
ABSTRACT
We examined nongenomic signaling and functional effects (cell proliferation; prolactin release) of R-equol (R-eq), a synthetically produced metabolite of the phytoestrogen daidzein that preferentially binds to estrogen receptor-α, across a broad concentration range (10–16 to 10–7M). Membrane estrogen receptor-α, via which nongenomic signaling occurs, is enriched in the GH3/B6/F10 pituitary tumor cells used in these studies. We previously reported that other phytoestrogens, including daidzein, are potent inducers of nongenomic signaling acting via membrane receptors for estrogens, resulting in changes to these same functional responses. In the present studies, R-eq activated typical rapid signaling pathways normally evoked by estrogens, but with some differences in response timings and functional outcomes. Levels of R-eq (at 15 nM, a blood level typical for dietary phytoestrogens) were compared to a female physiological level of estradiol (E2; 1 nM). Both estrogens activated multiple mitogen-activated protein kinases (ERK, JNK and p38) by phosphorylation within 2.5–15 min, with subsequent oscillations, as observed previously for other estrogens. Like E2, R-eq also augmented intracellular calcium levels and caused prolactin release; in contrast to E2, it did not produce a dose-dependent increase in cell proliferation, as estrogens that activate ERK often do. R-eq and E2, both alone and in combination, activated Gαi by GTP-charging. However, R-eq suppressed 1 nM E2-activated ERK, JNK and p38, as well as cell proliferation (most pronounced at typical phytoestrogen blood levels of 10–10 to 10–7M). We conclude that R-eq, like E2, is a rapid activator of nongenomic signals, but when combined with E2, can interfere with E2-induced nongenomic estrogenic effects .
Introduction
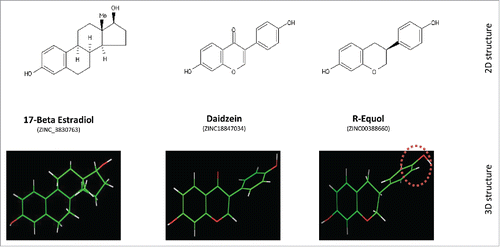
Soy-based foods are a major source of human exposure to phytoestrogensCitation1,2 that structurally resemble endogenous estrogens like estradiol (E2), as well as a variety of synthetic estrogens. Daidzein is a plentiful and common phytoestrogen available from soy-based foodsCitation3,4 that can be converted into 2 chiral forms of equol; gut micro-organisms produce S(-) equol (S-eq).Citation5-7 The core ring structures of the phytoestrogens daidzein and R(+)-equol (R-eq; the chiral form used in this study) closely resemble those of E2 (see ) and other estrogens. The binding affinity of S-eq to estrogen receptor-β (ERβ) is 13 times higher than that of R-equol (Ki 50nM), whereas R-eq showed a preference for binding to estrogen receptor-α (ERα) 4 times higher than that of S-eq; both have higher affinity for ERs than daidzein.Citation8 Therefore, it is not surprising that phytoestrogens can act as selective estrogen receptor (ER) modulators (SERMs), acting differentially as agonists or antagonists for ERs depending upon the cell type involved.Citation9-12 Modifications in both the compounds’ skeletal structure and the end hydroxyls (red-white moieties in ) can affect the affinity for ERα, as well as subsequent interactions of the liganded receptor. For example, daidzein sulfation at the 4’ position suppresses estrogen agonist activity, while at the 7’ position it increases estrogen agonist activity.Citation13 Differences in patterns of estrogenic signaling, and disturbances of E2-induced signaling when challenged with a phytoestrogen, can cause alterations of important estrogen-driven physiological actions.Citation14-16 Estrogenic activity via the nuclear forms of ERs α and β reflect their different binding affinities,Citation9 but differential binding and activities at membrane forms of ERs (mERs) that drive nongenomic signaling actions are largely unknown.
Figure 1. Structure of E2, daizein and R-eq. Upper Row: Two-dimensional (2D) structure comparisons of 17β estradiol, daidzein, and R-eq. Note hydroxyl groups at opposite ends of the molecules that are thought to be essential for binding into the ligand-binding pocket on ERα.Citation8 Lower Row: Three-dimensional (3D) structures showing differences in the spatial arrangements of these hydroxyl groups (red-white) in the same compounds. The asymmetric center of R-eq is shown at the rightmost position (red dotted circle)Citation9,10.
Phytoestrogens such as coumestrol, daidzein, genistein, and trans-resveratrol have been suggested as possible safe post-menopausal hormonal replacement alternatives,Citation17 though they may be riskier for breast cancer patients due to potential promotion of the growth of already established mammary tumors.Citation2,18,19 Asians eating diets high in soy are less susceptible to the disease consequences of estrogen overexposure (affecting initial induction of cancers of reproductive tissues)Citation2,20-22 or underexposure (causing osteoporosis, hot flashes, heart disease, and depression).Citation2,22-24 Interestingly, the incidences of these diseases in the Asian population become similar to Westerners when they adopt a Western diet.Citation2,20-23,25-30 So there are likely some physiology- and disease-modifying activities of such dietary estrogens.
Estrogens, including phytoestrogens, can activate both the slower (hours to days) classical genomic pathway of transcriptional actions, and the non-classical rapid (seconds to minutes) nongenomic signaling mechanismsCitation31 using a wide variety of signaling processes and protein mediators.Citation32,33 Our laboratory has previously investigated the lesser-known non-genomic signaling activities of phytoestrogens such as daidzein, coumestrol, genistein, and trans-resveratrol,Citation15,16,34 as well as a number of other estrogen mimetics.Citation31,33,35,36 We found phytoestrogens to be active at or below concentrations generally provided by dietary exposures. However, the daidzein equol metabolites have not previously been studied for actions through this non-classical pathway, and have interesting potential for dissimilar actions because of differential metabolic availability.
Daidzein serves as the microbial substrate for equols (mainly S-eq) when commonly introduced from various food sources such as soy beans, tofu, food additives, and milk substitutes. The range of equol concentrations in the plasma of humans varies widely depending upon dietary habits, geographic location, plant active constituents, gut microbiomes, and the use of commercially available supplement formulations.Citation3-8,37 However, human genetic variations prevalent in some subpopulations or individuals can result in dissimilar pharmacokinetics that affect attainable blood levels of S-eq. Only ∼35% of humans are high S-eq producers from daidzein metabolism; therefore, body fluid concentrations can vary significantly even with equivalent consumption.Citation3,37 Because of this, and because the chiral forms are differentially active and differentially produced by gut microflora, equols might be more easily controlled by direct dietary supplementation (instead of providing the precursor daidzein). It is also important to study the effects of equols over broad concentration ranges to fully consider their possible therapeutic uses or dangers, because equols can be present in humans at such widely varying doses due to both ingestion and metabolism. In addition, compounds that mimic hormones, like the hormones themselves, often display non-monotonic concentration dependence,Citation15,33,35,38-41 making examination of detailed concentration curves necessary to fully understand their health consequences.
Because R-eq preferentially binds to ERα, and S-eq to ERβ,Citation8 we evaluated R-eq for non-genomic effects in these studies using GH3/B6/F10 pituitary tumor cells that express relatively high levels of mERα.Citation42,43 We examined a variety of cellular signaling responses known to be elicited by estrogens, and 2 functional responses induced by estrogens in the pituitary.
