
Abstract
Higher alcohols are not only the key flavor substance but also the precursor of other flavor substances in Baijiu. In the metabolic pathway of higher alcohols, alcohol dehydrogenase (ADH) plays an important role and its genes can be used as a marker gene for analyzing the producer of higher alcohols. In this study, the degenerate primers of Zinc-dependent long-chain ADH gene (L.adh) were designed and applied to investigate the diversity of L.adh in different Daqu (HT, SC and SD). The results showed that the degenerate primers were able to amplify the fragment of L.adh and had good coverage in high-throughput sequencing. The diversity and composition of L.adh in the three kinds of Daqu were different and the diversity of L.adh in SC was the highest while HT was the lowest. The predominant L.adh in SC and HT were form Bacillus (54%–78%) and Aspergillus (64%–66%), respectively, and the predominant L.adh in SD were from Aspergillus (7%–51%) and Enterobacter (2%–47%). In addition, L.adh from bacteria and fungi could be clustered into one evolutionary branch and divided into 10 clusters. The findings of this study will provide a new perspective for evaluating the potential microorganisms associated with higher alcohol synthesis in Daqu and reference for selecting suitable Daqu in the production of Baijiu.
REVIEWING EDITOR:
Introduction
Baijiu, a traditional alcoholic drink in China, contains thousands of flavor substances (alcohols, aldehydes, acids, and esters, etc.) (Xu et al., Citation2012) and these flavor substances are synthesized by the mixed microorganism in the fermentation process. Higher alcohols, a key flavor substance in Baijiu, have mellow sweetness, aroma-assisting functions and are also the precursor of other flavor substances (esters and aldehydes) (Fang et al., Citation2018; Liu et al., Citation2018a). When the content of higher alcohols in Baijiu is moderate, Baijiu tastes harmonious and mellow. However, if the content of higher alcohols is too high, it will make Baijiu taste strong and pungent, and may cause health problems after drinking (Gerong et al., Citation2019; Sun et al., Citation2020). Therefore, how to control the synthesis of higher alcohols in the process of Baijiu fermentation is the focus of researchers in this field.
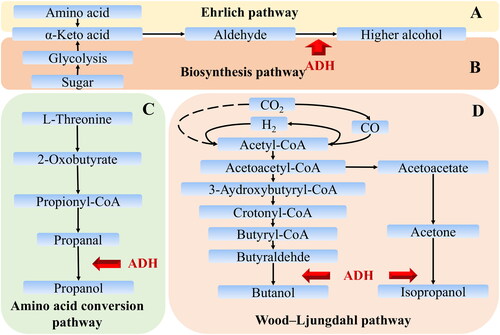
The study about higher alcohol synthesis in traditional fermented food can be divided into culture-dependent and culture-independent methods. In the study based on the culture-dependent method showed that higher alcohols can be synthesized by amino acid (Ehrlich pathway, ) and sugar metabolism (Biosynthesis pathway, ) in Saccharomyces cerevisiae (Hazelwood et al., Citation2008; Holt et al., Citation2019). Some bacteria, such as the genus of Clostridium can synthesis propanol, butanol, and isobutanol by amino acid conversion pathway () and Wood-Ljungdahl pathway () (Whitham et al., Citation2016). In addition, Lactobacillus (Lu et al., Citation2019), Bacillus (Wang et al., Citation2017; Zhao et al., Citation2017), Staphylococcus, Proteus, and Brachybacterium (Deetae et al., Citation2007) can synthesize propanol, amyl alcohol, isoamyl alcohol and isobutanol. Some non-S.cerevisiae yeast, including Brettanomyces, Saccharomycodes, Torulopsis, Hansenula, Geotrichum, Zygosaccharomyce, and Candida also have the ability for producing higher alcohol (Fang et al., Citation2019; Zhu et al., Citation2016). Therefore, the metabolism of higher alcohols is participated by a variety of microorganisms in the process of mixed fermentation process.
Figure 1. The metabolic pathway of higher alcohol in the microorganism.
(A) Ehrlich pathway and Biosynthesis pathway, (B) Amino acid transformation pathway, and (C) Wood-Ljungdahl pathway. ADH is the abbreviation of alcohol dehydrogenase.
Although the culture-dependent method can obtain the microorganisms to study the metabolic pathway of higher alcohols in individual microorganisms, it can`t fully reveal the synthesis mechanism of higher alcohols in the mixed microorganism process. Compared with the culture-dependent method, the culture-independent method can comprehensively reveal the microbial composition in mixed microorganism process. Moreover, the relationship between microorganisms and higher alcohols can be established by statistical analysis to trace the microorganisms associated with higher alcohols in the fermentation process. The studies show that Pediococcus and Proteus are positively correlated with isobutanol in the fermentation process of Huangjiu (an alcoholic beverage in China) (Sun et al., Citation2020), and multiple genera of bacteria and fungi are associated with higher alcohols in the Baijiu fermentation process (Liu & Miao, Citation2020; Pang et al., Citation2018). However, the relationship between the richness of microorganism and the concentration of higher alcohols established by correlation analysis is a statistical relationship, not functional correlation. In addition, our previous studies find that some strain can`t produce higher alcohols, but can reduce the content of higher alcohol and the alcohol dehydrogenase produced by the isolate can convert alcohols to aldehydes (Hao et al., Citation2021). This result indicates that some microorganisms containing alcohol dehydrogenase has an important effect on the content of higher alcohols in the process of mixed microbial fermentation.
