
ABSTRACT
5-Methylcytosine (m5C) is a widespread post-transcriptional RNA modification and is reported to be involved in manifold cellular responses and biological processes through regulating RNA metabolism. However, its regulatory role in antiviral innate immunity has not yet been elucidated. Here, we report that NSUN2, a typical m5C methyltransferase, negatively regulates type I interferon responses during various viral infections, including SARS-CoV-2. NSUN2 specifically mediates m5C methylation of IRF3 mRNA and accelerates its degradation, resulting in low levels of IRF3 and downstream IFN-β production. Knockout or knockdown of NSUN2 enhanced type I interferon and downstream ISGs during various viral infection in vitro. And in vivo, the antiviral innate response is more dramatically enhanced in Nsun2+/− mice than in Nsun2+/+ mice. The highly m5C methylated cytosines in IRF3 mRNA were identified, and their mutation enhanced cellular IRF3 mRNA levels. Moreover, infection with Sendai virus (SeV), vesicular stomatitis virus (VSV), herpes simplex virus 1 (HSV-1), or Zika virus (ZIKV) resulted in a reduction of endogenous NSUN2 levels. Especially, SARS-CoV-2 infection (WT strain and BA.1 omicron variant) also decreased endogenous levels of NSUN2 in COVID-19 patients and K18-hACE2 KI mice, further increasing type I interferon and downstream ISGs. Together, our findings reveal that NSUN2 serves as a negative regulator of interferon response by accelerating the fast turnover of IRF3 mRNA, while endogenous NSUN2 levels decrease during SARS-CoV-2 and various viral infections to boost antiviral responses for effective elimination of viruses.
Introduction
RNA modification is an important post-transcriptional modification process. To date, more than 100 types of chemical modifications to various types of RNAs have been recorded [Citation1]. Among these RNA modifications, N6-methyladenosine (m6A) and 5-methylcytosine (m5C) are ubiquitous, and have led to an increasing appreciation that RNA methylation can functionally regulate gene expression and cellular activity [Citation2–4]. The methyltransferase (writer), demethylase (eraser), and effector (reader) play coordinating roles in RNA metabolism, such as splicing, degradation, and translation [Citation5–9]. Recently, it was found that m6A methylation could negatively regulate interferon response by inducing IFNB1 mRNA degradation [Citation10,Citation11]. It was reported that m6A RNA-modification-mediated downregulation of the OGDH-itaconate pathway reprogrammes cellular metabolism to inhibit viral replication [Citation12]. Another study demonstrated that ALKBH5, an m6A demethylase, could be recruited by DDX46 and then erase the m6A modification in MAVS, TRAF3, and TRAF6 transcripts, thereby enforcing their retention in the nucleus and leading to their decreased translation, resulting in inhibited type I interferon production [Citation13]. Moreover, it is reported that the MAVS mRNA undergoes m6A modification through METTL14, which leads to a fast turnover of MAVS mRNA [Citation14]. Additionally, nuclear hnRNPA2B1 facilitates m6A modification and nucleocytoplasmic trafficking of CGAS, IFI16, and STING mRNAs, resulting in amplification of the innate immune response to DNA viruses [Citation15]. At present, m5C is not well studied compared to m6A. The primary writers for m5C methylation of mRNA in animals have been proposed to be NSUN2 and TRDMT1 (DNMT2) [Citation16,Citation17]. NSUN2 is reported to regulate the expression of numerous genes by methylating their mRNAs and thereby affecting their degradation or translation [Citation18–21]. Another report emphasized the transcriptome-wide role of NSUN2 as a major methyltransferase of the m5C epitranscriptomic mark and presented compelling evidence for the functional interdependence of mRNA m5C methylation and mRNA translation [Citation22]. Furthermore, it was reported that ALYREF and YBX1 served as potential m5C readers that could recognize m5C-modified mRNA and mediate mRNA export from the nucleus or affect the stability of their target mRNAs [Citation23–27]. Nevertheless, the demethylases responsible for removing m5C methylation on mRNA have not been clearly identified. Moreover, whether the m5C modification participates in the regulation of antiviral innate immunity, similarly to m6A modification, and especially in regulating the production of type I interferon responses, remains to be defined.
Elicitation of type I interferons (IFNs) by viruses or other pathogens plays an extremely critical role in innate immunity. The induction of type I interferons is primarily controlled at the level of gene transcription, wherein the interferon regulatory factor (IRF) family of transcription factors plays a central role [Citation28–31]. Interferon regulatory factor 3 (IRF3) acts as a master transcription factor responsible for the induction of type I interferons and is essential for the establishment of antiviral innate immunity [Citation32,Citation33]. After viral infection, IRF3 is phosphorylated by the kinases TBK1 and IKKϵ on its C-terminal and undergoes a conformational change and homodimerization, which leads to its translocation to the nucleus and subsequent association with the interferon-stimulated response elements of target genes [Citation34,Citation35]. Because of its pivotal role in the induction of type I interferons, the transcription factor IRF3 requires sophisticated regulation in order to effectively maintain immune homeostasis after viral infection. It has been reported that a great deal of regulators of IRF3 participate in maintaining the appropriate amounts of type I interferons stimulated by viral infection [Citation36–39]. The reported regulators of IRF3 mostly induce changes in the phosphorylation levels or quantity of IRF3 protein, which then affects type I interferon responses and downstream ISGs. Most reports mainly focus on the regulation of IRF3 at the protein level. However, there are few reports about the regulation of IRF3 at the mRNA level, especially involving epitranscriptomic modification.
Herein, we revealed that NSUN2, a typical RNA m5C methyltransferase, serves as a negative regulator of type I interferon responses during various viral infections, including SARS-CoV-2, especially. By combining RNA-seq, LACE-seq, bisulfite RNA sequencing and m5C-MeRIP-seq, we found that NSUN2 could specifically mediate m5C methylation of IRF3 mRNA and accelerate its degradation, and that knockout or knockdown of NSUN2 could elevate both mRNA and protein levels of IRF3 and thus amplify type I interferon responses and downstream ISGs expression after viral infection. The highly m5C-methylated cytosines in IRF3 mRNA were identified and the mutation of these cytosines could enhance the IRF3 levels and IFN-β production. Various viral infections resulted in a reduction in endogenous levels of NSUN2. Especially, SARS-CoV-2 infection (wild-type strain) also decreased endogenous levels of NSUN2 in COVID-19 patients. In K18-hACE2 KI mice, SARS-CoV-2 infection, including wild-type strain and BA.1 omicron variant, reduced endogenous mRNA levels of Nsun2 in lung tissues but increased mRNA levels of Irf3, Ifnb1 and downstream ISGs, Isg15 and Cxcl10, to resist SARS-CoV-2 infection. These results indicated that NSUN2 serves as a negative regulator of interferon response, while endogenous NSUN2 levels decrease during various viral infections to boost antiviral responses for the effective elimination of viruses. We outline a paradigm of innate immune responses to viral infection in which genes are ingeniously regulated by epitranscriptomic modification.
Materials and methods
Viruses, cells, and reagents
The SARS-CoV-2 WT strain (IVCAS 6.7512) was provided by the National Virus Resource, Wuhan Institute of Virology, Chinese Academy of Sciences. The SARS-CoV-2 BA.1 omicron variant (BA1-HB00004) was from Hubei Provincial Center for Disease Control and Prevention. Sendai virus (SeV), herpes simplex virus 1 (HSV-1), and vesicular stomatitis virus carrying a GFP reporter gene (VSV-GFP) were kindly provided by Dr. Hong-Bing Shu. Zika virus (ZIKV) was kindly provided by Dr. Bo Zhang. Vesicular stomatitis virus (VSV) was kindly provided by Dr. Ming-Zhou Chen. A549 IFNAR1−/− cell was kindly provided by Dr. Ying Zhu [Citation40]. Human colorectal adenocarcinoma (Caco-2), HEK293T, HeLa, Vero and A549 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) and THP-1 was maintained in RPMI 1640, with 10% fetal bovine serum, 100 U/mL penicillin and 100 µg/mL streptomycin, at 37°C in 5% CO2 incubator. Plasmids were transfected using Lipofectamine 3000 (Invitrogen) or Neofect (Neofect, TF201201) following the manufacturer’s instructions, and siRNAs (RiboBio, Guangzhou) were transfected using RNAiMAX (Invitrogen) following the manufacturer’s instructions. Ruxolitinib and actinomycin D were from MCE (MedChemExpress).
Mice
Nsun2+/− C57BL/6J mice and K18-hACE2 knock-in (KI) mice were obtained from Gempharmatech Co.,Ltd (Nanjing, China) and housed and bred in specific pathogen-free conditions. All animal experiments were conducted in accordance with the Regulations of Hubei Province Laboratory Animal Management and approved by Wuhan University Animal Experiment Ethics Committee.
SARS-CoV-2 infection K18-hACE2 KI mice models
For the establishment of SARS-CoV-2 infection mouse model, 8-week-old K18-hACE2 KI mice were lightly anesthetized with isoflurane and intranasally with 250 plaque-forming units (PFU) of SARS-CoV-2 WT strain or 8000 PFU of SARS-CoV-2 BA.1 omicron variant in 50 μL DMEM. Mice were monitored and weighted daily. Two days post infection, mice were euthanized and lung tissues was taken.
Preparation of bone marrow-derived dendritic cells (BMDCs) and bone marrow-derived macrophages (BMDMs)
Bone marrow cells were isolated from C57BL/6J mouse tibia and femur and then cultured for 7–9 days in 10% FBS DMEM containing mouse GM-CSF (50 ng/mL, Peprotech) for BMDCs or M-CSF (50 ng/mL, Peprotech) for BMDMs.
Preparation of bronchoalveolar lavage fluid (BALF) and RNA-seq library construction and sequencing
The methods were described previously [Citation41]. NSUN2 expression analysis in COVID-19 patients compared with healthy individuals was obtained from the analysis of previous results (https://github.com/zhouyulab/ncov/).
Plasmids and RNA interference
NSUN2 was cloned into both the pCAGGS and pGEX6P-1 vector. The sequences of siRNAs were si-h-NSUN2#1: GAGATCCTCTTCTATGATC; si-h-NSUN2#2: GGAGAACAAGCTGTTCGAG; si-h-TRDMT1: GCGATATGCTCTTCT GTTA; si-h-METTL3: CTGCAAGTATGTTCACTATGA; si-h-METTL14: AAGGATGAGTTAATAGCTAAA; si-h-ALKBH5: GTCGGGACTGCATAATTAA; si-m-Nsun2#1: GGAAGAATGGACAACCTT; si-m-Nsun2#2: GCTGGAAAGTCAGATGATA. The knockdown efficiency was checked 36–48 h after transfection using immunoblot analysis or qPCR.