Results
Time dependence of 1 nM E2 vs. 15 nM R-eq MAPK activations
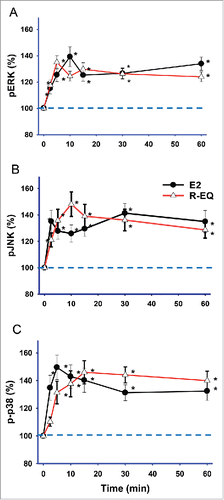
E2 at a female physiological concentration (1 nM) caused rapid activation of ERKs (extracellular signal regulated kinases), initiating a response by 2.5 min that peaked at 10 min, then declined slightly, and rose again at 60 min (), similar to what we observed previously.Citation44-46 R-eq at a concentration that approximates physiological S-eq concentrations resulting from dietary exposure to daidzein (15 nM) also significantly increased pERK by 2.5 min, peaking at 5 min, and sustaining this activation through 60 min with minor fluctuations. E2 significantly activated JNKs (jun N-terminal kinases) () at all time points (2.5 min – 60 min), with peaks at 2.5 min and 30 min. Treatment with R-eq caused a slightly delayed JNK activation, peaking at 10 min and sustained through 60 min. Finally, E2 significantly activated p38 kinase () by 2.5 min, peaking at 5 min, followed by a slight decline. Similarly, though not identically, R-eq activated p38 kinase by 5 min, peaking at 15 min, and this activity was sustained up to 60 min.
Figure 2. Time-dependent effects of E2 and R-eq on MAPK activations. Cells were exposed to physiologic levels of E2 (1nM), and dietary levels of R-eq (15nM) for 2.5 - 60 min. (A) Effects of E2 and R-eq on ERK phosphorylation (pERK). For E2 n = 56 spread over 6 experiments; for R-eq n=56 over 4 experiments. (B) Effects of E2 and R-eq on JNK phospho-activation (pJNK). For E2 or R-eq, n = 38 over 3 experiments. (C) Effects of E2 and R-eq on p38 phospho-activation (p-p38). For E2 n = 38 over 4 experiments; for R-eq n = 46 over 4 experiments. * = significant at p< 0.05 vs. control (vehicle) group.
Concentration dependence of E2 vs. R-eq treatment on MAPK activations at 5 min
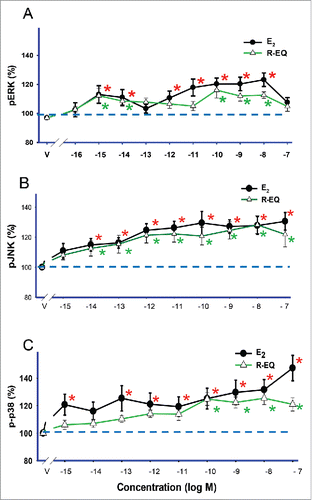
Cells were exposed to log incremental concentrations (10−16 to 10−7 M) of E2 or R-eq for 5 min (). E2 activated ERK () with a pattern similar to that we observed previously, showing a non-monotonic response with 2 different dose optima (10−15 and 10−8 M) and a midpoint non-effective dose (10−13 M). E2 did not produce significant activation at the highest dose (10−7 M). R-eq also activated ERK with 2 dose optima (10−15 and 10−10 M), with ineffective doses in between and at the highest concentration. E2 and R-eq significantly increased pJNK across the same concentration ranges, with a simpler concentration-dependent pattern at all doses of 10−14M and above (). E2 treatment caused the activation of p38 at all concentrations tested (10−15-10−7 M) except 10−14 M. R-eq also increased p-p38 (phospho-p38), but only at concentrations above 10−10 M (). So both a typical female physiological concentration (1nM) of E2 and a common dietary concentration for naturally occurring phytoestrogens (15nM) of R-eq activated all 3 kinases (ERK, JNK, and p38), with ERK activation being non-monotonic for both estrogens. We recognize that preparing concentrations at such a low levels can be more error-prone. In the past some experiments have shown significant results at this lowest concentration, and some did not. We have inspected our individual experiments to see if non-monotonic patterns are usually revealed, and we see them often, though not in every case. We have also seen non-monotonic dose-dependence patterns in many of our past published studies.Citation15,33,35,44-62
Figure 3. Dose-dependent effects of E2 and R-eq on MAPK activations. Cells were exposed for 5 min to various concentrations of E2 and R-eq. A), B) and C) line graphs demonstrating the effects of E2 and R-eq on pERK, pJNK and p-p38, respectively. * = p< 0.05 vs. control (vehicle) group.
Dose-dependent effects of adding R-eq to E2-induced phosphorylation of MAPKs
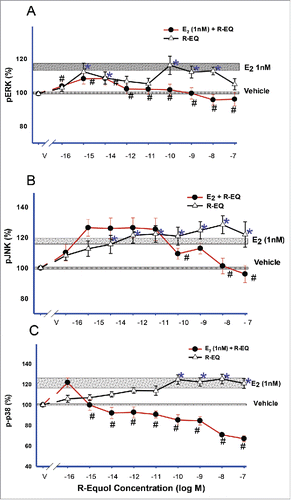
Next we wanted to determine if adding R-eq on top of already existing physiological E2 (1 nM) would alter the response to E2. Treating cells for 5 min with either 1nM E2 (see horizontal bars, ) or R-eq (10−16 to 10−7 M) alone caused phosphorylation of all MAPKs (ERK, JNK and p38). R-eq elicited a non-monotonic concentration pattern. However the addition of varying concentrations of R-eq to 1 nM E2 suppressed the E2-induced increase in ERK (), JNK (), and p38 phosphorylation (), at higher concentrations. The activated state of p38 was the most sensitive to these effects, showing a suppression at as little as 10−15 M R-eq.
Figure 4. Dose-dependent effects of added R-eq on E2-induced MAPK activities. Cells were exposed for 5 min to a mixture of E2 (1nM, physiological concentration) and R-eq (range 10−16 to 10−7M). pERK (A), pJNK (B) and p-p38 (C) were measured compared to the activity level of the vehicle-treated controls (*= p< 0.05 vs. control) and the 1nM E2-induced (see horizontal stippled bars) pERK, pJNK and p-p38 levels (# = p < 0.05 vs E2).
Effects of E2 and R-eq on Gαi activation
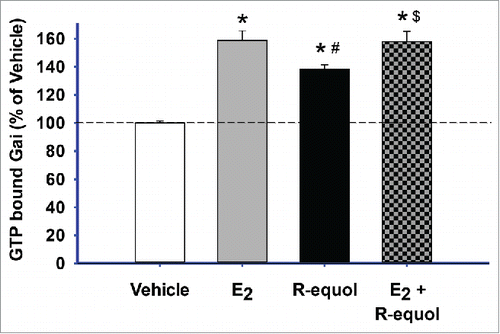
We then tested R-eq for another recently demonstrated rapid estrogenic signaling effect, activation of Gαi protein by GTP chargingCitation56 (). With the addition of GTPγS to block dissociation (by irreversible binding of this analog), we measured the amount of GTP-bound G protein that accumulated over 5 min. Both E2 (1nM) and R-eq (15nm) significantly increased GTP-charged Gαi compared to vehicle controls, though R-eq did so less. The mixture of the 2 estrogens activated Gαi to the same extent as had E2 alone.