Functional gene in metabolic pathways can be applied as marker gene for characterizing metabolic function (Yilmaz et al., Citation2011) and studying the potential producer of metabolite in the process of mixed microorganism process (Gu et al., Citation2017; Sheng et al., Citation2019). Based on the literature, alcohol dehydrogenase (ADH) is a key enzyme in microbial higher alcohol metabolic pathways () and Zinc-dependent long-chain alcohol dehydrogenase (L.ADH) is the main ADH. Moreover, two relatively conservative regions are found by comparing the amino acid sequence of L.ADH in microorganisms of Baijiu in our previous study. Therefore, the gene of L.ADH (L.adh) could be applied as a marker gene to study the producer of higher alcohol in mixed microorganism process of Baijiu.
Daqu, the starter of Baijiu, contains multiple microorganisms and enzymes (Li et al., Citation2018; Shen et al., Citation2018; Xiu et al., Citation2012; Zhang et al., Citation2014; Zou et al., Citation2018) and is a key source of microorganisms in the fermentation process of Baijiu. Therefore, the microbial composition in Daqu has an important effect on the quality of Baijiu. Analysis the diversity of key enzyme gene in metabolic pathway of higher alcohols in Daqu and trace the microorganisms directly related to higher alcohol metabolism in Daqu is the basis for regulating the synthesis of higher alcohol in the mixed microorganism process of Baijiu by selecting different Daqu. Based on the above, a pair of degenerate primers was designed and amplifies the L.adh fragments in different Daqu to investigate the diversity of L.adh and trace the potential producer of higher alcohol in this study.
Materials and methods
Sample collection
Daqu (HT, SC and SD) was collected from the manufacturer located in Inner Mongolia, Sichuan and Shandong province, respectively. Three types of Daqu were all made from wheat and the temperatures for cultivating Daqu are 58 °C–63 °C. Five-point sampling method was applied to sample the samples form one Daqu in triplicate.
Design of L.adh primer
The domain of L.ADH was predicted by Pfam (http://pfam.xfam.org/) (Mistry et al., Citation2021) and the amino acid sequence of L.ADH was obtained from Uniport (https://www.uniprot.org/) (UniProt Consortium, Citation2021). Fifty-one amino acid sequences of L. ADH (including bacterial and fungal; S1 Table) was aligned by Cluster W in Bioedit (8.1.0) (Helge-Friedrich, Citation2004) and the degenerate primer was designed by j-CODEHOP (https://4virology.net/virology-ca-tools/j-codehop/) (Boyce et al., Citation2009). The main parameters were as follows: Clamp length by temp was 15, Max degeneracy was 100, and Core length was 3. The quality of degenerate primer was evaluated by Oligo Calc (http://biotools.nubic.northwestern.edu/OligoCalc.html) (Kibbe, Citation2007) and Primer-Blast.
Amplification and sequencing
The microbial genome in Daqu was extracted by the indirect extraction method. 7.0 g Daqu was washed by 0.1 mol/L PBS to obtained microorganisms and genome DNA was extracted using Omega EZNA™ (Wang et al., Citation2018). The amplification conditions (20 μL) were 4 μL of 5 × FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.4 μL of FastPfu Polymerase, 0.8 μL of Primer, 10 ng of template DNA and ddH2O. The PCR conditions were pre-denaturation (5 min at 95 °C), denaturation (30 s at 95 °C), annealing (30 s at 55 °C), extension (45 s at 72 °C), cycling (35 times) and final extension (10 min at 72 °C). The amplicon was purified and sequenced by Illumina platform (Shanghai BIOZERON Co., Ltd). The sequences were spliced by FLASH (1.2.11) (Magoč & Salzberg, Citation2011). Based on similarity of 100% and 97%, the sequences were clustered into operational taxonomic units (OTU) by USEARCH (version 10.0) (Cole et al., Citation2009). OTU was functional annotation and taxonomic classification by BlastX and the filtering principle is E value < 1 · E-10. Composition and diversity of OTU were evaluated by R (vegan and picante). MEGAX (Kumar et al., Citation2018) was applied to analyze the evolutionary of L.adh fragments and the phylogenetic tree was viewed in iTOL (https://itol.embl.de/) (Letunic & Bork, Citation2021, 2018). The raw reads were deposited into the NCBI Sequence Read Archive (SRA) database (Accession Number: SRP335287, https://www.ncbi.nlm.nih.gov/sra/?term=SRP335287).