Antibodies and immunoblot analysis
The antibodies used were as follows: rabbit anti-NSUN2 (Proteintech, 20854-1-AP), rabbit anti-Phospho-IRF-3-Ser396 (CST, 83611S), rabbit anti-IRF3 (Proteintech, 11312-1-AP), rabbit anti-phospho-TBK1/NAK-Ser172 (CST, 14590S), rabbit anti-TBK1/NAK (CST, 38066S), mouse anti-HA (Sigma, H6908), rabbit anti-HA (Sigma, H3663), mouse anti-Flag (Proteintech, 66008-3-Ig), rabbit anti-Flag (Sigma, SAB4301135), mouse anti-m5C antibody (Abcam, ab10805), mouse anti-GAPDH (Proteintech, 60004-1-Ig), mouse anti-β-actin (Proteintech, 66009-1-Ig). Cells were washed once with PBS and lysed in RIPA lysis buffer (50 mM Tris, pH 7.6, 1% NP-40, 150 mM NaCl, 0.1% SDS). 5× SDS loading buffer was added to the protein sample and boiled for 10 min. Samples were resolved on SDS-PAGE and transferred onto nitrocellulose membrane (GE Healthcare), followed by blocking with TBS containing 0.1% Tween-20 (TBST) and 5% non-fat powdered milk or bovine serum albumin (BSA) and probing with different antibodies.
Co-immunoprecipitation and RNA-binding protein immunoprecipitation (RIP-qPCR)
HEK293T cells were seeded onto 6 cm dishes and transfected as illustrated above. Thirty-six hours after transfection, cells were lysed in RIPA buffer (50 mM Tris, pH 7.6, 1% NP-40, 150 mM NaCl, 0.1% SDS) containing protease inhibitors and phosphatase inhibitors, if necessary. The cell lysates were incubated overnight at 4°C with HA-tag rabbit mAb beads (Sepharose Bead Conjugate, 3956S, CST) or Flag-tag rabbit mAb beads (Sepharose Bead Conjugate, 70569S, CST). The beads were washed five times with cold PBS and then mixed with SDS loading buffer and boiled for 10 min prior to SDS-PAGE and immunoblot analysis. For RNA-binding protein immunoprecipitation, HEK293T cells were transfected and lysed with lysis buffer (20 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% Triton-X100, 1 mM EDTA, 0.1% SDS, and 2 mM DTT, RNase free) and incubated overnight at 4 °C with HA-tag rabbit mAb beads. Beads were washed for five times with lysis buffer and divided in half for RNA extraction and qPCR analysis or for immunoblot analysis.
RNA isolation and qPCR
Total RNA was isolated using TRIzol reagent (Invitrogen) following the manufacturer’s instructions. The isolated RNA was reverse transcribed to cDNA using PrimeScript RT Reagent Kit (Takara, RR037A). Real-time quantitative PCR was carried out through ABI 7500 Real Time PCR System by SYBR Green Master Mix (YEASEN, 11199ES03). GAPDH was used in normalization via the ΔΔCt method. Primer sequences are shown in Supplementary Table S1.
Protein expression and purification
Escherichia coli BL21 cells were transformed with pGEX-6p-1-GST-NSUN2 and cultured in 10 mL Luria broth medium at 37°C for 6 h. The culture was then transferred to 1000 mL Luria broth medium and grown at 37°C to an absorbance of 0.6–0.8 as measured at 600 nm. IPTG was added to the culture to achieve a final concentration of 0.2 mM and induced at 16°C for 16–20 h. Cell cultures were harvested by centrifugation and then lysed by lysozyme and ultrasonication. GST-tagged NSUN2 proteins were purified by affinity chromatography using reduced glutathione resin (GenScript, L00206) following the manufacturer's instructions. Finally, the recombinant proteins were eluted through incubation for 30 min at 4°C with 100 μL of 50 mM Tris (pH 8.0), 2 mM DTT and 10 mM reduced glutathione and 8% glycerine was added for snap-freezing in liquid nitrogen and storage at −80°C until use. The purity and quantity of the recombinant proteins were assessed by SDS-PAGE followed by staining with Coomassie blue and immunoblot analysis.
In vitro transcription assays
The cDNA of HeLa cells was used as a template for PCR amplification of each segment of IRF3, which were then used as templates for in vitro transcription following the manufacturer’s instructions (Invitrogen, 00612295). All 5’ primers of the segments contained the T7 promoter sequence (TAATACGACTCACTATAGGG). The transcription reaction was performed at 30°C for 16 h. The transcribed RNA was precipitated and identified by agarose gel electrophoresis.
In vitro methylation assays
Reaction mixtures (50 µL) containing 0.2 nM recombinant GST-tagged NSUN2, 0.01 nM in vitro transcribed fragments of mRNA, 1 µCi of S-adenosyl [methyl-3H] methionine (0.5 μCi/μl, PerkinElmer) in reaction buffer (500 mM Tris–HCl (pH 7.5), 5 mM EDTA, 10 mM dithiothreitol, 20 mM MgCl2) and 40 units of RNase inhibitor were incubated for 60 min at 37°C, as described [Citation21]. The 3H-labeled products were isolated using DEAE-Sephadex A-50 columns and quantitated by liquid scintillation counting (PerkinElmer). Non-isotopic methylated RNA fragments were prepared using cold SAM (Biolabs, 0991410) and in vitro transcribed RNA fragments under similar conditions.
In vitro translation assays
For in vitro translation assays, a cell-free translation system (Promega) with rabbit reticulocyte lysate (RRL) was used. Luc-IRF3 fragments were amplified by PCR by using primer pairs. The Luc-IRF3 fragments then were in vitro transcribed and further methylated by NSUN2 in vitro with or without SAM. The methylated and non-methylated transcripts (0.01 nM) were then purified and used for in vitro translation assays following the instructions. The translation efficiency was determined by measuring the activity of firefly luciferase with substrate.
Quantification of ribosome loading on to IRF3 transcripts
Ribosome-loaded RNA was isolated essentially as previously described [Citation42,Citation43]. Briefly, wild type and NSUN2 knockout HEK293T cells were seeded on 6-well plate and incubated for 48 h. Cells were incubated with media containing 100 µg/mL of cycloheximide for 10 min at 37°C, then washed in ice-cold PBS containing 100 µg/ mL cycloheximide. Ribosome lysis buffer (10 mM Tris–HCl pH 7.4, 5 mM MgCl2, 100 mM KCl, 1% Triton X, Protease inhibitor, 2 mM DTT, 100 mg/mL cycloheximide and RNase inhibitor) was used to lyse cells. Lysate was sheared by passage through a 26-gauge needle, slowly. One-tenth of the lysate was then used for RNA isolation for the “input” sample. The rest of the lysates were centrifuged at 2,000 × g for 5 min to pellet nuclei and large debris, then centrifuged at 13200 rpm for 5 min to remove smaller debris. After centrifuging, the supernatants were brought up to 4 mL in lysis buffer, and overlaid on a 30% sucrose cushion, then centrifuged at 164,000 g for 2 h at 4°C. RNAs were then extracted from the ribosomal pellet using TRIzol. RNA isolated from the input and ribosome loaded fractions then underwent reverse transcription followed by qPCR to quantify IRF3 and GAPDH mRNA levels.
Analysis of m5C/C ratio using LC-MS/MS
The LC-MS/MS analysis process was referred to the previous report [Citation44]. The digestion mixture contains 1 μg total RNA, 1 U nuclease P1, 10 mM NaCl and 2 mM ZnCl2 in a final volume of 30 μL. The mixture was incubated at 37°C for 3 h. Then 1 U shrimp alkaline phosphatase and 2.5 μL ammonium bicarbonate (1 M) were added into the mixture for another 2 h and diluted to 100 μL. 3 μL of the mixture was injection into the LC-MS/MS for m5C analysis. The nucleosides were separated by a C18 column and detected by triple-quadrupole MS (Shimadzu MS-8050 mass spectrometer, Tokyo, Japan). The amount of m5C in the samples were calculated by the stand curves.
RNA-seq
Total RNAs were extracted from indicated cells using TRIzol Reagent. DNA digestion was carried out after RNA extraction by DNaseI. RNA quality was determined by examining A260/A280 with Nanodrop. RNA Integrity was confirmed by 1.5% agarose gel electrophoresis. Qualified RNAs were finally quantified by Qubit 3.0 with QubitTM RNA Broad Range Assay kit (Life Technologies, Q10210). 2 µg total RNAs were used for stranded RNA sequencing library preparation using KCTM Stranded mRNA Library Prep Kit (Wuhan Seqhealth Co., Ltd. China, DR08402) for Illumina following the manufacturer’s instruction. PCR products corresponding to 200–500 bps were enriched, quantified and finally sequenced on Novaseq 6000 sequencer (Illumina) with PE150 model. The RNA-seq data has been deposited in the GSA database under the accession number: HRA002086 and CRA006300.
LACE-seq (Linear amplification of complementary DNA ends and sequencing)
LACE-seq service was provided by Wuhan Seqhealth Co., Ltd (Wuhan, China). Cells were crosslinked by UV-irradiation at 400 mJ/cm2. The crosslinked samples were resuspended in 2 mL lysis buffer 20 mM Tris-HCl, pH 8.0, 1 mM EDTA, 0.1% SDS, 400 mM NaCl, 1 mM DTT, 0.5% Triton X-100 (Invitrogen), Protease Inhibitor (Roche), 200 U/mL RNasin Plus (Promega) and 20 U/mL DNase I (NEB). RNAs in the supernatant were then fragmented with 0.1 U/µL MNase (Thermo Scientific, EN0181) for 10 min at 37°C. To perform the immunoprecipitation, the mixture was added 10 μg NSUN2-antibody (proteintech: 20854-1-AP) or HA-antibody (CST: 3724) and control IgG-antibody (CST: 2729) and rotating slowly at 4°C for 4 h after preclearing with 10 µL protein A/G magnetic beads (Thermo Scientific, 26162). The RNAs in the immunoprecipitate were then dephosphorylated for 10 min at 37°C by FastAP alkaline phosphatase (Thermo Scientific, EF0651). A linker DNA (AGATCGGAAGAGCACACGTCT) was added to the 3’end of RNAs by T4 RNA ligase 2, truncated (NEB, M0242), followed by reverse transcription. The first-strand cDNAs were recovered by phenol–chloroform extraction, followed by ethanol precipitation. The purified cDNAs were applied to sequencing library preparation using KC-Digital Stranded RNA Library Prep Kit for Illumina (Wuhan Seqhealth Co., Ltd. China, DR08502), omitting the ligation and reverse transcription steps. In brief, the Illumina P5 adapter was ligated to the cDNAs, followed by PCR amplification and purification. The PCR products between 130 and 300 bp were enriched, quantified and finally sequenced on Novaseq 6000 sequencer (Illumina) with PE150 model.
Raw reads were filtered by fastp (version 0.23.1). Clean Reads were further treated to eliminate duplication bias introduced in library preparation and sequencing. De-duplicated consensus sequences mapped to rRNAs were removed with SortMeRNA (version 4.3.3). The filtered reads were used for protein binding site analysis which were mapped to Human reference genome hg38 using STAR (version 2.7.6a) with default parameters. The RSeQC (version 2.6) was used for reads distribution analysis. The macs2 (version 2.2.7.1) software was used for peak calling. Peaks were annotated using bedtools (version 2.30.0). The R (version 3.6.1) was used for peak distribution visualization. The differentially binding peaks were identified by QNB (version 1.1.11). Sequence motifs enriched in peak regions were identified using Homer (version 4.10).
