
ABSTRACT
Bacillus paranthracis, a Gram-positive conditional pathogen of Bacillus cereus group species, is capable of causing foodborne and waterborne illnesses, leading to intestinal diseases in humans characterized by diarrhea and vomiting. However, documented cases of B. paranthracis infection outbreaks are rare in the world, and the genomic background of outbreak strains is seldom characterized. This study retrospectively analyzed strains obtained from a outbreak in schools, as well as from water systems in peri-urban areas, China, in 2020.In total, 28 B. cereus group isolates were retrieved, comprising 6 from stool samples and 22 from water samples. Epidemiological and phylogenetic investigations indicated that the B. paranthracis isolate from drinking water as the causative agent of the outbreak. Genomic comparison revealed a high degree of consistency among 8 outbreak-related strains in terms of antimicrobial resistance gene profiles, virulence gene profiles, genomic content, and multilocus sequence typing (MLST). The strains related to the outbreak show highly similar genomic ring diagrams and close phylogenetic relationships. Additionally, this study shed light on the pathogenic potential and complexity of B. cereus group through its diversity in virulence genes and mice infection model. The findings highlight the usefulness of B. paranthracis genomes in understanding genetic diversity within specific environments and in tracing the source of pathogens during outbreak situations, thereby enabling targeted infection control interventions.
Disclaimer
As a service to authors and researchers we are providing this version of an accepted manuscript (AM). Copyediting, typesetting, and review of the resulting proofs will be undertaken on this manuscript before final publication of the Version of Record (VoR). During production and pre-press, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal relate to these versions also.Introduction
The Bacillus cereus group of bacteria consists of a complex of very closely related species with varying pathogenic potential. Notably, it consists of three main species: B. cereus sensu stricto (s.s.), which has been associated with numerous illnesses, including foodborne emetic intoxication, diarrheal toxicoinfection, anthrax-like illness, and nongastrointestinal infections, are significant examples; B. anthracis, an etiologic agent of anthrax; and B. thuringiensis, an entomopathogen that is well-known for its use as a biopesticide[2].
It is important to note that phenotypic characteristics may vary within and among the B. cereus group, leading to inconsistent species identification. A previous study indicated that some whole genome sequencing (WGS)-based species classification methods pertaining to the B. cereus group can be easily misinterpreted[3]. Current species definitions of the B. cereus group result in overlapping genomospecies clusters, with 66.2% of genomes belonging to multiple genomospecies at a conventional 95 average nucleotide identity (ANI) threshold, while a ≈92.5 ANI threshold reveals distinct genomospecies clusters with minimal overlap[4].
The clinical presentation of pathogenic B. cereus group infections raises serious public health concerns, due to its high pathogenicity [8, 9]. B. cereus group-associated foodborne outbreaks accounted for 19% of cases in the United States from 1998 to 2008 [10]. In France, B. cereus ranks second, following Staphylococcus aureus, as the most frequent bacterial agent responsible for food-borne outbreaks [11]. The European Union (EU) identified B. cereus as the second most common cause of food-borne outbreaks in 2016 [12]. In China, B. cereus was responsible for 4% of all food poisoning cases between 2011 and 2017, ranking the fourth most frequent cause of bacterial food poisoning [13].
Whole-genome sequencing technology is increasingly utilized in public health laboratories for surveillance and outbreak programs of infectious diseases. Its high discriminatory power allows for large-scale genomic epidemiological studies on international level [16, 17]. However, interpreting WGS results from B. cereus group is challenging; insights derived from WGS may conflict with information provided by traditional microbiologic assays.
Bacillus paranthracis, first identified in 2017, was historically referred to as B. cereus or group III B. cereus and encompasses B. cereus group strains capable of causing emetic and/or diarrheal foodborne illness[4]. Here, we conducted a retrospective study on a water-borne outbreak caused by B. paranthracis and the contamination status of paranthracis in the surrounding water systems. To our knowledge, this is the first description of a B. paranthracis outbreak in which WGS was employed to characterize outbreak-associated isolates. Through systematic evolutionary analysis and comparative genomics analysis, we determined the genetic relatedness and diversity of these isolates. In addition, this study evaluates the sanitary risk potential of waterborne B. paranthracis by analyzing virulence genes and their potential pathogenicity.
Materials and methods
Epidemiological investigation
On September 19, 2020, a clustered outbreak of diarrhea was reported in schools situated in a county of Shandong Province, China. In response to this notification, the Shandong Provincial Center for Disease Control and Prevention (Shandong CDC) conducted field epidemiological investigations and collected samples. An outbreak of diarrhea caused by B. cereus group was confirmed when six B. cereus group isolates were identified from stool samples of affected individuals. In this study, a suspected case was defined as any person who vomited more than once or having diarrhea more than three times in one day, and a confirmed case was a suspected case with a positive stool sample containing B. cereus group isolates. To identify the source of the outbreak and search potential cases, a questionnaire was conducted with suspected cases through face-to-face or telephone interview. This survey mainly focused on general health, clinical presentation, diet history, and hospital visits of all participants. Our investigations found that during September 16 to 19, 2020, several teachers (n = 6) and students (n = 31) exhibited symptoms suggestive of food poisoning, such as fever, nausea, vomiting, diarrhea, or abdominal pain. Prior to commencing the study, ethics approval was obtained from the Ethical Review Committee of the Shandong CDC.