TA clone
The template-independent base "A" was added to the amplicon by applying the A-Tailing kit (Sangon Biotech Co., Ltd). After purification (SanPrep Column DNA Gel Extraction Kit, Sangon Biotech Co., Ltd), the product was ligated and transformed by pEASY-T1 Cloning Kit (TransGen Biotech Co., Ltd). After transformation, it was screened and amplified for identification by Sangon Biotech Co., Ltd. (Shanghai, China).
Results
Design of degenerate primers
Several degenerate primers were designed based on the conservativeness of 51 L.ADH amino acid sequences () and L.adh-F and L.adh-R were selected as the primers to amplify L.adh () after checking by Oligo Calc and Primer-Blast. It was found that the primers could amplify the target lengths (426 bp) in gel electrophoresis and there were different lengths of amplification (). In order to ensure the accuracy of L.adh in the amplified fragments, samples in same group (100–200 bp, 200–400 bp and 400-600) were mixed into 1 and 3 different libraries were constructed for each Daqu to high-throughput sequencing (). The fragments larger than 600 bp were verified by TA cloning and the results showed that they did not belong to L.adh.
Figure 2. Design and verification of degenerate primers.
(A) WebLogo of the amino acid sequences of 51 L.ADH, (B) Degenerate primers, and (C) the electrophoretogram of the amplification by degenerate primer.
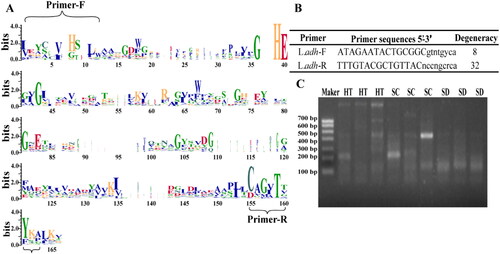
Table 1. Sequencing library and fragment length.
Diversity of L.adh in Daqu
After amplification by degenerate primers and sequencing, 45,269 reads were obtained from 3 Daqus. The reads were clustered into 8,415 OTUs and 2,570 OTUs were annotated as L.adh by BlastX with 100% similarity (). Three kinds of L.adh fragments (library 1:100–200 bp, library 2:200–400 bp and library 3:400–600bp) were found in Daqu of SD and two kinds of L.adh fragments (library 2:200–400 bp and library 3:400–600bp) were found in HT and SC Daqu, respectively. But, the number of L.adh OUT in SC Daqu was 2063 and it was higher than HT (730) and SD (941). This indicates that there were more potential microorganisms associated with higher alcohol synthesis in SC Daqu. As showed in , the Shannon and Chao 1 indices of L.adh in SC was the highest (library 2:6.416 and 1,346; library 3: 6.128 and 1,087) and it indicated that the diversity and species number of potential microorganisms associated with higher alcohol synthesis in SC was higher than HT and SD.
Table 2. The diversity of the L.adh gene fragments of Daqu.
The coverage index was applied to evaluate the sequencing coverage and the coverage index of L.adh was between 0.73-0.95, which partly reflected the real situation of L.adh sequencing (). Therefore, the designed degenerate primers could effectively amplify the target fragments and achieve good coverage in high-throughput sequencing.
Composition of L.adh in Daqu
After functional annotation and taxonomic classification, L.adh in Daqu of HT, SC and SD mostly belong to the genus of Bacillus, Enterobacter, Aspergillus, and Mycobacterium (). The results indicated that the composition of potential microorganisms associated with higher alcohol synthesis in three Daqu was different.
Figure 3. Annotation of the potential producers of higher alcohol of different Daqu in genus level.
(A) Library 1, (B) Library 2, and (C) Library 3.
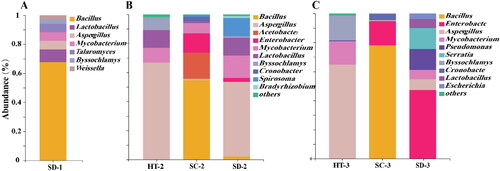
In library 1, L.adh of SD-1 was mainly from Bacillus (). In library 2, L.adh of HT-2 and SD-2 was from Aspergillus, L.adh of SC-2 was from Bacillus with more than 50% relative abundance. Acetobacter derived L.adh was only detected in SC-2 and its relative abundance was greater than 10% (). In library 3, L.adh was mainly from Aspergillus and its relative abundance were higher than 60% in HT-3. In SC-3, L.adh was mainly from Bacillus and had more than 70% relative abundance. In SD-3, L.adh was mainly from the genus of Enterobacter and Serratia and with abundances of 40% and 10%, respectively ().
The dominant L.adh were same in different fragment lengths in same Daqu, but the origin of L.adh in different Daqu was diverse. Specifically, L.adh from the genus of Bacillus and Enterobacter were not found in HT but were dominant in SC and SD.
