
ABSTRACT
Seasonal influenza, causes hundreds of thousands of deaths annually, posing a severe threat to human health. Currently available influenza vaccines are targeted only at specific strains or conserved epitopes; however, these vaccines are not completely efficacious because influenza viruses can undergo mutation during circulation, leading to antigenic mismatch between recommended strains and circulating strains and elusion from the immune system. Therefore, developing an influenza vaccine that is quick, effective, and broadly protective has become crucial, and the integral part of hemagglutinin (HA) remains an ideal target for vaccine development. This study developed a lipid nanoparticle-encapsulated nucleoside-modified mRNA vaccine (mRNA-LNPs) encoding a consensus full-length HA sequence (H1c) and evaluated its protective efficacy and immunogenicity through in vitro and in vivo assays. Following two intramuscular immunizations (2, 10 µg, or 20 µg) at a 3-week interval in BALB/c mice, H1c-mRNA-LNP vaccine induced strong antibodies as shown in the hemagglutination-inhibition test and protective neutralizing antibodies against numerous heterologous H1N1 influenza viruses as shown in the microneutralization assay. Additionally, both Th1- and Th2-biased cellular immune responses were elicited, with the Th1-biased response being stronger. Two doses of the H1c-mRNA-LNP vaccine could neutralize a panel of heterologous H1N1 influenza viruses and could confer protection in mice. Taken together, these findings suggest that the H1c-mRNA-LNP vaccine encoding a consensus full-length HA is a feasible strategy for developing a cross-protective vaccine against a panel of heterologous H1N1 influenza viruses.
Introduction
Influenza is caused by the influenza virus and has always been a threat to human health. Globally, seasonal influenza causes approximately 0.29–0.65 million respiratory-related deaths annually, and most of the affected people are aged above 65 years among adults and under 5 years among children[Citation1]. Vaccination has been considered as an optimal and highly efficient strategy for preventing influenza.
Seasonal influenza virus includes two influenza A subtypes (H1N1 and H3N2) and two influenza B lineages (Victoria and Yamagata), and current seasonal influenza vaccines contain these viruses as constituents[Citation2]. Among all types of seasonal influenza viruses, the H1N1 subtype has been an important agent in several pandemics, leading to substantial burden on the economy and society[Citation3, Citation4]. Currently available licensed seasonal influenza vaccines specifically target the strains recommended by the World Health Organization (WHO) each year. However, influenza viruses are likely to undergo mutation during circulation, which leads to an antigenic mismatch between the recommended strains and the circulating strains, thereby decreasing vaccines’ efficacy. According to the Centres for Disease Control and Prevention, influenza vaccines have only achieved a protection rate of 19%−52% in recent decades [Citation5]. Therefore, developing an influenza vaccine that is potent, quick, and effective and affords cross-protection has become a top priority.
Influenza viruses are members of Orthomyxoviridae family, whose genome includes eight negative-sense single-stranded RNA segments encoding viral structural proteins and nonstructural proteins such as neuraminidase, hemagglutinin (HA), polymerase complex (PB2, PB1, and PA), matrix proteins 1 and 2 (M1 and M2), nonstructural proteins 1 (NS1), nuclear export protein (NEP), and nucleoprotein (NP)[Citation2]. The surface glycoprotein HA, which is responsible for viral adsorption and invasion, contains the largest number of neutralizing epitopes and therefore serves as the main immunogen for influenza vaccines[Citation6]. However, when the virus circulates in the body, the head of HA tends to accumulate additional mutations, which facilitates the virus to elude from the immune system. Several studies have focused on the conserved regions in NP[Citation7], M1,[Citation7] M2e[Citation8], and stem part of HA[Citation9], which have the potential to serve as targets for universal vaccines. Despite many encouraging results in mouse models, vaccines based on these targets are incapable of inducing neutralizing antibodies and remain ineffective in preventing influenza infection. Therefore, the integral region, namely, HA, can serve as an ideal target for developing influenza vaccines against a wide range of viral subtypes. Several cross-protective vaccines have recently been developed using full-length HA sequences [Citation10–12], but these platforms were not optimal.
Currently, various platforms are being applied for developing influenza virus vaccines, that is, live-attenuated vaccines, inactivated vaccines, recombinant subunit vaccines, viral vector vaccines, peptide vaccines, virus-like particles, DNA vaccines, and mRNA vaccines[Citation13]. Among them, mRNA vaccine is a newly developed platform in recent years and has previously been used in the prevention of infectious diseases caused by rabies virus[Citation14], human immunodeficiency virus[Citation15], respiratory syncytial virus[Citation16], Zika virus[Citation17], or SARS-CoV-2[Citation18]. To date, two mRNA vaccines (BNT162b2 and mRNA-1273) for COVID-19 have been licensed by the US Food and Drug Administration, and many others are being tested in clinical trials. When it comes to the application of the influenza vaccine, the mRNA vaccine has demonstrated numerous advantages over other vaccine types as a novel platform. First and foremost, the design is quick and flexible, and the production period is short [Citation19]. Second, unlike DNA vaccines, mRNA vaccines are safe, with no risk of integrating into the host cell genome, and no infectious elements are used in their production[Citation20]. Third, unlike inactivated and attenuated vaccines, mRNA vaccine production is not limited by egg supply and can be quickly standardized and scaled-up, improving response to newly emerging and reemerging outbreaks[Citation19, Citation21]. Finally, vaccination can elicit both humoral and cellular responses[Citation20, Citation22]. Therefore, mRNA vaccines are an excellent platform for influenza vaccines.
Several attempts have been made to develop influenza mRNA vaccines, but most of them are designed to target only a particular strain or are available as a single vaccine targeting conserved epitopes such as NP[Citation23, Citation24], M1[Citation23, Citation24], M2e[Citation25], and stem of HA[Citation24, Citation25]. Only a few cross-protective mRNA vaccines targeting the full-length HA sequence are available. The present study developed a lipid nanoparticle (LNP)-encapsulated nucleoside-modified mRNA vaccine (mRNA-LNPs) that encodes a consensus full-length HA sequence, and its immunogenicity and protective efficacy were assessed through in vitro and in vivo assays. The findings of the study showed that two doses of intramuscular immunization with H1c-mRNA-LNPs elicited robust cellular and humoral immunity, which conferred cross-protection from a panel of heterologous H1N1 influenza viruses.