The LACE-seq data has been deposited in the GSA database under the accession number: HRA002556.
MeRIP-seq
The m5C-MeRIP-seq was provided by Cloudseq Biotech Inc. (Shanghai, China). Briefly, m5C RNA immunoprecipitation was performed with the GenSeq m5C RNA IP Kit (GenSeq, China) by following the manufacturer’s instructions. Both the input samples without immunoprecipitation and the m5C IP samples were used for RNA-seq library geneMouseion with NEBNext Ultra II Directional RNA Library Prep Kit (NEB, USA). The library quality was evaluated with BioAnalyzer 2100 system (Agilent Technologies, USA). Library sequencing was performed on an illumina Novaseq 6000 instrument with 150 bp paired-end reads. The m5C-MeRIP-seq data has been deposited in the GSA database under the accession number: HRA002087.
Reporter gene assays
Cells were seeded into 24-well plates (2 × 105 cells per well) and transfected with 100 ng of luciferase reporter plasmid together with a total of 0.5 μg of expression plasmid or empty control plasmid. Twenty nanograms of pRL-TK Renilla luciferase reporter plasmid was also transfected to normalize the transfection efficiency. For the knockdown system, siRNAs were first transfected and 36 h later, luciferase reporter plasmid and pRL-TK Renilla luciferase reporter were subsequently transfected. Luciferase activity in total cell lysates was measured using a dual-luciferase reporter assay system (Promega).
VSV plaque assay
Vero cells were seeded into 24-well plates to about 80%–90% density before infection. The supernatants containing VSV then were serially diluted for infection of Vero cells. Two hours later, supernatants were removed, and PBS was used to wash the infected Vero cells. The DMEM containing 2% methylcellulose and 10% FBS was overlaid onto the cells. Two days later, cells were fixed and stained with formaldehyde (4%) and crystal violetin (0.2%) for 6 h followed by washing with water. Finally, plaques were counted, and the results were averaged and multiplied by the dilution factor for calculation of viral titers as PFU/mL and statistical analyses were performed.
Endogenous IRF3 mRNA pull down
The four IRF3 CHIRP probes were as follows: CTTTATCATTCTTTGGGTAACA, AACTCGTAGATTTTATGTGGGT, AGATGGTCTGCTGGAAGACTTG, and AGGAACCAGTTTATTGGTTGAG. All the probes were 3’biotin-TEG-modified (Sangon Biotech). Ten × 10 cm dishes of cells were used for total RNA extraction for each group. The total RNA was dissolved in 600 µL hybrid buffer (350 mM NaCl, 0.5% SDS, 25 mM Tris–HCl, 1 mM EDTA, 7.5% formamide, pH 7.5), and 5 µL IRF3 probes (100 μM) were added and incubated at 65°C for 5 min followed by 37°C while rotating for 2 h. Then, 100 µL Dynabeads M-280 streptavidin (ThermoFisher, 11205D) was added followed by rotating at 37°C for 1 h. Six hundred microliters of wash buffer (2× SSC buffer, 0.5% SDS, RNase inhibitor) was used to wash the beads 5 times for 5 min at 4°C. RNase-free water (20 μL) was added for elution followed by incubation at 75°C for 5 min. After centrifuging at 1000 g for 3 min, the pulled down RNA was got in the eluate supernatant.
M5C dot blot analysis
Equal amounts mRNA was denatured at 65°C for 10 min followed by immediate chilling on ice. mRNA was mixed with RNA loading buffer and then carefully spotted onto a Hybond-N + membrane (GE Healthcare), followed by UV crosslinking. The membranes were washed with TBST 2 times and blocked with 5% BSA in TBST for 2 h. The anti-m5C antibody (Abcam, ab10805) was diluted 1:500 and incubated with the membranes at 4 °C overnight. Membranes were washed 3 times with TBST for 10 min and then incubated with goat anti-mouse IgG-HRP for 1 h at room temperature. Membranes were washed 3 times with TBST for 5 min followed by chemiluminescence. Equal RNA loading was verified by methylene blue (MB) staining.
m5C-Methylated RNA immunoprecipitation qPCR (m5C-RIP-qPCR)
200 μg of total RNA was incubated with anti-m5C antibody in 800 µL of IPP buffer (150 mM NaCl, 0.1% NP-40, 10 mM Tris–HCl, pH 7.4) for 2 h at 4°C. The mixture was then incubated with 30 µL proteinA/G beads overnight. The beads were then washed 5 times with IPP buffer, followed by RNA extraction and qPCR analysis.
Bisulfite RNA sequencing
The adaptor sequences used were Adaptor-F: AGGTCTGGCTGAAGTTGA; Adaptor-R: ATACCTCCGTGACCATTT. The sequencing primers were Adaptor-F-mut: AGGTTTGGTTGAAGTTGA; Adaptor-R-mut: ATACCTCCATAACCATTT. Bisulfite RNA sequencing was performed to identify the m5C methylation site within an RNA fragment as previously described [Citation45,Citation46]. Briefly, 10 μg in vitro methylated RNA fragment (methylated by NSUN2 using cold SAM or unmethylated) was dissolved in 10 µL of RNase-free water and denatured at 65°C for 10 min followed by immediate chilling on ice. Samples were then mixed with 42.5 µL of 5 M sodium bisulfite mix (Epitect) and 17.5 µL DNA protection buffer (Epitect) and incubated at 70°C for 5 min then 60°C for 1 h, and this process was repeated for 4 cycles, followed by desalting using Micro Bio-spin 6 Chromatography Columns (Biorad, 732-6200). Then, the RNA adducts were desulfonated by adding 1 volume of Tris–HCl (pH 9.0) at 37°C for 1 h. Next, 0.3 M sodium acetate (pH 5.2), 20 μg glycogen (Beyotime, D0812) and 3 volumes of 100% ethanol were added for precipitation. The RNA was precipitated at −80°C for at least 5 h and then centrifuged. The bisulfite-converted RNA was reverse-transcribed using Adaptor-R-mut primer and random primer and subjected to PCR with Es Taq DNA polymerase (CW0688S) using Adaptor-mut primer pairs. The PCR products were inserted into the pGEM-T Easy Vector System (Promega, A1360) following the manufacturer’s instructions. The plasmids purified from single clones were sequenced by T7 promoter. The sequencing results were checked by alignment with the corresponding original IRF3 mRNA sequence, and the retained cytosines (C) were considered to be methylated by NSUN2. The unmethylated cytosines (C) were converted to uracils (U) on RNA segments.
Lentiviral package and infection
A lentiviral system was utilized to obtain NSUN2 knockout cells or stable cell lines in Irf3−/−Irf7−/− MEFs. For this, lentiviral backbone (2 µg), psPAX2 (1 μg), and pMD2.G (1 μg) were transiently transfected into HEK293T cells which were plated on 6-well plates. Forty-eight hours later, supernatants were collected and filtered using a 0.45 μm filter to infect target cells with polybrene (8 µg/mL). Cells were infected twice to get a higher transduction efficiency. Then, puromycin was used to screen positive cells.
Construction of knockout cell line by CRISPR/Cas9
The gRNAs were NSUN2-gRNA-1: F-CACCGACGCGGAGGATGGCGCCGA and R-AAACTCGGCGCCATCCTCCGCGTC; NSUN2-gRNA-2: F-CACCACCGTG GCGTTTCAGCGGTT and R-AAACAACCGCTGAAACGCCACGGT. The gRNAs were constructed in lentiCRISPR-v2 plasmid (Addgene). The lentiviral package and infection were the same as above, followed by seeding into 96-well plates (1 cell per well). After two weeks’ cultivation, single clones were selected following enlarged cultivation with puromycin selection. Single clones were identified by immunoblot analysis, and genomic DNA was extracted followed by PCR and sequencing.
Results
NSUN2 negatively regulates type I interferon responses
To explore the function of RNA methyltransferase or demethylases involved in type I interferon responses, we knocked down different RNA methyltransferases or demethylases in HEK293T cells using small interfering RNAs (siRNAs) and detected the endogenous levels of IFNB1 mRNA. We found that compared with other methyltransferases or demethylases, knockdown of NSUN2 could more dramatically enhance endogenous IFNB1 mRNA levels (a). To confirm the impact of NSUN2 on type I interferon responses, we examined the effect of exogenous NSUN2 expression and found that it could inhibit the activation of IFN-β promoter activity induced by Sendai virus (SeV) in a dose-dependent manner (b). Exogenous NSUN2 expression could also inhibit the activation of IFN-β promoter activity induced by different stimulants (c). In NSUN2 knockdown HEK293T cells, the SeV-induced increase in endogenous IFNB1 mRNA levels was dramatically enhanced as was the mRNA levels of downstream ISG15 and CXCL10 (d). SeV-induced type I interferon responses were also significantly enhanced in NSUN2 knockout HEK293T cells (e and Supplementary Fig. S1a) and A549 cells (Supplementary Fig. S2a and Fig. S1b). Moreover, knockdown of NSUN2 also promoted SeV-induced type I interferon responses in HeLa, THP-1, bone marrow-derived dendritic cells (BMDCs) and bone marrow-derived macrophages (BMDMs) (f,g). Of note, knockdown of NSUN2 consistently promoted type I interferon responses in SARS-CoV-2 infected Caco-2 cells, which revealed that NSUN2 also plays an important regulatory role in the infection of SARS-CoV-2 (h).
Figure 1. NSUN2 negatively regulates antiviral innate type I interferon responses. (a) qPCR analysis of IFNB1 mRNA in HEK293T cells transfected with siControl or siRNAs targeting different RNA methyltransferases or demethylases for 36 h, with or without infection by SeV for another 8 h. (b) Dual-luciferase assay analysing IFN-β promoter activity (IFN-β-Luc) in HEK293T cells in 24-well plates transfected for 24 h with 100 ng IFN-β-Luc plasmid and 20 ng Renilla luciferase plasmid (RL-TK) along with vector or increasing amounts (0, 0.1, 0.2, and 0.5 μg) of plasmid encoding NSUN2, with or without infection by SeV, for another 10 h. (c) Dual-luciferase analysis of IFN-β-Luc in HEK293T cells in 24-well plates transfected for 24 h with vector (Vec) or NSUN2, with or without infection by SeV or VSV for another 10 h, or transfected with poly (I:C) (1 µg/mL) for another 10 h. (d) qPCR analysis of IFNB1, ISG15, CXCL10 and NSUN2 mRNA in HEK293T cells transfected with siControl or siRNAs targeting NSUN2, with or without infection by SeV for 8 h. Immunoblot analysis shows knockdown efficiency of siRNAs targeting NSUN2. (e) qPCR analysis of IFNB1, ISG15 and CXCL10 mRNA in wild-type HEK293T cells or NSUN2−/− HEK293T cells, with or without infection by SeV for 8 h.