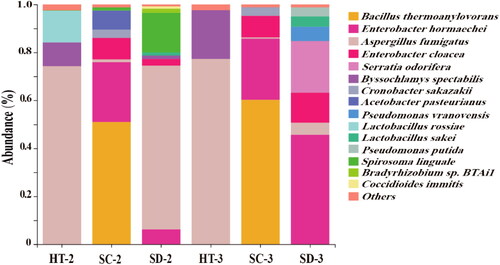
Further analysis revealed that 81.31% of L.adh could be a trace in species level () by taxonomic classification. In general, the abundance of L.adh fragments at the species level was similar with the genus level. There are 15 microorganisms with relative abundance greater than 1% at the species level, among which the dominant microorganisms with higher alcohol-converting potential were Aspergillus fumigatus, Bacillus thermoamylovorans, Enterobacter cloacae and Enterobacter hormaechei. A. fumigatus was the dominant microorganisms with higher alcohol-converting potential in HT-2, HT-3 and SD-2. B. thermoamylovorans was only found in SC-2 and SC-3 with a relative abundance of greater than 50%. E. cloacae and E. hormaechei were not found in HT-2 and HT-3, while found in other Daqu. Moreover, E. hormaechei was the dominant of potential microorganisms associated with higher alcohol synthesis in SD-3 with more than 40% relative abundance (). In each Daqu, the sum of abundances of the four dominant microorganisms derived from L.adh gene was exceeded 60%. Therefore, A. fumigatus, B. thermoamylovorans, E. cloacae, and E. hormaechei were the dominant microorganisms with higher alcohol-converting potential in this study. In summary, the degenerate primers could amplify L.adh gene fragment and there were differences in dominant microorganisms with higher alcohol-converting potential in different Daqu.
Figure 4. Annotation of the potential producers of higher alcohol of different Daqu in species level.
Evolutionary analysis of L.adh in Daqu
The results of evolutionary analysis showed that the different L.adh from the microorganisms with higher alcohol-converting potential could be cluster into one evolutionary branch (). L.adh of HT could be further divided into 5 categories (L.adh 1, L.adh 2, L.adh 3, L.adh 4, and L.adh 5) and the dominant microorganisms with higher alcohol-converting potential in L.adh 1, L.adh 2, L.adh 3 and L.adh 4 were Lactobacillus, Mycobacterium, Byssochlamys and Aspergillus, respectively. L.adh 5 included the genus of Anacromyxobacter, Brevundimonas, Pseudomonas and Spirosoma (). There were 3 categories of L.adh in SC (L.adh 6, L.adh 7, and L.adh 8), and they were from the genus of Enterobacter, Acetobacter, and Bacillus, respectively (). Moreover, L.adh in SD can be divided into 5 categories (L.adh 4, L.adh 2, L.adh 1, L.adh 9, and L.adh 10) were mostly from the genus of Aspergillus, Mycobacterium, Lactobacillus, and Enterobacter, respectively. L.adh 10 was from the genus of Bacillus, Byssochlamys and Tatumella ().
Figure 5. Phylogenetic tree of L.adh gene fragments of Daqu.
(A) HT, (B) SC, and (C) SD.
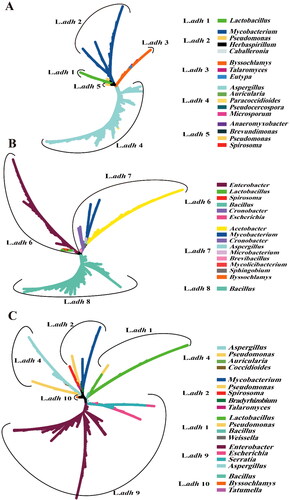
There were evolutionary branches of L.adh from Lactobacillus, Aspergillus and Mycobacterium in HT and SD with different properties. In HT, L.adh 1 included the genus of Lactobacillus, but included Pseudomonas and Lactobacillus in SD. In HT and SD, L.adh 2 include Mycobacterium and Pseudomonas, and L.adh 4 included Aspergillus and Auricularia. The microorganisms with higher alcohol-converting potential in the L.adh 2 are bacteria and in the L.adh 4 branch is fungi in HT. In SD, L.adh 2 included Talaromyces and L.adh 4 included Pseudomonas, indicating that bacteria and fungi coexist in this branch. Based on the above, a total of 13 evolutionary branches were obtained by the evolution analysis of L.adh fragment and divided into 10 types of potential higher alcohol-producing microorganisms ().
Cluster of L.adh in Daqu
In order to lay a foundation for further analysis, the OTUs of L.adh were further clustered with 97% similarity and 54 new OTUs were obtained (named L97-1∼L97-54). The results showed that L.adh fragments from the same genus could produce multiple new OTUs, such as Aspergillus, Bacillus, Enterobacter, Lactobacillus, Acetobacter, and Cronobacter (). L.adh that belongs to 9 fungi were clustered into 2 categories (L97-1 and L97-39). L.adh of Bacterial belongs to the genus of Escherichia and Serratia (L97-33), Caballeronia and Pseudomonas (L97-44) were clustered into 1 category, respectively. Therefore, we propose that the similarity of L.adh in fungi is higher than in bacteria. In addition, taxonomic classification of L.adh 4 and L.adh 3 was consistent with the results of clustering L97-1 and L97-39 in the evolutionary analysis (). It indicated that L.adh 9 contained the L97-33 and was gathered in one branch of L.adh 9 ().