Figure 5. E2 and R-eq, alone or in combination – effects on GTP-bound Gαi. Cells were exposed to E2 (1nM) or R-eq (15nM) or both for 5 min, using the irreversible binding of the GTPγS analog to measure cumulative binding. GTPγS-bound Gαi levels were detected using an Ab specific for the complex. Vehicle n = 36; E2 n = 20, and R-Eq n = 36, over 2 experiments. * = significance at p < 0.05 vs. vehicle; # = significance vs. E2; $ = significance vs. R-eq.
Effects of E2 and R-eq on intracellular free calcium levels and PRL release
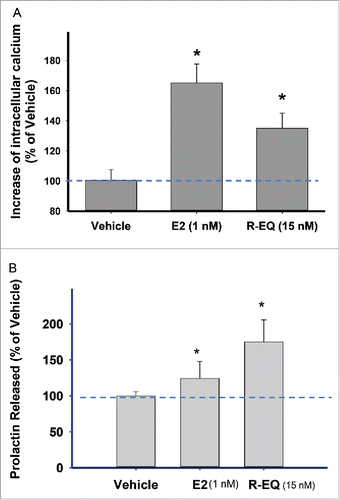
We next examined another pathway of rapid estrogenic signaling responses, changes in intracellular calcium levels (), to see if they would be affected by R-eq in a dietary phytoestrogen concentration range in comparison to a physiologic level of E2 (1nM). Exposure to either compound for 1 min significantly increased the intracellular calcium levels compared to controls, though E2 was more effective (). We then tested a functional consequence of elevated calcium levels in these cells, PRL release (). A 1 min exposure to 1nM E2 significantly increased PRL secretion into the medium by ∼15%, as we have seen previously.Citation15,53 However, R-eq applied at a 15nM typical phytoestrogen dietary concentration increased secreted PRL by ∼70%.
Figure 6. Effects of E2, R-Eq, and their combination on intracellular calcium concentrations and PRL release. Cells were exposed to E2 (1nM) or R-eq (15nM) or both for 1 min. (A) Effects of E2 and R-eq on intracellular calcium levels compared to vehicle control. Vehicle n = 142; E2 n = 72; R-eq n = 25, over 4 experiments. (B) Effects of E2 and R-eq on PRL release. The concentration of PRL released into the medium was measured by radioimmunoassay. Vehicle n = 36; E2 n = 30 and R-eq n = 30, over 3 experiments. *=significance vs. vehicle, #=significance vs. E2 at p<0.05.
Dose dependence of cell proliferation on E2, R-eq and the mixture
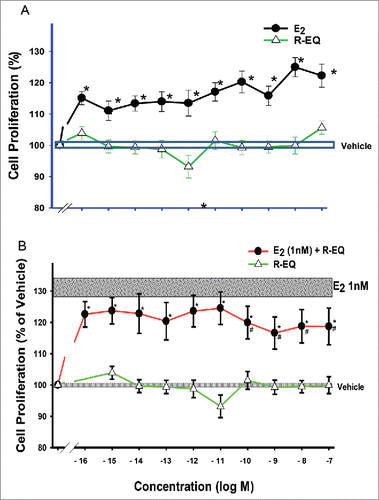
Cells treated for 3 d with E2 (10−16 to 10−7 M) showed increases in cell number, as we observed previously.Citation34,46,54,63 However, R-eq treatment did not change cell numbers over the entire concentration range tested. Addition of this entire range of R-eq concentrations to a 1nM physiological E2 level (shaded horizontal bar) suppressed the E2-induced increase in cell number ().
Figure 7. Dose-dependent effects of E2, R-eq, and their combination on cell proliferation. Cells were exposed to various doses of E2, R-eq and a mixture of E2 and R-eq for 3 d. (A) E2 vs. R-eq at a range of doses for each (10−16 to 10−6 M). For E2 n = 42 over 7 experiments; for R-eq n = 30 over 5 experiments. All E2 treated cells were significantly different than vehicle controls or R-eq at p< 0.05 (*). (B) A mixture of various concentration of R-eq (10−16 to 10−7 M) with a physiological E2 concentration (1nM). For E2 + R-eq n = 24 in 3 experiments; for R-eq n = 30 over 5 experiments. * = p < 0.05 vs. control (vehicle) group. # = p < 0.05 vs E2.
Discussion
R-eq increased the activities of all MAPKs, elevated intracellular calcium, activated Gαi, and elicited PRL release in time frames consistent with rapid nongenomic signaling pathway actions; it thus joins the ranks of phytoestrogens known to act via these nonclassical (extranuclear) pathways. Yet R-eq had no effect on cell proliferation, and inhibited the proliferative effects of E2, making it promising for therapeutic interventions without exacerbating the growth of tumor tissues for which estrogens can be a risk factor. This constellation of different effects on estrogen-inducible signaling pathways illustrates how complex is estrogen signaling in general, how estrogens can act differentially, and how the control of cell proliferation involves multiple estrogenic signaling inputs.
Many physiological estrogens and xenoestrogens can act potently via liganded mERα in multiple tumor cell types (breast,Citation48,64-66 pituitary,Citation15,47,58,67-69 prostate,Citation19,60,70 pheochromocytomaCitation71). We previously reported the ability of the R-eq precursor daidzein to activate nongenomic actions in mER-α enriched pituitary tumor cells.Citation15,34 Here we document that the metabolite R-equol can also initiate rapid (< 15 min) non-genomic signaling in pituitary cells at concentrations as low as femtomolar. All signaling pathways tested (3 types of MAPK activations, calcium influx, G protein activation) were actuated, as we saw previously with other phytoestrogens,Citation15,34,48 environmental estrogensCitation33,49,58,63,71 and their metabolites,Citation57 and physiological estrogens.Citation31,46,50,53,54,72 Though R-eq evoked signals in all of these pathways, it did not exactly duplicate the timing and concentration-dependence patterns elicited by E2. The R-eq concentration dependence of ERK activations was non-monotonic, though not as strikingly as for some other xenoestrogens previously examined.Citation15,33,44,49,53,63,71,72 These data show once again that different estrogens are unique in the signaling profiles they induce.
One of the most rapid nongenomic actions we have studied to date (occurring within 15 seconds) is the charging of Gαi regulatory subunit with its activating nucleotide GTP, while mERα physically associates with Gαi proteins in the cell membrane of these pituitary cells.Citation56 Though R-equol increased GTP binding to Gαi, combining a common dietary phytoestrogen level of R-eq with E2 did not blunt the E2-induced response, as it did for other responses measured (MAPKs activations). The lack of an effect at one dose could reflect previous observations where some concentrations of a xenoestrogen used to challenge the physiological estrogens E2, estrone, and estriol enhanced their actions, while others (often higher) blunted the response, and some had no effect.Citation34,45,53,54,58,71 However, if R-eq does act on this particular signaling pathway with a non-monontonic dose-response pattern, only detailed concentration range studies may reveal any enhancing or inhibiting effects at other possible exposure levels.
We measured 2 functional responses, and found them to be associated with the expected signaling responses, though not perfectly matching with the expected dose and signaling contributions. Secretion of peptide hormones like PRL is thought to be triggered by increases in cellular calcium levels, which both R-eq and E2 elicited. However, R-eq caused a larger PRL release, while evoking a smaller change in intracellular calcium compared to E2. We and others have seen similar deviations from simple correlations of peptide secretion magnitude with calcium levels. There are certainly other controls over the final secreted peptide levels, such as voltage-dependent calcium channel activity, vesicle docking and membrane fusion, protein kinases A and C activities, and nucleotide levels (ATP, cAMP and GTP)Citation72,73 which we have not tested here. PRL is also known as a growth factor,Citation74 but its release by R-eq apparently did not provoke cell proliferation in this system.