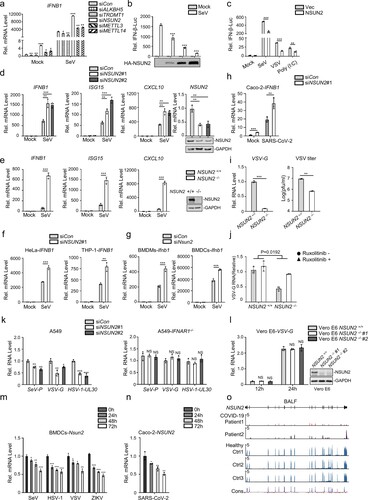
We next investigated whether NSUN2 is involved in antiviral responses during vesicular stomatitis virus (VSV) infection. Knockout of NSUN2 in HEK293T significantly inhibited the replication of VSV carrying a green fluorescent protein (GFP) reporter (VSV-GFP) (i). The same results were also obtained in NSUN2-knockout A549 cell lines compared with wild-type A549 cells (Supplementary Fig. S2b-e). These results indicate that knockout of NSUN2 results in cells being less vulnerable to VSV-GFP infection compared to wild-type cells. To further confirm that the inhibition of VSV replication in the NSUN2-deficient cells was indeed due to more potent type I interferon responses, we tested whether inhibition of interferon pathway affected VSV propagation. For this, we used ruxolitinib, a potent and selective JAK 1/2 inhibitor that blocks signalling downstream of type I interferon receptors. As shown in (j), the inhibition of VSV propagation in NSUN2-knockout cells could be rescued by ruxolitinib treatment. Furthermore, knockdown of NSUN2 inhibited the propagation of SeV, VSV and herpes simplex virus 1 (HSV-1) in A549 wild-type cells but not in A549 IFNAR1−/− cells (k). We also generated NSUN2 knockout cell lines in Vero E6 cells which are deficient in interferon secretion. As shown in (l) and Supplementary Fig. S1c, knockout of NSUN2 did not affect the VSV propagation in Vero E6 cells. These results confirmed that the enhanced antiviral effects due to NSUN2 loss mainly depended on the increased expression of type I interferon. To summarize, these results strongly suggest that NSUN2 is a negative regulator of type I interferon responses and that NSUN2 deficiency prominently enhances antiviral innate responses and, thus, inhibits virus propagation.
To further investigate the biological role of NSUN2 during viral infection, we observed that the Nsun2 mRNA indeed decreased with the progression of time following infection of BMDCs by SeV, HSV-1, VSV, or Zika virus (ZIKV), which reveals the potential function of NSUN2 during viral infections (m). Of note, we found that SARS-CoV-2 infection could also significantly reduce NSUN2 mRNA levels in Caco-2 cells (n). We further carried out transcriptome sequencing of the RNAs isolated from the bronchoalveolar lavage fluid (BALF) of two COVID-19 patients [Citation41]. NSUN2 mRNA was consistently reduced in COVID-19 patients compared with healthy individuals (o). Taken together, the results indicate that NSUN2 may serve as a negative regulator of type I interferon responses, and that expression of NSUN2 is dramatically reduced to enhance antiviral type I interferon responses during infection with different viruses, including SARS-CoV-2.
NSUN2 inhibits type I interferon responses by regulating IRF3 expression levels
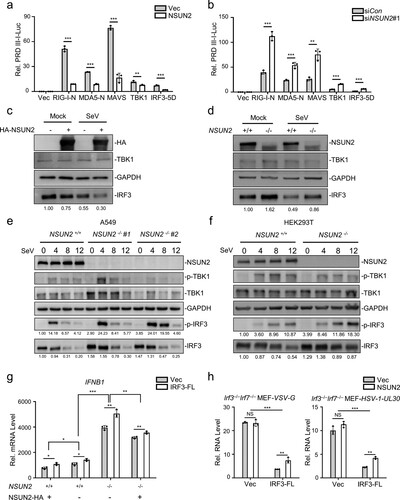
To investigate the mechanism of NSUN2 in the regulation of type I interferon responses, exogenous NSUN2 expression markedly suppressed the PRDIII-I-luc activity induced by upstream activators, including RIG-I, MDA5, MAVS, TBK1, and the constitutively active phosphorylation mimetic IRF3-5D (a), while knockdown of NSUN2 had the opposite effect (b). Since IRF3 is the final factor in the process of initiation of type I interferon responses, we speculated that NSUN2 may exert its function at IRF3 node. As shown in (c), immunoblot analysis revealed that exogenous NSUN2 expression could inhibit the expression of endogenous IRF3 and that the levels of endogenous IRF3 were enhanced in NSUN2-knockout cells compared with those in wild-type HEK293T (d) and A549 (e) cells. By contrast, endogenous TBK1 protein levels did not show significant change. Moreover, knockout of NSUN2 promoted levels of IRF3 Ser396 phosphorylation but not TBK1 Ser172 phosphorylation (e,f). These results demonstrate that NSUN2 deletion could enhance the overall levels of IRF3 protein as well as its phosphorylation. Moreover, qPCR analysis of IFNB1 mRNA in Nsun2+/+ and Nsun2−/− cells with co-transfection of NSUN2 or IRF3-FL (full length, which contains CDS and UTR) under SeV stimulation suggested that IRF3 was the predominant downstream target resulting in the regulation of interferon responses by NSUN2 (g). Therefore, we further used Irf3−/−Irf7−/− MEFs with co-transfection of NSUN2 and IRF3-FL with VSV or HSV-1 infection, and found that the promotion of viruses’ propagation by NSUN2 depended on the presence of IRF3 (h). To summarize, these results reveal that NSUN2 could specifically inhibit the expression of IRF3 and therefore negatively regulate type I interferon responses following virus infection.
Figure 2. NSUN2 inhibits the expression level of IRF3. (a) Dual-luciferase assay analysing a luciferase reporter plasmid for the IRF3-responsive promoter containing positive regulatory domains III and I of the IFN-β promoter (PRDIII-I-Luc) in HEK293T cells in 24-well plates transfected for 36 h with the RIG-N, MDA5-N, MAVS, TBK1, and IRF3-5D expression plasmids, as indicated, with co-transfection with empty vector or NSUN2. (b) Dual-luciferase analysis of PRDIII-I-Luc in HEK293T cells in 24-well plates transfected for 36 h with the indicated RIG-N, MDA5-N, MAVS, TBK1, and IRF3-5D expression plasmids with co-transfection with siControl or siNSUN2-1. (c) Immunoblot analysis in HEK293T cells transfected with vector or NSUN2 for 36 h, with or without infection by SeV for another 12 h. (d) Immunoblot analysis in wild-type HEK293T cells or NSUN2−/− HEK293T cells with or without infection by SeV for 12 h. (e) Immunoblot analysis in wild-type A549 cells or NSUN2−/− A549 cells with infection by SeV for 0, 4, 8, and 12 h. (f) Immunoblot analysis in wild-type HEK293T cells or NSUN2−/− HEK293T cells, with infection by SeV for 0, 4, 8, and 12 h. (g) qPCR analysis of IFNB1 mRNA in wild-type HEK293T cells or NSUN2−/− HEK293T cells transfected with vector or NSUN2, with co-transfection with vector or IRF3-FL, as indicated, with infection by SeV for 12 h. (h) qPCR analysis of VSV-G or HSV-1-UL-30 RNA in Irf3−/−Irf7−/− MEFs transfected with vector or NSUN2, with co-transfection with vector or IRF3-FL, as indicated, with infection by VSV or HSV-1 for 12 h. Data are representative of three independent experiments and were analysed by two-tailed unpaired t test (or by two-factor ANOVA test for 2 h and 2i). Graphs show the mean ± SD (n = 3) derived from three independent experiments. NS, not significant for P > 0.05, *P < 0.05, **P < 0.01, ***P-< 0.001.
NSUN2 catalyzes m5C methylation of IRF3 mRNA
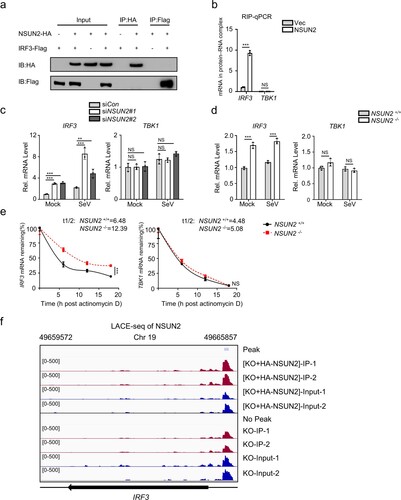
Since NSUN2 has been reported to regulate a number of genes by methylating their mRNAs and affecting RNA fate or function [Citation18–21], we speculated that it might physically interact with IRF3 mRNA. Firstly, co-immunoprecipitation followed by immunoblot analysis showed that there was no interaction between NSUN2 and IRF3 protein in HEK293T (a). We further overexpressed and immunoprecipitated NSUN2 protein in SeV-stimulated HEK293T cells and subjected it to RNA extraction and qPCR. The results revealed that NSUN2 indeed binds with endogenous IRF3 mRNA, while endogenous TBK1 mRNA did not interact with NSUN2 (b). However, the other m5C methyltransferase TRDMT1 (DNMT2) did not bind with endogenous IRF3 or TBK1 mRNA (Supplementary Fig. S3). Furthermore, knockdown or knockout of NSUN2 boosted endogenous IRF3 mRNA levels while endogenous TBK1 mRNA levels were not affected (c,d, Supplementary Fig. S4). We then detected the half-life of endogenous IRF3 mRNA in wild-type and NSUN2−/− HEK293T cells following treatment of actinomycin D (ActD) which inhibits mRNA transcription in mammalian cells. The results showed that knockout of NSUN2 significantly increased the half-life of IRF3 mRNA from 6.48 h in wild-type cells to 12.39 h in NSUN2 knockout cells (e), while the half-life of TBK1 mRNA had no significant difference, from 4.48 h in wild-type cells to 5.08 h in NSUN2 knockout cells. Consistent results were also found in A549 cells, as shown in Supplementary Fig. S5. Of note, we adopted LACE-seq (Linear amplification of complementary DNA ends and sequencing, a new method for unbiasedly mapping the binding sites of multiple RBPs [Citation47]) to identify transcriptome-wide NSUN2-bound RNAs. We used HA-tag antibody to precipitate HA-NSUN2 protein from NSUN2-/- HEK293T with reconstitution of exogenous HA-NSUN2 (f). The result demonstrated that NSUN2 protein binds with endogenous IRF3 mRNA and the binding site is mainly located in 5’UTR region. We also used NSUN2 antibody to precipitate endogenous NSUN2 protein from NSUN2-/- cells followed by LACE-seq. The binding peak located in 5’UTR region disappeared in this group, which served a negative control. The data illustrated high consistency in NSUN2-/- HEK293T with reconstitution of exogenous HA-NSUN2 group but not in NSUN2-/- HEK293T group, as demonstrated by the correlation and clustering analysis (Supplementary Fig S6). Together, these results indicated that NSUN2 decreased IRF3 protein levels mainly by binding to IRF3 mRNA and accelerating its degradation.