Materials and methods
Ethics statement
All BALB/c mice used in this study were raised in separately ventilated cages and had access to sterile food and water. All animals were treated according to the provisions of the Regulations for the Administration of Affairs Concerning Experimental Animals of China. The study protocols received approval from the Animal Experimentation Ethics Committee of Wuhan Institute of Biological Products Co. Ltd. (WIBP-A II312022001).
Cells, viruses, and animals
The human embryonic kidney cell line HEK293 T (ATCC, CRL-3216) was maintained in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific, USA) supplemented with 10% fetal bovine serum (FBS). Additionally, Madin-Darby canine kidney (MDCK) cells (ATCC, CRL-2935) were cultured and maintained in the Virus Production (VP, Thermo Fisher Scientific, USA) medium supplemented with 1% GlutaMAX (Thermo Fisher Scientific, USA) and 5% FBS. The aforementioned cell lines were incubated at 37 °C and 5% CO2.
The influenza A viruses A/Singapore/GP1908/2015, A/Victoria/2570/2019, A/Guangdong-Maonan/SWL1536/2019, A/California/07/2009, A/Victoria/2454/2019, and A/Christchurch/16/2010 were propagated in embryonated eggs and preserved in the Viral Vaccine Research Department of the Wuhan Institute of Biological Products.
Female BALB/c mice (6–8 weeks old) of specific-pathogen-free grade were provided by the Laboratory Animal Department of the Wuhan Institute of Biological Products Co. Ltd.
Target sequence design
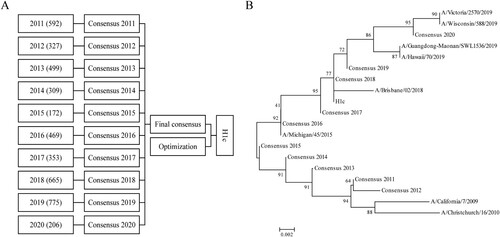
HA protein sequences of the human H1N1 influenza virus were obtained from the National Centre for Biotechnology Information (NCBI) influenza virus database (January 2011 to June 2020). After collapsing identical sequences, 4,367 sequences were obtained, which were divided into 10 groups depending on the year of isolation. Then, online alignment was performed in each group, which yielded 10 primary consensus sequences. Then, VectorNTI software was used to perform a second round of comparison, followed by optimization, and an eventual consensus sequence (namely, “H1c”) with the most frequently occurring amino acid was acquired. As for H1N1 influenza vaccines, their full-length HA protein sequences recommended by the WHO from 2011 to 2020 were obtained from the Global Initiative on Sharing All Influenza Data (gisaid.org). MEGA5.0 software was used to perform the phylogenetic analysis.
Preparation and characterization of H1c-mRNA-LNPs
H1c nucleotide sequence was optimized and synthesized by GENEWIZ (Suzhou, China) and was cloned into a pBlueScript II SK (+) vector containing one poly(A) tail along with 5′- and 3′-untranslated regions. mRNA encoding H1c was prepared with T7 RNA polymerase (Novoprotein, China) and Cap 1 Capping system (Novoprotein, China) from a linearized plasmid template in vitro, wherein uridine triphosphate (UTP) was replaced with N1-methylpseudouridine-5′-triphosphate (Trilink, USA). The products were purified using magnetic beads (Vazyme, China) and stored at −80 °C. NanoDrop 2000 (Thermo Fisher Scientific, USA) and Agilent 5200 Fragment Analyzer (Agilent, USA) were used to assess the purified mRNA for its content and integrity, respectively.
LNPs were prepared using a self-assembly procedure as previously described [Citation26]. Briefly, lipid mixture containing ionizable lipid, PEGylated lipid, 1,2-distearoyl-sn-glycero-3-phosphocholine, and cholesterol at a 46.3:1.6:9.4:42.7 molar ratio was first dissolved in absolute ethanol and then mixed with citrate buffer (20 mM, pH 4.0) containing mRNA at 1:3 ratio using a microfluidic device. After diafiltration and sterile filtration, a final formulation was prepared. The average particle size was determined through dynamic light scattering, using Nano-ZS90 (Malvern, UK). The RiboGreen assay (Thermo Fisher Scientific, USA) was performed to measure the encapsulation efficiency of the nanoparticles.
mRNA transfection
HEK293 T cells (500,000/well) were seeded in 6-well plates, and the plates were incubated at 37 °C and 5% CO2. After incubation, once the cell confluence reached approximately 80%, the cells were transfected with Lipofectamine™ MessengerMAX™ Reagent (Thermo Fisher Scientific, USA) according to specific protocols. Complexes containing 2.5 µg of H1c-mRNA and 7.5 µL of MessengerMAX™ Reagent were prepared in the Opti-MEM™ Reduced Serum Medium (Thermo Fisher Scientific, USA) and added gently to the cells. Lipofectamine™ MessengerMAX™ Reagent without mRNA was set as the negative control.
Western blotting assay
Twenty-four hours after transfection, radioimmunoprecipitation assay lysis buffer (Sangon Biotech, China) was added to the harvested cells to lyse the cells. The lysates were mixed with sample buffer and boiled for 10 min, after which the proteins in the sample were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. Next, phosphate-buffered saline (PBS) with 0.1% Tween-20 (PBST) containing 5% nonfat dry milk powder was added to the membrane as a blocking solution. H1c expression was detected initially with anti-H1c rabbit polyclonal antibody (1:2000 dilution, in-house reagent) and then with horseradish peroxidase (HRP)-labeled goat anti-rabbit secondary antibody (1:5000 dilution, Biodragon, China). Later, the HRP substrate luminol reagent (Merck, Germany) was added to develop the exposure, and Amersham ImageQuant™ 800 system (Cytiva, Japan) was used for image capturing.
Indirect immunofluorescence assay
After being transfected for 24 h, HEK293 T cells were fixed for 30 min in 4% paraformaldehyde, followed by permeabilization for 10 min with a cell permeabilization solution. After blocking the membrane for 1 h with 1% bovine serum albumin, the cells were incubated for another 1 h with anti-H1c rabbit polyclonal antibody (1:1000 dilution, in-house reagent) at 37 °C and for another 1 h with Alexa Fluor® 488 goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody (1:500 dilution, Thermo Fisher Scientific, USA) at 37 °C in the dark. Images were captured by fluorescence microscopy.