Table 3. Sequencing results of 97% similarity of the L.adh gene fragments of Daqu.
Discussion
In this study, L.adh was used as the maker gene to evaluate the diversity and composition of the microorganisms with higher alcohol-converting potential in Daqu. Due to low conservation of functional gene, primer design was the key to amplify and sequencing of functional gene. Degenerate primers based on amino acid conservative sequences have been successfully applied in many research fields. pah and nifH gene was amplified by degenerate primers to analyze its diversity in oil field, mangrove forests and seawater (Jayakumar et al., Citation2017; Liang et al., Citation2019) and the soil microbial enzyme genes associated with phosphorus mineralization, dissolution and absorption was amplified by degenerate primers to study its diversity (Bergkemper et al., Citation2016). The degenerate primers were also designed for the hppd gene to investigate the evolution of HPPD bacterial communities in herbicides (Thiour-Mauprivez et al., Citation2020) and it has also been used to study species composition and diversity of lactate dehydrogenase (L-LDH/LDH) in traditional fermented vinegar (Sheng et al., Citation2019). Based on conservative amino acid sequences, the degenerate primers (L.adh-F/L.adh-R) of the L.adh gene were successfully designed and obtain target fragments. The results showed that most of the L.adh gene fragments were bacterial, while a few were derived from fungi. The dominant microorganisms with higher alcohol-converting potential were fungi and bacteria in HT and SC, respectively.
The microbial composition in different Daqu was quite different (Cai et al., Citation2021; Deng et al., Citation2020) and this difference was mainly determined by producing environment. In the three Daqu, Bacillus, Enterobacter, Aspergillus and Mycobacterium were the dominant genera with higher alcohol-converting potential and B. thermoamylovorans, E. cloacae, E. hormaechei, and A. fumigatus were the dominant species with higher alcohol-converting potential. The presence of ADHs in animals, plants and microorganisms has previously been reported (Hans et al., Citation1999), 7 medium-chain ADHs were found in S. cerevisiae (de Smidt et al., Citation2008) and alcohol-consuming ADHs could be detected in Pichia (Karaoğlan et al., Citation2020). However, S. cerevisiae and Pichia were not found in this study and Saccharomyces and Pichia are non-dominant in Daqu (Li et al., Citation2016; Citation2018; Wang et al., Citation2021).
The results showed that the fungus (Aspergillus) was the dominant microorganisms with higher alcohol-converting potential in HT and SD and it was reported that Aspergillus flavus contain alcohol dehydrogenases (ADHI) (Woloshuk & Payne, Citation1994). In addition, ADH has been successfully obtained from Bacillus (Çelik & Aktaş, Citation2013; Dowds et al., Citation1988; Kato et al., Citation2001; Robinson et al., Citation1994), Mycobacterium smegmatis (Galamba et al., Citation2001), Acetobacter (Chowdhury et al., Citation2021; Masud et al., Citation2011), and Lactobacillus (Chen et al., Citation2010; Rauter et al., Citation2015) by genetic engineering method. Lactobacillus reuteri has five ADHs with zinc-dependent properties (Hu et al., Citation2019) and Lactobacillus were the dominant genus in the brewing process of different flavor Baijiu that can metabolize and produce n-propanol (Lu et al., Citation2019). These results indicated that the metabolism of higher alcohols in mixed fermentation was participated by many microorganisms and the existence of different microorganisms related to higher alcohol metabolism may have an important impact on the content of higher alcohol in the fermentation process of Baijiu.
Based on evolutionary analysis, Bacillus, Enterobacter, Aspergillus, and Mycobacterium were the dominant microorganisms with higher alcohol-converting potential in L.adh 8, L.adh 6, L.adh 4, and L.adh 2, respectively. The L.adh in Daqu could be classified into 10 evolutionary types and different genus of microorganisms with higher alcohol-converting potential has co-existed in the same evolutionary branch. The evolutionary relationships among different genera of fungal are mostly different (Thompson et al., Citation2018). The results showed that different L.adh fragments had the same evolutionary relationship and the evolutionary relationship in bacteria and fungi were similar.