The MAPKs have been strongly implicated in influencing cell number changes,Citation75 and R-eq caused the activation of all 3 kinase classes assessed here similarly, though not identically to E2.Though it is generally thought that ERK activation should predict a proliferative response, this is not always the case, as weCitation34,46,51,58,59 and othersCitation76,77 have shown previously, and here. Some of the additional downstream consequences of ERK and p38 activation, such as the induction of reactive oxygen species,Citation78 can lead to cell death and so reduce cell numbers. In addition, the alterations in the balance of ERK, JNK (usually eliciting apoptosis), and p38 responses at certain timing interval oscillations may be more predictive. Clearly each signaling or functional endpoint must be assessed independently and in detail to build a composite mingled signaling profile of xenoestrogen actions on a given cell type. How they may interfere with different endogenous estrogen actions on the same endpoints will predict their effects in a hormone-replete animal or human.
The sensitivity of our model system to physiological as well as other estrogens is high. We sometimes see no effects at the 10−15 to 10−16 M levels,Citation34,51 and at other times we do.Citation45,46,54,58,64 That is why we have examined concentration ranges at increasingly lower levels recently. We think it is very rare that femtomolar sensitivity is tested by others, for several reasons: 1) There has not been general recognition of such potent responses because most researchers do not have experimental systems that allow such a wide and detailed range of concentrations to be tested at the same time; the relatively high through-put nature of our assay allows that. 2) Along with that, there has been an under-appreciation of often non-monotonic concentration dependence caused by steroids and their mimics. When a non-responsive dose is found, few ever test responses below that dose. 3) Our well-controlled in vitro settings that allow sensitivities like these to be observed involve exhaustive charcoal stripping of our sera to remove endogenous steroids (see Methods). This allows us to observe previously unrecognized non-monotonic patterns involving these much lower concentrations where few have measured responses before. Non-monotonic response patterns are very prevalent in these rapid signaling responses in our hands and in a cell model that was selected for the robust expression of mERα.Citation42
Cell proliferation is a complex endpoint that weighs a variety of upstream inputs before it executes. It is not surprising that this includes both nongenomic and genomic signaling pathways of estrogens, and cannot be predicted by any one signaling stream. Estrogen-mediated cell proliferation occurs over a longer time frame (stably measured by 3 d after exposureCitation34,46,75), and the cell-cycle checkpoints must meet many requirements before allowing the cell to divide.Citation79 Our study showed that R-eq was itself unable to cause cell proliferation, but could suppress E2-induced pituitary cell proliferation. By contrast, daidzein, the precursor of R-eq, was able to increase cell proliferation at 10−7 to 10−6 M, an achievable blood level.Citation80 Some individuals do not metabolize daidzein to S-eq (for which we do not yet have these types of data). R-eq, as a cell proliferation-inert compound that inhibits the growth-enhancing properties of other estrogens, but is not produced naturally in humans, may potentially be a therapy against estrogen-fueled tumor growth. This would require providing the synthesized metabolite directly as a dietary supplement, while limiting dietary exposures from daidzein. We note that R-eq was found to reduce palpable rodent mammary tumors in one study.Citation81 If S-eq were to be found in the future to have any of these cancer-cell proliferation capabilities, then it might be a natural preventative for those who consume daidzein and produce S-eq naturally.
Though phytoestrogens in general have been touted as therapeutic alternatives for replacing missing physiological estrogens after surgical or natural menopause, not much is known about the mechanistic effects of many individual compounds or their combinations with levels of endogenous estrogens such as developing/young adult women and men would experience. As we have shown here, and in our past studies,Citation45,53,54,71 either enhancements or disruptions are possible, depending upon the kind of response examined. It will be essential to understand the mechanisms driving biological effects in each type of cell or tissue, for each type of phytoestrogen and its metabolites, so that unique patterns of therapeutic or harmful actions can be understood. Because levels of physiological estrogens vary with age and sex, the presence of R-eq and other phytoestrogens could cause alterations (either enhancements or interference) differentially in different sex and life-stage categories of people. That is why the consumption of a phytoestrogen or precursor such as daidzein by infants and children as milk substitutes may present an entirely different situation when compared to adults or the elderly, or those worried about estrogen-driven tumor cell proliferation, or pregnant women. Our study speaks to the issue of R-eq interactions with only one physiological estrogen at a concentration representing a mid-cycle adult woman (E2, 1nM). Understanding the features of phytoestrogens that drive these disruptive vs. enhancing mechanisms will allow us to choose or redesign exogenous estrogen sources that exclude estrogenic or anti-estrogenic activities when needed. Perhaps we may eventually be more successful at predicting outcomes based on details of estrogens’ chemical structures, but we will need to train such an analysis system with many more examples of the multiplicity of signaling pathways affecting specific endpoints before this can be truly useful in preventive and therapeutic medicine.
Materials and methods
We purchased phenol red-free Dulbecco's Modified Eagle Medium (DMEM) from Mediatech, horse serum from Gibco BRL, and defined supplemented calf sera and fetal bovine sera from Hyclone. Paraformaldehyde and glutaraldehyde were purchased from Fisher Scientific. Antibodies (Abs) used in the ERK, JNK, and p38 phosphorylation studies (individually described below) were purchased from Cell Signaling. All other materials were purchased from Sigma (now Merck). R-eq (Santa Cruz Biotechnology) and 17-β E2 were dissolved in absolute ethanol and this stock was then diluted to the desired concentration with DMEM media containing 1% charcoal-stripped serum. The final concentration of ethanol vehicle was 0.001%.
GH3/B6/F10 (mERα-enriched) rat pituitary tumor cell sublinesCitation42 were routinely cultured in DMEM containing 12.5% horse serum, 2.5% defined supplemented calf serum, and 1.5% fetal calf serum. They were then switched to hormone-free media in Corning multi-well plates prior to our experiments, as described below. Cells were used between passages 10 and 20.
Hormone images
ZINC is a free database of commercially available compounds for virtual screening. It gives a unique external ID for a particular compound (ZINC ID's). The 3D visualization files were obtained from PubChem (an open chemistry database). 3D visualization tools (RASMOL, JMOL etc) were used to draw 3D structures of R-eq, E2, and daidzein. The ZINC ID for R-eq is ZINC00388660 (PubChem CID: 6950272).