Mouse immunization and virus challenge
Female BALB/c mice (6–8 weeks old) were intramuscularly immunized with 2 µg (n = 9), 10 µg (n = 9), or 20 µg (n = 9) of H1c-mRNA-LNPs at a 3-week interval. Mice in the control groups were given 15 g of the H1N1 split vaccine (n = 9) or PBS (n = 9) under the same schedule. Serum was harvested from mice on days 21 and 42 after initial immunization for the detection of hemagglutination-inhibition (HAI) antibodies, neutralizing antibodies, and IgG subclasses (IgG1 and IgG2a).
For the measurement of cellular response, pro-inflammatory cytokines, and protective efficacy, female BALB/c mice (6–8 weeks old) were intramuscularly immunized with 10 µg of H1c-mRNA-LNPs (n = 36) or PBS (n = 36) at a 3-week interval. Serum was harvested from mice at 0, 3, 6, 12, 24, 48, 72, and 96 h after initial immunization for the measurement of pro-inflammatory cytokines. Spleen was separated on day 28 after initial immunization to evaluate the cellular response. For challenge studies, on day 49 after initial immunization, mice immunized with 10 µg of H1c-mRNA-LNPs or PBS were challenged intranasally with 100 µL of each of the following viruses: A/Singapore/GP1908/2015 (107.33 CCID50/0.1 mL), A/Guangdong-Maonan/SWL1536/2019 (107.23 CCID50/0.1 mL), A/Victoria/2570/2019 (105.48 CCID50/0.1 mL), or A/California/07/2009 (107.00 CCID50/0.1 mL). The body weight of the animals and survival rate were determined every day for 14 consecutive days. The animals were humanely euthanized once the body weight was reduced by more than 75% of the initial weight or for three days continuously once the body weight was reduced by over 20% of the initial weight. Four days after the challenge, four mice were humanely euthanized, and their lung tissues were obtained for hematoxylin-and-eosin (HE) staining of the sections for pathological examination and viral load detection.
Hemagglutination-inhibition test
The serum was treated with a receptor-destroying enzyme (Merck, Germany), and the resultant solution was incubated for 16–18 h at 37 °C. The solution mixture was first inactivated for 1 h at 56 °C and then diluted with PBS to an initial dilution of 1:10, after which serial dilution (twofold) was performed. The HAI assay was performed in V-bottomed 96-well plates, wherein 25 µL of the serially diluted serum (solution mixture) was mixed with an equivalent volume of standardized influenza virus (4 HA units/25 µL) and allowed to react for 40 min at room temperature. Finally, 25 µL of 1% chicken erythrocytes were added to the plates, and the plates were incubated for 30 min at ambient temperature. The HAI titre was calculated as the reciprocal of the greatest serum dilution at which agglutination was inhibited.
Microneutralization assay
MDCK cells (20,000 cells/well) were seeded into 96-well plates (100 µL/well), followed by the overnight culture at 37 °C and 5% CO2. The monolayer MDCK cells were then washed with PBS. The serum was subjected to inactivation for 30 min at 56 °C, followed by dilution with the VP medium supplemented with 1% penicillin–streptomycin (PS) solution (Thermo Fisher Scientific, USA) and 2 µg/mL L-(tosylamido-2-phenyl) ethyl chloromethyl ketone (TPCK)-treated trypsin (Merck, Germany) to an initial dilution of 1:10, which was later diluted to twofold to a total volume of 60 µL in a 96-well plate with VP medium. The serum was first serially diluted and then mixed with an equivalent volume of virus containing 100× median tissue culture infectious dose (TCID50), and the mixture solution was incubated for 1 h at 37 °C. After incubation, the serum-virus mixture solution (100 µL) was added to the monolayer MDCK cells, and the cells were cultured for 96 h at 37 °C and 5% CO2. The titre was measured with the supernatant through hemagglutination assays. Briefly, 25 µL of 1% chicken erythrocytes were added to the culture supernatant (25 µL) and incubated for 30 min at ambient temperature, and hemagglutination was observed. Microneutralization (MN) titre was calculated as the reciprocal of the greatest serum dilution at which agglutination was inhibited.
Indirect enzyme-linked immunosorbent assay
Indirect enzyme-linked immunosorbent assay (ELISA) was performed to analyze IgG subclasses. Ninety-six-well polystyrene plates were pre-coated with recombinant H1c antigen (5 µg/mL) in carbonate–bicarbonate buffer, and the plates were incubated overnight at 4 °C. The next day, after washing thrice with PBST, the plates were subjected to blocking for 1 h with the blocking solution (5% bovine serum albumin in PBST) at 37 °C. The serum was twofold diluted with the blocking solution, and the diluted serum was transferred to the plates. After incubation for 1 h at 37 °C, the plates were rinsed thrice with PBST. Next, the samples were incubated with HRP-labeled goat anti-mouse IgG or IgG1 secondary antibody (1:5000, Bethyl, Germany) or HRP-conjugated goat anti-mouse IgG2a secondary antibody (1:5000, Bersee, China) for 1 h at 37 °C. After washing the plates with PBST five times, the 3,3′,5,5′-Tetramethylbenzidine substrate was added in all the wells, and the plates were incubated for 10 min in the dark at an ambient temperature. After incubation, the stop solution (2 M H2SO4) was added to terminate the reaction, and the microplate reader (Thermo Fisher Scientific, USA) was used to measure absorbance values (optical density) at 450 nm. Titres of antigen-specific IgG, IgG1, and IgG2a antibodies were calculated as the reciprocal of the greatest serum dilution at which the absorbance value S/N was >2.1.
