
ABSTRACT
Rickettsia heilongjiangensis, a tick-borne obligate intracellular bacterium and causative agent of spotted fever in China, has attracted increasing concern regarding its capability in causing human rickettsiosis. Here, we conducted a genomic analysis of a new R. heilongjiangensis strain B8 (B8) isolated from the serum of a patient who had been bitten by a Haemaphysalis longicornis tick in Anhui Province, China. The present study sought to identify exclusive genes that might be associated with the pathogenicity of B8 using comparative genomics. Specifically, the sequences of B8 were assembled into one circular chromosome of 1,275,081 bp and predicted to contain 1447 genes. Comparative genome analyses were performed based on the genome of B8 and 28 spotted fever group (SFG) rickettsial genomes deposited in NCBI. Phylogenomic analyses indicated the B8 strain was clustered within the R. heilongjiangensis species; however, a sum of 112 and 119 B8-unique genes was identified when compared with R. heilongjiangensis and R. japonica strains, respectively. Functional annotation analyses revealed that these B8-unique genes were mainly annotated to defence mechanisms, lipid transport and metabolism, cell wall/membrane/envelope biogenesis. These data indicate B8 rather represents a previously undescribed human-pathogenic SFG rickettsia lineage, which may be an intermediate lineage of R. heilongjiangensis and R. japonica. Overall, this study isolated a new strain of R. heilongjiangensis in East-Central China for the first time, and provided potential B8-unique genetic loci that could be used for the discrimination of B8 from other R. heilongjiangensis and closely related SFG Rickettsial strains.
Introduction
Spotted Fever Group of rickettsiae (SFGR) are a neglected and understudied group of exclusively intracellular pathogens belonging to the genus Rickettsia, and are agents of a large number of emerging infectious pathogens with a worldwide distribution [Citation1,Citation2]. SFGR causes diseases called spotted fever, which is zoonotic and is mainly transmitted to the host by the bite of ticks, posing a serious threat to human health. Once SFGRs invade the host, it primarily targets and multiplies in small vascular endothelial cells and reticulocyte system, leading to increased vascular permeability [Citation3], causing vascular endothelial damage and inflammation. This insult to the vasculature leads to oedema of the tissue around the lesion and reduced blood volume of the local microcirculation, resulting in vasculitis, perivascular inflammation, and organ lesions. Patients develop rash, ulceration, fever, local lymph node enlargement, skin bruising necrosis, and finger gangrene [Citation4,Citation5]. Disease progression can be fatal with reported complications, such as hypotensive shock, acute renal failure, pulmonary oedema, meningitis, and vital organ dysfunction [Citation6].
Rickettsia heilongjiangensis is antigenically defined as spotted fever group rickettsia and causes a rickettsiosis termed Far-Eastern spotted fever (FESF) in humans. Strain 054, the type strain of R. heilongjiangensis, was first isolated from Dermacentor silavrum ticks in the Heilongjiang Province, China, in 1982 [Citation7]. Previous research shown that the R. heilongjiangensis is originally classified as one of the subgroup of R. japonica [Citation8]. Prior to this, the epidemic zone of R. heilongjiangensis was reported mainly in the northeastern of China [Citation9]. However, it strongly required to identify whether R. heilongjiangensis is epidemic in other regions of China. Therefore, the novel SFG Rickettsia isolates acquired from patients or ticks in different geographic locations are highly important to be studied, especially for the investigation of genomic and functional information, evolutionary transitions, and interaction between the host and R. heilongjiangensis.
In the present study, a newly isolated R. heilongjiangensis (B8) was sequenced and the molecular functions were investigated based on the whole-genome sequence (WGS). In addition, a comprehensive comparison among the genome of B8 and other SFG rickettsial strains was performed to better characterize the genomic divergences among these inter-/intra-species and to identify genetic traits and targets that can be used to distinguish the B8 strain from other SFG Rickettsia in the different geographic locations of China. The whole-genome sequencing and pan-genome analysis performed here were pivotal in inferring the evolutionary relationships among inter-/intra-Rickettsia species, contributing to the foci surveillance, and providing the ideal genome regions that can use as PCR targets of B8 to differentiate from other R. heilongjiangensis and related SFG Rickettsia.
Materials and methods
Isolation, purification, and genome extraction of Rickettsia
The B8 strain was isolated from human blood sample collected by the People’s Hospital of Hanshan County, Anhui province, central-eastern part of China. In order for propagation, the bacterium was cultivated in Vero cells (ATCC® CCL-81™) and incubated in modified Eagle’s minimal essential medium supplemented with 4% foetal bovine serum and 2 mM L-glutamine at 34°C in an incubator with 5% CO2 for consecutive seven days as previously described [Citation9,Citation10]. The infected cells were disrupted by vortexing with glass beads, followed by centrifugation at 500 × g for 10 min to remove host cell debris. The supernatants were centrifuged at 10,000 × g for 30 min to recapture bacterial cells. The obtained pellets were resuspended in 1× PBS and then layered on 20–50% linear Renografin gradient and centrifuged at 197,000 × g. The rickettsial layer was collected and washed using 1× PBS for three times and then suspended in culture media. The viability at ∼95% of the purified bacteria was measured using a bacterial kit (LIVE/DEAD BacLight Bacterial Viability Kits, Invitrogen). The rickettsial genomic DNA extraction was extracted a QIAamp DNA minikit (Qiagen, Germany) according to manufacturer’s protocols, eluted in a final volume of 100 µl, and stored at −80°C until use.