Environmental health investigation
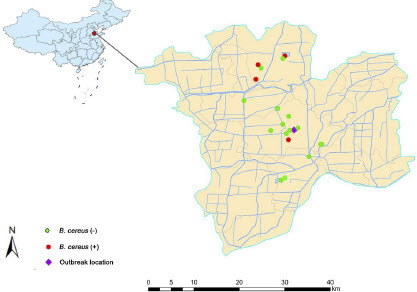
To identify the causative agent of the outbreaks and investigate the mode of transmission, we also carried out a hygiene investigation of the school dining room and overall sanitation within the school. In addition, a water quality survey was conducted in the surrounding outbreak areas over the following week to assess the potential contamination of specific bodies of water, including drinking water, reservoir water, and tap water from water-works (), by B. cereus group.
Figure 1. Geographical location of sampling points of Epidemiological investigation.
Sample collection and bacterial identification
Before initiating the epidemiological investigation, the Shandong CDC had conducted bacterial aetiology identification on 10 stool samples of diarrhea cases received. After the etiologic agent of infection was determined, in order to investigate the origin of infection and search for possible source samples, investigators also collected 38 food samples from school canteen, nine environmental samples from school canteen, and 26 drinking water samples within the school. Furthermore, we collected 19 additional drinking water samples, six source water samples from reservoirs, and six tap water samples from waterworks in two drinking source watersheds.
For all stool samples, bacterial isolation was performed to detect Salmonella, Shigella, Vibrio parahaemolyticus, B. cereus, Yersinia enterocolitica, Campylobacter and diarrheagenic Escherichia coli (DEC) according to the protocols outlined in the Chinese technical procedures of diarrheal pathogenic spectrum surveillance (i.e., WS271-2007, WS287-2008, and WS289-2008)[19].
Food samples (25 grams each) were homogenized in 225 ml of phosphate-buffered saline (PBS) for homogenization. To determine contamination by B. cereus, fecal samples, treated food samples, and environmental samples were inoculated onto MYP agar (Luqiao, Beijing, China), respectively, and incubated at 37 °C for 24 h. Water samples were filtered through sterile polycarbonate membranes with a pore size of 0.22 μm (Millipore, Billerica, MA), and the membranes were then transferred onto MYP agar for overnight culture. Identification of all suspected B. cereus group isolates using matrix assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) and species identification via WGS, followed by storage in 25% glycerol at -80 °C. To assess the risk of food poisoning due to the food intake or water intake, the plate count method was used for quantitative determination of B. cereus in positive samples.
Antibiotics susceptibility testing
The following antimicrobial agents were used: ampicillin, penicillin, oxacillin, erythromycin, clindamycin, ciprofloxacin, daptomycin, compound sulfamethoxazole, vancomycin, tetracycline, chloramphenicol, gentamicin, cefoxitin, imipenem, and erythromycin/clindamycin. The dose range for each antimicrobial can be found in Table S1. The minimum inhibitory concentrations (MICs) were determined by the broth microdilution method. The results were based on the breakpoints defined by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) and Clinical & Laboratory Standards Institute (CLSI). We used Staphylococcus aureus ATCC 29213 as a quality control strain for the above experiments.
Whole-genome sequencing
The genomic DNA of the outbreak-associated isolates underwent sequencing using Illumina HiSeq platform and PacBio platforms. The quality of the fastqc reads was evaluated with FastQC. Subsequently, the reads were assembled with the CLC Assembly Cell, and annotation was carried out using the RAST annotation pipeline. Virulence factors were identified by screening the genes, and this identification process utilized the Virulence Factor Database (VFDB). Genes exhibiting a subject coverage of ≥80% and an identity of ≥80% were categorized as virulence factor encoding genes. The esxL gene sequences were aligned utilizing the MUSCLE3.7 software, and an evolutionary tree for those genes was constructed through the neighbor-joining method in MEGA6.33. The sequence types of isolates were determined using the MultiLocus Sequence Typing (MLST) web-based service of the Center for Genomic Epidemiology (CGE). To ascertain the presence of antibiotic resistance genes, ResFinder 2.1 was employed. Insertion sequence (IS) elements were predicted using ISfinder, CRISPR elements were identified using CRISPRdigger, and prophages were predicted using PhiSpy. Comparative genomic analysis was conducted on 19 isolates using the BLAST Ring Image Generator (BRIG), with the B. cereus isolate GCF-025917685.1 serving as the reference for the analysis. The sequencing data have been submitted and deposited into the NCBI Sequence Read Archive database under the BioSample accession numbers SAMN40211118-SAMN40211145.
In vivo infection
Mice were used as animal models to test the in vivo pathogenicity of B. paranthracis isolates as previously described [23]. Cultures were grown in BHI medium to an OD600 of 0.8, and resuspended with PBS after centrifugation. Twenty mice were randomly divided into four groups, and were injected intraperitoneally with 200 μL 2 × 107 CFU/mL B. paranthracis, with one mouse in each group as a negative control. After infection for 12, 24, and 48 h, mice were sacrificed through cervical dislocation and their liver, kidney, and spleen were aseptically removed to obtain bacterial reproductive capacity in organs (CFU/g). Furthermore, the survival of mice in the final group was monitored closely.