Flow cytometry
To assess cellular immune responses, the spleen was removed from immunized mice, and an individual splenocyte suspension was prepared in RPMI 1640 medium (Thermo Fisher Scientific, USA) supplemented with 1% PS and 10% FBS. Subsequently, a total of 1,000,000 cells per sample were stimulated with recombinant H1c antigen (5 µg/mL) at 37 °C and 5% CO2 and incubated for 6 h. Brefeldin A solution (BioLegend, USA) was added after stimulation for 2 h. Cells were stained using the Zombie Aqua™ Fixable Viability kit (BioLegend, USA), followed by rinsing with PBS and re-staining with APC/Cyanine7-labelled anti-mouse CD3ϵ, PerCP/Cyanine5.5-labelled anti-mouse CD4, and FITC-labelled anti-mouse CD8a (BioLegend, USA). Next, the fixation buffer (BioLegend, USA) was added to fix the cells. The cells were rinsed with intracellular staining permeabilization wash buffer (BioLegend, USA) twice and re-stained using PE-labelled anti-mouse interleukin (IL)−2, Brilliant Violet 421™–labeled anti-mouse interferon (IFN)-γ, and APC-labelled anti-mouse tumour necrosis factor (TNF)-α (BioLegend, USA). The stained cells were subsequently subjected to flow cytometry (FCM) detection. CytExpert software was used for data analysis.
The LEGENDplex™ mouse inflammation panel (BioLegend, USA) was used to detect and measure pro-inflammatory cytokines according to specific protocols. Briefly, serum was mixed with capture beads in 96-well V-bottomed plates under shaking for 2 h at 800 rpm at an ambient temperature. After centrifuging and rinsing the plates, detection antibodies were added under shaking for 1 h at 800 rpm at an ambient temperature. After centrifuging and resuspending, a flow cytometer (Beckman, USA) was used to quantify the samples. LEGENDplexTM data analysis software (BioLegend, USA) was used for data analysis.
Plaque assay
MDCK cells (100,000 cells/well, 2 mL/well) were seeded into 6-well plates, and the plates were incubated overnight at 37 °C and 5% CO2. Lung tissues were homogenized in 1 mL of VP medium, and the homogenate was centrifuged for 10 min at 12,000 rpm. The supernatants were then diluted with VP medium supplemented with TPCK-treated trypsin (2 µg/mL; Merck, Germany) and 1% PS solution (Thermo Fisher Scientific, USA) to an initial dilution of 1:10, which was later serially diluted by tenfold. When the MDCK cells reached above 90% confluence, PBS was added to rinse the plates twice, after which infection was carried out using each dilution of the supernatant (200 µL). Following the 1-hour incubation at 37 °C (under shaking every 20 min), samples were eliminated and substituted by VP medium (2 mL) containing TPCK-treated trypsin (2 µg/mL; Merck, Germany), 1% PS solution (Thermo Fisher Scientific, USA), and 1.6% agarose (Lonza, Swiss). The plates were then incubated for 72 h at 37 °C and 5% CO2. Next, the neutral red staining solution (Beyotime, China) was added to all wells, and the plates were incubated for another 4 h at 37 °C and 5% CO2. Finally, the staining solution was discarded, and the viral plaques were counted. The lung viral load was calculated and expressed as plaque forming unit (PFU) per millilitre.
Histopathology assay
Lung tissues were collected from mice in challenge studies, and 4% PFA was added to fix the tissues over a 48-hour period, followed by paraffin embedding, sectioning, and HE staining. A digital slide scanner (3DHISTECH, Hungary) was used to scan images.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.0 software. Data were presented as mean ± standard deviation. Multiple comparisons were performed using one-way or two-way analysis of variance, and two groups were compared using an unpaired t-test. p < 0.05 indicated statistical significance.
Results
Antigen design
HA, a crucial surface antigen on influenza virus, contains the major neutralizing epitope and mediates viral invasion. HA blockade can effectively prevent influenza infection. Therefore, in this study, we chose HA as the immunogen for constructing an influenza mRNA vaccine. To acquire a broadly reactive HA sequence, we first retrieved the HA protein sequences of human H1N1 influenza viruses from the NCBI influenza database (January 2011 to June 2020). After collapsing identical sequences, 4,367 sequences were obtained, which were then sorted into 10 groups according to the year of isolation. After online alignment by each group, ten primary consensus sequences were generated. VectorNTI software was used to perform a second round of comparison, and a final consensus sequence was acquired (A). Meanwhile, a phylogenetic tree was built using the neighbor-joining approach for the ten primary consensus sequences and the HA protein sequences of H1N1 influenza vaccines for strains recommended by the WHO from 2011 to 2020 (Table S1). The results showed that sequences identified from 2011 to 2015 were located on one branch and sequences identified after 2015 were located on another branch (B). Considering the prevalent trends of the virus, the consensus sequence was further optimized, which was termed “H1c” (A). Phylogenetic tree analysis demonstrated that H1c has the highest antigenic similarity with sequences identified in 2018 (B).
Figure 1. Generation of the consensus sequence H1c. (A) A total of 4,367 sequences were obtained from the NCBI influenza database (January 2011 to June 2020) and classified into ten groups according to the year of isolation. Online alignment was performed for each group, and ten primary consensus sequences were generated. VectorNTI software was used for the second round of comparison, and a final consensus sequence was obtained after optimization. (B) A phylogenetic tree was constructed for the consensus sequence H1c and HA protein sequences of H1N1 influenza vaccines recommended by the WHO from 2011 to 2020 on the basis of the neighbor-joining method using MEGA5.0 software.
Preparation and characterization of H1c-mRNA-LNPs
The in vitro enzymatic synthesis of the nucleotide-modified mRNA encoding the full-length codon-optimized H1c was carried out using a linearized plasmid as the template (A). A single uniform peak was observed on capillary electrophoresis analysis, indicating that the synthesized mRNA had good integrity (B). To verify antigen expression, H1c-mRNA was transfected in HEK293 T cells. Western blotting results showed a significant expression of the H1c antigen (C). The results of the immunofluorescence assay further demonstrated that H1c could be recognized by H1c-specific polyclonal antibodies (D).
Figure 2. Design and identification of mRNA encoding H1c. (A) Sketch map showing the mRNA expressing H1c. (B) Integrity of the H1c-mRNA determined using Agilent 5200 Fragment Analyzer. (C) H1c expression in HEK293 T cells detected through WB assay. H1c-mRNA was transfected into HEK293 T cells using Lipofectamine™ MessengerMAX™ Reagent. The cells were harvested at 24 h after incubation, after which they were lysed and detected through the WB assay. (D) H1c expression confirmed by immunofluorescence staining. HEK293 T cells transfected with H1c-mRNA were fixed with 4% paraformaldehyde after incubation for 24 h, after which they were evaluated by immunofluorescence analysis. (E) Size distribution of H1c-mRNA-LNPs measured using Nano-ZS90 through the dynamic light scattering method. WB, western blotting.