Ticks collection, DNA isolation, and PCR screening for positive R. heilongjiangensis
Ticks were collected by flagging over vegetation in the territory of Hanshan County near the patient lived, and the collection sites were either large forests or grasses near woodlands. The ticks were kept alive in 50 ml plastic tubes until identification and further testing in the laboratory. The ticks were firstly dipped in 75% ethanol two times (10 min for each time) and then washed twice in distilled water and air-dried prior to homogenization in 1.5 mL Eppendorf tubes with disposable pestles and subsequent DNA extraction using the QIAamp® DNA Blood Mini Kit (Qiagen, Germany) following the manufacturer’s instruction [Citation11,Citation12]. Rickettsia heilongjiangensis was identified by PCR screening with four pairs premiers ().
Table 1. The information of public Rickettsia species (retrieved from NCBI).
Whole-genome sequencing, assembly, and annotation of B8
The B8 strain isolated in this study was subjected for next-generation sequencing by using the Nextera XT DNA Sample Preparation kit (Illumina, San Diego, CA, USA) and the Illumina MiSeq platform. After filtering adaptors and low-quality reads by Fastp (version 0.20.1) using default settings [Citation13], then we performed de novo assembly of the genome sequence by using SPAdes v 3.12.0 as previously described [Citation14]. The taxonomic information of this genome sequence was identified by using Kraken2 against RefSeq annotation based on k-mers strategy. The genome sequence is available at the NCBI’s GenBank under the accession ID: CP112971.
The circular map of the genome was generated and represented using CGView server (http://cgview.ca/) [Citation15]. Protein-coding regions in the assembled sequences were predicted using Prodigal [Citation16]. In addition, the estimation of the number of tRNA and rRNA sequences were identified by tRNA-scan and RNAmmer, respectively. The genomes of B8 and other publicly downloaded SFGRs were annotated by a command line software: Prokka (v 1.14.1) [Citation17]. For functional annotation, the genome sequences were examined using several protein databases. In brief, the function of predicted protein CDSs was determined against the NCBI non-redundant (NR), Gene Ontology (GO), eggnog 5.0 [Citation18], and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases with E-value cut-off set to 1e−5 and subsequent filtering for the best hit. In addition, the whole genome was blasted against VFDB and CARD for screening virulence genes and resistance genes with ABRicate (https://github.com/tseemann/abricate).
Prophage regions were predicted, and figures were generated using PHAST (http://phast.wishartlab.com) [Citation19]. As submitted to NCBI, the whole chromosome was uploaded to analysis using PHAST to predict the completeness of the prophage regions, and to identify and map their elements. Insertion sequence (IS) elements were predicted using IS Finder software (http://issaga.biotoul.fr/issaga_index.php) [Citation20]. Genomic islands were predicted using IslandPath and SIGI-HMM, part of software IslandViewer4 [Citation21], to identify regions with evidence of horizontal transfer in that of the B8 genome. Predicted surface proteins were carried out using Signal-P−5.0 [Citation22].
Phylogenetic analysis and average nucleotide identity (ANI) calculation
To perform a comparative analysis, 28 most closely related species and representative SFGRs were retrieved from the NCBI (). Rickettsial representative markers, groEL, gltA, 17-kD genes, and 16S rRNA gene sequences, and whole genomes were used to construct phylogenetic trees. Firstly, before the B8 genome sequenced, the sequences of groEL, gltA, 17-kD, and 16S rRNA marker genes were obtained by paired primers amplification. Next, all amplicons were cleaned via Qiagen QIAamp mini spin columns (Qiagen Inc., Valencia, CA) and the amplification products were sent to GENERAL BIOL (Anhui, China) for Sanger sequencing (3730 DNA ANALYZER) (Table S1) [Citation23]. In addition, the groEL, gltA, 17-kD genes, and the 16S rRNA gene of other strains were extracted from the genomic sequences by using blast and Barrnap [Citation24], respectively. The phylogenetic tree of Rickettsia spp. based on the above-mentioned marker gene sequences was constructed using the NJ method in MEGA v10.0 with 500 bootstraps replications [Citation25]. However, phylogeny based on only one common gene would result in bias; therefore, we performed the phylogenetic analysis based on the concatenated sequences of groEL, gltA, 17-kD, and 16S rRNA gene sequences. The multiple sequence alignments of single-copy genes were performed using MUSCLE and the phylogenetic tree was generated using the NJ method in MEGA v10.0 with 500 bootstraps replications. Furthermore, the phylogenetic tree based on core genome of incorporated Rickettsia strains was also constructed in our study according to previous [Citation26,Citation27]. In brief, multiple sequence alignments for these genomes were created using MUSCLE. The resulting alignments were used for phylogenetic analysis by employing Maximum-Likelihood (ML) with 100 bootstrap replicates. Evolutionary distances for ML were calculated leveraging Jukes-Cantor model [Citation28] and visualized by EvolView [Citation29]. The average nucleotide identity (ANI) values between the newly sequenced B8 genome and the representative genomes of other 28 public SFGRs were calculated using fastANI v1.2 based on a BLAST algorithm [Citation30].