Genome analysis and phylogenetic comparison
Phylogenetic and molecular evolutionary analyses of isolates during the outbreak were conducted by using different methods as described previously [24, 25]. A cgMLST scheme for B. cereus has been defined in the cgMLST database and the assemblies of the 28 genomes were loaded into BioNumerics 8.0 software. Comparatively, the cgMLST scheme utilizes 3,571 target genes, offering a heightened discriminatory resolution in comparison to the conventional MLST, which relies on seven housekeeping genes [26]. The genetic distance calculations based on the allele number of each loci between isolates were used to identify clusters. Subsequently, a minimal spanning tree (MST) was constructed to visually represent this analysis using BioNumerics 8.0. SNPs analysis was then conducted to establish the phylogenetic relationship between B. cereus group isolates (including 28 isolates in this study and 758 isolates obtained from various geographical locations through public database). This analysis included isolates of B. paranthracis (177 isolates), B. anthracis (341 isolates), B. albus (16 isolates), B. luti (3 isolates), B. cereus (100 isolates), and B. pseudomycoides (121 isolates). It was important to mention that all existing genomes of B. paranthracis, B. albus, B. luti, and B. pseudomycoides from databases were included in this study. Based on the core genomes of B. cereus group, kSNP3 analysis software was used for single nucleotide polymorphism (SNP) analysis. The kmer_length parameter was fixed at 13 (k = 13) for SNP analysis, while default values were retained for all other parameters. The visualization of the phylogenetic trees was facilitated by the online tool EvolView.
Results
Epidemiological investigation of the potential source of outbreak isolates
The primary and initial clinical manifestations among these 37 cases encompassed diarrhea (37 cases, 100.0%), abdominal pain (33 cases, 89.2%), myalgia (13 cases, 35.1%), nausea/vomiting (8 cases, 21.6%), and fever (2 cases, 5.4%). Seventeen patients with severe and moderate symptoms were admitted for further management. Following rehydration and antibiotics treatment, their overall health condition improved gradually, and no instances of critical cases or fatalities were recorded. The earliest reported symptom onset was September 16 at 4:00 p.m, while the latest was September 19 at 6:00 p.m. The median incubation period was found to be 5 hours, with a range spanning from 1.5 to 7 hours. As a precautionary measure against further outbreak propagation, this school were closed on September 20. The majority of patients (excluding teachers) were in the age range of 14-15 years, with a male to female ratio of 1:0.7. Among the various grade levels, Grade 9, situated on the first floor of teaching buildings, exhibiting the highest incidence of cases (29 cases, 93.5%), suggesting a potential link to the source of infection. Remarkably, six unwell teachers’ offices were also located on this floor. The investigation found that the drinking water treatment equipment on this floor was outdated, and the membrane filtration systems had not undergone timely replacement. In light of this, we speculated that the sterilization efficacy of this equipment had deteriorated, contributing to a decline in water quality (resulting in elevated bacterial levels) and triggering the outbreak. After a thorough inquiry, the hygiene conditions of the school's dining area and overall indoor environment were deemed satisfactory. All staff members in the school’s dining facility possessed valid health certificates, and no issues pertaining to unclear sourcing of food ingredients or sensory degradation were identified on-site.
Laboratory investigation
In the present study, a total of 28 B. cereus group strains were isolated from various types of samples collected. Among these, 6 strains originated from stool samples and 22 from water samples. To traceback the source for this outbreak and identify B. cereus contamination in the drinking water within this region, we collected a total of 19 residential water samples, 6 tap water samples from the waterworks, and 6 environmental water samples from the reservoir. The sampling locations are illustrated in . Among these samples, a total of 4 B. cereus group strains were confirmed. 6 strains of fecal origin and 2 strains of water origin were identified as B. paranthracis by WGS, and the other strains species are shown in . The above results suggest that a potential role of waterborne transmission for the outbreak. Among the 22 positive water samples, there were 18 (water samples within school) with colony counts between 1.2 × 105 CFU/ml and 5.0 × 105 CFU/ml, and 4 (water samples outside the school) with colony counts between 1.5 × 102 CFU/ml and 1.0 × 104 CFU/ml. In accordance with the test rules given by Chinese Diagnostic Crieria for Food Poisoning of B. cereus group, the presence of B. cereus group in foods/waters with ≥ 105 cfu/g or ml is capable of inducing food poisoning in humans [27]. Hence, the levels of B. cereus group in drinking water exceeds the food poisoning exposure limits for food poisoning.
Figure 2. Phylogenetic trees, B. cereus group species, source, virulence genes, multilocus sequence typing (MLST), antimicrobial resistance and resistance genes distribution of B. cereus group isolates. Virulence genes positivity was indicated in red, resistance genes positivity was indicated in purple, resistance is indicated in pink, and susceptibility is indicated in grey. AMP, ampicillin; PEN, penicillin; OXA, oxacillin; ERY, erythromycin; CLI, clindamycin; CIP, ciprofloxacin; DAP, daptomycin; SXT, compound sulfamethoxazole; VAN, vancomycin; TET, tetracycline; CHL, chloramphenicol; GEN, gentamicin; CFX, cefoxitin; IMP, imipenem, and E/C, erythromycin/clindamycin.

