralizing antibodies against Ebola virus infection. Vaccine. 2007 Jan 22;25(6):993–999.
- Chang LY, King CC, Hsu KH, et al. Risk factors of enterovirus 71 infection and associated hand, foot, and mouth disease/herpangina in children during an epidemic in Taiwan. Pediatrics. 2002 Jun;109(6):e88.
- Jin J, Ma H, Xu L, et al. Development of a Coxsackievirus A16 neutralization assay based on pseudoviruses for measurement of neutralizing antibody titer in human serum. J Virol Methods. 2013 Feb;187(2):362–367.
- Mizuta K, Abiko C, Murata T, et al. Frequent importation of enterovirus 71 from surrounding countries into the local community of Yamagata, Japan, between 1998 and 2003. J Clin Microbiol. 2005 Dec;43(12):6171–6175.
- Lee MS, Chang LY. Development of enterovirus 71 vaccines. Expert Rev Vaccines. 2010 Feb;9(2):149–156.
- Liu Q, Shi J, Huang X, et al. A murine model of coxsackievirus A16 infection for anti-viral evaluation. Antiviral Res. 2014 May;105:26–31.
- Hu Z, Zhao J, Zhao Y, et al. Hemagglutinin-specific non-neutralizing antibody is essential for protection provided by inactivated and viral-vectored H7N9 avian influenza vaccines in chickens. Front Vet Sci. 2020 Jan 9;6: 482.
- Gunn BM, Yu WH, Karim MM, et al. A role for Fc function in therapeutic monoclonal antibody-mediated protection against ebola virus. Cell Host Microbe. 2018 Aug 8;24(2):221–233. e5.
- Nimmerjahn F, Gordan S, Lux A. Fcγr dependent mechanisms of cytotoxic, agonistic, and neutralizing antibody activities. Trends Immunol. 2015 Jun;36(6):325–336.
- Bournazos S, Gupta A, Ravetch JV. The role of IgG Fc receptors in antibody-dependent enhancement. Nat Rev Immunol. 2020 Oct;20(10):633–643.
- DiLillo DJ, Tan GS, Palese P, et al. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat Med. 2014 Feb;20(2):143–151.
- Ilinykh PA, Huang K, Santos RI, et al. Non-neutralizing antibodies from a marburg infection survivor mediate protection by Fc-effector functions and by enhancing efficacy of other antibodies. Cell Host Microbe. 2020 Jun 10;27(6):976–991. e11.
- Li C, Wang H, Shih SR, et al. The efficacy of viral capsid inhibitors in human enterovirus infection and associated diseases. Curr Med Chem. 2007;14(8):847–856.
- Shi J, Huang X, Liu Q, et al. Identification of conserved neutralizing linear epitopes within the VP1 protein of coxsackievirus A16. Vaccine. 2013 Apr 19;31(17):2130–2136.
- Wang X, Peng W, Ren J, et al. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat Struct Mol Biol. 2012 Mar 4;19(4):424–429.
- Plevka P, Lim PY, Perera R, et al. Neutralizing antibodies can initiate genome release from human enterovirus 71. Proc Natl Acad Sci U S A. 2014 Feb 11;111(6):2134–2139.
- Xu L, He D, Yang L, et al. A broadly cross-protective vaccine presenting the neighboring epitopes within the VP1 GH loop and VP2 EF loop of enterovirus 71. Sci Rep. 2015 Aug 5;5: 12973.
- Plevka P, Perera R, Cardosa J, et al. Crystal structure of human enterovirus 71. Science. 2012 Jun 8;336(6086):1274.
- Liu CC, Chou AH, Lien SP, et al. Identification and characterization of a cross-neutralization epitope of Enterovirus 71. Vaccine. 2011 Jun 10;29(26):4362–4372.
- Xu L, He D, Li Z, et al. Protection against lethal enterovirus 71 challenge in mice by a recombinant vaccine candidate containing a broadly cross-neutralizing epitope within the VP2 EF loop. Theranostics. 2014 Feb 18;4(5):498–513.
- Kiener TK, Jia Q, Lim XF, et al. Characterization and specificity of the linear epitope of the enterovirus 71 VP2 protein. Virol J. 2012 Feb 24;9: 55.
- Breschkin AM, Ahern J, White DO. Antigenic determinants of influenza virus hemagglutinin. VIII. topography of the antigenic regions of influenza virus hemagglutinin determined by competitive radioimmunoassay with monoclonal antibodies. Virology. 1981 Aug;113(1):130–140.