Comparative genomics analyses
Whole-genome sequences of 29 Rickettsia strains were obtained for pan-genome analysis. Among which, 28 were publicly retrieved from National Center for Biotechnology Information (NCBI) database (). We leveraged Bacterial Pan Genome Analysis tool (BPGA) pipeline (v1.3) with default parameters for pan-/core-genome analysis [Citation31], and all of orthologous among 29 Rickettsia spp. in this study were identified. The pan genome is referred to the whole set of genes in the test genomes. However, the core genome is the set of shared genes that is present in all tested strains. In addition, the accessory genome is the set of genes shared with more than two but not all tested strains. Furthermore, the unique genes are the set of genes in each strain not shared with other strains [Citation32]. The details of the strains were showed in . A structural comparison via multiple genome alignment was performed using Mauve software [Citation33]. Motif searches, for detecting conserved or variable motifs were performed using MEME [Citation34], and the phylogenetic tree, conserved motifs were visualized using Tbtools software [Citation35]. Gene sequences alignment and polymorphic sites identification were performed and visualized in JalView [Citation36].
Anti-Rickettsia japonica OmpB polyclonal antibody production
In this study, the anti-Rickettsia japonica OmpB polyclonal antibody was generated according to the previous report [Citation37]. In brief, the sequence in the C-terminal of OmpB (OmpBC, 480 bp), was amplified by PCR from the genomic DNA of Rickettsia japonica deposited in our lab. Then, OmpBC was cloned into the pET28a plasmid with an N-terminal 6×His tag. Production of 6×His-OmpBC protein was induced in Escherichia coli str. BL21 (DE3) by the addition of 0.1 mM isopropyl-β-d-thio-galactoside (IPTG) for 4 h at 37°C. Bacterial cells were harvested and resuspended in lysis buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, and 10 mM imidazole) and stored at −80°C. For protein purification, the bacteria were thawed, lysozyme was added to 1 mg ml−1 (Sigma, L4919) and lysis was carried out by sonication. The lysate containing 6×His-OmpBC was incubated with Ni-NTA resin (Qiagen) and the bound proteins were eluted with 300 mM imidazole (in lysis buffer). The fractions were analysed by SDS-PAGE and those with the highest concentrations of 6×His-OmpBC were pooled before desalting, using PD-10 desalting columns (GE Healthcare, cat. no. 17085101) and equilibrated with lysis buffer lacking imidazole. Next, the OmpBC protein was fully emulsified with an equal volume of freund’s complete adjuvant at a dose of 0.8 mg/each by subcutaneous injection twice of Beijing white rabbits at two-week intervals. Finally, the polyclonal antibody was generated by the isolation of rabbits’ serum and the titre was determined by indirect ELISA.
Immunofluorescence analysis and electron microscopy
Human microvascular endothelial cell (HMEC-1) cells were cultured in 12-well dishes containing coverslips coated with poly-L-lysine. The cells were fixed for 15 min on ice in 4% formaldehyde in PBS, followed by incubation with 0.1% Triton X-100 and then incubated for 2 h with primary antibodies anti-Rickettsia japonica ompB. After washing with 1× PBS for three times, cells were incubated for 1 h with Cy3 conjugated secondary antibodies (goat anti-Rabbit IgG, Beyotime) at 1:1000 in PBS containing 5% horse serum and 0.1% Triton X-100. DNA was stained with 0.5 µg/mL DAPI. Confocal images were acquired with LSM880 microscope (ZEISS) equipped with 60× Plan Apochromat VC objective and airyscan detector. For the transmission electron microscopy, rickettsia-infected HMEC in each time point were harvested by centrifugation and fixed using 2.5% glutaraldehyde for 3 h, followed by washing with 1× PBS for three times. The next step in sample preparation before imaging is post-staining. Typically, the sections were stained in 2% aqueous uranyl acetate for 20 min, washed well with distilled water, followed by staining in Reynold’s lead citrate for 15 min, and washed again with distilled water to enhance the contrast [Citation38]. After air-drying, TEM images of the specimens were captured with Tecnai G2 transmission electron microscope at an accelerating voltage of 120 kV.
Ethical statement
Only patient serum-isolated bacteria were used in this study. No human subjects participated in this study. Animals used in this work were approved by the ethics committee of Anhui Medical University Biomedical Ethics Committee (Approval No. 2021H021).
Results
Identification of B8 by clinical presentation, immunofluorescent, and morphological characterization
On 17 April 2020, a patient was hospitalized and complained of a febrile illness with fever at 39.0° and other clinical signs, including asthenia, anorexia, nausea, and skin rashes all over the body. The patient confirmed that she had been bitten by ticks under the armpit five days ago when picking tea on a mountain. Physical examination identified a 1.3 × 1.1 cm2 erythematous rashes around the tick-bite eschar, and vesicles were seen surrounding the bite site. A papular rash with papules ∼0.1–0.3 cm in diameter was found all over the body. Blood samples were collected, and routine laboratory and haematologic assays were performed. Because of the rashes found during physical examination, infection with a spotted fever group Rickettsia was suspected. Therefore, the patient was given doxycycline on the day of admission. The patient was successfully cured and discharged in three days.
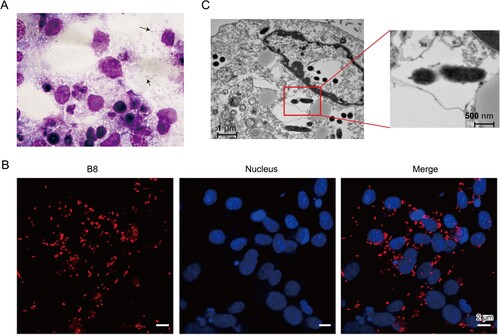
Afterwards, the patient’s blood specimens collected at the first day of admission were inoculated onto THP-1 and Vero cells. Cytopathic effects were observed for both cell lines after two weeks of cultivation. Firstly, the Diff-Quick stained smears of Vero cells showed Rickettsia-like bacilli in the cytoplasm ((A)). Then, Immuno-fluorescence assays were then performed using a polyclonal antibody against Rickettsia japonica ompB every two days, which exhibited rod-shaped particles in the inoculated cultures ((B)). In addition, transmission electron microscopy revealed a typical ultrastructure of Rickettsia bacteria with dimensions of approximately 0.2 μm × 1.0 μm ((C)).