Drug susceptibility phenotypes and drug-resistant genotype of B. cereus group isolates
Analysis of the genomic data revealed some isolates in this study harbor several antimicrobial resistance genes, including vancomycin resistance related genes (vanR and vanS), penicillin resistance gene blaZ, and fosfomycin resistance gene fosB1. The results from the antimicrobial susceptibility test indicated that all isolates (28 strains) displayed 100% resistant to ampicillin, penicillin, oxacillin, compound sulfamethoxazole, and cefoxitin (). B. cereus is known to generally produce a constitutive β-lactamase, and its resistance to penicillin, ampicillin, and cephalosporins is attributed to the impact of trimethoprim. This mechanism clarifies the discordance observed between the drug susceptibility phenotypes and the drug-resistant genotypes within the isolates under investigation [28]. The phenotypic resistance of the BC013 strain to vancomycin could potentially be linked to the phosphorylation of the VanR protein through the action of the VanS protein, ultimately triggering the transcription of the vanHAX gene cluster [29].
Virulence characteristics of B. paranthracis isolates
Through whole-genome analysis, it was demonstrated that among 28 isolates examined, a total of 34 virulence genes were identified. The eight isolates linked to the outbreak shared an identical virulence gene profile, encompassing 16 specific genes located on chromosomes. These included the non-hemolytic enterotoxin NHE gene cluster (nheA, nheB, and nheC), five virulence genes associated with iron uptake (dhbA, dhbB, dhbC, dhbE, and dhbF), two FtsK/SpoIIIE family protein genes (yukB and yueL), the acyltransferase gene required for hemolysin toxin activation (hlyC), fatty acid biosynthesis enzymes gene (cylI), and WXG100 protein gene (esxL). Notably, pivotal biofilm formation-related genes (clpC, clpE, clpV) were identified in the outbreak-associated isolates. The non-outbreak isolates diverged markedly from those of the outbreak isolates. Virulence genes connected with iron uptake (ilsA) was present in 50% (10/20) of non-outbreak isolates, while immune inhibitor A precursor genes (inhA) was present in 45% (9/20) of nonoutbreak isolates. ClpC, clpE, lplA1, yscN, hal, asbA, asbB, asbC, asbE, nheA, nheB, and nheC were identified in 30% (6/20) of non-outbreak isolates. In addition, four key genes (clpC, clpE, clpV, and cytK) involved in biofilm formation were identified in non-outbreak isolates. In a broader context, it was observed that strains sharing close genetic relationships or belonging to the same sequence type (ST) tended to exhibit similar or identical virulence gene profiles ().
Sequence analyses and phylogenetic of the esxL gene unveiled intriguing findings. Within the BC001 strains, representative of the outbreak, our investigation revealed three distinct esxL genes. Remarkably, these genes diverged into separate evolutionary branches in comparison to other esxL genes, constituting a distinct cluster of their own (A). Moreover, multiple copies of similar esxL genes were found in eight outbreak isolates such as BC001, which generally these strains might diverge a separate subspecies. Sequence comparison between the only esxL sequences (accession No. NC_000962) in the NCBI database from Mycobacterium tuberculosis and those sequences in this study showed that the homology was 77.1%-91.7%. The homology with six screened esxL sequences in this study was between 82.8% and 99.6%, indicating a higher degree of homology between these gene sequences (B). The mice infection assay revealed the presence of B. paranthracis in the organs of all examined mice at three different time points, with the highest bacterial load consistently observed in the kidneys, followed by the spleen and liver. Following 6 hours post-infection, a decline in motor activity was noted among the infected mice, and mortality commenced 3 days post-infection. All infected mice succumbed to the infection within 5 days of exposure, whereas the uninfected control mice survived beyond the 7-day observation period ().
Figure 3. (A) The phylogenetic analysis of B. paranthracis esxL gene sequencing. (B) The percentage of B. paranthracis esxL nucleotide sequence identity.
Figure 4. (A) Mice was infected with B. paranthracis BC002 (upper panel: death) and not infected with B. paranthracis (lower panel: control). (B, C, D) Dissemination of B. paranthracis in mice tissues. Measure the recovery rate of bacteria in tissues at different time points. The results represent the average of five parallel samples in each group.

Genome analysis and phylogenetic comparison of B. cereus group isolates
The complete genome of outbreak isolates BC002 consists of a circular chromosome and two circular plasmids (named Plas1 and Plas2) of 307.87 kp and 73.37 kp, respectively. All 16 detected virulence genes are situated on the chromosome. Plas1 and Plas2 each harbor 1 prophage, with Plas1 additionally containing 1 CRISPR and 1 ISfinder (Figure S1). Utilizing core genome MLST, the isolates featured in this study were effectively categorized into distinct clusters: two prominent clusters (designated as: Cluster A and Cluster B), as well as two smaller clusters characterized by a genetic divergence spanning 0 to 1 allele. This analysis also identified 10 individual, distinct isolates (). Within this grouping, eight ST205 B. paranthracis isolates (six from fecal samples and the other two from domestic water) collectively constituted Cluster A. These strains exhibited an incredibly tight genetic relationship, differing by a mere 0 to 2 alleles. This remarkable similarity strongly suggests a shared origin, potentially stemming from a singular clone. A more intricate resolution of the strains within the same ST type was accomplished through SNP analysis. By scrutinizing variations in base pair positions within the genomic DNA, SNP-based analysis aligned coherently with the cgMLST approach in elucidating the interrelationships among strains. Notably, the eight ST205 isolates were closely clustered together by SNP analysis with their core genome differing only by a few SNPs (ranging from 0 to 10 SNPs), further indicating that they were closely associated with the outbreaks. Six ST378 Bacillus sp. isolates, three ST1149 B. albus isolates, two ST476 B. luti isolates, and two unknown type B. cereus isolates in this study, with both phylogenetic analyses yielding separate clusters. In addition, we employed SNP analysis to compare genome sequences of these isolates with selected B. cereus group isolates from various sources such as food, environment, water, soil, air, animal and human sources, available in public databases. As expected, each species within the B. cereus group clade clustered together. ST205 outbreak-related isolate BC001 was closely related to the Dairy equipment filters -derived sample isolate SAMN12529906 from Australia in 2017, displaying a divergence of 1583 SNPs. In contrast, the remaining 176 isolates were the more distantly related to the isolates examined within this study ().
