
ABSTRACT
The development of novel culture-independent techniques of microbial identification has allowed a rapid progress in the knowledge of the nasopharyngeal microbiota and its role in health and disease. Thus, it has been demonstrated that the nasopharyngeal microbiota defends the host from invading pathogens that enter the body through the upper airways by participating in the modulation of innate and adaptive immune responses. The current COVID-19 pandemic has created an urgent need for fast-track research, especially to identify and characterize biomarkers to predict the disease severity and outcome. Since the nasopharyngeal microbiota diversity and composition could potentially be used as a prognosis biomarker for COVID-19 patients, which would pave the way for strategies aiming to reduce the disease severity by modifying such microbiota, dozens of research articles have already explored the possible associations between changes in the nasopharyngeal microbiota and the severity or outcome of COVID-19 patients. Unfortunately, results are controversial, as many studies with apparently similar experimental designs have reported contradictory data. Herein we put together, compare, and discuss all the relevant results on this issue reported to date. Even more interesting, we discuss in detail which are the limitations of these studies, that probably are the main sources of the high variability observed. Therefore, this work is useful not only for people interested in current knowledge about the relationship between the nasopharyngeal microbiota and COVID-19, but also for researchers who want to go further in this field while avoiding the limitations and variability of previous works.
1. Microflora, microbiota, microbiome, and metagenome: similar terms but different meanings
The lack of consensus in the different fields that study the microorganisms that are present in animals and plants has driven scientists to use the terminology in a confusing manner, including terms which are used arbitrarily with different meanings depending on the authors. Therefore, we will first clarify such terminology for a better understanding of the present work, according to the most accepted definitions nowadays. Starting with the oldest term, microflora, although this word has been used in the scientific and medical literature for decades, it is a misnomer as its definition and etymological origin make it clear that this term refers to plants and not microbes [Citation1]. The fact that some dictionaries now include a new definition for the word microflora, “the aggregate of bacteria, fungi, and other microorganisms normally occurring on or in the bodies of humans and other animals: intestinal flora”, could lead us astray, but it is only the result of over one century using this term incorrectly [Citation1]. Thus, the word microflora with this newly added definition should be replaced by microbiota, first used by Lederberg and McCray [Citation2], that can be defined as the set of ecological communities of microorganisms present inside and on the surface of all plants and animals [Citation1]. Moreover, the term microbiota is also appropriate to describe the composition and abundance of microbial communities in the environment [Citation1]. Another common cause of confusion is that microbiota and microbiome are often used interchangeably albeit both terms are different, since microbiome refers to the aggregate of all the different microbiotas that reside in an organism, including the microorganisms, their genomes, and the surrounding environmental conditions [Citation1,Citation3]. Finally, the term metagenome is commonly used in the context of genomics and refers to the collective genomes and genes from the members of a microbiota [Citation1].
2. The human microbiome: diversity, anatomical distribution, and abundance
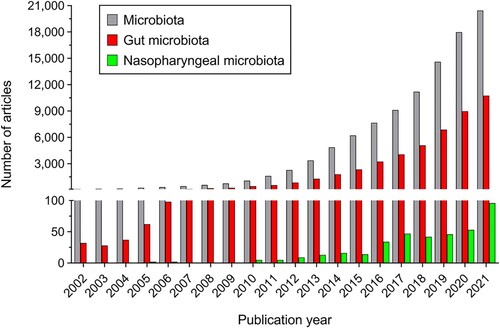
Bacteria, archaea, fungi, viruses, and protists have been found to be part of the microbiota in the different locations of the healthy human body, where they form complex ecological communities and influence the human physiology [Citation4]. Human microbiota is present and biologically relevant in multitude of anatomical areas, including the gastrointestinal tract, skin, vagina, male reproductive tract, oral cavity, uterus, placenta, ovarian follicle, urogenital tract, eye, external ear, mucous membranes, and respiratory tract [Citation5,Citation6]. Among these places, the gut has long been considered as the main location of human microbiota as this habitat harbors the largest collection of microorganisms by far [Citation7]. From a quantitative point of view, the number of microorganisms that populate our body is astronomical, the gut microbiota being the best example of this since it comprises over 40,000 different bacterial species and reaches a concentration of 1010 microorganisms per gram of stool in the colon [Citation8,Citation9]. However, other more conservative studies defend that the number of different bacterial species present in the human gut would be approximately 1,000 [Citation6]. In any case, numbers are even more impressive if we refer to the amount of bacterial genetic material present in the human body, since it is estimated to be at least 2,000,000 different bacterial genes only in the human gut [Citation10]. Given this relevance of the gut microbiota compared to that of other anatomical areas, and in a context in which annual publications on the microbiota have multiplied by more than 170 in the last 20 years, publications on the gut microbiota are the ones that have grown the most in the same period, exceeding 52% of the total publications on the microbiota in 2021 (10,747 out of 20,441) ().
Figure 1. Number of items retrieved from Pubmed database for the last 20 years (between 2002 and 2021) using the word “microbiota” (grey bars); using the words “gut” AND “microbiota” (red bars); and using the words “nasopharyngeal” AND “microbiota” (green bars).
As high-throughput sequencing based investigations into the diversity and composition of the human microbiota have focused almost exclusively on bacteria, particularly on the phyla Bacteroidetes and Firmicutes which are predominant in the majority of the anatomical areas explored, these microorganisms have been the most studied components of the human microbiota [Citation11]. Unlike bacteria, which are present in all niches where human microbiota has been studied and showing a huge variety, archaea diversity is much lower and they have been found mainly in the gastrointestinal tract [Citation12], but also in low numbers in the oral cavity [Citation13], respiratory tract [Citation14], and skin [Citation15]. Although the dominant group of archaea in the human gut are the methanogens, curiously only about 50% of people are colonized by these microorganisms [Citation12]. Fungi are even less abundant and more anatomically restricted than archaea, as only about 200 species (most of them in the Candida genus) have been found to be present in the gastrointestinal tract, and in low numbers on the skin, of healthy individuals [Citation16]. The most notable exception is C. albicans, the most clinically relevant Candida species, since it is part of the normal microbiota of the mucous membranes of the respiratory, gastrointestinal, and female genital tracts [Citation17]. Regarding viruses, although their number is not well known in part because new members of this group are continuously being identified in all the human anatomical areas studied, including the oral cavity, gut, skin, lungs and blood, recent studies suggest that there may be at least ten times more viruses than bacterial cells in the human body [Citation10]. Finally, the human body, particularly the intestinal tract, is also home to many eukaryotes besides fungi, including protists (e.g. flagellates and amoebae) and animals (e.g. helminths), which have been investigated for decades from a parasitological point of view [Citation18]. Even though eukaryotic microbes in the gut have generally been considered as parasites and have long been associated to host morbidity and mortality, recent studies have shown that many of them are part of the healthy human microbiota as commensals and play relevant roles as probiotics or cellulose degraders [Citation18]. Importantly, the Human Microbiome Project, launched by the United States National Institutes of Health (NIH) in 2007, has been essential for identifying and characterizing the human microbiota and, in a second phase launched in 2014, for generating resources to characterize the human microbiome and elucidating the roles of microorganisms in health and disease [Citation19,Citation20].