Figure 1. Morphological and biochemical characterization of Rickettsia heilongjiangensis strain B8. (A) Diff-Quick stained smears of Vero cells showed Rickettsia-like bacilli in the cytoplasm. The black arrow points to an individual bacterium. (B) Immunofluorescence microscopy of B8-infected Vero cells. DAPI (blue) was used to stain host cell nucleus; polyclonal anti-ompB antibody followed by Cy3 conjugated secondary antibody (red) was used to stain B8. (C) Scanning electron microscope (SEM) micrograph showing numerous Rickettsia-like organisms parasitizing in the cytoplasm of infected Vero cells.
Local epidemic zone investigation of R. heilongjiangensis and taxonomic, genomic characterization of B8
Firstly, to identify the local natural foci, adult ticks, including 12 Ixodes persulcatus, 175 Haemaphysalis longicornis, and 21 Dermacentor silvarum, were collected in vegetation near where the patient lived (Figure S1(A)). DNA was extracted from these ticks. The 17-kDa, 16S rRNA, groEL, and gltA genes were amplified using the corresponding primers (Table S1). The specificity of these primers was examined by agarose gel electrophoresis of the PCR product (Figure S1(B)). As a result, R. heilongjiangensis was identified in 11 of 175 H. longicornis ticks and 0 of the other ticks tested (Table S2).
Subsequently, DNA was extracted from purified bacteria propagated by culturing in Vero cells. Based on the immunofluorescence results, we tentatively determined that B8 could be a new strain of Rickettsia japonica or another SFGR strain. Firstly, we amplified several marker genes, such as 17-kDa, 16S rRNA, groEL, and gltA with specific primers (Table S1), and then delivered to sanger sequencing. We concatenated the sequences of these genes and constructed a phylogenetic tree, shown that the amplified sequences from B8 clustered into the same clade as R. heilongjiangensis ((A)). Next, WGS of B8 yields a total of 2,038,576 quality-filtered reads, 604 Mb of sequence data. After subjected to de novo assembly, we found the draft genome of B8 consists of 82 contigs ranging in size from 1108 to 11,183 bases, resulting in a circular chromosome of 1,275,081 bp, with an overall 32.4% GC content. A total of 1441 coding sequences (CDS) were predicted to be protein coding ((B), Table S3). Based on the whole-genome annotation against bacterial RefSeq database by Kraken software, B8 was verified as belonging to R. heilongjiangensis. This was further corroborated with the phylogenetic analysis based on the WGS using core-genome (Figure S1(C)). According to the results obtained from WGS-ANI (average nucleotide identity) analysis using fastANI, the genome of B8 was nearly identical to R. heilongjiangensis and R. japonica clusters, and the values of ANI between each two strains was more than 99.35% and 99.22%, respectively ().
Figure 2. Genome structures and phylogenetic relatedness of B8. (A) Phylogenetic tree (Neighbour-joining) construction based on the concatenated multiple-sequence alignment (MSA) of four representative genes (gltA, groEL, 17-kD, and 16S rDNA). B8 highlighted in red was clustered with five other R. heilongjiangensis strains with green background. (B) Circular genome maps of B8. The four circles (outer to inner) show the following. Circle 1 and 2 exhibit CDS on the forward and reverse strands, as well as rRNA, tRNA, and assembly gap. Circle 3 represents the GC skew ((C − G)/(C + G)) curve (positive GC skew, dark green; negative GC skew, violet). Circle 4 shows the GC content.
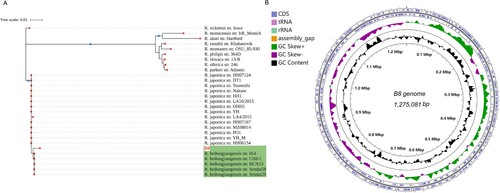
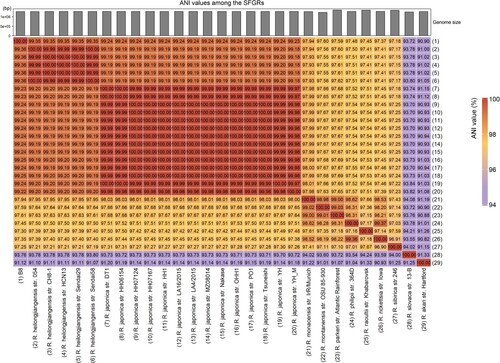
Figure 3. Heat-map of Average Nucleotide identity (ANI) values amongst different lineages representative strains of SFGR and B8. The bar in the head of heat-map shows the size of genome.
However, none plasmids were detected in the B8 genome, which is consistent with the genomic feature of type strain R. heilongjiangensis 054 [Citation8], and also no IS elements were identified. In the present study, however, two incomplete prophage regions covering a sum of 5 most likely prophage-like proteins were discovered in the genome of B8 (Figure S2, Table S4). Accumulating evidences suggest that the surface proteins of rickettsial organisms play a crucial role in mediating the direct Rickettsia-host interaction. Thus, we profiled the putative surface proteome of the B8 strain using Signal-P (v5.0) which detects cell membrane (Table S5). Altogether, 63 genes were predicted as signal peptide secreted in general secretory pathway and cleaved by signal peptidase I (Sec/SPI), and 35 genes were predicted to be associated with lipoproteins and cleaved by signal peptidase II (Sec/SPII) annotated.