MAPK assays
We originally developed this assay to assess activated ERK 1 and 2 levels in fixed GH3/B6/F10 cells,Citation47 and subsequently adapted it to equivalent assays for p38s and JNKs.Citation34,82 Briefly, cells were plated at a density of 10,000 cells/poly-D-lysine-coated well of 96-well plates. The following day growth media were replaced with DMEM containing 1% charcoal-stripped (4×) serum (to deprive cells of steroids) for 48 hr. For time-dependent experiments, cells were then treated with physiological/dietary levels of estrogens (either 1 nM E2 or 15 nM R-Eq or vehicle) for 2.5 to 60 min. For dose-dependent experiments cells were treated with a wide range (10−16 to 10−7 M) of E2 or R-eq doses (or vehicle) for 5 min. The cells were then fixed with 2% paraformaldehyde/0.2% picric acid in phosphate-buffered saline (PBS) at 4°C for 48 hr. After fixation, the cells were permeabilized with PBS containing 0.2% fish gelatin and 0.1% Triton X-100 for 1 hr at RT (room temperature), and washed 3X with PBS. Primary Abs against phosphorylated ERKs (p-Thr202/Tyr204) or phosphorylated JNK (p-Thr183/Tyr185), or phosphorylated p38 (p-Thr180/Tyr182) were added at 1:500 dilutions. After overnight incubation at 4°C, the cells were washed 3X with PBS; a biotin-conjugated secondary Ab (Vector Labs, 1:300) in PBS/0.2% fish gelatin was then added for 1 hr at RT. The cells were again washed 3X with PBS, incubated with Vectastain ABC-AP solution (Vector Labs) for 1 hr at RT, and again washed 3X with PBS, followed by addition of Vectastain alkaline phosphatase substrate plus levamasole (an endogenous phosphatase inhibitor). Plates were incubated in the dark for 30 min at 37°C, and the signal for the product of the phosphatase action (para-nitrophenol) was read at A405 in a model 1420 Wallac microplate reader (Perkin Elmer). The number of cells in each well was estimated by the crystal violet (CV) assayCitation64,82,83 and used to normalize the antigen levels of these phosphorylated enzymes to cell numbers in individual wells. Briefly, the cells were stained with 0.1% CV for 30 min and destained with distilled water. The dye was released with 10% acetic acid at RT for 30 min, and the A590 signal of the extract was read in the microplate reader. The experiments were repeated at least 3 times using different passages of cells on different days.
Cytosolic free Ca2+ imaging
GH3/B6/F10 cells were plated at a density of 15,000/well in black-sided, clear bottom 96-well tissue culture plates. The next day, the growth media were changed to 1% 4X charcoal-stripped fetal calf serum for 48 hrs. Cai2+ release was determined using a Fluo-4 No-wash Calcium Assay Kit (cat# F36205, Molecular Probes, Inc.) following the company's protocol. Briefly, culture media were removed and 50 μL of the dye loading solution (containing 2.5 mM probenecid) was added to each well. Plates were incubated for 60 min at 37oC and an additional 60 min at RT in the dark. Fluorescence (excitation at 485 nm and emission at 525 nm) was measured with a Flexstation 3 instrument (Molecular Devices). A baseline was established for each well during the initial segment of each run for 17 sec, followed by addition of 50 μL of 2X concentrated treatment agents (estrogens or vehicle). Fluorescence was recorded every 1.7 second for 60 sec. Maximum peak height was determined for each well with Flexstation software (SoftMax Pro 5.2).
Gαi activation assay
GH3/B6/F10 cells were plated in poly-D-lysine-coated 96-well plates (10,000/well) and the next day incubated with media containing 1% charcoal-stripped FBS, continuing for 48 hrs. Cells were then incubated with DMEM medium alone for 2 hrs. The cells were then permeabilized with saponin (0.03mg/ml) and GTPyS (100μM) was added for 10 min at RT. Following a 5 min estrogen treatment the cells were fixed with 4% paraformaldehyde, centrifuged at 500 xg for 2 min and then incubated for an additional 8 min at RT. Cells were then blocked for 1 hr at RT with PBST (PBS with 0.1% Tween 20) + 1% gelatin. Next the cells were incubated with a mouse monoclonal Ab to the active (GTP-charged) form of Gαi, (1:1000; New East Biochemicals) overnight at 4°C. Cells were then washed 3X with PBS, and a biotin-conjugated secondary Ab (Vector Labs, 1:300) in PBS/1% fish gelatin was added for 1 hr at RT. The cells were again washed 3X with PBS, incubated with Vectastain ABC-AP solution (Vector Labs) for 1 hr at RT, and again washed 3X with PBS. Plates were incubated in the dark for 30 min at 37°C, and the signal for the product of the phosphatase action (para-nitrophenol) was read at A405 in a model 1420 Wallace microplate reader (Perkin Elmer). The data were normalized to cell number in each well as estimated by the CV assay.Citation64,82,83
Cell proliferation assays
Subconfluent cells growing in serum-containing medium were seeded into 96-well plates (5000 cells/well) coated with poly-D-lysine, allowed to attach overnight, and the next day treated with E2 or R-eq in DMEM medium containing 1% 4× charcoal-stripped FBS for 3 d. Cell numbers were assessed by the CV assay (described above) to compare the proliferative effects of E2 or R-eq at different concentrations. We used ∼6 samples for each treatment group, and experiments were repeated at least 3 times using different passages of cells. We chose a day 3 assessment time point based on our previous studies.Citation34,46
Prolactin (PRL) release
Cells (0.5–0.7 × 106) were plated in poly-D-lysine-coated 6-well plates. After serum deprivation in ITS defined medium [5 μg/mL insulin, 5 μg/mL transferrin, 5 ng/mL sodium selenite, and 0.1% bovine serum albumin (BSA)] for 48 h, this medium was replaced with DMEM/0.1% BSA for a 1-hr incubation at 37°C. The cells were treated with E2 or R-eq for 1 min and centrifuged at 4°C (350×g for 5 min); the supernatant was then collected and stored at -20°C. The PRL concentrations were determined using components of the rat PRL RIA kit from the National Institute of Diabetes and Digestive and Kidney Disease and the National Hormone and Pituitary Program. Briefly, RIA buffer [80% PBS, 20% DMEM, 2% normal rabbit serum], 100 μL cold standard (rat PRL-RP-3) or unknown sample, rPRL-s-9 antiserum (final dilution of 1:437,500 in RIA buffer), and [125I]-rat-PRL (PerkinElmer; using 15,000 counts per tube diluted in RIA buffer) were combined and incubated with shaking overnight at 4°C. Anti-rabbit IgG (R-0881; Sigma) was added to a final dilution of 1:9, and the samples were incubated with shaking at RT for 2 hr. One mL of polyethylene glycol solution [1.2 M polyethylene glycol (P-6667; Sigma), 50 mM Tris, pH 8.6] was then added; the samples were incubated with shaking at RT for 15 min, then centrifuged at 4,000 × g for 10 min at 4°C. The supernatant was decanted, and the pellet counted in a Wizard 1470 Gamma Counter (PerkinElmer). The PRL concentration was then calculated and normalized to the CV values that estimated the number of cells in each well,Citation82,83 as described above. These measurements were done in at least 3 different experiments on different days.
Statistical analysis
Data from all experiments were analyzed by one-way analysis of variance (ANOVA) followed by multiple comparisons versus control group (Holm-Sidak method). The Sigma Stat 3 program (Systat Software, Inc.) was used for all statistical analysis, and significance was accepted at p < 0.05.
Abbreviations
| Abs | = | antibodies |
| ANOVA | = | one-way analysis of variance |
| ATP | = | adenosine tri-phosphate |
| BSA | = | bovine serum albumin |
| cAMP | = | cyclic adenosine mono-phosphate |
| DMEM | = | Dulbecco's Modified Eagle Medium |
| E2 | = | estradiol |
| ER | = | estrogen receptor |
| ERK | = | extracellular-regulated kinase |
| Gαi | = | G protein α subclass i |
| GTP | = | guanine nucleotide tri-phosphate |
| GTPγS | = | guanine nucleotide tri-phosphate γS |
| JNK | = | jun N-terminal kinase |
| mER | = | membrane estrogen receptor |
| PBS | = | phosphate-buffered saline |
| PRL | = | prolactin |
| R-eq | = | R-equol |
| S-eq | = | S-equol |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. David Konkel (UTMB Institute for Translational Sciences) for critically editing the manuscript.