Figure 3. NSUN2 interacts with IRF3 mRNA and induces its degradation. (a) Coimmunoprecipitation (IP) and immunoblot (IB) analysis of HEK293T cells transfected with plasmids encoding HA-NSUN2 and Flag-IRF3. (b) Immunoprecipitation by HA-Tag-conjugated beads and immunoblot analysis of HEK293T cells transfected with plasmids encoding HA-NSUN2, with SeV infection for 8 h, followed by RNA extraction and qPCR analysis of combined IRF3 mRNA. (c) qPCR analysis of IRF3 mRNA and TBK1 mRNA in HEK293T cells transfected with siControl or siRNAs targeting NSUN2, with or without infection by SeV for 8 h. (d) qPCR analysis of IRF3 mRNA and TBK1 mRNA in wild-type HEK293T cells or NSUN2−/− HEK293T cells, with or without infection by SeV for 8 h. (e) Stability analysis of IRF3 mRNA and TBK1 mRNA in wild-type HEK293T cells or NSUN2−/− HEK293T cells with treatment of actinomycin D (ActD) for 0, 6, 12, and 18 h. (f) The piled reads of IRF3 mRNA from NSUN2-LACE-seq from SeV-infected cells: NSUN2−/− HEK293T (lower) and NSUN2−/− HEK293T with reconstitution of exogenous HA-NSUN2 (upper). Y-axis represents the normalized signals along the gene. Data are representative of three independent experiments and were analysed by two-tailed unpaired t test. Graphs show the mean ± SD (n = 3) derived from three independent experiments (or two independent experiments for 3f). NS, not significant for P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
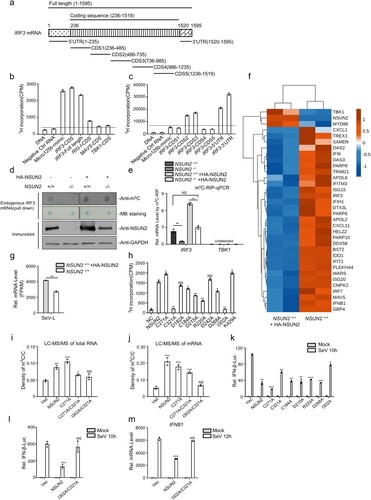
Since NSUN2 is a typical RNA methyltransferase catalyzing the formation of m5C in coding and non-coding RNAs, we speculated that NSUN2 might catalyze the formation of m5C in IRF3 mRNA and then induce its degradation. Therefore, we prepared RNA segments of RIG-I, MAVS, TBK1, and IRF3, the four key signalling molecules that determine the innate immune response to viral infection, by in vitro transcription. Micro-125b, which can be methylated by NSUN2, served as a positive control [Citation48]. The RNAs were used for in vitro methylation assays using recombinant GST-NSUN2 and 3H-labeled S-adenosyl methionine (SAM). The transcribed IRF3 mRNA could be highly methylated by NSUN2 compared with transcripts of RIG-I, MAVS, and TBK1 (a,b). The data suggest that NSUN2 could efficiently mediate the methylation of IRF3 mRNA in vitro compared with RIG-I, MAVS and TBK1 mRNA. To determine which region might be methylated, we divided IRF3 mRNA into seven parts, including 5’UTR (1–235 nt), CDS1 (236–485 nt), CDS2 (486–735 nt), CDS3 (736–985 nt), CDS4 (986–1235 nt), CDS5 (1236–1519 nt), and 3’UTR (1520–1595 nt) (a). As is demonstrated in (c), IRF3 5’UTR, 3’UTR, CDS2, and CDS3 were highly methylated by NSUN2 compared with other segments. To further verify whether endogenous IRF3 mRNA could be methylated by NSUN2 in vivo, we pulled down endogenous IRF3 mRNA using specific IRF3 CHIRP probes which were 3’biotin-TEG-modified. Equal amounts of endogenous IRF3 mRNA were loaded on the membrane, and the levels of m5C were assayed. As is shown in (d), the m5C methylation level of IRF3 mRNA in NSUN2 knockout cells was markedly lower than that of wild-type cells. Reconstitution of exogenous NSUN2 into NSUN2 knockout cells restored the m5C methylation levels of endogenous IRF3 mRNA. Consistent with this, the results of m5C-RIP-qPCR showed that the levels of endogenous m5C methylated IRF3 mRNA in NSUN2 knockout cells was significantly lower than in wild-type cells, and exogenous NSUN2 expression could dramatically enhance the levels of endogenous m5C-methylated IRF3 mRNA (e). We further performed RNA-seq to systematically analyse the expression levels of mRNAs in HEK293T cells or HEK293T cells with NSUN2 overexpression. From these results, IRF3, IFNB1 and its downstream ISGs were consistently down-regulated in NSUN2 overexpression cells (f), while the corresponding SeV replication was enhanced (g). However, the upstream signalling factors, such as RIG-I (DDX58), TBK1 or MAVS, didn’t show consistent or significant regulation by NSUN2. Together with (d,e), these results revealed that NSUN2-mediated m5C methylation of IRF3 mRNA regulated IRF3 mRNA levels and IRF3-mediated IFNB1 and downstream ISGs levels. The above results demonstrate that NSUN2 could mediate m5C methylation of IRF3 mRNA both in vitro and in vivo, and that the four highly methylated regions in IRF3 mRNA are the major targets of NSUN2. This methylation might result in the degradation of IRF3 mRNA and, thereby, decreased levels of IRF3 protein.
Figure 4. NSUN2 catalyzes the formation of m5C methylation of IRF3 mRNA both exogenously and endogenously. (a) Schematic diagram of the IRF3 mRNA segments used for in vitro methylation assays and bisulfite RNA sequencing. (b) In vitro m5C methylation assays using recombinant GST-NSUN2 and the in vitro transcripts. (c) In vitro m5C methylation assays using recombinant GST-NSUN2 and the in vitro transcribed segments of IRF3 mRNA depicted in (a). (d) m5C dot blot analysis of endogenous IRF3 mRNA (200 ng) pulled down by IRF3 CHIRP probes in wild-type HEK293T cells or NSUN2−/− HEK293T cells with or without exogenous NSUN2 overexpression. Equal IRF3 mRNAs were also loaded and verified by methylene blue (MB) staining. (e) The m5C-RIP-qPCR analysis of the m5C methylated IRF3 mRNA immunoprecipitated by m5C antibody from wild-type HEK293T cells or NSUN2−/− HEK293T cells, with or without exogenous NSUN2 expression. TBK1 was used as a negative control. (f-g) RNA-seq of HEK293T cells or HEK293T cells with NSUN2 overexpression, with infection by SeV. The heatmap (f) shows the expression levels of ISGs and several signalling molecules. The genes that we focus on are labelled with asterisks. The column diagram (g) shows the SeV replication levels. (h) In vitro m5C methylation assays using recombinant GST-NSUN2 and different mutant proteins.
To further confirm whether m5C methyltransferase activity of NSUN2 is the determining factor that results in the inhibition of interferon responses, we generated different NSUN2 methyltransferase mutants, including C271A and C321A, which are reported to be the key sites whereby their mutation may inhibit NSUN2 m5C methyltransferase activity [Citation23], as well as several predicted inactivating mutations. The in vitro methylation results show that the NSUN2 mutants, including C184A, D215A, R220A, and D268A, had partially decreased methylation activity, while C321A and I302A mutations almost completely abolished catalytic activity. However, C271A resulted in mildly increased catalytic activity of NSUN2 (h). We further detected the m5C methylation levels of total RNAs (i) or mRNAs (j) in NSUN2−/− HEK293T cells transfected with NSUN2 or its mutants using LC-MS/MS. In accordance with the above results, I302A/C321A double mutation resulted in almost complete loss of m5C methyltransferase activity. Of note, we investigated the relationship between the methylation activities and the stimulation of IFN-β pathway in an SeV-triggered IFN-β-Luc reporter system. As shown in (k), some of inhibition ability in SeV-induced-IFN-β luciferase assay was lost following overexpression of either I302A or C321A compared with wild-type NSUN2, while C271A could enhance the inhibition ability compared with wild-type NSUN2. Moreover, we found that the double mutant I302A/C321A had totally lost its inhibition ability in terms of both function (l) and effects on IFNB1 mRNA levels (m) in NSUN2−/− HEK293T cells. Moreover, ALYREF has been characterized as an m5C reader in the nucleus, facilitating the export of m5C-modified mRNAs [Citation23]. The negatively regulation of type I interferon responses by NSUN2 may also occur in collaboration with ALYREF, which has also been independently observed to negatively regulate type I interferon responses, as depicted in Supplementary Fig. S7. We subsequently checked whether the translation efficiency of IRF3 mRNA was regulated by NSUN2-mediated m5C methylation. We used the in vitro translation assay to check the translation efficiency of IRF3 mRNA methylated or unmethylated by recombinant NSUN2 protein, and found that methylation on IRF3 mRNA gently inhibited its translation efficiency (Supplementary Fig. S8a). However, in ribosome loading assay, there was no obvious change of ribosome loading on to IRF3 mRNA in both wild-type and NSUN2−/− cells (Supplementary Fig. S8b).
Moreover, RNA-seq of A549 wild-type cells or A549 NSUN2 knockout cells. The GO enrichment biological process analysis showed that knockout of NSUN2 contributed to the dramatic elevation of immune responses, which further demonstrates the critical role of NSUN2 in antiviral innate immunity responses (a). IRF3, IFNB1 and its downstream ISGs were consistently up-regulated in A549 NSUN2 knockout cells while the corresponding SeV replication was inhibited (b,c). However, the upstream signalling factors, such as RIG-I (DDX58), TBK1 or MAVS, didn’t show consistent regulation by NSUN2. Meanwhile, the levels of endogenous m5C methylated IRF3 mRNA in A549 NSUN2 knockout cells was significantly lower than in A549 wild-type cells (d). However, there was no significant signal of m5C modified mRNAs of RIG-I (DDX58), TBK1 or MAVS. To summarize, NSUN2 could catalyze the formation of m5C modification of IRF3 mRNA and accelerate its fast turnover and regulate IRF3-mediated type I interferon responses. Of note, this regulation by NSUN2 is dependent on its m5C methyltransferase activity.