3. The nasopharyngeal microbiota and its role in health and disease: the case of COVID-19
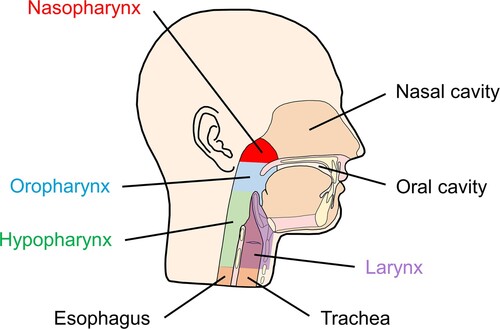
The human upper respiratory tract comprises the respiratory system parts that are located outside the thorax, which are the anterior nares, nasal cavity, sinuses, nasopharynx, Eustachian tube, middle ear cavity, oral cavity, oropharynx, and larynx [Citation21]. Among them, the nasopharynx is anatomically unique since it presents a common meeting place for the ear, nose, and mouth cavities () [Citation21]. Although lungs are constantly exposed to microorganisms from inhaled air and from the upper respiratory tract, they were long believed to be sterile. This has been the main cause that the respiratory microbiota, including that of the nasopharynx, has barely been studied until very recent times, in contrast to the most studied gut microbiota () [Citation22]. However, novel culture-independent techniques of microbial identification, such as 16S ribosomal RNA (rRNA) gene sequencing or metagenomic sequencing, have revealed that the respiratory tract is a dynamic ecosystem and has aroused the interest of the scientific community in the role of the respiratory microbiota in health and disease [Citation22].
Figure 2. Diagram showing the different anatomical areas of the upper respiratory tract, including the nasopharynx.
The upper respiratory tract microbiota, which participates in vital functions spanning from metabolic functions to immune system activities, is a major contributor to respiratory health as the commensal microorganisms shape the local immune system to establish a “healthy balance” between the host and the microbes [Citation23]. In the context of disease, the upper respiratory tract is the major portal of entry and infection site for aerosol-transmitted microorganisms or infectious droplets and, once these invading pathogens have entered, the nasopharyngeal microbiota has been shown to play a crucial role in defending the host from bacterial and viral infections [Citation24]. Thus, besides the local immune system and the epithelial layer, the nasopharyngeal microbiota is also essential for maintaining mucosal homeostasis [Citation23,Citation25]. In fact, the nasopharyngeal microbiota directly interacts with the nasopharyngeal-associated lymphoid tissue (NALT), which presents a large variety and number of immune cells such as macrophages, lymphocytes, and dendritic cells, and collaborates in its development [Citation26]. Moreover, the nasopharyngeal microbiota also participates in the regulation of adaptive responses such as the activation of both cell-mediated and humoral immune responses [Citation27,Citation28]. It has been demonstrated that the microbiota of the upper and lower respiratory tracts is altered by respiratory viruses, resulting in an enriched abundance of opportunistic pathogens which ultimately increase the severity of the disease in the infected subjects [Citation29,Citation30]. Interestingly, multiple studies have found unquestionable correlations between the nasopharyngeal microbiota composition and the susceptibility of patients to different viral infections, such as influenza A and B, rhinoviruses (HRVs), and respiratory syncytial virus (RSV), among others [Citation22]. Indeed, it has been recently shown that individuals infected with metapneumovirus, influenza A and B viruses, RSVs, and HRVs, had in their nasopharynx a reduced abundance of anaerobic bacteria and an invasion of pathogenic bacteria, including Corynebacterium pseudodiphtheriticum, Staphylococcus aureus, Streptococcus pneumoniae, Haemophilus influenzae, Dolosigranulum pigrum, and Moraxella catarrhalis [Citation31]. Therefore, characterizing the nasopharyngeal microbiota of patients could help predict the severity of respiratory infections in some cases [Citation23,Citation30].
The novel betacoronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is the causative agent of the coronavirus disease 2019 (COVID-19) [Citation32], penetrates the host through the upper airways [Citation33]. Expression of the enzyme ACE2, which acts as the SARS-CoV-2 receptor to enter human cells [Citation34], and the serine protease TMPRSS-2, which activates the coronavirus by priming its Spike (S) protein [Citation35], has been found to be enriched in the epithelial lining of the upper respiratory tract [Citation36]. In addition, ACE2 could be modulated by the respiratory microbiota as it is an interferon-stimulated gene [Citation37]. Although their role in health and disease had been increasingly studied in recent years, the nasopharynx and its microbiota have gained a sudden and special prominence since the moment the SARS-CoV-2 virus and the consequent outbreak of the current COVID-19 pandemic came on the scene () [Citation32]. Importantly, in both, symptomatic and asymptomatic COVID-19 patients, nasopharyngeal swabs present higher viral loads than nasal swabs, suggesting the relevance of the nasopharyngeal epithelium as a portal for initial SARS-CoV-2 infection and transmission [Citation38]. This is the reason why nasopharyngeal swabs are considered “gold standard” for the diagnosis of SARS-CoV-2 infections [Citation38]. Furthermore, the recently proposed “epithelial barrier hypothesis’ defends that a disruption of the epithelial barrier caused by different offending substances, such as microplastics, particulate matter, diesel exhaust, ozone, nanoparticles, and allergens could cause tissue inflammation and microbial dysbiosis and play a role in the development and exacerbation of many diseases, including COVID-19 [Citation39,Citation40]. This means that environmental factors could also be relevant for the nasopharyngeal microbiota composition of SARS-CoV-2 infected subjects, adding even more complexity to this field. Therefore, unfortunately, the role of the nasopharyngeal microbiota in COVID-19 is not well understood yet, and results from many studies on this are contradictory in some respects, as we will discuss in this work aiming to shed light on this issue.