Funding
These studies were supported by NIH grant R01ES15292.
References
- Fletcher RJ. Food sources of phyto-oestrogens and their precursors in Europe. Br J Nutr 2003; 89 Suppl 1:S39-43; PMID:12725655; http://dx.doi.org/10.1079/BJN2002740
- Cornwell T, Cohick W, Raskin I: Dietary phytoestrogens and health. Phytochemistry 2004; 65(8):995-1016
- Setchell KD, Brown NM, Desai P, Zimmer-Nechemias L, Wolfe BE, Brashear WT, Kirschner AS, Cassidy A, Heubi JE. Bioavailability of pure isoflavones in healthy humans and analysis of commercial soy isoflavone supplements. J Nutri 2001; 131:1362s-75s; PMID:11285356
- Setchell KD, Brown NM, Desai PB, Zimmer-Nechimias L, Wolfe B, Jakate AS, Creutzinger V, Heubi JE. Bioavailability, disposition, and dose-response effects of soy isoflavones when consumed by healthy women at physiologically typical dietary intakes. J Nutr 2003; 133(4):1027-35; PMID:12672914
- Setchell KD, Clerici C. Equol: history, chemistry, and formation. J Nutr 2010; 140(7):1355S-62S; PMID:20519412; http://dx.doi.org/10.3945/jn.109.119776
- Setchell KD, Clerici C. Equol: pharmacokinetics and biological actions. J Nutr 2010; 140(7):1363S-8S; PMID:20519411; http://dx.doi.org/10.3945/jn.109.119784
- Setchell KD, Brown NM, Summer S, King EC, Heubi JE, Cole S, Guy T, Hokin B. Dietary factors influence production of the soy isoflavone metabolite s-(-)equol in healthy adults. J Nutr 2013; 143(12):1950-8; PMID:24089421; http://dx.doi.org/10.3945/jn.113.179564
- Muthyala RS, Ju YH, Sheng S, Williams LD, Doerge DR, Katzenellenbogen BS, Helferich WG, Katzenellenbogen JA. Equol, a natural estrogenic metabolite from soy isoflavones: convenient preparation and resolution of R- and S-equols and their differing binding and biological activity through estrogen receptors α and β. Bioorg Med Chem 2004; 12(6):1559-67; PMID:15018930; http://dx.doi.org/10.1016/j.bmc.2003.11.035
- Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der BB, Gustafsson JA. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor β. Endocrinology 1998; 139(10):4252-63; PMID:9751507
- Shinkaruk S, Carreau C, Flouriot G, Bennetau-Pelissero C, Potier M. Comparative effects of R- and S-equol and implication of transactivation functions (AF-1 and AF-2) in estrogen receptor-induced transcriptional activity. Nutrients 2010; 2(3):340-54; PMID:22254026; http://dx.doi.org/10.3390/nu2030340
- Dutertre M, Smith CL. Molecular mechanisms of selective estrogen receptor modulator (SERM) action. J Pharmacol Exp Ther 2000; 295(2):431-7; PMID:11046073
- Brzezinski A, Debi A. Phytoestrogens: the "natural" selective estrogen receptor modulators? Eur J Obstet Gynecol Reprod Biol 1999; 85(1):47-51; PMID:10428321; http://dx.doi.org/10.1016/S0301-2115(98)00281-4
- Pugazhendhi D, Watson KA, Mills S, Botting N, Pope GS, Darbre PD. Effect of sulphation on the oestrogen agonist activity of the phytoestrogens genistein and daidzein in MCF-7 human breast cancer cells. J Endocrinol 2008; 197(3):503-15; PMID:18492816; http://dx.doi.org/10.1677/JOE-07-0384
- Schug TT, Janesick A, Blumberg B, Heindel JJ. Endocrine disrupting chemicals and disease susceptibility. J Steroid Biochem Mol Biol 2011, 127(3-5):204-15; PMID:21899826; http://dx.doi.org/10.1016/j.jsbmb.2011.08.007
- Jeng YJ, Kochukov MY, Watson CS. Membrane estrogen receptor-α-mediated nongenomic actions of phytoestrogens in GH3/B6/F10 pituitary tumor cells. J Mol Signal 2009; 4:2; PMID:19400946; http://dx.doi.org/10.1186/1750-2187-4-2
- Jeng YJ, Kochukov M, Nauduri D, Kaphalia BS, Watson CS. Subchronic exposure to phytoestrogens alone and in combination with diethylstilbestrol - pituitary tumor induction in Fischer 344 rats. NutrMetab (Lond) 2010; 7:40; http://dx.doi.org/10.1186/1743-7075-7-40
- Ruggiero RJ, Likis FE. Estrogen: physiology, pharmacology, and formulations for replacement therapy. J Midwifery Womens Health 2002; 47(3):130-8; PMID:12071379; http://dx.doi.org/10.1016/S1526-9523(02)00233-7
- Bilal I, Chowdhury A, Davidson J, Whitehead S. Phytoestrogens and prevention of breast cancer: The contentious debate. World J Clin Oncol 2014; 5(4):705-12; PMID:25302172; http://dx.doi.org/10.5306/wjco.v5.i4.705
- Hwang KA, Choi KC. Anticarcinogenic effects of dietary phytoestrogens and their chemopreventive mechanisms. Nutr Cancer 2015; 67(5):796-803; PMID:25996655; http://dx.doi.org/10.1080/01635581.2015.1040516
- Hedelin M, Klint A, Chang ET, Bellocco R, Johansson JE, Andersson SO, Heinonen SM, Adlercreutz H, Adami HO, Gronberg H, et al. Dietary phytoestrogen, serum enterolactone and risk of prostate cancer: the cancer prostate Sweden study (Sweden). Cancer Causes Control 2006; 17(2):169-80; PMID:16425095; http://dx.doi.org/10.1007/s10552-005-0342-2
- Hedelin M, Lof M, Andersson TM, Adlercreutz H, Weiderpass E: Dietary phytoestrogens and the risk of ovarian cancer in the women's lifestyle and health cohort study. Cancer Epidemiol Biomarkers Prev 2011; 20(2):308-17; PMID:21098648; http://dx.doi.org/10.1158/1055-9965.EPI-10-0752
- Sirtori CR, Arnoldi A, Johnson SK: Phytoestrogens: end of a tale? Ann Med 2005; 37(6):423-38; PMID:16203615; http://dx.doi.org/10.1080/07853890510044586
- Ziegler RG, Hoover RN, Pike MC, Hildesheim A, Nomura AM, West DW, Wu-Williams AH, Kolonel LN, Horn-Ross PL, Rosenthal JF, et al. Migration patterns and breast cancer risk in Asian-American women. J Natl Cancer Inst 1993; 85(22):1819-27; PMID:8230262; http://dx.doi.org/10.1093/jnci/85.22.1819
- Albert A, Altabre C, Baro F, Buendia E, Cabero A, Cancelo MJ, Castelo-Branco C, Chantre P, Duran M, Haya J, et al. Efficacy and safety of a phytoestrogen preparation derived from Glycine max (L.) Merr in climacteric symptomatology: a multicentric, open, prospective and non-randomized trial. Phytomedicine 2002; 9(2):85-92; PMID:11995954
- Bhakta D, Higgins CD, Sevak L, Mangtani P, Adlercreutz H, McMichael AJ, dos Santos Silva I. Phyto-oestrogen intake and plasma concentrations in South Asian and native British women resident in England. Br J Nutr 2006; 95(6):1150-8; PMID:16768838; http://dx.doi.org/10.1079/BJN20061777