Figure 5. Knockout of NSUN2 decreased m5C methylation on IRF3 mRNA but increased IRF3 and IFNB1 and downstream ISGs mRNA levels. (a) RNA-seq analysis from SeV-infected A549 wild-type cells or A549 NSUN2 knockout cells. GO-term functional enrichment by biological process of up-regulated genes. Data show the top 10 biological processes. (b) The heatmap shows the expression levels of ISGs and several signalling molecules. The genes that we focus on are labelled with asterisks. (c) The column diagram shows the SeV replication levels. (d) The m5C-RIP-qPCR analysis of the m5C methylated mRNA levels immunoprecipitated by m5C antibody from A549 wild-type cells or A549 NSUN2 knockout cells. Data are representative of three independent experiments and were analysed by two-tailed unpaired t test. Graphs show the mean ± SD (n = 3) derived from three independent experiments. NS, not significant for P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
The methylated cytosines of IRF3 mRNA were identified to regulate RNA levels
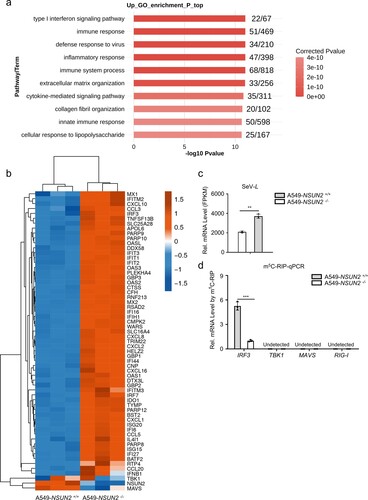
Furthermore, we performed m5C-MeRIP-seq to transcriptome-widely analyse the endogenous m5C sites in cellular RNAs in uninfected or SARS-CoV-2-infected Caco-2 cells (a). Of note, the m5C signals of IRF3 mRNA were both detected in uninfected and SARS-CoV-2-infected Caco-2 cells and the m5C signals located in the 5’UTR of IRF3 mRNA were especially noteworthy. We also add the systematic analysis of two groups by the correlation and clustering analysis (Supplementary Fig S9), which indicated the repeatability and stability of m5C-MeRIP-seq data. Further, we aimed to identify the exact methylation cytosines in IRF3 mRNA. Using bisulfite sequencing assays (b), we identified a methylated cytosine in IRF3 mRNA as major site of methylation that were highly methylated by recombinant NSUN2 protein in vitro: C169 in 5’UTR (c). In addition to this, we also identified another three highly methylated cytosines: C1569 in 3’UTR, C556 in CDS2 (486–735), and C815 in CDS3 (736–985), which is consistent with the four high methylation regions observed earlier in IRF3 mRNA (c and Supplementary Fig S10a-b).
Figure 6. IRF3 m5C methylation site mutation results in enhanced IRF3 expression and antiviral response. (a) Visualization of m5C-MeRIP-seq results shows regions of enrichment for m5C immunoprecipitation (upper) over input (lower) from uninfected or SARS-CoV-2-infected Caco-2 cells. The positive m5C site identified by in vitro bisulfite RNA sequencing (c) are marked in red solid triangle and the control sites are marked in black hollow triangle which are not identified by in vitro bisulfite RNA sequencing and identified as no biological function (e–g). (b) Schematic depiction of in vitro bisulfite RNA sequencing to distinguish m5C (cytosine methylated by NSUN2) from C (cytosine not methylated). (c) Identification of m5C modification on cytosines of 5’UTR of IRF3 mRNA by bisulfite RNA sequencing. Data are expressed as the ratio of m5C to (C + m5C) from three independent replicates. (d) Wild-type HEK293T cells or NSUN2−/− HEK293T cells were transfected with pGL.3.0-CMV-Luc or pGL3.0-CMV-IRF3-CDS-Luc or pGL3.0-CMV-IRF3-5’UTR-Luc or pGL3.0-CMV-IRF3-3’UTR-Luc, together with Renilla luciferase (RL-TK). Forty-eight hours later, firefly luciferase activity against Renilla luciferase activity was analysed. (e) In vitro m5C methylation assays of the IRF3 segments or the cytosines mutated segments using recombinant GST-NSUN2. (f-g) qPCR analysis of IRF3 mRNA or Ifnb1 mRNA in Irf3−/−Irf7−/− MEFs transfected with plasmid encoding NSUN2 along with wild-type IRF3 full length (IRF3-FL-WT) or various cytosine-mutated IRF3-FLs for 48 h, as indicated, with stimulation by SeV for 8 h. Data are representative of three independent experiments and were analysed by two-tailed unpaired t test. Graphs show the mean ± SD (n = 3) derived from three independent experiments (or two independent experiments for 6e). NS, not significant for P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
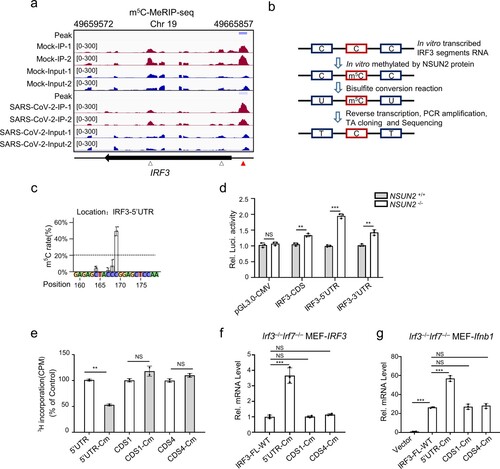
To further investigate the biological function of m5C methylation of IRF3 mRNA by NSUN2, we constructed pGL3-derived reporters bearing either IRF3-5’UTR, IRF3-CDS, or IRF3-3’UTR. We tested the activity of these reporters in NSUN2-knockout HEK293T cells compared with those in wild-type HEK293T. As shown in (d), knockout of NSUN2 could increase the luciferase activity of reporter pGL3-IRF3-5’UTR, pGL3-IRF3-CDS, and pGL3-IRF3-3’UTR. The increase in the luciferase activity of 5’UTR is especially noteworthy compared with CDS and 3’UTR. We then tested whether these four identified highly methylated cytosines are indeed methylated and involved in the regulation of IRF3 mRNA by NSUN2 protein. It was observed that mutations C169 (C to G) in the 5’UTR, C1569 (C to G) in the 3’UTR, C556 (C to T, synonymous mutation) in CDS2 (486–735) and C815 (C to A, synonymous mutation) in CDS3 (736–985) reduced the methylation level by half in biochemical assays with recombinant NSUN2 (e and Supplementary Fig S10c). We then constructed expression plasmids containing either wild type IRF3 full length (IRF3-FL, 1–1595nt) or various site-mutated IRF3-FLs. We observed that mutations of the four cytosines could consistently enhance the expression levels of IRF3 mRNA in Irf3−/−Irf7−/− MEFs compared to wild type IRF3 full length (IRF3-FL-WT) (f and Supplementary Fig S10d). Correspondingly, the IRF3-mediated Ifnb1 mRNA levels were also remarkably elevated upon SeV infection (g and Supplementary Fig S10e). In contrast, we also tested another two cytosines located in CDS1 (C303) or CDS4 (C1135) (a) which were not identified by bisulfite sequencing and found that these two cytosines had no effect on the function (f and g). We then utilized the lentiviral system to generate stable IRF3 cell lines in Irf3−/−Irf7−/− MEFs. The IRF3 mRNA levels in the IRF3-FL-Mut (IRF3-FL-5’&3’UTR-CDS2&3-Cm) stable cell line was 15-fold higher than that in the IRF3-FL-WT stable cell line. Moreover, exogenous NSUN2 expression significantly reduced IRF3 mRNA levels in the IRF3-FL-WT stable cell line, while IRF3 mRNA levels in the IRF3-FL-Mut stable cell line were mildly decreased, indicating that methylation of these four cytosines in IRF3 mRNA might predominantly influence its stability (Supplementary Fig S10f). To confirm this, we measured the stability of these transcripts and found that the IRF3-FL-Mut transcript was remarkably higher than the IRF3-FL-WT transcript, indicating that methylation of these four cytosines by NSUN2 is indeed critical for regulating IRF3 mRNA stability (Supplementary Fig S10g). Taken together, our results demonstrate that the loss of m5C modification could lead to increased stability of IRF3 mRNA and enhanced IFN-β production, thus facilitating a stronger antiviral response, and that the highly methylated cytosines in IRF3 mRNA play a critical role in NSUN2-mediated regulation of antiviral responses.
Pivotal role of NSUN2 in the induction of type I interferon and antiviral response in vivo
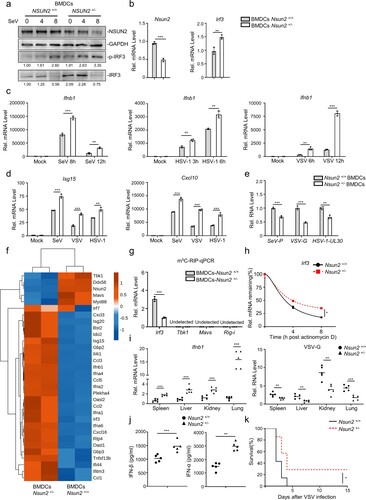
To determine the role of NSUN2 in antiviral response in vivo, we created targeted deletions of NSUN2 in mice by removing 10 bp in exon 3 of Nsun2 genome by CRISPR/Cas9, which resulted in a frameshift mutation (Supplementary. Fig S11). However, we found that Nsun2−/− mice died in utero. We found that Nsun2+/− progeny could reach adulthood, so we chose Nsun2+/− mice as “NSUN2-knockdown mice.” As expected, the Nsun2 expression in Nsun2+/− mice did reduce by half than their wild-type littermates (a,b). The endogenous mRNA levels and protein levels of IRF3 were both enhanced in BMDCs from Nsun2+/− mice than in those from their wild-type littermates following infection with SeV (a,b). Furthermore, as shown in (c), the production of Ifnb1 mRNA was more dramatically enhanced in BMDCs from Nsun2+/− mice than wild-type mice with SeV, HSV-1 or VSV. The IFN-β mediated downstream Isg15 and Cxcl10 were also significantly enhanced in BMDCs from Nsun2+/− mice (d). Accordingly, the propagation levels of SeV, HSV-1 or VSV were markedly inhibited in BMDCs from Nsun2+/− mice (e). RNA-seq of BMDCs from Nsun2+/− mice or Nsun2+/+ mice showed that Irf3, Ifnb1 and downstream ISGs were consistently elevated in BMDCs from Nsun2+/− mice (f). However, the upstream signalling factors, such as Rig-i (Ddx58), Tbk1 or Mavs even decreased in BMDCs from Nsun2+/− mice, which requires further investigation. Meanwhile, the levels of endogenous m5C methylated Irf3 mRNA in BMDCs from Nsun2+/− mice was lower than in BMDCs from Nsun2+/+ mice (g). However, there was no significant signal of m5C modified mRNAs of Rig-i, Tbk1 or Mavs. The stability of endogenous IRF3 mRNA in BMDCs from Nsun2+/− mice was higher (h). We then investigated innate antiviral responses in wild-type mice and Nsun2+/− mice. We found a higher IFN-β production and a lower viral burden of VSV in various organs of Nsun2+/− mice than in wild-type mice at the mRNA levels (i). Furthermore, we observed significantly higher IFN-β and IFN-α production in the serum of Nsun2+/− mice after intraperitoneal injection of VSV by ELISA (j). We also compared the survival rates after intraperitoneal injection of VSV. The results indicate that wild-type mice were more vulnerable to VSV-triggered mortality than were Nsun2+/− mice (k).