Figure 5. Minimum-spanning tree of cgMLST of 28 B. cereus group isolates. Allele distances between isolates are indicated, cluster with allele difference ≤ 1 is shaded in grey.
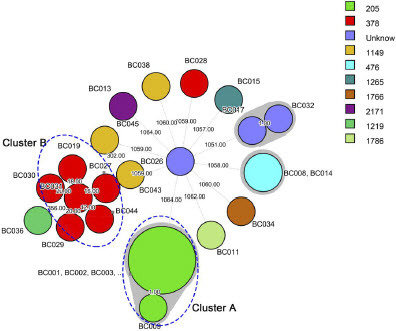
Figure 6. Phylogenetic tree of 28 B. cereus group isolates this study and another 758 B. cereus group isolates in NCBI database.
Phylogenetic grouping of B. cereus group isolates based on BRIG
The BLAST Ring Image Generator (BRIG) tool was used to compare the whole genomes of the reference strain GCF-025917685.1 with isolates representing phylogenetic groups A (8 outbreak isolates), B (6 Bacillus sp. isolates), C (3 B. albus isolates), and D (2 B. luti isolates), all carefully selected from the isolates within this study. The results revealed a remarkable consistency in nucleic acid sequences across the four groups, exceeding a 99% similarity level. Consequently, the genomic ring diagrams from each group displayed subtle variations, indicating a high degree of genetic resemblance. BRIG-BLAST method analysis showed the differences between these groups mainly in the mobile genetic elements of genomic islands and phage (Figure S2).
Discussion
In the present study, we present findings concerning a B. paranthracis outbreak that occurred via waterborne transmission in Binzhou City, China, during September 2020. The epidemiological investigation a potential link between drinking water contamination with B. paranthracis and malfunction of water treatment devices. Strikingly, both the genomic ring diagrams and the highly close phylogenetic relationships of B. paranthracis strains extracted from the drinking water and infected patients displayed remarkable similarity. The genetic differences within outbreak genomes ranged from 0 to 10 pairwise SNP differences, and the distinctions based on cgMLST were limited to 0 to 2 alleles. Through a synergistic approach, we melded the insights gleaned from the epidemiological inquiry with high-resolution comparative genomics to decisively establish that the source of the outbreak was the B. paranthracis present in the drinking water. To prevent further spread of the outbreak, the school implemented a series of rigorous measures, including temporary school closure, the overhaul and replacement of drinking water treatment devices, and extensive environmental cleaning and disinfection of water pipelines. Furthermore, no new cases were reported within a year’s time after the outbreak.
B. cereus exhibits the capability to produce heat-labile diarrheal enterotoxin, while certain strains can additionally produce heat-stable emetic enterotoxin, while B. paranthracis encompasses many environmental and food isolates that are incapable of causing emetic illness. The diarrhea syndrome caused by B. cereus group can be attributed to several enterotoxins and hemolysins, such as nonhemolytic enterotoxin (Nhe), hemolysin BL (Hbl), and cytotoxin K (CytK) [31]. The combination of the Nhe protein complex with cells may also lead to the accumulation of Ca2+ and reactive oxygen species (ROS) in the cytoplasm. This, in turn, activates the Fas pathway, blocks the cell cycle in G0/G1 phase, and induces apoptosis [32]. In this study, we identified the presence of the NHE gene cluster (nheA, nheB, and nheC) in the outbreak strain, suggesting that this gene cluster is likely the primary cause of diarrhea in patients. Interestingly, two non-outbreak isolates, one from school drinking water and one from residential drinking water, carry the virulence gene of cytK, which codes for cytotoxin K. Furthermore, one non-outbreak isolate from residential drinking water harbored the emetic toxin-encoding gene cesC. CytK belongs to the family of β-barrel pore-forming toxins is considered to be a potentially fatal factor in B. cereus food poisoning. The ces gene encodes the emetic toxin cereulide, an emetic toxin known to cause emetic food poisoning in humans, and in severe cases, can lead to fulminant liver failure [33]. Consuming a significant amount of cereulide can have deadly consequences, particularly for children with sensitive constitution [34]. The presence of highly pathogenic virulence genes in residential drinking water implies the necessity of implementing transmission-based preventive measures in residential areas to mitigate the risk of potential B. paranthracis infections. The esxL gene encodes one of several WXG100 proteins (the prototypical substrates of bacterial type VII secretion systems) [35]. ESX is a specialized secretion system used by bacteria to deliver virulence factor and other important molecules to target cells, which is considered to be important for virulence and survival of Bacteria [36].