Unravelling the gene function in the genome of R. heilongjiangensis strain B8
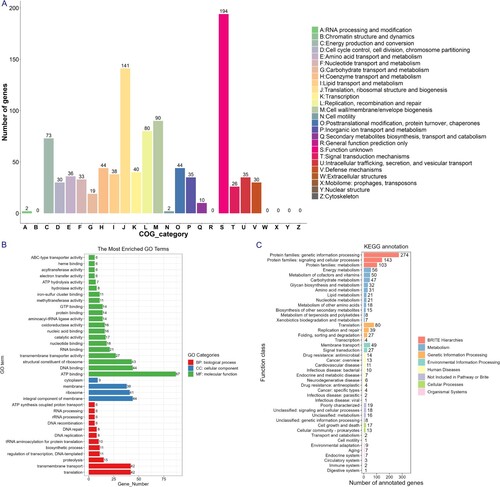
The genes of B8 showed a wide range of functional categories when annotated to COG, GO, and KEGG databases, mainly encompassing the categories representing cellular process, metabolic process, and catalytic activity ((A–C)). In addition, according to the annotation results of CAZy, we found that the B8 genome contains five major families of glycoside hydrolase (GH), glycosyl transferase (GT), carbohydrate-binding enzyme (CBM), auxiliary active enzyme (AA), and polysaccharide lyase (PL), in which the GT family is the most abundant CAZy genes (Figure S3).
Figure 4. Functional category analysis of the B8 genome annotated to the COG, GO, and KEGG databases. (A) COG annotation assignments. The COG functional annotations were divided into 26 categories. The COG categories are shown on the X-axis as alphabets, with category names on the right. (B) GO annotation distribution (level 2). The GO assignments were divided into three categories (level 1) namely, biological process (red), cellular process (blue), and molecular function (green). (C) KEGG annotation distribution. The KEGG orthologies were categorized into eight major categories: Brite Hierarchchies, Metabolism, Genetic Information Processing, Environmental Information Processing, Human Diseases, Not Included in Pathway or Brite, Cellular Processes and Organismal Systems.
Virulence factors determination
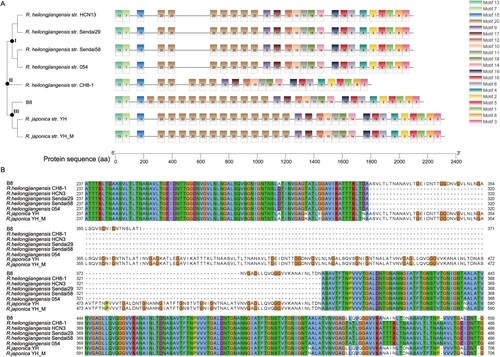
In terms of VFDB analysis, 30 predicted genes in the genome of B8 were aligned with 30 virulence genes using R. heilongjiangensis 054 as reference. The virulence genes could be divided into four classes, in which the Actin-based motility (RickA, Sca2), adherence and invasion (rOmpA/Sca0, rOmpB/Sca5), enzyme and secretion system (T4SS) were presented in all strains of representative Rickettsia spp. Particularly, we found phospholipase A2 (pat2) was specific in the B8 genome in comparison with reference genome of R. heilongjiangensis strain 054 (Table S6). In order to reveal the existence of difference in virulent gene structure within R. heilongjiangensis strains, the predicted ompA gene was selected as representative for motif search and phylogenetic analysis by the N-J method with 500 bootstrap replicates. Twenty motifs were identified with duplicates in terms of ompA gene structure. Interestingly, the tree is divided into three clades (clade I, II and, III). Clade I involves four strains within R. heilongjiangensis (Sendai58, 054, Sendai29, and HCN13), clade III contains R. heilongjiangensis strain B8 and 2 strains within R. japonica (YH and YH_M), and R. heilongjiangensis strain CH8-1 belonging to clade II ((A)). These differences are mostly single nucleotide variation and verified by the amino acid sequences alignment with MUSCLE using Jalview tool ((B)). These results revealed that there are several disparities in the genome of B8 compared to its intra- or interspecies Rickettsia.
Figure 5. Analyses of ompA gene variations among B8, other 5 R. heilongjiangensis and 2 R. japonica strains. (A) Phylogenetic analysis of ompA, constructed by the neighbour-joining method in MEGA 10.0 software, and identification of conserved motif by MEME online tool. The tree and motifs distribution were visualized by TBtools. (B) For a portion of the polymorphic sites, showing sequence variation of ompA gene in the B8 genome among the selected SFGRs. The sequences were aligned with MUSCLE algorithm and visualized with Jalview.
Multiple comparative genomic analysis across the B8 and other SFG Rickettsia
To expand the insight into the difference in genomic composition and structure of B8 across other inter-/intra-SFGR species, a comparative genome analysis was performed to find orthologs among the genome of B8 and additional 28 available but completed SFGR genomes from NCBI. In total, we found 5,117 orthologs in the pan-genome of 29 SFGRs, containing 254 core genes and 4863 (95.03%) accessory genes (Figure S4(A)). Notably, the pan-genome experienced a dramatic increase when strains of R. akari, R. monacensis, R. montanen, R. parkeri, R. philipii, R. raoultii, R. sibirica, and R. slovaca were taken into analysis (Figure S4(B)). Furthermore, multiple genome alignment demonstrated the consistency or discordance among the B8, R. heilongjiangensis, R. japonica, and other SFGRs using the Mauve tool (Figure S5).