4. Changes in the nasopharyngeal microbiota of SARS-CoV-2-infected individuals
To date, numerous studies have attempted to identify and characterize alterations in the diversity or composition of the nasopharyngeal microbiota of SARS-CoV-2-infected subjects, as well as any potential correlation between such alterations and the severity or outcome of the COVID-19 disease () (Table S1). The analysis of the published research articles on this subject clearly evinces that reported results are highly variable, even from works with apparently similar experimental designs, with the existence of studies that do not find any significant changes while others show clear differences that could be relevant from a biological and biomedical point of view (Table S1). Since it is known that the microbiota changes in the different anatomical areas of the upper respiratory tract although they are nearby places [Citation41,Citation42], we will focus just on the nasopharyngeal microbiota in order to avoid confusion. Therefore, only those studies that clearly state that they used nasopharyngeal swabs for their analyses will be included and discussed in the present work, whereas other works, which are not of lesser quality nor their results less interesting, but which refer in a more general way to the upper respiratory tract and which collected samples from other anatomical areas such as midturbinate swabs, nasal/oropharyngeal swabs, bronchoalveolar lavage fluid, nasopharyngeal/oropharyngeal swabs, throat swabs, sputum, saliva, among others, will not be included nor discussed here. Once the different anatomical origins of the samples have been eliminated as one of the possible sources of variability, other potential causes of variability that might be affecting the consistency of the results will be discussed later.
4.1. Diversity changes in the nasopharyngeal microbiota of SARS-CoV-2 infected subjects
Diversity measurements are a central topic in microbiome data analysis. The two main ways for estimating diversity are alpha diversity, which summarizes the distribution of species abundances in a given sample into a single number that depends on species richness and evenness, and beta diversity, which quantifies (dis-)similarities between communities (samples) [Citation43]. Despite the aforementioned data variability observed in this field, the majority of studies analyzing whether there is an association between the nasopharyngeal microbiota and SARS-CoV-2 infection have in common that they found statistically significant changes in nasopharyngeal microbiota diversity and/or composition when comparing infected patients to uninfected healthy controls (Table S1). However, there are a couple of notable exceptions: De Maio and colleagues did not observe any differences in terms of neither microbiota diversity nor bacterial composition between SARS-CoV-2-infected patients compared to uninfected controls [Citation44]. Unfortunately, sample sizes for this study were low (including only 18 infected patients) and samples were collected from patients with different characteristics but always at the moment of COVID-19 diagnosis, when all of them had a mild disease, without studying infected patients at longer times (Table S1) [Citation44]. Results obtained by Braun and colleagues were similar, as they were also unable to detect any differences when comparing the nasopharyngeal microbiota of SARS-CoV-2 positive and negative individuals [Citation45]. Moreover, they found that samples from a same individual clustered together independently on whether such samples were positive or negative for SARS-CoV-2, reinforcing their conclusion that SARS-CoV-2 does not have a strong effect on the nasopharyngeal microbiota [Citation45]. Sample sizes were also an important limitation in this case, with a total of only 33 subjects included in the study (Table S1) [Citation45]. Besides these two publications that did not find any differences at all, including microbiota diversity and composition, there are other studies which were also unable to observe significant differences in nasopharyngeal microbiota alpha and/or beta diversity, although in these cases they showed that the nasopharyngeal microbiota composition was altered in SARS-CoV-2 positive subjects compared to uninfected controls (Table S1) [Citation46-50]. Interestingly, while most of the studies that observed significant differences in the nasopharyngeal microbiota diversity reported a decrease in SARS-CoV-2 positive subjects compared to uninfected individuals [Citation51,Citation52], or in the most severe cases of COVID-19 disease compared with milder cases [Citation53-56], the only work exploring the nasopharyngeal microbiota profile of pregnant women with SARS-CoV-2 infection, conducted by Crovetto and colleagues, found that alpha diversity was higher in pregnant infected women compared to uninfected (Table S1) [Citation57]. Unfortunately, as there were no previous data on the nasopharyngeal microbiota during pregnancy, and this study did not include samples from women in a nonpregnant status, it was not possible to determine whether changes were a consequence only of the pregnancy status itself (Table S1) [Citation57]. Undoubtedly, these surprising data from pregnant women will deserve further research, ideally with higher sample sizes and including nonpregnant women and SARS-CoV-2 positive subjects with different severities of COVID-19 disease.
4.2. Compositional structure alterations in the nasopharyngeal microbiome of SARS-CoV-2 infected individuals
While an important part of the research in this field, especially at the initial moments of the COVID-19 pandemic, has focused mainly on comparing the nasopharyngeal microbiota of SARS-CoV-2 positive individuals to uninfected controls independently on the symptomatology of those patients or the severity of their disease [Citation44-49,Citation51,Citation52,Citation57-63], many of the most recent studies have gone further and have examined the possible association between the nasopharyngeal microbiota and the COVID-19 disease severity or outcome (Table S1) [Citation50,Citation53-56,Citation64-70]. The publications which only compared the nasopharyngeal microbiota composition of asymptomatic or paucisymptomatic SARS-CoV-2 positive subjects to negative healthy controls generally found little or no differences (Table S1) [Citation44,Citation63,Citation65]. Thinking of the other extreme in respect to the severity of the COVID-19 disease, a couple of studies included only critically ill COVID-19 patients: on the one hand, Llorens-Rico and colleagues exclusively enrolled patients who were admitted to the intensive care unit for a variable period (Table S1) [Citation60]. This work revealed that the nasopharyngeal microbiota of all those patients was dominated by the bacterial genera Staphylococcus and Corynebacterium, but woefully the study did not include any less severely ill, asymptomatic, or uninfected individuals as controls with whom to compare (Table S1) [Citation60]. On the other hand, consistent with these results, the work by Bai and colleagues, which included 37 critically ill COVID-19 patients and 20 uninfected controls, showed a reduction in commensal bacteria and higher abundance of pathogenic bacteria in patients compared to healthy uninfected controls (Table S1) [Citation52]. But even more interesting are the approaches analyzing the nasopharyngeal microbiota composition of COVID-19 patients with different levels of disease severity within the same study, since most of these works observed significant differences when comparing symptomatic subjects to asymptomatic/paucisymptomatic individuals and/or uninfected controls (Table S1) [Citation46,Citation50,Citation54-56,Citation64-66,Citation68-70]. Among the studies belonging to this group, Nardelli and colleagues, who were the first authors to select a homogeneous group of COVID-19 patients sharing that all of them were symptomatic, revealed that Proteobacteria and Fusobacteria relative abundances decreased in symptomatic COVID-19 patients compared to uninfected controls, particularly the genera Leptotrichia, Haemophilus, and Fusobacterium (Table S1) [Citation46]. Another work by Kolhe and colleagues, which compared the nasopharyngeal microbiota of symptomatic COVID-19 patients not only with that of uninfected healthy controls, but also with that of asymptomatic SARS-CoV-2 positive individuals, found higher abundance of Cutibacterium and Lentimonas and lower abundance of Prevotellaceae, Luminiphilus, Flectobacillus, Comamonas, and Jannaschia, in symptomatic compared to asymptomatic infected individuals (Table S1) [Citation64]. Unfortunately, the fact that this study only enrolled subjects between 49 and 78 years of age represents an important limitation for the robustness of its conclusions, which cannot be extrapolated to other stages of life (Table S1) [Citation64]. Ventero and colleagues also observed a clear association between changes in the nasopharyngeal microbiota composition and the COVID-19 disease severity, as their results showed that the presence of the bacterial genera Selenomonas, Filifactor, Actinobacillus, and Chroococcidiopsis, was associated with a reduction of more than 90% of invasive mechanical ventilation, regardless of age, gender, or comorbidity (Table S1) [Citation54].