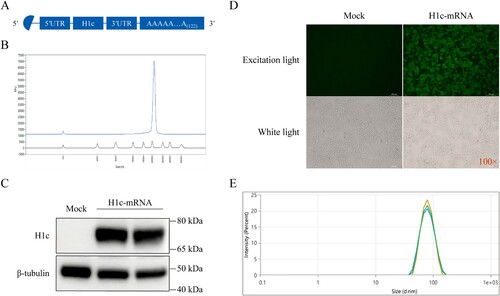
LNPs are the most extensively investigated delivery system for nucleic acid vaccines[Citation27]. In this study, a self-assembly process was applied to prepare H1c-mRNA-LNPs. Briefly, the ethanolic phase containing various lipid components was rapidly mixed with citrate buffer (20 mM, pH 4.0) containing H1c-mRNA using a microfluidic device at 1:3 ratio. The final formulation was obtained after ultrafiltration and sterile filtration. The average particle size of the prepared H1c-mRNA-LNPs was 77.09 ± 0.38 nm (E), with a polydispersity index (PDI) of 0.045 ± 0.004 and an encapsulation efficiency of more than 94% (Table S2).
H1c-mRNA-LNPs elicit broadly reactive antibody responses in mice
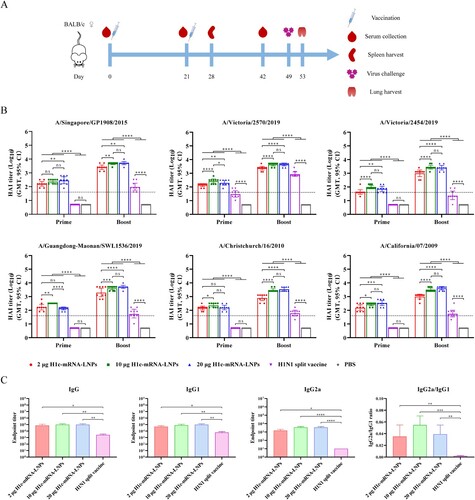
After successful in vitro expression, the immunogenicity of H1c-mRNA-LNPs was evaluated in BALB/c mice. Briefly, BALB/c female mice received intramuscular immunizations with 2, 10, or 20 µg of H1c-mRNA-LNPs as the prime-boost regimen at a 3-week interval; sera were collected on days 21 and 42 after initial immunization (A). Mice in the control groups received 15 µg of the H1N1 split vaccine or PBS under the same schedule. The levels of antibodies against HA were characterized in the HAI assay, a common criterion for assessing the immunogenicity of influenza vaccines[Citation28]. In the present study, six H1N1 influenza viruses from similar or different phylogenetic groups (A/Singapore/GP1908/2015, A/Victoria/2570/2019, A/Guangdong-Maonan/SWL1536/2019, A/Victoria/2454/2019, A/California/07/2009, and A/Christchurch/16/2010) were selected to evaluate the HAI titres of H1c-mRNA-LNP vaccines. Three weeks after the first immunization, all doses of H1c-mRNA-LNP vaccines elicited protective HAI titres against all six viruses (≥1:40), and the titres significantly increased after the booster. However, H1N1 split vaccine only induced remarkably HAI titres to the homologous virus with little to no protective HAI titres toward other viruses even after the booster dose. HAI antibodies were induced at similar levels when the mice were immunized with 10 and 20 µg of H1c-mRNA-LNPs, and these levels were significantly higher than those induced with 2 µg of H1c-mRNA-LNPs. All mice that received PBS were determined to be seronegative (B).
Figure 3. Cross-reactive antibody response of H1c-mRNA-LNPs in mice. (A) Flow chart of the immunization, sampling, and challenge protocol. Each BALB/c female mouse received intramuscular immunization with 2 µg (n = 9), 10 µg (n = 9), or 20 µg (n = 9) of H1c-mRNA-LNPs, followed by a booster at the same dose on day 21. Mice received 15 µg of the H1N1 split vaccine (n = 9) or PBS (n = 9) as control. Serum was harvested on days 21 and 42 after the initial immunization. (B) Antigen-specific antibodies were determined by hemagglutination-inhibition test. (C) Antigen-specific IgG, IgG1, and IgG2a were evaluated through indirect ELISA (n = 4). Two-way and one-way ANOVA were adopted to compare differences for (B) and (C), respectively. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant. HAI, hemagglutination-inhibition; ANOVA, analysis of variance; IgG, immunoglobulin G; PBS, phosphate-buffered saline.
To further assess the neutralizing antibody elicited by the H1c-mRNA-LNP vaccine, the MN assay was performed using the six selected viruses. The results showed that all doses of H1c-mRNA-LNPs could induce protective neutralizing antibodies (≥1:40) after immunization with the prime-boost regimen, with a significant increase noted after immunization with the booster. MN titres in the 10 and 20 µg groups were significantly greater than that in the 2 µg group after the booster. Mice receiving the H1N1 split vaccine showed protective MN titres only to homologous viruses with no cross-protective titres to other viruses. All mice immunized with PBS were determined to be seronegative (Figure S1).
IgG subclasses are a common indicator that reflects the bias of T helper (Th) cells[Citation29]. In this study, indirect ELISA was adopted for determining the IgG subclasses in the booster-immunized sera. The results indicated that HA-specific levels of total IgG, IgG1, and IgG2a significantly increased after immunization with H1c-mRNA-LNPs when compared with those with the H1N1 split vaccine (C). The value of IgG2a/IgG1 ratio indicated that H1c-mRNA-LNPs induce a stronger Th1-biased cellular immune response than that of the H1N1 split vaccine.
Overall, our findings indicate that two doses of H1c-mRNA-LNPs can elicit a robust antibody response and can neutralize a wide range of heterologous H1N1 influenza viruses.