B. cereus group can form biofilms due to its ability to adhere to surfaces, produce extracellular polymeric substances (EPS), employ quorum sensing, and form spores [15]. Biofilm formation allows B. cereus to survive in hostile environments, resist antibiotics, and persist within various settings [37]. Currently, many genes related to the biofilm formation have been identified, and these genes are involved in various cellular processes, such as motility, adhesion, metabolism and stress response [38]. Four biofilm formation-related virulence genes (clpC, clpE, clpV, and cytK) were identified from the B. paranthracis isolates in this study. ClpC and ClpE promote the expression of genes that promote biofilm formation by degrading the transcription factor SinR. ClpV is thought to be involved in the degradation of extracellular DNA, which is an important component of the biofilm matrix [39, 40]. CytK has been shown to facilitate cell detachment from the biofilm, thereby allowing for the dispersal of the biofilm and the colonization of new surfaces [41]. Bacterial biofilm formation is a critical factor in the establishment of bacteria in water pipes and the occurrence of waterborne outbreaks. In water distribution systems, biofilms can form on the inner surfaces of pipes, providing a protective environment for bacteria to grow and multiply. When biofilms in water pipes contain pathogenic bacteria, they can break off and enter the water supply, leading to waterborne disease outbreaks [42-44].
The outbreak of infectious diseases caused by pathogenic bacteria through drinking water as a medium is a serious public health problem. It is worth noting that waterborne outbreaks caused by B. cereus group are relatively rare compared to other types of waterborne illness. This study identified the presence of B. paranthracis in drinking water from the water supply in the local area, suggesting a potential link with its biofilm formation. Nevertheless, B. paranthracis exhibits a high resistance to disinfectants and can swiftly regrow quickly, making it a formidable challenge to prevent the formation of biofilms in water distribution systems. As a result, it becomes imperative to routinely employ effective biofilm-targeting disinfectants such as chlorine dioxide or ozone for cleaning and maintaining the water supply system, thereby contributing to biofilm control.
Currently, as the two most common way to compare genomes, cgMLST and SNP analyses have been widely used to describe the relatedness of outbreak strains [17, 18]. Currently, the MLST typing of B. cereus group was based on seven housekeeping genes (glp, gmk, ilv, pta, pur, pyc, and tpi) commonly used for B. cereus[45]. As depicted in , the 28 B. cereus group strains in this study were divided into 9 STs while the ST types of the remaining three strains remained unidentified. Specifically, all 8 outbreak strains belong to the ST205 category, which is consistent with the SNPs analysis results. Conversely, other B. cereus group strains encompass ST1149, ST378, ST1219, ST1766, ST1786, ST1265, ST476, and ST2171. Phylogenetic analysis encompassing cgMLST and SNPs, indicated that strains belonging to the same ST within this study form coherent clusters. The identification of specific STs can be useful in tracking outbreaks and understanding the genetic diversity of the bacteria. In addition to the outbreak strain ST205 in this study, the other currently described isolates of B. paranthracis related to human pathogenicity have STs of 26 and 1076[2, 45]. ST1149, ST378, ST1219, ST1766, ST1786, ST1265, ST476, and ST2171 were isolated from drinking water and environmental water, suggesting that strains carrying these STs may exhibit a remarkable resilience to external conditions and possess a high degree of adaptability to diverse environments. The coexistence of multiple STs in this study underscores the considerable genetic diversity among these strains, which may contribute to their ability to adapt to particular environments and their resistance to disinfectants. Consequently, it holds significant importance to monitor the presence of these strains in water sources and take adequate preventive measures to prevent their multiplication and transmission.
In this study, our research unequivocally establishes that contaminated drinking water containing B. paranthracis was the source of this outbreak through comprehensive genomic analyses. We provide valuable insight into the phylogenetic relations among diverse isolates by employing various phylogenetic methodologies and thoroughly assessing the pathogenic potential of the B. cereus group isolates. Moreover, WGS method employed for B. cereus group strain characterization should be interpreted with caution; a core-genome based SNPs analysis is warranted. Lastly, this study underscores the imperative for the ongoing monitoring of both the drinking water system and its source to proactively avert waterborne disease outbreaks triggered by B. paranthracis.
temi_a_2348498_sm6320.zip
Download Zip (11.4 MB)temi_a_2348498_sm6324.docx
Download MS Word (15.7 KB)temi_a_2348498_sm6311.docx
Download MS Word (107.9 KB)Acknowledgements
We are grateful to all participants of this programme.
Disclosure statement
No potential conflict of interest was reported by the author (s).
Additional information
Funding
Reference
- Fox D, Mathur A, Xue Y, Liu Y, Tan WH, Feng S, Pandey A, Ngo C, Hayward JA, Atmosukarto, II et al: Bacillus cereus non-haemolytic enterotoxin activates the NLRP3 inflammasome. Nat Commun 2020, 11(1):760.
- Carroll LM, Matle I, Kovac J, Cheng RA, Wiedmann M: Laboratory Misidentifications Resulting from Taxonomic Changes to Bacillus cereus Group Species, 2018-2022. Emerg Infect Dis 2022, 28(9):1877-1881.
- Carroll LM, Cheng RA, Wiedmann M, Kovac J: Keeping up with the Bacillus cereus group: taxonomy through the genomics era and beyond. Crit Rev Food Sci Nutr 2022, 62(28):7677-7702.
- Carroll LM, Pierneef R, Mathole A, Atanda A, Matle I: Genomic Sequencing of Bacillus cereus Sensu Lato Strains Isolated from Meat and Poultry Products in South Africa Enables Inter- and Intranational Surveillance and Source Tracking. Microbiol Spectr 2022, 10(3):e0070022.
- Buss da Silva N, Baranyi J, Carciofi BAM, Ellouze M: From Culture-Medium-Based Models to Applications to Food: Predicting the Growth of B. cereus in Reconstituted Infant Formulae. Front Microbiol 2017, 8:1799.