Interestingly, contradictory results regarding the abundance of the genus Corynebacterium in the nasopharynx of COVID-19 patients associated to the severity of their disease have been reported. While some studies suggest an association between its increased abundance or its prevalence in the nasopharynx and a more severe disease [Citation60,Citation68], others observed that the genus Corynebacterium was less abundant in SARS-CoV-2 positive subjects compared to uninfected controls [Citation49], or that its abundance decreased as the severity of the COVID-19 disease was higher (Table S1) [Citation65,Citation69]. Thus, besides the previously mentioned results showing that Corynebacterium, together with Staphylococcus, dominates the nasopharyngeal microbiota of severe COVID-19 patients who were admitted to the intensive care unit [Citation60], Tchoupou Saha and colleagues also detected that the relative abundance of a member of the Corynebacterium genus, C. propinquum, was increased in symptomatic COVID-19 patients compared to asymptomatic (Table S1) [Citation68]. On the contrary, a study by Shilts and colleagues, which included 20 uninfected controls and 83 SARS-CoV-2 positive subjects with different COVID-19 severities, ranging from asymptomatic to very severe cases, found that changes in the nasopharyngeal microbiota were associated to the disease severity, and in particular, Corynebacterium clearly decreased as COVID-19 severity increased (Table S1) [Citation65]. Finally, and consistent with these last data, another work analyzed the nasopharyngeal microbiota of hundreds of children, adolescents, and young adults under 21 years of age, including COVID-19 patients with different severities and uninfected controls, and revealed that the prevalence of respiratory symptoms in SARS-CoV-2 infected individuals with Corynebacterium/Dolosigranulum-dominant microbiota profiles was lower than in infected cases with other microbiota profiles (Table S1) [Citation69]. Therefore, further studies establishing clear criteria to stratify COVID-19 patients by severity and ideally by age too, with similar and high sample sizes for the different groups resulting from such stratification, are required to elucidate this issue.
Another controversial question is whether COVID-19 patients could present an increased abundance of pathobionts in the nasopharynx that may result in a higher incidence of secondary bacterial infections and aggravate pneumonia. Regarding this, although Chen and colleagues, who analyzed nasopharyngeal swabs from 64 hospitalized COVID-19 patients with different disease severities, found that severe patients had lower abundance of opportunistic pathogens compared to mild patients, including the bacterial genera Actinomyces, Prevotella, Rothia, Streptococcus, and Veillonella (Table S1) [Citation56], the majority of studies analyzing this issue support the idea that SARS-CoV-2 infection alters the symbiosis between commensal bacteria and opportunistic pathogens that are present in the nasopharynx, resulting in an over-proliferation of pathogenic bacteria that predispose to the development of comorbidities (Table S1) [Citation50,Citation52,Citation55,Citation66,Citation71]. In this regard, Bai and colleagues reported an increased abundance of pathogenic bacteria in critically ill COVID-19 patients compared to uninfected subjects (Table S1) [Citation52]. Moreover, Qin and colleagues demonstrated that pathobiontic bacterial genera, such as Staphylococcus, Corynebacterium, and Acinetobacter, were present in the nasopharynx of 40.7% of severe patients while of 10.8% of mild patients (Table S1) [Citation55]. Consistent with this, Giuglian and colleagues revealed a correlation between high SARS-CoV-2 burden and the abundance of super-pathogenic bacterial species such as Staphylococcus aureus, Klebsiella pneumoniae, Streptococcus pneumoniae, Pseudomonas aeruginosa, and Acinetobacter baumannii (Table S1) [Citation66]. In addition, a recent work by Gauthier and colleagues showed an increased abundance of Enterobacteriaceae among hospitalized COVID-19 patients, suggesting that medical interventions in this group such as intubation or antibiotic exposure might be playing a role [Citation50]. Finally, preexisting asthma seems to also affect the nasopharyngeal microbiome of COVID-19 patients, since asthmatic patients exhibited reduced abundance of the bacterial genera Porphyromonas, Haemophilus, Alloprevotella, Moraxella, Facklamia, Campylobacter, and Janibacter compared with patients without preexisting asthma [Citation72].
Even though it has been demonstrated that the coinfection with other viruses is one of the factors that affects COVID-19 patient prognosis [Citation73], the nasopharyngeal virome of SARS-CoV-2 positive individuals, whose knowledge would be essential to assess the possible association of such coinfections with disease severity, have scarcely been studied. Undoubtedly, the study that has gone deeper into this question was the one conducted by Ferravante and colleagues, which used metagenomic sequencing to analyze the nasopharyngeal virome composition of 89 SARS-CoV-2 positive subjects with different COVID-19 disease severities (Table S1) [Citation67]. Although coinfections with the Coronaviridae, Retroviridae, Herpesviridae, Poxviridae, Pneumoviridae, Pandoraviridae, and Anelloviridae families were detected, only 2.2% of the cases (2/89) identified respiratory viruses, suggesting that respiratory viral coinfections seem to be not closely associated with SARS-CoV-2 infection or disease severity (Table S1) [Citation67]. Unfortunately, this study had several limitations that compromise the robustness of its conclusions and the possibility to extrapolate them: (1) it involved COVID-19 patients only from a certain small Italian region, Campania; (2) COVID-19 patients came from three different pandemic waves, making probable that they were infected by different SARS-CoV-2 variants which could differentially affect the nasopharyngeal virome; (3) authors used two different sequencing platforms, NextSeq 500 (Illumina) and NovaSeq 6000 (Illumina), increasing the risk of introducing technical bias; (4) despite the fact that the global sample size of the study (N = 89) was not particularly low, the classification of the COVID-19 patients by severity resulted in some groups with very low sample sizes, such as the groups of moderate (N = 6) or severe (N = 10) patients. Moreover, there was a large group of patients whose severity was unknown (N = 34); and (5) the study did not enroll any uninfected healthy controls, which would have made it possible to make interesting comparisons (Table S1) [Citation67]. Another more recent work from the same authors used metagenomic sequencing to study the phageome of SARS-CoV-2 positive subjects with different disease severities, and observed a very high abundance of the bacteriophage families Siphoviridae and Myoviridae as well as some evidence suggesting that age and disease severity could be associated with the bacteriophage abundance in COVID-19 patients (Table S1) [Citation70]. Unfortunately, this study did not include any uninfected healthy controls to compare with and, as the experimental design is mostly shared with the previously discussed work also from Ferravante and colleagues (Table S1) [Citation67], both approaches share the same already commented limitations. Therefore, new approaches without such limitations are required to elucidate the potential relationship between the nasopharyngeal virome composition and the severity of the COVID-19 disease, as well as the possible relevance of viral coinfection in SARS-CoV-2 positive individuals. Interestingly, a recent study compared the nasopharyngeal microbiota of individuals infected with SARS-CoV-2, flu A, flu B, and uninfected controls, finding that while Streptococcus, Prevotella, Veillonella, and Fusobacterium were the most abundant bacterial genera in uninfected healthy controls, Enterobacteriaceae was predominant in influenza patients, and Staphylococcus and Pseudomonas in the SARS-CoV-2 positive subjects (Table S1) [Citation62]. However, coinfection cases were not studied.