H1c-mRNA-LNPs stimulate the secretion of pro-inflammatory cytokines
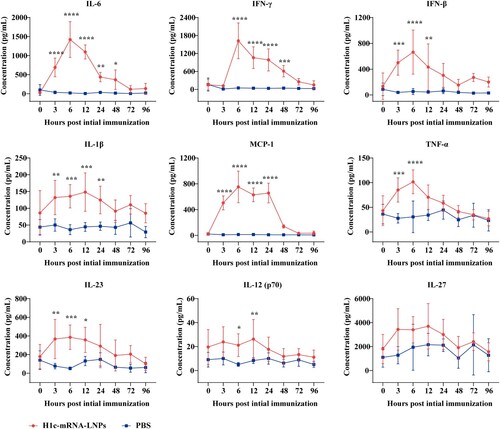
Pro-inflammatory factors such as IL-1β, IL-12, IL-23, IL-27, monocyte chemoattractant protein (MCP)−1, IL-6, TNF-α, IFN-β, and IFN-γ were measured in the serum of mice at 0, 3, 6, 12, 24, 48, 72, and 96 h after initial immunization with 10 µg of H1c-mRNA-LNPs. The results showed that IL-1β, IL-12, IL-23, MCP-1, IL-6, TNF-α, IFN-γ, and IFN-β levels remarkably elevated after 3 h, peaked at 6 h, and gradually declined to levels that were comparable to those of PBS control within 72 h. However, no salient change was noted in the levels of IL-27 ().
Figure 4. Profiles of pro-inflammatory cytokines. Serum was harvested from mice immunized with H1c-mRNA-LNPs (10 µg), and pro-inflammatory cytokines were measured at 0, 3, 6, 12, 24, 48, 72, and 96 h after the initial immunization. Serum pro-inflammatory factors, including IL-1β, IL-12, IL-23, IL-27, MCP-1, IL-6, TNF-α, IFN-β, and IFN-γ were determined as a panel of mouse inflammatory factors by flow cytometry (n = 5). Differences were compared using two-way ANOVA. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant. ANOVA, analysis of variance; IFN, interferon; IL, interleukin; MCP, monocyte chemoattractant protein; TNF, tumour necrosis factor.
H1c-mRNA-LNPs trigger antigen-specific cellular immunity
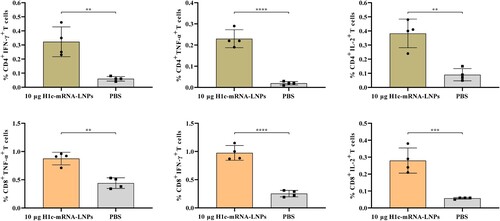
To evaluate T-cell response, splenocytes were first isolated from mice that received the two-dose immunization and then stimulated with recombinant H1c antigen, after which the tissues were stained with fluorescent-conjugated monoclonal antibodies for FCM analysis. The results showed that H1c-mRNA-LNPs induced a remarkably higher number of HA-specific CD4+ and CD8+ T cells that principally produced factors, including TNF-α, IFN-γ, and IL-2 (). Together, these results indicated that H1c-mRNA-LNPs could elicit a Th1-biased response, conforming to the obtained IgG2a/IgG1 ratio.
Figure 5. T-cell responses resulting from H1c-mRNA-LNPs in mice. Mice were initially immunized with H1c-mRNA-LNPs (10 µg) as a two-dose regimen. Splenocytes were harvested on day 28 following initial immunization. CD4+ T cells and CD8+ T cells that secreted TNF-α, IFN-γ, and IL-2 were analyzed through flow cytometry (n = 4). The results are presented as mean ± SD. Differences were compared using an unpaired t-test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant. CD, cluster of differentiation; IFN, interferon; IL, interleukin; SD, standard deviation; TNF, tumour necrosis factor.
Protection of H1c-mRNA-LNPs against challenge with multiple H1N1 influenza viruses
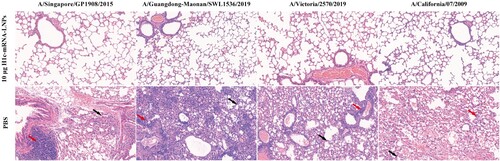
To further assess the in vivo protective efficacy of the vaccine, a series of challenge experiments were performed with BALB/c mice after immunization with 10 µg of H1c-mRNA-LNPs at two doses. On day 49 following the initial immunization, 100 µL of each of the viruses, namely, A/Singapore/GP1908/2015 (107.33 CCID50/0.1 mL), A/Guangdong-Maonan/SWL1536/2019 (107.23 CCID50/0.1 mL), A/Victoria/2570/2019 (105.48 CCID50/0.1 mL), or A/California/07/2009 (107.00 CCID50/0.1 mL), was inoculated into each mouse intranasally. The body weight of the animals and survival rate were recorded every day for 14 consecutive days. The results showed that all mice immunized with H1c-mRNA-LNPs survived when challenged with multiple heterologous H1N1 influenza viruses, with no or only slight weight loss, but animals that received PBS displayed remarkable weight loss and a high mortality rate (A and B). All mice immunized with H1c-mRNA-LNPs showed significantly reduced lung viral loads when compared with animals that received PBS (C). Histopathology images revealed characteristic lung lesions with features of alveolar septal thickening and inflammatory cell infiltration in lung sections from mice that received PBS; however, no significant pathological alteration was observed in H1c-mRNA-LNP- immunized animals (). In conclusion, two doses of the H1c-mRNA-LNP vaccine could effectively confer protection from H1N1 influenza viruses in mice.
Figure 6. Protective efficacy of the H1c-mRNA-LNP vaccine in mice following challenge with various H1N1 influenza viruses. Mice were immunized with H1c-mRNA-LNPs (10 µg) in two doses in a 3-week interval. Forty-nine days after initial immunization, each mouse received intranasal inoculation (100 µL) of each of the following viruses: A/Singapore/GP1908/2015 (107.33 CCID50/0.1 mL), A/Guangdong-Maonan/SWL1536/2019 (107.23 CCID50/0.1 mL), A/Victoria/2570/2019 (105.48 CCID50/0.1 mL), or A/California/07/2009 (107.00 CCID50/0.1 mL). (A) Body weight of the animals measured daily for 14 days (n = 8 each group). Data are presented as proportion of original weight. (B) Survival rates monitored daily for 14 days (n = 8 each group). (C) Viral loads in the lung tissues measured through plaque assay 4 days after the challenge (n = 4 each group). The results are presented as mean ± SD. PFU, plaque forming unit; SD, standard deviation.