Figure 7. Pivotal role for NSUN2 in the induction of type I interferon and antiviral response in vivo. (a) Immunoblot analysis in BMDCs from Nsun2+/+ mice or Nsun2+/− mice with infection by SeV for 0, 4, and 8 h. (b) qPCR analysis of Nsun2 or Irf3 mRNA in BMDCs from Nsun2+/+ mice or Nsun2+/− mice. (c) qPCR analysis of Ifnb1 mRNA in BMDCs from Nsun2+/+ mice or Nsun2+/− mice, with or without infection by SeV for 8 and 12 h, HSV-1 for 3 and 6 h, or VSV for 6 and 12 h. (d) qPCR analysis of Isg15 and Cxcl10 in BMDCs from Nsun2+/+ mice or Nsun2+/− mice, with or without infection by SeV, HSV-1, or VSV. (e) qPCR analysis of SeV-P, VSV-G or HSV-1-UL-30 RNA in BMDCs from Nsun2+/+ mice or Nsun2+/− mice, with infection by SeV, VSV or HSV-1 for 12 h. (f) RNA-seq analysis from SeV-infected BMDCs from Nsun2+/+ mice or Nsun2+/− mice. The heatmap shows the expression levels of ISGs and several signalling molecules. The genes that we focus on are labelled with asterisks. (g) The m5C-RIP-qPCR analysis of the m5C methylated mRNA levels immunoprecipitated by m5C antibody from BMDCs from Nsun2+/+ mice or Nsun2+/− mice. (h) Stability analysis of Irf3 mRNA in BMDCs from Nsun2+/+ mice or Nsun2+/− mice, with treatment of actinomycin D (ActD) for 0, 4, and 8 h. (i) qPCR analysis of Ifnb1 mRNA and the corresponding VSV-G RNA in different organs from Nsun2+/+ mice (n = 5) or Nsun2+/− mice (n = 5), injected intraperitoneally for 16 h with VSV (4 × 107 PFU per mouse).
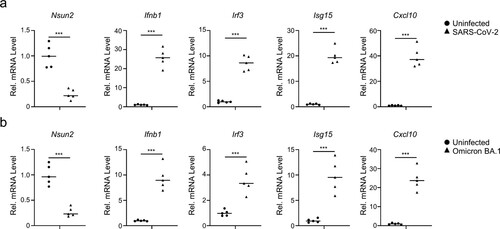
We further investigated the innate immunity responses and NSUN2 expression levels in SARS-CoV-2 infected K18-hACE2 knock-in (KI) mice models. We found that infection of SARS-CoV-2 WT strain (a) or BA.1 omicron variant (b) activated the innate immunity response in K18-hACE2 KI mice models. Endogenous Nsun2 mRNA levels significantly decreased in SARS-CoV-2 WT strain or BA.1 omicron variant infected groups compared to uninfected groups (a and b). These results were consistent with SARS-CoV-2 infected Caco-2 cells models and COVID-19 patients and together revealed that NSUN2 plays an important regulatory role in the infection of SARS-CoV-2 (n and o). Meanwhile, endogenous mRNA levels of Irf3, Ifnb1 and downstream ISGs including Isg15 and Cxcl10 notably increased in SARS-CoV-2 WT strain or BA.1 omicron variant infected groups.
Figure 8. SARS-CoV-2 infection decreases Nsun2 expression levels and activates innate immunity responses in K18-hACE2 KI mice. (a) qPCR analysis of Nsun2, Irf3, Ifnb1, Isg15 and Cxcl10 mRNA in lungs from SARS-CoV-2 WT strain infected K18-hACE2 KI mice (n = 5) or uninfected K18-hACE2 KI mice (n = 5), injected intranasally for 48 h with SARS-CoV-2 WT strain (250 PFU per mouse). (b) qPCR analysis of Nsun2, Irf3, Ifnb1, Isg15 and Cxcl10 mRNA in lungs from SARS-CoV-2 BA.1 omicron variant infected K18-hACE2 KI mice (n = 5) or uninfected K18-hACE2 KI mice (n = 5), injected intranasally for 48 h with SARS-CoV-2 WT strain (8000 PFU per mouse). NS, not significant for P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
Together, our findings reveal that NSUN2 serves as a negative regulator of interferon response by accelerating the fast turnover of IRF3 mRNA. NSUN2 is constitutively expressed in resting cells and that IRF3 expression is maintained at a relatively low level. During various viral infections, including SARS-CoV-2, the endogenous expression level of NSUN2 is dramatically reduced, via unknown mechanism, to enhance IRF3 levels and downstream antiviral type I interferon responses.
Discussion
Antiviral innate immunity involves sophisticated signalling pathways for sensing pathogens and initiating innate immune responses against infection, which requires ingenious regulation at different levels including transcriptional, translational, and post-translational. It is known that IRF3, which plays a vital role in the initiation of type I interferon responses after infection, is regulated by multiple modifications, such as phosphorylation, ubiquitination, and acetylation, which function in maintaining immune homeostasis [Citation39,Citation49,Citation50]. Recently, the m6A machinery has been reported to be involved in immune responses via epitranscriptomic modification. For example, it has been reported that the m6A machinery could inhibit the innate immune response to infection by directly dictating the fast turnover of IFNB1 mRNAs and consequently facilitating viral propagation [Citation10]. Another study demonstrated that ALKBH5 could erase the m6A modification of MAVS, TRAF3, and TRAF6 mRNAs, enforce their retention in the nucleus and result in their decreased translation and inhibited type I interferon production [Citation13]. Moreover, deficiency of METTL14 increases MAVS mRNA stability, and downstream phosphorylation of TBK1/IRF3 and interferon response to RNA viruses [Citation14]. The effects of m6A modification on interferon responses may vary because of the different systems and different readers and precise downstream regulation. However, there are few reports of m5C modification regulating antiviral innate immunity.
Here, firstly, we revealed a novel mechanism by which the m5C machinery functions in innate immune responses via the methylation of IRF3 mRNA to negatively regulate type I interferon responses [Citation51], indicating that the m5C and m6A machineries may have different specificities with respect to regulating multiple signalling molecules involved in antiviral innate immune responses. Recently on October 2022, Zhang et al. also reported that depletion of NSUN2 enhanced type I interferon response and inhibited virus replication, which is dependent on RIG-I but not MDA5 [Citation52]. But the mechanism they reported is different from what we found, which implies the important function of m5C modification in innate immune regulation during various viral infections. In this study, we demonstrated that NSUN2 could specifically methylate IRF3 mRNA via four major cytosine sites. The mutation of these four major cytosines enhanced the stability and expression of IRF3 mRNA () and, thereby, interferon responses. Moreover, in our system, the m6A machinery was also found to be involved in regulating interferon responses (a), but the overall effect was not significant compared with the m5C machinery, which may be because the m6A machinery regulates other signalling molecules with different effects, as mentioned above. However, we do not preclude the possibility that other mechanisms beyond an elevation in IRF3 mRNA stability may contribute to the stronger type I interferon responses following knockout of NSUN2. We may speculate that the mRNAs of some other signalling molecules may also be m5C-modified and regulated by NSUN2, such as is the case with m6A modification. Future work is required to demonstrate how m5C methylation and its downstream recognition and regulation collaboratively and precisely function in antiviral innate immunity.
Moreover, we found that the regulation of type I interferon responses by NSUN2 was dependent on its m5C methyltransferase activity. According to our results (i–n), the NSUN2 I302A/C321A mutant had almost completely lost its m5C methyltransferase activity and ability to regulate type I interferon responses, which is in contrast with the reports of C271A/C321A mutation of NSUN2 [Citation23,Citation53]. In our study, the C271A mutation maintained m5C methyltransferase activity in biochemical assays and could still negatively regulate interferon responses. The discrepancy in the key sites of NSUN2 methyltransferase activity may be due to the different systems and the different roles NSUN2 plays in multiple physiological processes. Further work is required to uncover the structure of NSUN2 protein and the key sites that determine its m5C methyltransferase activity and regulation activity in multiple physiological processes.
NSUN2 and TRDMT1 (DNMT2) are two m5C methyltransferases reported in animals, but the identity of the m5C demethylase remains unknown [Citation16,Citation17]. In our study, TRDMT1 did not show significant regulation of interferon responses or bind with IRF3 unlike NSUN2 (a, Supplementary. Fig S3). ALYREF has earlier been characterized as an m5C reader in the nucleus involved in facilitating the export of m5C-modified mRNAs [Citation23]. In our results, exogenous NSUN2 expression could dramatically inhibit IFN-β production, and exogenous ALYREF expression could also (a-c, Supplementary. Fig S7), which is interesting, and may also indirectly imply that m5C modification is involved in regulating type I interferon responses. Because ALYREF recognizes the m5C modified RNAs and regulates their nuclear exportation, the mechanism of IFN-β inhibition by exogenous ALYREF expression needs further investigation. YBX1 was identified as another m5C reader that could maintain the stability of its target mRNA by recruiting ELAVL1 [Citation24]. In our study, NSUN2 could directly methylate IRF3 mRNA and accelerates its degradation, which seems to contradict the function of the NSUN2–YBX1–ELAVL1 axis. These two seemingly opposing mechanisms may uncover the different roles that m5C modification play in various biological processes. Different m5C readers might have different functions and play different roles. For example, YTH family members have been reported to serve as m6A readers that recognize m6A-modified RNA and further regulate mRNA splicing, translation, or degradation [Citation54–57]. We have also used in vitro translation and ribosome loading assay to check the translation efficiency of m5C methylated IRF3 mRNA. However, only slight inhibition of m5C methylated IRF3 mRNA was observed in translation assay (Supplementary Fig. S8a). Therefore, our results suggested that the major function of m5C methylation on IRF3 mRNA is to accelerate its degradation ( and ), although we could not exclude the possibility of the influence on translation efficiency, which requires further investigation. The specific degradation mechanism induced by m5C and m6A modification has not yet been clarified clearly and requires more investigation. Further work is required to delineate these different mechanisms and the different roles that m5C readers play. The m5C demethylase, which may maintain balance in the m5C modification level in various biological processes, must also be clearly clarified.
In terms of several m5C methylation modification sequencing methods, including bisulfite sequencing and m5C-MeRIP-seq, each method has limitations. We also performed RNA-Bis-seq at transcriptome level, but it is a pity that m5C sites on IRF3 mRNA was not detected. We speculated that it might be related to the expression abundance of IRF3 mRNA, and bisulfite treatment caused great damage to RNA. We then compared RNA-Bis-seq sequencing data of several reports [Citation23,Citation24,Citation58], and found that m5C methylation modification sites in the results varied a lot. But we found that there was a study in which RNA-Bis-seq data showed m5C methylation modification sites on Irf3 mRNA in many mouse tissues [Citation23]. Together with our sequencing data of m5C modified Cs in human mRNA and biological roles of NSUN2-mediated regulation of IRF3, these results demonstrates that both human IRF3 mRNA and mouse Irf3 mRNA are m5C modified and involved in IRF3-mediated IFN responses. In our study, we combined m5C-MeRIP-seq, LACE-seq and in vitro bisulfite sequencing to identify the m5C methylated modification sites on IRF3 mRNA. We also used in vitro methylation system and in vivo functional experiments to verify physiological functions of these sites. The highly m5C methylated modification sites identified on IRF3 mRNA were demonstrated to have physiological functions. We do not exclude the existence of other m5C methylation modification sites on IRF3 mRNA, which requires more ingenious detection systems for further verification.