- Adlercreutz H, Hamalainen E, Gorbach SL, Goldin BR, Woods MN, Dwyer JT: Diet and plasma androgens in postmenopausal vegetarian and omnivorous women and postmenopausal women with breast cancer. Am J Clin Nutr 1989; 49(3):433-42; PMID:2923075
- Adlercreutz H: Western diet and Western diseases: some hormonal and biochemical mechanisms and associations. Scand J Clin Lab Invest Suppl 1990; 201:3-23; PMID:2173856; http://dx.doi.org/10.1080/00365519009085798
- Adlercreutz H, Mazur W. Phyto-oestrogens and Western diseases. Ann Med 1997; 29(2):95-120; PMID:9187225; http://dx.doi.org/10.3109/07853899709113696
- Cross HS, Kallay E, Lechner D, Gerdenitsch W, Adlercreutz H, Armbrecht HJ. Phytoestrogens and vitamin D metabolism: a new concept for the prevention and therapy of colorectal, prostate, and mammary carcinomas. J Nutr 2004; 134(5):1207S-12S; PMID:15113973
- Heinonen SM, Hoikkala A, Wahala K, Adlercreutz H. Metabolism of the soy isoflavones daidzein, genistein and glycitein in human subjects. Identification of new metabolites having an intact isoflavonoid skeleton. J Steroid Biochem Mol Biol 2003; 87(4-5):285-99; PMID:14698210; http://dx.doi.org/10.1016/j.jsbmb.2003.09.003
- Watson CS, Bulayeva NN, Wozniak AL, Finnerty CC. Signaling from the membrane via membrane estrogen receptor-α: estrogens, xenoestrogens, and phytoestrogens. Steroids 2005; 70(5-7):364-71; PMID:15862819; http://dx.doi.org/10.1016/j.steroids.2005.03.002
- Watson CS, Gametchu B. Membrane-initiated steroid actions and the proteins that mediate them. Proc Soc Exp Biol Med 1999; 220(1):9-19; PMID:9893163; http://dx.doi.org/10.3181/00379727-220-44338
- Watson CS, Jeng YJ, Kochukov MY. Nongenomic signaling pathways of estrogen toxicity. Toxicol Sci 2010; 115(1):1-11; PMID:19955490; http://dx.doi.org/10.1093/toxsci/kfp288
- Jeng YJ, Watson CS. Proliferative and anti-proliferative effects of dietary levels of phytoestrogens in rat pituitary GH3/B6/F10 cells - the involvement of rapidly activated kinases and caspases. BMC Cancer 2009; 9:334; PMID:19765307; http://dx.doi.org/10.1186/1471-2407-9-334
- Vinas R, Jeng YJ, Watson CS. Non-genomic effects of xenoestrogen mixtures. Int J Environ Res Public Health 2012; 9(8):2694-714; PMID:23066391; http://dx.doi.org/10.3390/ijerph9082694
- Watson CS, Jeng Y-J, Guptarak J. Endocrine disruption via estrogen receptors that participate in nongenomic signaling pathways. J Steroid Biochem Mol Biol 2011; 127(1-2):44-50; PMID:21300151; http://dx.doi.org/10.1016/j.jsbmb.2011.01.015
- Lampe JW, Karr SC, Hutchins AM, Slavin JL. Urinary equol excretion with a soy challenge: Influence of habitual diet. Proc Society Exp Biol Med 1998; 217(3):335-9; PMID:9492344; http://dx.doi.org/10.3181/00379727-217-44241
- Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR, Jr, Lee DH, Shioda T, Soto AM, vom Saal FS, Welshons WV et al. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev 2012; 33(3):378-455; PMID:22419778; http://dx.doi.org/10.1210/er.2011-1050
- Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM. Bisphenol-A and the great divide: a review of controversies in the field of endocrine disruption. Endocr Rev 2009; 30(1):75-95; PMID:19074586; http://dx.doi.org/10.1210/er.2008-0021
- Vandenberg LN. Low-dose effects of hormones and endocrine disruptors. Vitam Horm 2014; 94:129-65; PMID:24388189; http://dx.doi.org/10.1016/B978-0-12-800095-3.00005-5
- Rhomberg LR, Goodman JE. Low-dose effects and nonmonotonic dose-responses of endocrine disrupting chemicals: has the case been made? Regul Toxicol Pharmacol 2012; 64(1):130-3; PMID:22750031; http://dx.doi.org/10.1016/j.yrtph.2012.06.015
- Pappas TC, Gametchu B, Watson CS. Membrane estrogen receptor-enriched GH3/B6 cells have an enhanced non-genomic response to estrogen. Endocrine 1995; 3:743-9; PMID:21153164; http://dx.doi.org/10.1007/BF03000207
- Campbell CH, Watson CS. A comparison of membrane vs. intracellular estrogen receptor-α in GH(3)/B6 pituitary tumor cells using a quantitative plate immunoassay. Steroids 2001; 66(10):727-36; PMID:11522334; http://dx.doi.org/10.1016/S0039-128X(01)00106-4
- Bulayeva NN, Watson CS. Xenoestrogen-induced ERK-1 and ERK-2 activation via multiple membrane-initiated signaling pathways. Environ Health Perspect 2004; 112(15):1481-7
- Vinas R, Watson CS. Mixtures of xenoestrogens disrupt estradiol-induced nongenomic signaling and functions in pituitary cells. BMC Environmental Health 2013; 12:26
- Watson C, Jeng YJ, Kochukov M. Nongenomic actions of estradiol compared with estrone and estriol in pituitary tumor cell signaling and proliferation. FASEB J 2008; 22(9):3328-36; PMID:18541692; http://dx.doi.org/10.1096/fj.08-107672
- Bulayeva NN, Gametchu B, Watson CS. Quantitative measurement of estrogen-induced ERK 1 and 2 activation via multiple membrane-initiated signaling pathways. Steroids 2004; 69(3):181-92; PMID:15072920; http://dx.doi.org/10.1016/j.steroids.2003.12.003
- Zivadinovic D, Watson CS. Membrane estrogen receptor-α levels predict estrogen-induced ERK1/2 activation in MCF-7 cells. Breast Cancer Res 2005; 7(1):R130-44; PMID:15642162; http://dx.doi.org/10.1186/bcr959
- Watson CS, Bulayeva NN, Wozniak AL, Alyea RA. Xenoestrogens are potent activators of nongenomic estrogenic responses. Steroids 2007; 72(2):124-34; PMID:17174995; http://dx.doi.org/10.1016/j.steroids.2006.11.002
- Watson CS, Alyea RA, Jeng YJ, Kochukov MY. Nongenomic actions of low concentration estrogens and xenoestrogens on multiple tissues. Mol Cell Endocrinol 2007; 274(1-2):1-7; PMID:17601655; http://dx.doi.org/10.1016/j.mce.2007.05.011
- Kochukov MY, Jeng YJ, Watson CS. Alkylphenol xenoestrogens with varying carbon chain lengths differentially and potently activate signaling and functional responses in GH3/B6/F10 somatomammotropes. Env Health Perspect 2009; 117(5):723-30; http://dx.doi.org/10.1289/ehp.0800182
- Alyea RA, Watson CS. Nongenomic mechanisms of physiological estrogen-mediated dopamine efflux. BMC Neurosci 2009; 10:59; PMID:19531209; http://dx.doi.org/10.1186/1471-2202-10-59