- Ehling-Schulz M, Lereclus D, Koehler TM: The Bacillus cereus Group: Bacillus Species with Pathogenic Potential. Microbiol Spectr 2019, 7(3).
- Glasset B, Herbin S, Granier SA, Cavalie L, Lafeuille E, Guerin C, Ruimy R, Casagrande-Magne F, Levast M, Chautemps N et al: Bacillus cereus, a serious cause of nosocomial infections: Epidemiologic and genetic survey. PLoS One 2018, 13(5):e0194346.
- Ramarao N, Lereclus D: The InhA1 metalloprotease allows spores of the B. cereus group to escape macrophages. Cell Microbiol 2005, 7(9):1357-1364.
- Wong CH, Jenne CN, Petri B, Chrobok NL, Kubes P: Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol 2013, 14(8):785-792.
- Kovac J, Miller RA, Carroll LM, Kent DJ, Jian J, Beno SM, Wiedmann M: Production of hemolysin BL by Bacillus cereus group isolates of dairy origin is associated with whole-genome phylogenetic clade. BMC Genomics 2016, 17:581.
- Gao T, Ding Y, Wu Q, Wang J, Zhang J, Yu S, Yu P, Liu C, Kong L, Feng Z et al: Prevalence, Virulence Genes, Antimicrobial Susceptibility, and Genetic Diversity of Bacillus cereus Isolated From Pasteurized Milk in China. Front Microbiol 2018, 9:533.
- Assoni L, Milani B, Carvalho MR, Nepomuceno LN, Waz NT, Guerra MES, Converso TR, Darrieux M: Resistance Mechanisms to Antimicrobial Peptides in Gram-Positive Bacteria. Front Microbiol 2020, 11:593215.
- Liu C, Yu P, Yu S, Wang J, Guo H, Zhang Y, Zhang J, Liao X, Li C, Wu S et al: Assessment and molecular characterization of Bacillus cereus isolated from edible fungi in China. BMC Microbiol 2020, 20(1):310.
- Shemesh M, Chai Y: A combination of glycerol and manganese promotes biofilm formation in Bacillus subtilis via histidine kinase KinD signaling. J Bacteriol 2013, 195(12):2747-2754.
- Alonso VPP, Harada AMM, Kabuki DY: Competitive and/or Cooperative Interactions of Listeria monocytogenes With Bacillus cereus in Dual-Species Biofilm Formation. Front Microbiol 2020, 11:177.
- Watts SC, Holt KE: hicap: In Silico Serotyping of the Haemophilus influenzae Capsule Locus. J Clin Microbiol 2019, 57(6).
- Lagos AC, Sundqvist M, Dyrkell F, Stegger M, Soderquist B, Molling P: Evaluation of within-host evolution of methicillin-resistant Staphylococcus aureus (MRSA) by comparing cgMLST and SNP analysis approaches. Sci Rep 2022, 12(1):10541.
- Gona F, Comandatore F, Battaglia S, Piazza A, Trovato A, Lorenzin G, Cichero P, Biancardi A, Nizzero P, Moro M, Cirillo DM: Comparison of core-genome MLST, coreSNP and PFGE methods for Klebsiella pneumoniae cluster analysis. Microb Genom 2020, 6(4).
- Yu H, Elbediwi M, Zhou X, Shuai H, Lou X, Wang H, Li Y, Yue M: Epidemiological and Genomic Characterization of Campylobacter jejuni Isolates from a Foodborne Outbreak at Hangzhou, China. Int J Mol Sci 2020, 21(8).
- Zou H, Berglund B, Xu H, Chi X, Zhao Q, Zhou Z, Xia H, Li X, Zheng B: Genetic characterization and virulence of a carbapenem-resistant Raoultella ornithinolytica isolated from well water carrying a novel megaplasmid containing bla(NDM-1). Environ Pollut 2020, 260:114041.
- Zhu Z, Shan L, Hu F, Li Z, Zhong D, Yuan Y, Zhang J: Biofilm formation potential and chlorine resistance of typical bacteria isolated from drinking water distribution systems. RSC Adv 2020, 10(52):31295-31304.
- Stepanovic S, Vukovic D, Dakic I, Savic B, Svabic-Vlahovic M: A modified microtiter-plate test for quantification of staphylococcal biofilm formation. J Microbiol Methods 2000, 40(2):175-179.
- Wang Y, Zhang J, Yuan Z, Sun L: Characterization of the pathogenicity of a Bacillus cereus isolate from the Mariana Trench. Virulence 2022, 13(1):1062-1075.
- Xu X, Chen Y, Pan H, Pang Z, Li F, Peng X, Ed-Dra A, Li Y, Yue M: Genomic characterization of Salmonella Uzaramo for human invasive infection. Microb Genom 2020, 6(7).
- Leong KWC, Kalukottege R, Cooley LA, Anderson TL, Wells A, Langford E, O'Toole RF: State-Wide Genomic and Epidemiological Analyses of Vancomycin-Resistant Enterococcus faecium in Tasmania's Public Hospitals. Front Microbiol 2019, 10:2940.
- Yu S, Yu P, Wang J, Li C, Guo H, Liu C, Kong L, Yu L, Wu S, Lei T et al: A Study on Prevalence and Characterization of Bacillus cereus in Ready-to-Eat Foods in China. Front Microbiol 2019, 10:3043.