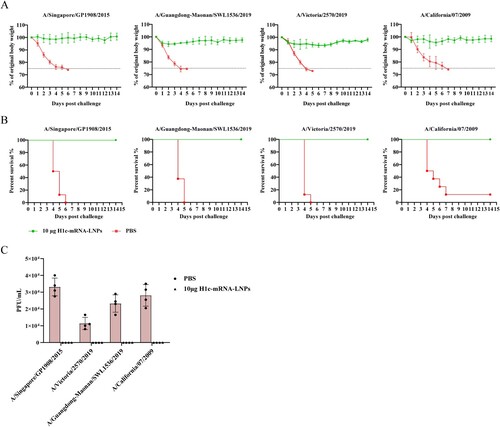
Figure 7. Lung pathology of the challenged mice. Paraformaldehyde (4%) was added to fix the lung tissues collected from challenged mice. The tissues were then paraffin-embedded, sectioned, and stained with hematoxylin-eosin. A digital slide scanner (3DHISTECH, Hungary) was used for image scanning. The black arrow indicates alveolar septal thickening, while the red arrow indicates inflammatory cell infiltration.
Discussion
In this study, 4,367 HA protein sequences were extracted from the NCBI influenza database and were aligned using an online tool and Vector NTI software. After alignment and optimization, a final consensus sequence (H1c) that included the most frequently occurring amino acid was generated. Then, the mRNA-LNP vaccine was constructed, and its immunogenicity and protective efficacy were evaluated in mice. The results demonstrated that the H1c-mRNA-LNP vaccine could elicit a robust antibody response that could neutralize a panel of heterologous H1N1 influenza viruses and provide protection when challenged with multiple heterologous H1N1 influenza viruses. A previous study based on a DNA vaccine that encoded the consensus HA sequence reported a broadly reactive response to heterologous H1N1 influenza viruses but not to pdm09 H1N1 influenza viruses[Citation30]. The present study confirmed that the antibodies induced by the H1c-mRNA-LNP vaccine can neutralize A/H1N1pdm09 viruses, but whether the vaccine can neutralize viruses at a much earlier stage still need further investigations. Additionally, IgG subclasses analysis showed that the H1c-mRNA-LNP vaccine elicits Th1-biased immunity together with Th2-biased immunity. As suggested by FCM results, antigen-specific CD4+ and CD8+ T cells showed a remarkable increase. These findings suggested that the H1c-mRNA-LNP vaccine triggered both cellular and humoral immunity.
Interestingly, even though there is only one amino acid difference between A/Guangdong-Maonan/SWL1536/2019 and A/Victoria/2454/2019, the antibodies induced by A/Guangdong-Maonan/SWL1536/2019 are nearly twice as much as that induced by A/Victoria/2454/2019, indicating that one amino acid mutation may affect the immunogenicity of the vaccine.
The serum profiles of nine pro-inflammatory cytokines were investigated at various intervals after the initial immunization. Compared with mice that received PBS, which exhibited little variation over time, animals that received the H1c-mRNA-LNP vaccine showed markedly elevated levels for most of the pro-inflammatory cytokines 3 h after vaccination, peaked at 6 h, and waned gradually to levels similar to those in the PBS group. However, no salient change was found in IL-27 levels. After 72 h, the levels of all pro-inflammatory cytokines declined to levels comparable to those in the PBS control group, implying vaccine safety.
For maximizing mRNA vaccine efficacy, an appropriate delivery system must be used. Starostina et al. constructed a nucleotide-modified trivalent influenza mRNA vaccine that encoded fragments of the stem of H1 and H3 and conserved protein M2, but they did not use a delivery system[Citation31]. After two doses of immunization in mice, only low antibody titres were reported, indicating that the delivery vehicle used is crucial. Currently, various materials have been used for mRNA delivery, such as protamine, polymers, LNPs, and lipidoid nanoparticles[Citation32]. Among them, lipid-based nanoparticles such as liposomes and LNPs have proved to be effective delivery systems. Recently, a mannose-modified cationic liposome-encapsulated mRNA vaccine encoding the influenza HA sequence has been tested in mice. The 12-µg mRNA vaccine was administered intranasally in two doses, and it successfully induced antigen-specific cellular and humoral immunity and complete protection against lethal dosages of influenza virus challenge[Citation33]. However, HAI titres elicited by this liposome-based vaccine were much lower than those elicited by H1c-mRNA-LNPs in this study, which may be attributed to the adjuvant effect of ionized lipids in H1c-mRNA-LNPs because ionized lipids have shown to stimulate the synthesis of IL-6, which, in turn, provokes a follicular T-helper cell (Tfh) response[Citation34]. Additionally, elevated levels of antibodies induced by the LNP formulation may potentially contribute to cross-protection.
The National Institute of Allergy and Infectious Diseases (NIAID) organized a seminar in 2017 on the topic “Pathway to a Universal Influenza Vaccine,” and proposed that developing a universal vaccine in incremental steps might be feasible[Citation35]. In this study, we created an LNP-encapsulated mRNA vaccine which could not only induce cross-protective neutralizing antibodies and cellular responses but also provide protection against a panel of heterologous H1N1 seasonal influenza viruses, laying the groundwork for developing a multivalent universal vaccine with broader influenza virus protection.
Supplemental Material
Download Zip (680.2 KB)Acknowledgements
We thank Shuyan Liang and Juan Zhang from Wuhan Biobank Co., Ltd for their kind help in the flow cytometric analysis of experimental samples.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Iuliano AD, Roguski KM, Chang HH, et al. Estimates of global seasonal influenza-associated respiratory mortality: a modelling study. Lancet (London, England). 2018 Mar 31;391(10127):1285–1300.
- Petrova VN, Russell CA. The evolution of seasonal influenza viruses. Nat Rev Microbiol. 2018;16(1):47–60.
- Dawood FS, Jain S, Finelli L, et al. Emergence of a novel swine-origin influenza a (H1N1) virus in humans. N Engl J Med. 2009;360(25):2605–2615.