Furthermore, we found that NSUN2 expression is decreased after infections with different viruses, including SeV (negative-strand RNA virus), HSV-1 (DNA virus), VSV (negative-strand RNA virus), ZIKV (positive-strand RNA virus), and especially SARS-CoV-2 (positive-strand RNA virus, beta-coronavirus), via unknown mechanism, which require further investigation for their elucidation. Notably, transcriptome sequencing of the RNAs isolated from the bronchoalveolar lavage fluid (BALF) of two COVID-19 patients revealed that NSUN2 expression was dramatically decreased after infection of SARS-CoV-2 compared with healthy individuals (n,o). Consistently, endogenous Nsun2 mRNA levels significantly decreased in SARS-CoV-2 WT strain or BA.1 omicron variant infected K18-hACE2 KI mice models compared to uninfected groups (a and b). These results revealed that NSUN2 also plays an important regulatory role in the infection of SARS-CoV-2 WT strain or BA.1 omicron variant. We can therefore propose a model whereby NSUN2 is constitutively expressed in resting cells and that IRF3 mRNA is modified by m5C and maintained at a relatively low level. During viral infection, endogenous NSUN2 expression levels decrease, so the m5C modification level of IRF3 mRNA decreased but the stability increased. Then IRF3 expression level would therefore be elevated to allow a stronger interferon response and the effective elimination of viruses. The evolution of such a significant host antiviral strategy to regulate the interferon responses suggests the important physiological significance of NSUN2-mediated m5C modification.
Supplemental Material
Download Zip (9.9 MB)Acknowledgement
We thank Dr. Yingle Liu and Dr. Mang Shi for providing BALF samples of COVID-19 patients. We thank Dr. Zheng-Li Shi for providing SARS-CoV-2, Dr. Hong-Bing Shu for providing SeV, HSV-1, VSV-GFP, Dr. Bo Zhang for providing ZIKV, Dr. Ming-Zhou Chen for providing VSV and Dr. Ying Zhu for providing A549 IFNAR1−/− cell. We thank Dr. Yun-Gui Yang and Dr. Cheng-Peng Fan for critical advice. Author Contributions: Y.C. and H.W. conceived the research and experiments. H.W., C.Z., J.F., L.Zhang., Z.F., J.J.L., J.L.L.and M.H. performed the major experiments and analysis. Y.Y.C. and L.Zhou. participated in mice experiments. Y.W. and X.Z. helped LC-MS/MS experiments. Y.Zhou., K.L., and D.W. analysed transcriptome sequencing data. H.T., A.J., D.G. and K.X. provided critical advice. H.W. and Y.C. wrote and revised the manuscript with contributions from all other authors.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Roundtree IA, Evans ME, Pan T, et al. Dynamic RNA modifications in gene expression regulation. Cell. 2017 Jun 15;169(7):1187–1200.
- Dezi V, Ivanov C, Haussmann IU, et al. Nucleotide modifications in messenger RNA and their role in development and disease. Biochem Soc Trans. 2016 Oct 15;44(5):1385–1393.
- Trixl L, Lusser A. The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark. Wiley Interdiscip Rev RNA. 2019 Jan;10(1):e1510.
- Chellamuthu A, Gray SG. The RNA methyltransferase NSUN2 and Its potential roles in cancer. Cells. 2020 Jul 22;9(8):1758.
- Yang Y, Hsu PJ, Chen YS, et al. Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018 Jun;28(6):616–624.
- Cao G, Li HB, Yin Z, et al. Recent advances in dynamic m6A RNA modification. Open Biol. 2016 Apr;6(4):160003.
- Yang J, Wang H, Zhang W. Regulation of virus replication and T cell homeostasis by N(6)-methyladenosine. Virol Sin. 2019 Feb;34(1):22–29.
- Han D, Liu J, Chen C, et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature. 2019 Feb;566(7743):270–274.
- Frye M, Harada BT, Behm M, et al. RNA modifications modulate gene expression during development. Science. 2018 Sep 28;361(6409):1346–1349.
- Winkler R, Gillis E, Lasman L, et al. M6a modification controls the innate immune response to infection by targeting type I interferons (vol 20, pg 173, 2018). Nat Immunol. 2019 Feb;20(2):243–243.
- Rubio RM, Depledge DP, Bianco C, et al. RNA m(6) A modification enzymes shape innate responses to DNA by regulating interferon beta. Genes Dev. 2018 Dec 1;32(23-24):1472–1484.
- Liu Y, You Y, Lu Z, et al. N6-methyladenosine RNA modification–mediated cellular metabolism rewiring inhibits viral replication. Science. 2019;365(6458):1171–1176.
- Zheng Q, Hou J, Zhou Y, et al. The RNA helicase DDX46 inhibits innate immunity by entrapping m(6)A-demethylated antiviral transcripts in the nucleus. Nat Immunol. 2017 Oct;18(10):1094–1103.
- Qin F, Cai B, Zhao J, et al. Methyltransferase-Like protein 14 attenuates mitochondrial antiviral signaling protein expression to negatively regulate antiviral immunity via N(6) -methyladenosine modification. Adv Sci (Weinh. 2021 Aug;8(15):e2100606.
- Wang L, Wen M, Cao X. Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science. 2019;365(6454):eaav0758.
- Squires JE, Patel HR, Nousch M, et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012 Jun;40(11):5023–5033.
- Tuorto F, Liebers R, Musch T, et al. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat Struct Mol Biol. 2012 Sep;19(9):900–905.
- Zhang X, Liu Z, Yi J, et al. The tRNA methyltransferase NSun2 stabilizes p16INK(4) mRNA by methylating the 3'-untranslated region of p16. Nat Commun. 2012 Mar 6;3:712.
- Mei L, Shen C, Miao R, et al. RNA methyltransferase NSUN2 promotes gastric cancer cell proliferation by repressing p57(Kip2) by an m(5)C-dependent manner. Cell Death Dis. 2020 Apr 24;11(4):270.
- Tang H, Fan X, Xing J, et al. NSun2 delays replicative senescence by repressing p27 (KIP1) translation and elevating CDK1 translation. Aging (Albany NY). 2015 Dec;7(12):1143–1158.
- Li Q, Li X, Tang H, et al. NSUN2-Mediated m5c methylation and METTL3/METTL14-mediated m6A methylation cooperatively enhance p21 translation. J Cell Biochem. 2017 Sep;118(9):2587–2598.
- Schumann U, Zhang HN, Sibbritt T, et al. Multiple links between 5-methylcytosine content of mRNA and translation. BMC Biol. 2020 Apr 15;18(1):40.
- Yang X, Yang Y, Sun BF, et al. 5-methylcytosine promotes mRNA export-NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 2017 May;27(5):606–625.
- Chen X, Li A, Sun BF, et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat Cell Biol. 2019 Aug;21(8):978–990.
- Yang Y, Wang L, Han X, et al. RNA 5-Methylcytosine facilitates the maternal-to-zygotic transition by preventing maternal mRNA decay. Mol Cell. 2019 Sep 19;75(6):1188–1202. e11.
- Zou F, Tu R, Duan B, et al. Drosophila YBX1 homolog YPS promotes ovarian germ line stem cell development by preferentially recognizing 5-methylcytosine RNAs. Proc Natl Acad Sci U S A. 2020 Feb 18;117(7):3603–3609.
- Eckwahl M, Xu R, Michalkiewicz J, et al. 5-Methylcytosine RNA modifications promote retrovirus replication in an ALYREF reader protein-dependent manner. J Virol. 2020 Jun 16;94(13):e00544-20.
- Honda K, Takaoka A, Taniguchi T. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006 Sep;25(3):349–360.
- Ablasser A, Hur S. Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat Immunol. 2019;21(1):17–29.
- Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell. 2020 Mar 19;180(6):1044–1066.
- Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. 2020 Sep;20(9):537–551.
- Honda K, Taniguchi T. IRFs: master regulators of signalling by toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006 Sep;6(9):644–658.
- Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. 2014;32:461–488.
- Tamura T, Yanai H, Savitsky D, et al. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584.
- Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545.
- Li S, Zhu M, Pan R, et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat Immunol. 2016 Mar;17(3):241–249.
- Mancino A, Natoli G. Specificity and function of IRF family transcription factors: insights from genomics. J Interferon Cytokine Res. 2016 Jul;36(7):462–469.
- Cao Y, Wang H, Yang L, et al. PTEN-L promotes type I interferon responses and antiviral immunity. Cell Mol Immunol. 2018 Jan;15(1):48–57.
- Zhou Y, Li M, Xue Y, et al. Interferon-inducible cytoplasmic lncLrrc55-AS promotes antiviral innate responses by strengthening IRF3 phosphorylation. Cell Res. 2019 Aug;29(8):641–654.
- Xia Z, Xu G, Yang X, et al. Inducible TAP1 negatively regulates the antiviral innate immune response by targeting the TAK1 complex. J Immunol. 2017 May 1;198(9):3690–3704.
- Xiong Y, Liu Y, Cao L, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect. 2020 Dec;9(1):761–770.
- Courtney DG, Tsai K, Bogerd HP, et al. Epitranscriptomic addition of m(5)C to HIV-1 transcripts regulates viral gene expression. Cell Host Microbe. 2019 Aug 14;26(2):217–227. e6.
- Subtelny AO, Eichhorn SW, Chen GR, et al. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature. 2014 Apr 3;508(7494):66–71.
- Li X, Xiong X, Wang K, et al. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat Chem Biol. 2016 May;12(5):311–316.
- Pollex T, Hanna K, Schaefer M. Detection of cytosine methylation in RNA using bisulfite sequencing. Cold Spring Harb Protoc. 2010 Oct 1;2010(10). pdb prot5505.
- Schaefer M, Pollex T, Hanna K, et al. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 2009 Feb;37(2):e12.
- Su R, Fan LH, Cao C, et al. Global profiling of RNA-binding protein target sites by LACE-seq. Nat Cell Biol. 2021 Jun;23(6):664–675.
- Yuan S, Tang H, Xing J, et al. Methylation by NSun2 represses the levels and function of microRNA 125b. Mol Cell Biol. 2014 Oct 1;34(19):3630–3641.
- Huai W, Liu X, Wang C, et al. KAT8 selectively inhibits antiviral immunity by acetylating IRF3. J Exp Med. 2019 Apr 1;216(4):772–785.
- Wang P, Zhao W, Zhao K, et al. TRIM26 negatively regulates interferon-beta production and antiviral response through polyubiquitination and degradation of nuclear IRF3. PLoS Pathog. 2015 Mar;11(3):e1004726.
- Wang H, Zhang L, Zeng C, et al. NSUN2-mediated m5c methylation of IRF3 mRNA negatively regulates type I interferon responses. bioRxiv. 2021:2021.07.09.451748.
- Zhang Y, Zhang LS, Dai Q, et al. 5-methylcytosine (m(5)C) RNA modification controls the innate immune response to virus infection by regulating type I interferons. Proc Natl Acad Sci U S A. 2022 Oct 18;119(42):e2123338119.
- Moon HJ, Redman KL. Trm4 and Nsun2 RNA:m5C methyltransferases form metabolite-dependent, covalent adducts with previously methylated RNA. Biochemistry. 2014 Nov 18;53(45):7132–7144.
- Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014 Jan 2;505(7481):117–120.
- Shi H, Wang X, Lu Z, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017 Mar;27(3):315–328.
- Wang X, Zhao BS, Roundtree IA, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015 Jun 4;161(6):1388–1399.
- Xiao W, Adhikari S, Dahal U, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016 Feb 18;61(4):507–519.
- Huang T, Chen W, Liu J, et al. Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat Struct Mol Biol. 2019 May;26(5):380–388.