- Gdoura-Ben Amor M, Siala M, Zayani M, Grosset N, Smaoui S, Messadi-Akrout F, Baron F, Jan S, Gautier M, Gdoura R: Isolation, Identification, Prevalence, and Genetic Diversity of Bacillus cereus Group Bacteria From Different Foodstuffs in Tunisia. Front Microbiol 2018, 9:447.
- Pitt TL, McClure J, Parker MD, Amezquita A, McClure PJ: Bacillus cereus in personal care products: risk to consumers. Int J Cosmet Sci 2015, 37(2):165-174.
- Szakacs TA, Kalan L, McConnell MJ, Eshaghi A, Shahinas D, McGeer A, Wright GD, Low DE, Patel SN: Outbreak of vancomycin-susceptible Enterococcus faecium containing the wild-type vanA gene. J Clin Microbiol 2014, 52(5):1682-1686.
- Egan K, Field D, Rea MC, Ross RP, Hill C, Cotter PD: Bacteriocins: Novel Solutions to Age Old Spore-Related Problems? Front Microbiol 2016, 7:461.
- Kilcullen K, Teunis A, Popova TG, Popov SG: Cytotoxic Potential of Bacillus cereus Strains ATCC 11778 and 14579 Against Human Lung Epithelial Cells Under Microaerobic Growth Conditions. Front Microbiol 2016, 7:69.
- Liu X, Ding S, Shi P, Dietrich R, Martlbauer E, Zhu K: Non-hemolytic enterotoxin of Bacillus cereus induces apoptosis in Vero cells. Cell Microbiol 2017, 19(4).
- Guinebretiere MH, Velge P, Couvert O, Carlin F, Debuyser ML, Nguyen-The C: Ability of Bacillus cereus group strains to cause food poisoning varies according to phylogenetic affiliation (groups I to VII) rather than species affiliation. J Clin Microbiol 2010, 48(9):3388-3391.
- Jovanovic J, Tretiak S, Begyn K, Rajkovic A: Detection of Enterotoxigenic Psychrotrophic Presumptive Bacillus cereus and Cereulide Producers in Food Products and Ingredients. Toxins (Basel) 2022, 14(4).
- Garufi G, Butler E, Missiakas D: ESAT-6-like protein secretion in Bacillus anthracis. J Bacteriol 2008, 190(21):7004-7011.
- Bachmann NL, Salamzade R, Manson AL, Whittington R, Sintchenko V, Earl AM, Marais BJ: Key Transitions in the Evolution of Rapid and Slow Growing Mycobacteria Identified by Comparative Genomics. Front Microbiol 2019, 10:3019.
- Gao T, Foulston L, Chai Y, Wang Q, Losick R: Alternative modes of biofilm formation by plant-associated Bacillus cereus. Microbiologyopen 2015, 4(3):452-464.
- Su T, Qiu Y, Hua X, Ye B, Luo H, Liu D, Qu P, Qiu Z: Novel Opportunity to Reverse Antibiotic Resistance: To Explore Traditional Chinese Medicine With Potential Activity Against Antibiotics-Resistance Bacteria. Front Microbiol 2020, 11:610070.
- Soria-Bustos J, Ares MA, Gomez-Aldapa CA, Gonzalez YMJA, Giron JA, De la Cruz MA: Two Type VI Secretion Systems of Enterobacter cloacae Are Required for Bacterial Competition, Cell Adherence, and Intestinal Colonization. Front Microbiol 2020, 11:560488.
- Kajfasz JK, Martinez AR, Rivera-Ramos I, Abranches J, Koo H, Quivey RG, Jr., Lemos JA: Role of Clp proteins in expression of virulence properties of Streptococcus mutans. J Bacteriol 2009, 191(7):2060-2068.
- Koo HJ, Ahn S, Chung HY, Kim S, Kim K, Ryu S, Lee JH, Choi SH, Kim H: Comparative genomic analysis reveals genetic features related to the virulence of Bacillus cereus FORC_013. Gut Pathog 2017, 9:29.
- Yan F, Yu Y, Gozzi K, Chen Y, Guo JH, Chai Y: Genome-Wide Investigation of Biofilm Formation in Bacillus cereus. Appl Environ Microbiol 2017, 83(13).
- Okshevsky M, Louw MG, Lamela EO, Nilsson M, Tolker-Nielsen T, Meyer RL: A transposon mutant library of Bacillus cereus ATCC 10987 reveals novel genes required for biofilm formation and implicates motility as an important factor for pellicle-biofilm formation. Microbiologyopen 2018, 7(2):e00552.
- Ahmad JI, Dignum M, Liu G, Medema G, van der Hoek JP: Changes in biofilm composition and microbial water quality in drinking water distribution systems by temperature increase induced through thermal energy recovery. Environ Res 2021, 194:110648.
- Matson MJ, Anzick SL, Feldmann F, Martens CA, Drake SK, Feldmann H, Massaquoi M, Chertow DS, Munster VJ: Bacillus paranthracis Isolate from Blood of Fatal Ebola Virus Disease Case. Pathogens 2020, 9(6).
- Zhang Y, Chen J, Feng C, Zhan L, Zhang J, Li Y, Yang Y, Chen H, Zhang Z, Zhang Y et al: Quantitative Prevalence, Phenotypic and Genotypic Characteristics of Bacillus cereus Isolated from Retail Infant Foods in China. Foodborne Pathog Dis 2017, 14(10):564-572.