- Kilbourne ED. Influenza pandemics of the 20th century. Emerging Infect Dis. 2006;12(1):9–14.
- CDC Seasonal Flu Vaccine Effectiveness Studies [Internet]. Georgia: Centers for Disease Control and Prevention, National Center for Immunization and Respiratory Diseases (NCIRD); [updated 2022 Aug 31; cited 2022 Dec 23]; Available from: https://www.cdc.gov/flu/vaccines-work/past-seasons-estimates.html.
- Wu NC, Wilson IA. Influenza hemagglutinin structures and antibody recognition. Cold Spring Harbor Perspect Med. 2020 Aug 3;10(8):a038778.
- Berthoud TK, Hamill M, Lillie PJ, et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA-NP + M1. Clin Infectious Diseases. 2011 Jan 1;52(1):1–7.
- Ong HK, Yong CY, Tan WS, et al. An influenza a vaccine based on the extracellular domain of matrix 2 protein protects BALB/C mice against H1N1 and H3N2. Vaccines (Basel). 2019 Aug 19;7(3):91.
- Darricarrère N, Qiu Y, Kanekiyo M, et al. Broad neutralization of H1 and H3 viruses by adjuvanted influenza HA stem vaccines in nonhuman primates. Sci Transl Med. 2021;13(583):eabe5449.
- Fadlallah GM, Ma F, Zhang Z, et al. Vaccination with consensus H7 elicits broadly reactive and protective antibodies against Eurasian and North American lineage H7 viruses. Vaccines (Basel). 2020;8(1):143.
- Reneer ZB, Skarlupka AL, Jamieson PJ, et al. Broadly reactive H2 hemagglutinin vaccines elicit cross-reactive antibodies in ferrets preimmune to seasonal influenza a viruses. mSphere. 2021 Mar 10;6(2):e00052-21.
- Sautto GA, Kirchenbaum GA, Ecker JW, et al. Elicitation of broadly protective antibodies following infection with influenza viruses expressing H1N1 computationally optimized broadly reactive hemagglutinin antigens. Immuno Horizons. 2018;2(7):226–237.
- Kim YH, Hong KJ, Kim H, et al. Influenza vaccines: past, present, and future. Rev Med Virol. 2022 Jan;32(1):e2243.
- Armbruster N, Jasny E, Petsch B. Advances in RNA vaccines for preventive indications: a case study of a vaccine against rabies. Vaccines (Basel). 2019;7(4):132.
- Gómez CE, Perdiguero B, Usero L, et al. Enhancement of the HIV-1-specific immune response induced by an mRNA vaccine through boosting with a poxvirus MVA vector expressing the same antigen. Vaccines (Basel). 2021;9(9):959.
- Espeseth AS, Cejas PJ, Citron MP, et al. Modified mRNA/lipid nanoparticle-based vaccines expressing respiratory syncytial virus F protein variants are immunogenic and protective in rodent models of RSV infection. NPJ Vaccines. 2020;5(1):16.
- Richner JM, Himansu S, Dowd KA, et al. Modified mRNA vaccines protect against Zika virus infection. Cell. 2017;168(6):1114–1125.e1110.
- Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N Engl J Med. 2020;383(27):2603–2615.
- Shartouny JR, Lowen AC. Message in a bottle: mRNA vaccination for influenza. J Gen Virol. 2022 Jul;103(7):001765.
- Kowalzik F, Schreiner D, Jensen C, et al. mRNA-based vaccines. Vaccines (Basel). 2021 Apr 15;9(4):390.
- Pardi N, Hogan MJ, Weissman D. Recent advances in mRNA vaccine technology. Curr Opin Immunol. 2020;65:14–20.
- Liu MA. Immunologic basis of vaccine vectors. Immunity. 2010;33(4):504–515.
- Magini D, Giovani C, Mangiavacchi S, et al. Self-amplifying mRNA vaccines expressing multiple conserved influenza antigens confer protection against homologous and heterosubtypic viral challenge. PloS One. 2016;11(8):e0161193.
- Freyn AW, da Silva R, Rosado J, et al. A multi-targeting, nucleoside-modified mRNA influenza virus vaccine provides broad protection in mice. Mol Ther: J Am Soc Gene Therapy. 2020;28(7):1569–1584.
- Hajam IA, Senevirathne A, Hewawaduge C, et al. Intranasally administered protein coated chitosan nanoparticles encapsulating influenza H9N2 HA2 and M2e mRNA molecules elicit protective immunity against avian influenza viruses in chickens. Vet. Res. 2020;51(1):37.
- Zhang NN, Li XF, Deng YQ, et al. A thermostable mRNA vaccine against COVID-19. Cell. 2020;182(5):1271–1283.e1216.
- Aldosari BN, Alfagih IM, Almurshedi AS. Lipid nanoparticles as delivery systems for RNA-based vaccines. Pharmaceutics. 2021;13(2):206.
- Zakay-Rones Z. Human influenza vaccines and assessment of immunogenicity. Expert Rev Vaccines. 2010;9(12):1423–1439.
- Snapper CM, Paul WE. Interferon-gamma and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science (New York, NY). 1987 May 22;236(4804):944–947.
- Ping X, Hu W, Xiong R, et al. Generation of a broadly reactive influenza H1 antigen using a consensus HA sequence. Vaccine. 2018;36(32 Pt B):4837–4845.
- Starostina EV, Sharabrin SV, Antropov DN, et al. Construction and immunogenicity of modified mRNA-vaccine variants encoding influenza virus antigens. Vaccines (Basel). 2021;9(5):452.
- Buschmann MD, Carrasco MJ, Alishetty S, et al. Nanomaterial delivery systems for mRNA vaccines. Vaccines (Basel). 2021;9(1):65.
- Zhuang X, Qi Y, Wang M, et al. mRNA vaccines encoding the HA protein of influenza A H1N1 virus delivered by cationic lipid nanoparticles induce protective immune responses in mice. Vaccines (Basel). 2020;8(1):123.
- Alameh MG, Tombácz I, Bettini E, et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity. 2021;54(12):2877–2892.e2877.
- Paules CI, Marston HD, Eisinger RW, et al. The pathway to a universal influenza vaccine. Immunity. 2017;47(4):599–603.