Currently, the study on synthesis mechanism of metabolite in mixed microorganisms is mainly based on the method of establishing the relationship between non-functional gene (16S rRNA or ITS) abundance and metabolite content or environmental factors (Liu et al., Citation2018b; Ma et al., Citation2020; Qu et al., Citation2020; Sun et al., Citation2020; Wang et al., Citation2018). However, this method can`t establish direct contact between microorganism and metabolite in the mixed fermentation process. Functional genes in metabolic pathways can be used as a marker gene for characterizing metabolic functions (Yilmaz et al., Citation2011) and study the potential producer of metabolite in the mixed microorganism process (Sheng et al., Citation2019; Chai et al., Citation2019). Moreover, the application of amplification technology can also improve the probability of low abundance gene detection. In this study, L.adh was applied as the maker gene to study the microorganisms with higher alcohol-converting potential in Daqu. The result showed that the degenerate primer can amplify the L.adh fragment and the microorganisms with higher alcohol-converting potential can be obtained by bioinformatics analysis. The differences of microorganisms related to higher alcohol metabolism in different Daqu can be investigated by analyzing the diversity and composition of L.adh. In addition, the fragments of L.adh in Daqu can be classified into different types by their evolutionary relationship and the types contained different genera of microorganism can further be applied as operating unit combined with statistical methods to study the synthesis mechanism of higher alcohols in mixed fermentation process.
Conclusions
Higher alcohols, as important flavor substances in Baijiu, are produced by microorganisms during fermentation. Daqu is an important source of microorganism for Baijiu production. In this study, L.adh, a key gene of higher alcohol metabolism, was applied as a marker gene to study the diversity and composition of microorganisms with higher alcohol-converting potential in Daqu. The results showed that the degenerate primers of L.adh were able to amplify the target gene and had good coverage in high-throughput sequencing. The diversity and composition of the microorganisms with higher alcohol-converting potential in the three kinds of Daqu were different and L.adh could be used as a marker gene to track the microorganisms with higher alcohol-converting potential in Daqu. L.adh had the potential to be used as a probe for tracing higher alcohol-producing microorganisms. Moreover, L.adh fragments in Daqu can be divided into multiple types according to the evolutionary relationship and the synthesis mechanism of higher alcohols can be further revealed by establish the relationship between the abundance of L.adh (OTUs) and higher alcohols in the fermentation process of Baijiu.
Authors’ contributions
Mandlaa, Ziyu Sun and Zhongjun Chen: Methodology, Writing-original draft, Writing- review and editing, Funding acquisition. Yuting Ren and Meiling Qiao: Methodology, data curation, Writing-original draft. Guojun Ren and Yuzhen Yang: Conceptualization, Resources.
Table S1.xlsx
Download MS Excel (11.8 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bergkemper, F., Kublik, S., Lang, F., Krüger, J., Vestergaard, G., Schloter, M., & Schulz, S. (2016). Novel oligonucleotide primers reveal a high diversity of microbes which drive phosphorous turnover in soil. Journal of Microbiological Methods, 125, 1–12. https://doi.org/10.1016/j.mimet.2016.04.011
- Boyce, R., Chilana, P., & Rose, T. (2009). iCODEHOP: A new interactive program for designing Consensus-degenerate hybrid oligonucleotide primers from multiply aligned protein sequences. Nucleic Acids Research, 37(Web Server issue), W222–228. https://doi.org/10.1093/nar/gkp379
- Cai, W., Wang, Y., Ni, H., Liu, Z., Liu, J., Zhong, J., Hou, Q., Shan, C., Yang, X., & Guo, Z. (2021). Diversity of microbiota, microbial functions, and flavor in different types of low-temperature Daqu. Food Research International (Ottawa, Ont.), 150(Pt A), 110734. https://doi.org/10.1016/j.foodres.2021.110734
- Çelik, A., & Aktaş, F. (2013). A new NADH-dependent, zinc containing alcohol dehydrogenase from Bacillus thuringiensis serovar israelensis involved in oxidations of short to medium chain primary alcohols. Journal of Molecular Catalysis B: Enzymatic, 89, 114–121. https://doi.org/10.1016/j.molcatb.2013.01.005