The nasopharyngeal metagenome, metatranscriptome and metabolome of SARS-CoV-2 infected individuals are also barely known. A total RNA-seq analysis using nasopharyngeal samples from COVID-19 patients, already recovered subjects, and uninfected controls revealed that genes coding for pyruvate carboxylase, adherent junction, tight junction, environmental information processing, carbohydrates metabolism, and oxidative phosphorylation had higher expression levels in the nasopharyngeal microbiome of COVID-19 patients compared to the microbiomes of recovered or uninfected subjects (Table S1) [Citation61]. Regarding the metabolome, Bai and colleagues, who analyzed nasopharyngeal samples from critically ill COVID-19 patients and uninfected healthy controls, identified a correlation between many metabolic pathways involved in vitamin K2 biosynthesis and both better respiratory status and lower inflammation level, suggesting that vitamin K2 could play a role in improving clinical outcome of COVID-19 patients (Table S1) [Citation52]. This is consistent with the reported correlations between vitamin K deficiency and severe COVID-19 outcome [Citation74], and its relevant role in immunomodulation [Citation75].
Finally, it is noteworthy that the experimental designs of the majority of these studies, with the collection of only one nasopharyngeal sample per infected subject at variable disease stages, make difficult to elucidate whether the different changes observed in the nasopharyngeal microbiota according to the COVID-19 severity reflect baseline differences prior to infection resulting in distinct levels of susceptibility to SARS-CoV-2 infection, or if those microbiota alterations are due to the existence of distinct COVID-19 severity levels, as consequence of other factors, that differentially alter the nasopharyngeal microbiota. Further studies are essential to shed light on this important pending issue, since if the different COVID-19 severities were consequence of the existence of distinct baseline nasopharyngeal microbiota compositions prior to infection, the nasopharyngeal microbiota of SARS-CoV-2 positive individuals could potentially be used to predict which patients may be more likely to progress to severe disease. In addition, if this were the case, novel strategies could be developed to modify the nasopharyngeal microbiota with the objective of decreasing COVID-19 severity.
4.3. Study limitations and potential sources of data variability
As previously mentioned, when results from the publications that study the changes in the nasopharyngeal microbiota of subjects infected with SARS-CoV-2 are analyzed in depth, the high variability observed in these data is striking. Regarding the methodology used in these studies, different sequencing technologies, namely 16S rRNA gene sequencing and metagenomic sequencing, have been used to analyze the possible alterations in the nasopharyngeal microbiota of SARS-CoV-2 positive subjects, as well as the potential correlations between such microbiota and the severity of the COVID-19 disease (Table S1). The advantages and disadvantages of both technologies are already well-known: in summary, while 16S rRNA profiling has higher sensitivity but lower taxonomic resolution and the need to assume PCR amplification bias, metagenomic sequencing presents higher taxonomic resolution and incorporates functional information without the need of extrapolation [Citation76]. Although the 16S rRNA gene sequencing approach to study the microbiota could introduce bias in the obtained data because this method does not allow the study of the whole microbiome, but only the genera amplified by PCR, it is the most commonly used technique to study microbiota in clinical samples. In addition, not all the studies using 16S rRNA gene sequencing amplify the same hypervariable regions of the 16S rRNA, whereas the approaches using metagenomic sequencing employ distinct sequencing platforms (Table S1). Importantly, it is not possible to identify any pattern showing a correlation between the results obtained in these studies and the sequencing technology used, suggesting that this is not one of the relevant causes of result variability (Table S1). However, one of the most common limitations of these publications, which could be an important source of variability and prevents robust conclusions from being obtained, is the low sample sizes, with several particularly remarkable cases of studies that include fewer than 25 subjects in total, of which the number of SARS-CoV-2 positive individuals is fewer than 10 (Table S1) [Citation49,Citation59,Citation61]. Note that, as commented above for the study conducted by Ferravante and colleagues (Table S1) [Citation67], even though some experimental designs do not enroll a low number of individuals, the sample sizes drastically decrease for the different groups resulting from the stratification of patients by severity, age, or any other parameter [Citation67,Citation72]. Although calculating the adequate sample size for a particular microbiome study depends on many factors, including the characteristics of the cohort, the experimental design, and the specific objectives of the study, among others, there are recent works which address this issue in a very detailed and rigorous way and that, therefore, could be of great help for the appropriate design of future studies in the field [Citation77]. Furthermore, another potential source of variability to consider here is the huge heterogeneity of the SARS-CoV-2 positive patients enrolled in many of these studies, due to multiple factors such as a very restricted geographical origin, the collection of samples from individuals with different COVID-19 severities or at different disease stages, the enrolment of subjects who have been treated with antibiotics and/or other drugs that could drastically alter the bacterial microbiota, the possibility that those who were selected as uninfected controls might have actually encountered the virus before the enrolment, the absence of multiple time-points for each case, the non-availability of information about comorbidities that have been associated with COVID-19 severity [Citation78] and that could influence the nasopharyngeal microbiota, the non-standardization of the sample and/or data collection in advance, and the lack of appropriate uninfected control subjects. It is also relevant that the studies aiming to identify and characterize potential associations between different nasopharyngeal microbiota compositions and distinct COVID-19 disease severities, usually do not use homogeneous or standardized criteria to stratify patients by severity. Something similar occurs with the stratification of subjects by age, even with studies that only include individuals between 49 and 78 years of age [Citation64] or subjects who are younger than 21 years old [Citation69], making difficult to extrapolate their results to other stages of life (Table S1).
Finally, and even more important, these studies did not consider the possibility that the SARS-CoV-2 positive individuals enrolled could be infected by distinct SARS-CoV-2 variants. Since it has already been described that the distinct SARS-CoV-2 variants of concern are able to elicit distinct responses in the host [Citation79,Citation80], it would not be surprising if these variants could differentially alter the nasopharyngeal microbiota. This is a particularly relevant issue to consider here, as some of these studies even state that their samples come from three different COVID-19 waves and from patients with different disease severities (Table S1) [Citation66,Citation67,Citation70], which makes it very likely that they may be infected by distinct variants of the virus and, therefore, the results from the different patients enrolled in these studies would not be comparable.