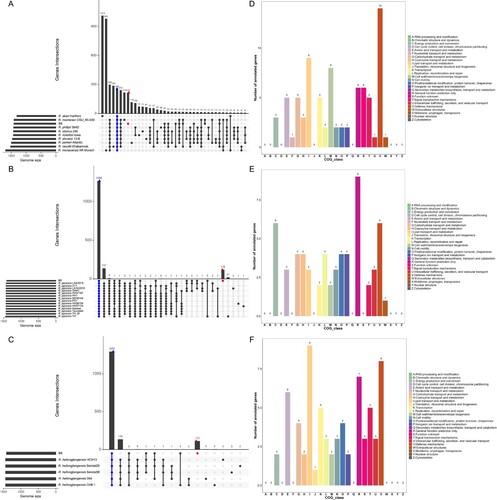
An open pan-genome could show the diverse response of the different taxa of Rickettsia to divergent environments. To gain a better understanding of the functional features of the pan-genome and identified specific genomic signature in the genome of B8. Comparative genomic analysis among the B8 and other nine SFGRs (not including 5 R. heilongjiangensis and 14 R. japonica strains) showed that a sum of 216 unique genes was found in the B8 genome ((A)). In addition, 119 and 112 genes were found exclusively in the genome of B8 when compared with 14 R. japonica and 5 R. heilongjiangensis strains ((B,C)). Collectively, COG annotations revealed that those B8-specific genes were mainly assigned to “[V] defense mechanism”, “[I] lipid transport and metabolism,” “[M] cell wall/membrane/envelope biogenesis,” “[C] Energy production and conversion,” and “[E] amino acid transport and metabolism” ((D–F)).
Figure 6. Genomic comparison amongst the B8 and additional SFGRs. (A–C) Upset diagram illustrating the number of core genome, shared orthologous and unique genes among B8 and 9 other SFGRs, B8 and other 14 R. japonica, as well as B8 and other 5 R. heilongjiangensis based on complete genome sequence of the selected strains in the present study. Vertical barplot indicates the number of overlapping genes (intersection size) for each intersection represented by the connected dots. The grey horizontal barplot, reported as “Gene Number,” indicate the total number of genes in each Strain. (D–F) COG annotation and categories assignment based on 216, 119, and 112 B8-unique genes.
Next, the phylogenetic relationships among B8, R. heilongjiangensis strain 054, R. japonica str. YH_M and nine other SFGRs were investigated. We found these 12 strains appear to have 255 ortho-groups in common and 36 specific homologous genes in the B8 genome (Figure S6(A)). However, the total number of specific genes in the B8 genome was less than in the genome of R. heilongjiangensis 054 (n = 58) and Rickettsia japonica YH_M (n = 43), though the taxonomy of B8 was classified as R. heilongjiangensis. Specifically, those 36 B8-specific genes were also predominantly classified into “[V] defense mechanisms” and “[I] lipid transport and metabolism” (Figure S6(B)).
Discussion
In recent years, the variety and distribution of SFGR in China have been showing a trend of diversification [Citation39–42]. Therefore, the precise identification and differentiation of SFGR and the elucidation of molecular mechanisms and pathogenesis are the focus of current research. It is well recognized R. heilongjiangensis 054 was first isolated from D. silvarum ticks in the Heilongjiang province, China. In this paper, we isolated a new strain of R. heilongjiangensis in Anhui Province, the central-eastern part of China, which suggests the epidemic zone of R. heilongjiangensis has been expanded or its occurrence has been detected in new regions. In addition, we confirmed the presence of R. heilongjiangensis in H. longicornis ticks collected near where the patient, from which the B8 strain was isolated, lived. This is a potentially new vector for R. heilongjiangensis and warrants further investigation. Haemaphysalis longicornis is reported widely distributed in China and around the world [Citation43], suggesting that we need to concentrate on the prevention and the control of the spread of R. heilongjiangensis in other tick hosts. In regarding to the WGS of B8, we first classified the isolate as a new strain of R. heilongjiangensis according to the reliable taxonomic annotation and precise phylogenetic analysis based on the core-genes of WGS, as well as morphological and immunofluorescence examination. We then deciphered the genomic architecture and functional information by complete genome sequencing. In terms of the genomic composition of B8, a total of 1411 CDSs were annotated. Next, we identified 1293 genes from the core genome of B8 and other five publicly R. heilongjiangensis strains from NCBI. Additionally, we noticed there were 112 unique genes in the B8 genome, which suggested the existence of genetic differences compared to other R. heilongjiangensis species. Overall, the present research revealed several divergences in the genomic composition of B8 as compared with other publicly R. heilongjiangensis and R. japonica strains. We thought B8 rather represents a previously undescribed human-pathogenic SFG rickettsia lineage, may be an intermediate of these lineages. Hence, the results suggested that we should gain a deep understanding of the genetic background of the R. heilongjiangensis strains before we know their biological potential, including their pathogenicity for humans and symbiotic relationship with the host, clarification of the metabolic pathways associated with intracellular parasitism.