- Chai, L.-J., Lu, Z.-M., Zhang, X.-J., Ma, J., Xu, P.-X., Qian, W., Xiao, C., Wang, S.-T., Shen, C.-H., Shi, J.-S., & Zheng-Hong, X. (2019). Zooming in on butyrate-producing clostridial consortia in the fermented grains of Baijiu via gene sequence-guided microbial isolation. Frontiers in Microbiology, 10, 1397. https://doi.org/10.3389/fmicb.2019.01397
- Chen, Q., Hu, Y., Zhao, W., Zhu, C., & Zhu, B. (2010). Cloning, expression, and characterization of a novel (S)-specific alcohol dehydrogenase from Lactobacillus kefir. Applied Biochemistry and Biotechnology, 160(1), 19–29. https://doi.org/10.1007/s12010-008-8442-6
- Chowdhury, N. P., Moon, J., & Müller, V. (2021). Adh4, an alcohol dehydrogenase controls alcohol formation within bacterial microcompartments in the acetogenic bacterium Acetobacterium woodii. Environmental Microbiology, 23(1), 499–511. https://doi.org/10.1111/1462-2920.15340
- Cole, J. R., Wang, Q., Cardenas, E., Fish, J., Chai, B., Farris, R. J., Kulam-Syed-Mohideen, A. S., McGarrell, D. M., Marsh, T., Garrity, G. M., & Tiedje, J. M. (2009). The ribosomal database project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Research, 37(Database issue), D141–145. https://doi.org/10.1093/nar/gkn879
- de Smidt, O., Du Preez, J. C., & Albertyn, J. (2008). The alcohol dehydrogenases of saccharomyces cerevisiae : A comprehensive review. FEMS Yeast Research, 8(7), 967–978. https://doi.org/10.1111/j.1567-1364.2008.00387.x
- Deetae, P., Bonnarme, P., Spinnler, H., & Helinck, S. (2007). Production of volatile aroma compounds by bacterial strains isolated from different surface-ripened French cheeses. Applied Microbiology and Biotechnology, 76(5), 1161–1171. https://doi.org/10.1007/s00253-007-1095-5
- Deng, L., Mao, X., Liu, D., Ning, X.-Q., Shen, Y., Chen, B., Nie, H.-F., Huang, D., & Luo, H.-B. (2020). Comparative analysis of physicochemical properties and microbial composition in high-temperature Daqu with different colors. Frontiers in Microbiology, 11, 588117. https://doi.org/10.3389/fmicb.2020.588117
- Dowds, B., Sheehan, M., Bailey, C., & McConnell, D. (1988). Cloning and characterization of the gene for a methanol-utilising alcohol dehydrogenase from Bacillus stearothermophilus. Gene, 68(1), 11–22. https://doi.org/10.1016/0378-1119(88)90594-X
- Fang, C., Du, H., Jia, W., & Xu, Y. (2018). Compositional differences and similarities between typical Chinese Baijiu and Western liquor as revealed by mass spectrometry-based metabolomics. Metabolites, 9(1), 2. https://doi.org/10.3390/metabo9010002
- Fang, G., Mu, X., & Jiang, Y. (2019). Correlation between structural shifts of bacterial community and organic acid formation during the fermentation of Zhejiang rosy vinegar. Food Sci, 40, 177–184. https://doi.org/10.7506/spkx1002-6630-20181012-098
- Galamba, A., Soetaert, K., Buyssens, P., Monnaie, D., Jacobs, P., & Content, J. (2001). Molecular and biochemical characterisation of Mycobacterium smegmatis alcohol dehydrogenase C. FEMS Microbiology Letters, 196(1), 51–56. https://doi.org/10.1111/j.1574-6968.2001.tb10539.x
- Gerong, Z., Huang, F., & Han, X. (2019). A method for determining associations between drinking discomforts and key higher alcohols in strong-aroma Baijiu. Food Ferment Ind, 45(14), 191–195. https://doi.org/10.13995/j.cnki.11-1802/ts.019118
- Gu, Y., D Van Nostrand, J., Wu, L., He, Z., Qin, Y., Zhao, F.-J., & Zhou, J. (2017). Bacterial community and arsenic functional genes diversity in arsenic contaminated soils from different geographic locations. PLoS One, 12(5), e0176696. https://doi.org/10.1371/journal.pone.0176696
- Hans, J., Jan, H., & Bengt, P. (1999). SDR and MDR: Completed genome sequences show these protein families to be large, of old origin, and of complex nature. FEBS Letters. 445, 261–264. https://doi.org/10.1016/S0014-5793(99)00130-1
- Hao, Y., Wang, D., Terigele, Y., Guo, H., & Chen, Z. (2021). Screening butanol-dehydrogenase producing microorganism and study on the enzymatic properties. Food Res Develop, 42, 197–204. https://doi.org/10.12161/j.issn.1005-6521.2021.02.032
- Hazelwood, L., Daran, J., Maris, A., Pronk, J., & Dickinson, J. (2008). The Ehrlich pathway for fusel alcohol production: a century of research on Saccharomyces cerevisiae metabolism. Applied and Environmental Microbiology, 74(8), 2259–2266. https://doi.org/10.1128/AEM.02625-07
- Helge-Friedrich, T. (2004). Analysis for free: Comparing programs for sequence analysis. Briefings in Bioinfor, 5, 82–87. https://doi.org/10.1093/bib/5.1.82
- Holt, S., Miks, M., de Carvalho, B., Foulquié-Moreno, M., & Thevelein, J. (2019). The molecular biology of fruity and floral aromas in beer and other alcoholic beverages. FEMS Microbiology Reviews, 43(3), 193–222. https://doi.org/10.1093/femsre/fuy041