5. Conclusions
To date, numerous studies have tried to determine whether SARS-CoV-2 infection or the COVID-19 disease severity are associated to changes in the nasopharyngeal microbiome. Diversity results, which are easy to interpret and compare, are controversial, since (1) there are studies that did not find any significant diversity differences when comparing the nasopharyngeal microbiota of SARS-CoV-2 infected subjects with that of uninfected controls; (2) other publications observed a decreased nasopharyngeal microbiota diversity in infected individuals when comparing those same groups; (3) a third group of studies revealed a decreased nasopharyngeal microbiota diversity in the most severe cases of COVID-19 disease compared to milder cases; and, finally, (4) even there is a study that, contrary to all the previously observed data, reported higher nasopharyngeal microbiota diversity in pregnant SARS-CoV-2 infected women compared to pregnant uninfected control women. Furthermore, nasopharyngeal microbiota composition results are also controversial and more difficult to compare between the different studies, as authors not always highlight or discuss changes in taxa that present the biggest differences, but frequently they focus on taxa which are of their interest because are related to their usual field of research. Independently on the high variability of these data, the majority of publications addressing this topic have in common that they found significant differences in the nasopharyngeal microbiota composition, either when comparing SARS-CoV-2 positive subjects (without attending to their disease severity) with uninfected controls, critically ill COVID-19 patients with asymptomatic patients and/or uninfected controls, or non-severe symptomatic COVID-19 patients with asymptomatic/paucisymptomatic individuals and/or uninfected controls. The most notable exceptions are the studies that compared asymptomatic/paucisymptomatic patients to uninfected healthy controls, which observed very similar nasopharyngeal microbiota compositions in both cases. Changes in the relative abundance of the bacterial genus Corynebacterium in the nasopharynx of SARS-CoV-2 infected subjects draws attention because, despite the heterogeneity of data, become the focus of multiple studies, suggesting that these changes are robust and could be biologically relevant. Unfortunately, not much has been reported about the possible association between changes in the nasopharyngeal virome and phageome and the infection by SARS-CoV-2 or the severity of the COVID-19 disease. Thus, the little data available on this suggest that respiratory viral coinfections seem to be not closely associated with SARS-CoV-2 infection or disease severity, and that age and disease severity could be associated with the bacteriophage abundance in COVID-19 patients. Similarly, little is known about the nasopharyngeal metagenome, metatranscriptome and metabolome of SARS-CoV-2 infected individuals, highlighting the finding that vitamin K2 could be playing a role in improving the clinical outcome of COVID-19 patients.
As previously mentioned, results from the different studies exploring the possible associations between changes in the nasopharyngeal microbiota and infection by SARS-CoV-2 or COVID-19 disease severity often present high variability, with contradictory data in some aspects. Here, we have identified and discussed many potential causes of that variability, including the different methodologies used, the low sample sizes, the heterogeneity of the enrolled subjects, or the lack of standardization of the distinct criteria used to stratify individuals, among others. It is understandable that most of these possible sources of variability were a consequence of the urgency to obtain new knowledge about the pathogenesis of a viral infection that was spreading rapidly around the world, but avoiding these problems should be an objective for the future research in the field. Among the multiple limitations observed in these studies, the fact that they have not taken into consideration which are the SARS-CoV-2 variants infecting the different individuals enrolled, even when they come from different COVID-19 pandemic waves, is not important only because the results obtained from those patients could be not comparable without stratifying them by their infecting variant, but also because it would be important to know whether each variant could differentially correlate with different alterations of the nasopharyngeal microbiota. If this were the case, characterizing which are the changes in the nasopharyngeal microbiota associated to the infection by each SARS-CoV-2 variant would be extremely relevant from a biomedical point of view.
In summary, (1) there is already a significant amount of information about the nasopharyngeal bacterial microbiota of SARS-CoV-2 positive subjects, including that of COVID-19 patients with different disease severities (albeit much of this information comes from studies which present important limitations); (2) almost nothing is known about the nasopharyngeal virome, metagenome, metatranscriptome, phageome, and metabolome of these individuals; and (3) absolutely nothing is known about their nasopharyngeal mycobiome. Therefore, further studies that avoid the limitations discussed here (especially with high sample sizes and standardized criteria to stratify patients) will be essential to fill these gaps in the knowledge of a field that could be crucial from a biomedical point of view, particularly with regard to the possibility of manipulating the nasopharyngeal microbiota, strategy that have already worked in the treatment of other diseases such as cancer, metabolic disorders, and virus infections [Citation81-83], in order to reduce the severity of COVID-19 patients and/or improve their disease outcome.
Table_S1_R1.pdf
Download PDF (148.7 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Marchesi JR, Ravel J. The vocabulary of microbiome research: a proposal. Microbiome. 2015;3:31.
- Kilian M, Chapple IL, Hannig M, et al. The oral microbiome - an update for oral healthcare professionals. Br Dent J. 2016;221(10):657–666.
- Berg G, Rybakova D, Fischer D, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome. 2020;8(1):103.
- Consortium HMP. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214.
- Lloyd-Price J, Abu-Ali G, Huttenhower C. The healthy human microbiome. Genome Med. 2016;8(1):51.
- Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14(8):e1002533.
- Guarner F, Malagelada JR. Gut flora in health and disease. Lancet. 2003;361(9356):512–519.
- Frank DN, Pace NR. Gastrointestinal microbiology enters the metagenomics era. Curr Opin Gastroenterol. 2008;24(1):4–10.
- Sansonetti PJ, Medzhitov R. Learning tolerance while fighting ignorance. Cell. 2009;138(3):416–420.
- Gilbert JA, Blaser MJ, Caporaso JG, et al. Current understanding of the human microbiome. Nat Med. 2018;24(4):392–400.
- Nemergut DR, Schmidt SK, Fukami T, et al. Patterns and processes of microbial community assembly. Microbiol Mol Biol Rev. 2013;77(3):342–356.
- Gaci N, Borrel G, Tottey W, et al. Archaea and the human gut: new beginning of an old story. World J Gastroenterol. 2014;20(43):16062–16078.
- Horz HP, Conrads G. Methanogenic Archaea and oral infections - ways to unravel the black box. J Oral Microbiol. 2011;3.
- Koskinen K, Reichert JL, Hoier S, et al. Massively parallel coincidence counting of high-dimensional entangled states. Sci Rep. 2018;8(1):1296.