The Majority of spotted fever group rickettsiae are transmitted to humans via a tick bite. Even if some Rickettsia species, such as R. rickettsii, are deleterious to host ticks infected by transovarial transmission in nature, a majority are known to persist and survive in infected vectors and lead to serious disease in humans and vertebrates [Citation44–46]. We found that the genome of R. heilongjiangensis strain B8 contains the common virulence factors which are conserved among SFG rickettsiae, suggesting a major role of the virulence genes in determining its pathogenicity. Previously reports have demonstrated the pivotal role of ompA (Sca0) gene in mediating the attachment and invasion of SFG rickettsiae, however, partially dependent on a conserved non-continuous RGD motif present in a predicted C-terminal “pertactin” domain in ompA [Citation47–49]. In this research, phylogenetic analyses, motif identification, and polymorphic sites investigation based on ompA gene sequence in the genome of B8 and other SFGRs showed obvious divergence, we concluded that there was minor gene flux between interspecies of R. heilongjiangensis and R. japonica. In addition, we found the virulent gene pat2 in the B8 genome, belonging to phospholipase A2, which was inconsistent with the type strains R. heilongjiangensis 054 and R. japonica YH. In light of previous research, rickettsial phospholipase A2 has been reported to mediate the entry into the host cell, escape from the phagosome, and cause damage to host cells via both typhus and spotted fever group rickettsiae [Citation50,Citation51]. These results together revealed the phylogenetic close relationships among the B8 and other R. heilongjiangensis strains, nevertheless, B8 also has different evolutionary patterns because of the two representative virulent genes, ompA and pat2. In addition, those B8-specific genes obtained from the pan-genome analysis offered an obvious hint that the genome evolution of B8 might go with the divergent geographical location and different tick hosts that reside in. Collectively, the genomic message of R. heilongjiangensis B8 delivered valuable information regarding the molecular function, pathogenicity, and genomic divergence when compared with the inter/intra-species Rickettsia. In addition, the variants of several genes, such as ompA and pat2, combined with a bulk of genes exclusive to B8 offered a substantial range of genetic markers for the detection and surveillance of new pathogenic R. heilongjiangensis, and also provides clues for subsequent studies on the bacterial pathogenicity.
Overall, the present study confirms the transmission of spotted fever R. heilongjiangensis with pathogenicity in East-Central China for the first time. This further suggests that H. longicornis might be a vector of R. heilongjiangensis or harbours it, which may broaden the variety of ticks as carrier of R. heilongjiangensis. As such, caution should be taken in districts with a distribution of the H. longicornis to avoid the infection of humans and animals by spotted fever R. heilongjiangensis. However, whether these B8-unique genes are associated with virulence and pathogenesis warranting further investigation.
Supplemental Material
Download Zip (4.6 MB)Acknowledgements
We gratefully acknowledge the Beijing Institute of Microbiology and Epidemiology, Academy of Military Medical Sciences for the isolation of B8 in the biosafety level 3 laboratory.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data Availability Statement
The complete genome sequence of Rickettsia heilongjiangensis strain B8 was deposited at the NCBI’s GenBank with the accession ID: CP112971.
Additional information
Funding
References
- Parola P, Paddock CD, Socolovschi C, et al. Update on tick-borne rickettsioses around the world: a geographic approach. Clin Microbiol Rev. 2013;26(4):657–702.
- Piotrowski M, Rymaszewska A. Expansion of tick-borne rickettsioses in the world. Microorganisms. 2020;8(12):1906.
- Walker DH, Valbuena GA, Olano JP. Pathogenic mechanisms of diseases caused by Rickettsia. Ann N Y Acad Sci. 2003;990:1–11.
- Helmick CG, Bernard KW, D’angelo LJ. Rocky mountain spotted fever: clinical, laboratory, and epidemiological features of 262 cases. J Infect Dis. 1984;150(4):480–488.
- Noguchi M, Oshita S, Yamazoe N, et al. Important clinical features of Japanese spotted fever. Am J Trop Med Hyg. 2018;99(2):466–469.
- Mansueto P, Vitale G, Cascio A, et al. New insight into immunity and immunopathology of Rickettsial diseases. Clin Dev Immunol. 2012;2012:967852.
- Zhang JZ, Fan MY, Wu YM, et al. Genetic classification of “Rickettsia heilongjiangii” and “Rickettsia hulinii,” two Chinese spotted fever group rickettsiae. J Clin Microbiol. 2000;38(9):3498–3501.
- Duan C, Tong Y, Huang Y, et al. Complete genome sequence of Rickettsia heilongjiangensis, an emerging tick-transmitted human pathogen. J Bacteriol. 2011;193(19):5564–5565.
- Kasama K, Fujita H, Yamamoto S, et al. Genomic features of Rickettsia heilongjiangensis revealed by intraspecies comparison and detailed comparison with Rickettsia japonica. Front Microbiol. 2019;10:2787.
- Killmaster LF, Zemtsova GE, Montgomery M, et al. Isolation of a Rickettsia slovaca-like agent from Dermacentor variabilis ticks in vero cell culture. Vector Borne Zoonotic Dis. 2016;16(1):61–62.
- Fischer T, Myalkhaa M, Krucken J, et al. Molecular detection of tick-borne pathogens in bovine blood and ticks from Khentii, Mongolia. Transbound Emerg Dis. 2020;67(Suppl 2):111–118.
- Guo WP, Wang YH, Lu Q, et al. Molecular detection of spotted fever group rickettsiae in hard ticks, northern China. Transbound Emerg Dis. 2019;66(4):1587–1596.
- Chen S, Zhou Y, Chen Y, et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–ii90.
- Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477.
- Grant JR, Stothard P. The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008;36(Web Server issue):W181–W184.
- Hyatt D, Chen GL, Locascio PF, et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11(1):1–11.
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069.