- Jeng YJ, Kochukov M, Watson CS. Combinations of physiologic estrogens with xenoestrogens alter calcium and kinase responses, prolactin release, and membrane estrogen receptor trafficking in rat pituitary cells. Environ Health 2010; 9:61; PMID:20950447; http://dx.doi.org/10.1186/1476-069X-9-61
- Jeng YJ, Watson CS. Combinations of physiologic estrogens with xenoestrogens alter ERK phosphorylation profiles in rat pituitary cells. Environ Health Perspect 2011; 119(1):104-12
- Watson CS, Jeng YJ, Guptarak J. Endocrine disruption via estrogen receptors that participate in nongenomic signaling pathways. J Steroid Bio Chem Mol Biol 2011; 127(1-2):44-50
- Watson CS, Jeng YJ, Hu G, Wozniak A, Bulayeva N, Guptarak J. Estrogen- and xenoestrogen-induced ERK signaling in pituitary tumor cells involves estrogen receptor-α interactions with G protein-alphai and caveolin I. Steroids 2011; 77(5):424-32; PMID:22230296
- Vinas R, Watson CS. Rapid estrogenic signaling activities of modified (chlorinated, sulfonated, and glucuronidated) endocrine disruptor bisphenol A. Endocri Disruptors 2013; 1(1):e26306; PMID:24795899; http://dx.doi.org/10.4161/endo.25411
- Vinas R, Watson C. Bisphenol s disrupts estradiol-induced nongenomic signaling in a rat pituitary cell line: effects on cell functions. Environ Health Perspect 2013; 121(3):352-8; PMID:23458715; http://dx.doi.org/10.1289/ehp.1205826
- Koong LY, Watson CS: Direct estradiol and diethylstilbestrol actions on early- vs. late-stage prostate cancer cells. Prostate 2014; 74(16):1589-603
- Koong LY, Watson CS. Xenoestrogen-induced signaling leading to cell viability changes on early- vs. late-stage prostate cancer cells. Endocrine Disruptors 2015; 3(1): e995003; http://dx.doi.org/10.4161/23273747.2014.995003.
- Alyea R, Laurence S, Kim S, Katzenellenbogen B, Katzenellenbogen J, Watson C. The roles of membrane estrogen receptor subtypes in modulating dopamine transporters in PC-12 cells. J Neuro Chem 2008; 106:1525-33
- Narita M, Miyagawa K, Mizuo K, Yoshida T, Suzuki T. Prenatal and neonatal exposure to low-dose of bisphenol-A enhance the morphine-induced hyperlocomotion and rewarding effect. Neuro Sci Lett 2006; 402(3):249-52
- Watson CS, Hu G, Paulucci-Holthauzen AA. Rapid actions of xenoestrogens disrupt normal estrogenic signaling. Steroids 2014; 81:36-42
- Zivadinovic D, Gametchu B, Watson CS. Membrane estrogen receptor-α levels in MCF-7 breast cancer cells predict cAMP and proliferation responses. Breast Cancer Res 2005; 7(1):R101-12; http://dx.doi.org/10.1186/bcr958
- Razandi M, Pedram A, Levin ER. Plasma membrane estrogen receptors signal to antiapoptosis in breast cancer. Mol Endocrinol 2000; 14(9):1434-47
- Marquez DC, Pietras RJ. Membrane-associated binding sites for estrogen contribute to growth regulation of human breast cancer cells. Oncogene 2001; 20(39):5420-30
- Zarate S, Jaita G, Zaldivar V, Radl DB, Eijo G, Ferraris J, Pisera D, Seilicovich A. Estrogens exert a rapid apoptotic action in anterior pituitary cells. Am J Physiol Endocrinol Metab 2009; 296(4):E664-71
- Christian HC, Morris JF. Rapid actions of 17beta-oestradiol on a subset of lactotrophs in the rat pituitary. J Physiol 2002; 539(Pt 2):557-66
- Otto C, Fuchs I, Altmann H, Klewer M, Schwarz G, Bohlmann R, Nguyen D, Zorn L, Vonk R, Prelle K, et al. In vivo characterization of estrogen receptor modulators with reduced genomic versus nongenomic activity in vitro. J Steroid BiochemMolBiol 2008; 111(1-2):95-100
- Lee HR, Hwang KA, Choi KC. The estrogen receptor signaling pathway activated by phthalates is linked with transforming growth factor-β in the progression of LNCaP prostate cancer models. Int J Oncol 2014; 45(2):595-602
- Alyea RA, Watson CS. Differential regulation of dopamine transporter function and location by low concentrations of environmental estrogens and 17beta-estradiol. Environ Health Perspect 2009; 117(5):778-83; http://dx.doi.org/10.1289/ehp.0800026
- Bulayeva NN, Wozniak A, Lash LL, Watson CS. Mechanisms of membrane estrogen receptor-{α}-mediated rapid stimulation of Ca2+ levels and prolactin release in a pituitary cell line. Am J Physiol Endocrinol Metab 2005; 288:E388-97
- Kits KS, Mansvelder HD. Regulation of exocytosis in neuroendocrine cells: spatial organization of channels and vesicles, stimulus-secretion coupling, calcium buffers and modulation. Brain Res Brain Res Rev 2000; 33(1):78-94
- Clevenger CV, Furth PA, Hankinson SE, Schuler LA. The role of prolactin in mammary carcinoma. Endocr Rev 2003; 24(1):1-27
- Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 2002; 12(1):9-18
- Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J 2008; 22(4):954-65
- Roskoski R, Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol Res 2012; 66(2):105-43
- Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J 2010; 277(1):2-21; PMID:19843174; http://dx.doi.org/10.1111/j.1742-4658.2009.07366.x
- Butt AJ, Caldon CE, McNeil CM, Swarbrick A, Musgrove EA, Sutherland RL. Cell cycle machinery: links with genesis and treatment of breast cancer. Adv Exp Med Biol 2008; 630:189-205; http://dx.doi.org/10.1007/978-0-387-78818-0_12
- Valentin-Blasini L, Blount BC, Caudill SP, Needham LL: Urinary and serum concentrations of seven phytoestrogens in a human reference population subset. J Expo Anal Environ Epidemiol 2003; 13(4):276-82; http://dx.doi.org/10.1038/sj.jea.7500278
- Brown NM, Belles CA, Lindley SL, Zimmer-Nechemias LD, Zhao X, Witte DP, Kim MO, Setchell KD. The chemopreventive action of equol enantiomers in a chemically induced animal model of breast cancer. Carcinogenesis 2010; 31(5):886-93
- Watson CS, Jeng YJ, Bulayeva NN, Finnerty CC, Koong LY, Zivadinovic D, Alyea RA, Midoro-Horiuti T, Goldblum RM, Anastasio NC, et al. Multi-well plate immunoassays for measuring signaling protein activations/deactivations and membrane vs. intracellular receptor levels. Methods Mol Biol 2014; 1204:123-33; http://dx.doi.org/10.1007/978-1-4939-1346-6_11
- Lottering ML, Haag M, Seegers JC. Effects of 17b-estradiol metabolites on cell cycle events in MCF-7 cells. Cancer Res 1992; 52:5926-32