- Moissl-Eichinger C, Probst AJ, Birarda G, et al. Human age and skin physiology shape diversity and abundance of Archaea on skin. Sci Rep. 2017;7(1):4039.
- Mukherjee PK, Sendid B, Hoarau G, et al. Mycobiota in gastrointestinal diseases. Nat Rev Gastroenterol Hepatol. 2015;12(2):77–87.
- Gow NAR, Yadav B. Microbe Profile: Candida albicans: a shape-changing, opportunistic pathogenic fungus of humans. Microbiology (Reading). 2017;163(8):1145–1147.
- Parfrey LW, Walters WA, Lauber CL, et al. Communities of microbial eukaryotes in the mammalian gut within the context of environmental eukaryotic diversity. Front Microbiol. 2014;5:298.
- Turnbaugh PJ, Ley RE, Hamady M, et al. The human microbiome project. Nature. 2007;449(7164):804–810.
- Team NHMPA. A review of 10 years of human microbiome research activities at the US National Institutes of Health, Fiscal Years 2007-2016. Microbiome. 2019;7(1):31.
- Sahin-Yilmaz A, Naclerio RM. Anatomy and physiology of the upper airway. Proc Am Thorac Soc. 2011;8(1):31–39.
- Dubourg G, Edouard S, Raoult D. Relationship between nasopharyngeal microbiota and patient's susceptibility to viral infection. Expert Rev Anti Infect Ther. 2019;17(6):437–447.
- Man WH, de Steenhuijsen Piters WA, Bogaert D. The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat Rev Microbiol. 2017;15(5):259–270.
- Salzano FA, Marino L, Salzano G, et al. Microbiota composition and the integration of exogenous and endogenous signals in reactive nasal inflammation. J Immunol Res. 2018;2018:2724951.
- Di Stadio A, Costantini C, Renga G, et al. The microbiota/host immune system interaction in the nose to protect from COVID-19. Life (Basel). 2020;10(12.
- Pabst R. Mucosal vaccination by the intranasal route. Nose-associated lymphoid tissue (NALT)—Structure, function and species differences. Vaccine. 2015;33(36):4406–4413.
- De Rudder C, Garcia-Tímermans C, De Boeck I, et al. Lacticaseibacillus casei AMBR2 modulates the epithelial barrier function and immune response in a donor-derived nasal microbiota manner. Sci Rep. 2020;10(1):16939.
- Dimitri-Pinheiro S, Soares R, Barata P. The Microbiome of the Nose-Friend or Foe? Allergy Rhinol (Providence). 2020 2020 Jan-Dec;11:2152656720911605.
- Hanada S, Pirzadeh M, Carver KY, et al. Respiratory viral infection-induced microbiome alterations and secondary bacterial pneumonia. Front Immunol. 2018;9:2640.
- Kumpitsch C, Koskinen K, Schöpf V, et al. The microbiome of the upper respiratory tract in health and disease. BMC Biol. 2019;17(1):87.
- Edouard S, Million M, Bachar D, et al. The nasopharyngeal microbiota in patients with viral respiratory tract infections is enriched in bacterial pathogens. Eur J Clin Microbiol Infect Dis. 2018;37(9):1725–1733.
- Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–733.
- Tay MZ, Poh CM, Rénia L, et al. The Trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363–374.
- Zipeto D, Palmeira JDF, Argañaraz GA, et al. Ace2/ADAM17/TMPRSS2 interplay May Be the main risk factor for COVID-19. Front Immunol. 2020;11:576745.
- Rahman N, Basharat Z, Yousuf M, et al. Virtual screening of natural products against type II transmembrane serine protease (TMPRSS2), the Priming Agent of Coronavirus 2 (SARS-CoV-2). Molecules. 2020;25(10.
- Hou YJ, Okuda K, Edwards CE, et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell. 2020;182(2):429–446.e14.
- Ziegler CGK, Allon SJ, Nyquist SK, et al. SARS-CoV-2 receptor ACE2 Is an interferon-stimulated gene in human airway epithelial cells and Is detected in specific cell subsets across tissues. Cell. 2020;181(5):1016–1035.e19.
- Zhou Y, Leary O, J T. Relative sensitivity of anterior nares and nasopharyngeal swabs for initial detection of SARS-CoV-2 in ambulatory patients: Rapid review and meta-analysis. PLoS One. 2021;16(7):e0254559.
- Akdis CA. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat Rev Immunol. 2021;21(11):739–751.
- Fiorito S, Soligo M, Gao Y, et al. Is the epithelial barrier hypothesis the key to understanding the higher incidence and excess mortality during COVID-19 pandemic? The case of Northern Italy. Allergy. 2022;77(5):1408–1417.
- Hang J, Zavaljevski N, Yang Y, et al. Composition and variation of respiratory microbiota in healthy military personnel. PLoS One. 2017;12(12):e0188461.
- De R, Dutta S. Role of the Microbiome in the Pathogenesis of COVID-19. Front Cell Infect Microbiol. 2022;12:736397.
- Lemon KP, Armitage GC, Relman DA, et al. Microbiota-targeted therapies: an ecological perspective. Sci Transl Med. 2012;4(137):137rv5.
- De Maio F, Posteraro B, Ponziani FR, et al. Nasopharyngeal microbiota profiling of SARS-CoV-2 infected patients. Biol Proced Online. 2020;22:18.
- Braun T, Halevi S, Hadar R, et al. SARS-CoV-2 does not have a strong effect on the nasopharyngeal microbial composition. Sci Rep. 2021;11(1):8922.
- Nardelli C, Gentile I, Setaro M, et al. Nasopharyngeal microbiome signature in COVID-19 positive patients: Can We definitively Get a role to fusobacterium periodonticum? Front Cell Infect Microbiol. 2021;11:625581.
- Nagy-Szakal D, Couto-Rodriguez M, Wells HL, et al. Targeted hybridization capture of SARS-CoV-2 and metagenomics enables genetic variant discovery and nasal microbiome insights. Microbiol Spectr. 2021;9(2):e0019721.
- Gupta A, Karyakarte R, Joshi S, et al. Nasopharyngeal microbiome reveals the prevalence of opportunistic pathogens in SARS-CoV-2 infected individuals and their association with host types. Microbes Infect. 2022;24(1):104880.
- Engen PA, Naqib A, Jennings C, et al. Nasopharyngeal microbiota in SARS-CoV-2 positive and negative patients. Biol Proced Online. 2021;23(1):10.
- Gauthier NPG, Locher K, MacDonald C, et al. Alterations in the nasopharyngeal microbiome associated with SARS-CoV-2 infection status and disease severity. PLoS One. 2022;17(10):e0275815.
- Zhang H, Ai JW, Yang W, et al. Metatranscriptomic characterization of coronavirus disease 2019 identified a host transcriptional classifier associated With immune signaling. Clin Infect Dis. 2021;73(3):376–385.