- Huerta-Cepas J, Szklarczyk D, Heller D, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47(D1):D309–DD14.
- Zhou Y, Liang Y, Lynch KH, et al. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39(Web Server issue):W347–W352.
- Varani AM, Siguier P, Gourbeyre E, et al. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 2011;12(3):R30.
- Langille MG, Brinkman FS. Islandviewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics. 2009;25(5):664–665.
- Armenteros JJA, Tsirigos KD, Sonderby CK, et al. Signalp 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. 2019;37(4):420–423.
- Li J, Hu W, Wu T, et al. Japanese spotted fever in eastern China, 2013. Emerg Infect Dis. 2018;24(11):2107–2109.
- Liu N, Zhu L, Zhang Z, et al. Draft genome sequence of a multidrug-resistant blaOXA-69-producing Acinetobacter baumannii L13 isolated from Tarim River sample in China. J Glob Antimicrob Resist. 2019;18:145–147.
- Kumar S, Stecher G, Li M, et al. MEGA x: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549.
- Naushad S, Barkema HW, Luby C, et al. Comprehensive phylogenetic analysis of bovine non-aureus staphylococci species based on whole-genome sequencing. Front Microbiol. 2016;7:1990.
- Glaeser SP, Doijad S, Hijazin M, et al. Varibaculum anthropi sp. nov. represented by three genetically different genomovars isolated from clinical material and emended description of the genus Varibaculum. Syst Appl Microbiol. 2016;39(8):546–552.
- Chor B, Hendy MD, Snir S. Maximum likelihood Jukes-Cantor triplets: analytic solutions. Mol Biol Evol. 2006;23(3):626–632.
- Subramanian B, Gao S, Lercher MJ, et al. Evolview v3: a webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019;47(W1):W270–W2W5.
- Jain C, Rodriguez RL, Phillippy AM, et al. High throughput ANI analysis of 90 K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9(1):5114.
- Chaudhari NM, Gupta VK, Dutta C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci Rep. 2016;6:24373.
- Tian X, Zhang Z, Yang T, et al. Comparative genomics analysis of Streptomyces species reveals their adaptation to the marine environment and their diversity at the genomic level. Front Microbiol. 2016;7:998.
- Darling AC, Mau B, Blattner FR, et al. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–1403.
- Bailey TL, Boden M, Buske FA, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server issue):W202–W208.
- Chen C, Chen H, Zhang Y, et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–1202.
- Waterhouse AM, Procter JB, Martin DM, et al. Jalview version 2 – a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–1191.
- Engstrom P, Burke TP, Mitchell G, et al. Evasion of autophagy mediated by Rickettsia surface protein OmpB is critical for virulence. Nat Microbiol. 2019;4(12):2538–2551.
- Chen HM, Chan SC, Lee JC, et al. Transmission electron microscopic observations of membrane effects of antibiotic cecropin B on Escherichia coli. Microsc Res Tech. 2003;62(5):423–430.
- Fang LQ, Liu K, Li XL, et al. Emerging tick-borne infections in mainland China: an increasing public health threat. Lancet Infect Dis. 2015;15(12):1467–1479.
- Li H, Cui XM, Cui N, et al. Human infection with novel spotted fever group Rickettsia genotype, China, 2015. Emerg Infect Dis. 2016;22(12):2153–2156.
- Qi Y, Ai L, Jiao J, et al. High prevalence of Rickettsia spp. in ticks from wild hedgehogs rather than domestic bovine in Jiangsu province, Eastern China. Front Cell Infect Microbiol. 2022;12:954785.
- Qin XR, Han HJ, Han FJ, et al. Rickettsia japonica and novel Rickettsia species in ticks, China. Emerg Infect Dis. 2019;25(5):992–995.
- Zhao L, Li J, Cui X, et al. Distribution of Haemaphysalis longicornis and associated pathogens: analysis of pooled data from a China field survey and global published data. Lancet Planet Health. 2020;4(8):e320–e3e9.
- Parola P, Paddock CD, Raoult D. Tick-borne rickettsioses around the world: emerging diseases challenging old concepts. Clin Microbiol Rev. 2005;18(4):719–756.
- Wood H, Artsob H. Spotted fever group rickettsiae: a brief review and a Canadian perspective. Zoonoses Public Health. 2012;59(Suppl 2):65–79.
- Robinson MT, Satjanadumrong J, Hughes T, et al. Diagnosis of spotted fever group Rickettsia infections: the Asian perspective. Epidemiol Infect. 2019;147:e286.
- Li H, Walker DH. Rompa is a critical protein for the adhesion of Rickettsia rickettsii to host cells. Microb Pathog. 1998;24(5):289–298.
- Clark TR, Noriea NF, Bublitz DC, et al. Comparative genome sequencing of Rickettsia rickettsii strains that differ in virulence. Infect Immun. 2015;83(4):1568–1576.
- Stenos J, Walker DH. The rickettsial outer-membrane protein A and B genes of Rickettsia australis, the most divergent Rickettsia of the spotted fever group. Int J Syst Evol Microbiol. 2000;50(Pt 5):1775–1779.
- Rahman MS, Gillespie JJ, Kaur SJ, et al. Rickettsia typhi possesses phospholipase A2 enzymes that are involved in infection of host cells. PLoS Pathog. 2013;9(6):e1003399.
- Walker DH, Feng HM, Popov VL. Rickettsial phospholipase A2 as a pathogenic mechanism in a model of cell injury by typhus and spotted fever group rickettsiae. Am J Trop Med Hyg. 2001;65(6):936–942.