- Bai X, Narayanan A, Skagerberg M, et al. Characterization of the upper respiratory bacterial microbiome in critically Ill COVID-19 patients. Biomedicines. 2022;10(5.
- Mostafa HH, Fissel JA, Fanelli B, et al. Metagenomic next-generation sequencing of nasopharyngeal specimens collected from confirmed and suspect COVID-19 patients. mBio. 2020;11(6.
- Ventero MP, Moreno-Perez O, Molina-Pardines C, et al. Nasopharyngeal Microbiota as an early severity biomarker in COVID-19 hospitalised patients. J Infect. 2022;84(3):329–336.
- Qin T, Wang Y, Deng J, et al. Super dominant pathobiontic bacteria in the nasopharyngeal microbiota cause secondary bacterial infection in COVID-19 patients. Microbiol Spectr. 2022;10(3):e0195621.
- Chen J, Liu X, Liu W, et al. Comparison of the respiratory tract microbiome in hospitalized COVID-19 patients with different disease severity. J Med Virol. 2022;94(11):5284–5293.
- Crovetto F, Selma-Royo M, Crispi F, et al. Nasopharyngeal microbiota profiling of pregnant women with SARS-CoV-2 infection. Sci Rep. 2022;12(1):13404.
- Ventero MP, Cuadrat RRC, Vidal I, et al. Nasopharyngeal microbial communities of patients infected With SARS-CoV-2 that developed COVID-19. Front Microbiol. 2021;12:637430.
- Liu J, Liu S, Zhang Z, et al. Association between the nasopharyngeal microbiome and metabolome in patients with COVID-19. Synth Syst Biotechnol. 2021;6(3):135–143.
- Lloréns-Rico V, Gregory AC, Van Weyenbergh J, et al. Clinical practices underlie COVID-19 patient respiratory microbiome composition and its interactions with the host. Nat Commun. 2021;12(1):6243.
- Hoque MN, Sarkar MMH, Rahman MS, et al. SARS-CoV-2 infection reduces human nasopharyngeal commensal microbiome with inclusion of pathobionts. Sci Rep. 2021;11(1):24042.
- Rattanaburi S, Sawaswong V, Chitcharoen S, et al. Bacterial microbiota in upper respiratory tract of COVID-19 and influenza patients. Exp Biol Med (Maywood). 2022;247(5):409–415.
- Ferrari L, Favero C, Solazzo G, et al. Nasopharyngeal bacterial microbiota composition and SARS-CoV-2 IgG antibody maintenance in asymptomatic/paucisymptomatic subjects. Front Cell Infect Microbiol. 2022;12:882302.
- Kolhe R, Sahajpal NS, Vyavahare S, et al. Alteration in nasopharyngeal microbiota profile in aged patients with COVID-19. Diagnostics (Basel). 2021;11(9.
- Shilts MH, Rosas-Salazar C, Strickland BA, et al. Severe COVID-19 Is associated With an altered upper respiratory tract microbiome. Front Cell Infect Microbiol. 2021;11:781968.
- Giugliano R, Sellitto A, Ferravante C, et al. NGS analysis of nasopharyngeal microbiota in SARS-CoV-2 positive patients during the first year of the pandemic in the Campania Region of Italy. Microb Pathog. 2022;165:105506.
- Ferravante C, Sanna G, Melone V, et al. Nasopharyngeal virome analysis of COVID-19 patients during three different waves in Campania region of Italy. J Med Virol. 2022;94(5):2275–2283.
- Tchoupou Saha OF, Dubourg G, Yacouba A, et al. Profile of the nasopharyngeal microbiota affecting the clinical course in COVID-19 patients. Front Microbiol. 2022;13:871627.
- Hurst JH, McCumber AW, Aquino JN, et al. Age-related changes in the nasopharyngeal microbiome are associated with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and symptoms among children, adolescents, and young adults. Clin Infect Dis. 2022;75(1):e928–e937.
- Ferravante C, Arslan-Gatz BS, Dell'Annunziata F, et al. Dynamics of nasopharyngeal tract phageome and association with disease severity and age of patients during three waves of COVID-19. J Med Virol. 2022;94(11):5567–5573.
- Musuuza JS, Watson L, Parmasad V, et al. Multiple spatial reference frames underpin perceptual recalibration to audio-visual discrepancies. PLoS One. 2021;16(5):e0251827.
- Kim JG, Zhang A, Rauseo AM, et al. The nasopharyngeal and salivary microbiomes in COVID-19 patients with and without asthma. Allergy. 2022;77(12):3676–3679.
- Chen X, Liao B, Cheng L, et al. The microbial coinfection in COVID-19. Appl Microbiol Biotechnol. 2020;104(18):7777–7785.
- Linneberg A, Kampmann FB, Israelsen SB, et al. The association of Low vitamin K status with mortality in a cohort of 138 hospitalized patients with COVID-19. Nutrients. 2021;13(6.
- Ohsaki Y, Shirakawa H, Miura A, et al. Vitamin K suppresses the lipopolysaccharide-induced expression of inflammatory cytokines in cultured macrophage-like cells via the inhibition of the activation of nuclear factor κB through the repression of IKKα/β phosphorylation. J Nutr Biochem. 2010;21(11):1120–1126.
- Young RB, Marcelino VR, Chonwerawong M, et al. Key technologies for progressing discovery of microbiome-based medicines. Front Microbiol. 2021;12:685935.
- Ferdous T, Jiang L, Dinu I, et al. The rise to power of the microbiome: power and sample size calculation for microbiome studies. Mucosal Immunol. 2022;15(6):1060–1070.
- Andrés M, Leon-Ramirez JM, Moreno-Perez O, et al. Fatality and risk features for prognosis in COVID-19 according to the care approach - a retrospective cohort study. PLoS One. 2021;16(3):e0248869.
- Chiu CH, Chang YH, Chang FY, et al. Humoral, cellular and cytokine immune responses against SARS-CoV-2 variants in COVID-19 convalescent and confirmed patients With different disease severities. Front Cell Infect Microbiol. 2022;12:862656.
- Tyrkalska SD, Martinez-Lopez A, Arroyo AB, et al. Differential proinflammatory activities of Spike proteins of SARS-CoV-2 variants of concern. Sci Adv. 2022;8(37):eabo0732.
- Panebianco C, Latiano T, Pazienza V. Microbiota manipulation by probiotics administration as emerging tool in cancer prevention and therapy. Front Oncol. 2020;10:679.
- Boulangé CL, Neves AL, Chilloux J, et al. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016;8(1):42.
- Li N, Ma WT, Pang M, et al. The commensal microbiota and viral infection: A comprehensive review. Front Immunol. 2019;10:1551.