- Hu, Z., Jia, P., Bai, Y., Fan, T., Zheng, X., & Cai, Y. (2019). Characterisation of five alcohol dehydrogenases from Lactobacillus reuteri DSM20016. Process Biochemistry. 86, 73–79. https://doi.org/10.1016/j.procbio.2019.08.010
- Jayakumar, A., Chang, B. X., Widner, B., Bernhardt, P., Mulholland, M. R., & Ward, B. B. (2017). Biological nitrogen fixation in the oxygen-minimum region of the eastern tropical North Pacific ocean. The ISME Journal, 11(10), 2356–2367. https://doi.org/10.1038/ismej.2017.97
- Karaoğlan, M., Erden-Karaoğlan, F., Yılmaz, S., & İnan, M. (2020). Identification of major ADH genes in ethanol metabolism of Pichia pastoris. Yeast (Chichester, England), 37(2), 227–236. https://doi.org/10.1002/yea.3443
- Kato, T., Miyanaga, A., Haruki, M., Imanaka, T., Morikawa, M., & Kanaya, S. (2001). Gene cloning of an alcohol dehydrogenase from thermophilic alkane-degrading Bacillus thermoleovorans B23. Journal of Bioscience and Bioengineering, 91(1), 100–102. https://doi.org/10.1016/S1389-1723(01)80122-5
- Kibbe, W. (2007). OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Research, 35(Web Server issue), W43–46. https://doi.org/10.1093/nar/gkm234
- Kumar, S., Stecher, G., Li, M., Knyaz, C., & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35(6), 1547–1549. https://doi.org/10.1093/molbev/msy096
- Letunic, I., & Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Research, 49(W1), W293–W296. https://doi.org/10.1093/nar/gkab301
- Liang, C., Huang, Y., Wang, Y., Ye, Q., Zhang, Z., & Wang, H. (2019). Distribution of bacterial polycyclic aromatic hydrocarbon (PAH) ring-hydroxylating dioxygenases genes in oilfield soils and mangrove sediments explored by gene-targeted metagenomics. Applied Microbiology and Biotechnology, 103(5), 2427–2440. https://doi.org/10.1007/s00253-018-09613-x
- Li, P., Lin, W., Liu, X., Wang, X., & Luo, L. (2016). Environmental factors affecting microbiota dynamics during traditional solid-state fermentation of Chinese Daqu starter. Frontiers in Microbiology, 7, 1237. https://doi.org/10.3389/fmicb.2016.01237
- Liu, P., & Miao, L. (2020). Multiple batches of fermentation promote the formation of functional microbiota in Chinese miscellaneous-flavor Baijiu fermentation. Frontiers in Microbiology, 11, 75. https://doi.org/10.3389/fmicb.2020.00075
- Liu, M., Xu, Z., Zhong, Q., Liu, C., Li, X., & Yuan, X. (2018b). Comparison of the threshold determination method for Chinese liquor (Baijiu) flavor substances. J Chin I Food Sci Technol, 18, 253–260. https://doi.org/10.16429/j.1009-7848.2018.04.033
- Liu, F., Zhou, X., Chen, X., Chen, J., Du, G., & Fang, F. (2018a). Microbial community of fermented grains and its correlation with organic acids biosynthesis during Yanghe strong-aroma liquor manufacturing process. Acta Microbiol Sin, 58, 2087–2099. https://doi.org/10.13343/j.cnki.wsxb.20170616
- Li, J., Wang, Y., He, H., Liu, G., Li, X., & Chen, B. (2018). High-throughput sequencing revealed fungal community structures at high temperature Daqu and medium temperature Daqu. Food and Fermentation Industries 44, 52–59. https://doi.org/10.13995/j.cnki.11-1802/ts.018043
- Lu, J., Yang, F., Yang, J., Chen, L., Liu, Y., & Wang, H. (2019). Source tracing of 1-propanol-producing microorganisms during Baijiu fermentation. China Brewing, 38, 151–155. https://doi.org/10.11882/j.issn.0254-5071.2019.07.030
- Ma, B., Fan, E., Li, Z., Zhang, Y., Zhang, Z., & Guo, Y. (2020). Microbial diversity and its relationship with flavor compounds in the process of Daqu making of laobaigan- flavor Liquor. Food and Fermentation Industries, 46, 7–16. https://doi.org/10.13995/j.cnki.11-1802/ts.023902
- Magoč, T., & Salzberg, S. L. (2011). FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics (Oxford, England), 27(21), 2957–2963. https://doi.org/10.1093/bioinformatics/btr507
- Masud, U., Matsushita, K., & Theeragool, G. (2011). Molecular cloning and characterization of two inducible NAD(+)-adh genes encoding NAD(+)-dependent alcohol dehydrogenases from Acetobacter pasteurianus SKU1108. Journal of Bioscience and Bioengineering, 112(5), 422–431. https://doi.org/10.1016/j.jbiosc.2011.07.020
- Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., Tosatto, S. C. E., Paladin, L., Raj, S., Richardson, L. J., Finn, R. D., & Bateman, A. (2021). Pfam: The protein families database in 2021. Nucleic Acids Research, 49(D1), D412–D419. https://doi.org/10.1093/nar/gkaa913
- Pang, X.-N., Han, B.-Z., Huang, X.-N., Zhang, X., Hou, L.-F., Cao, M., Gao, L.-J., Hu, G.-H., & Chen, J.-Y. (2018). Effect of the environment microbiota on the flavour of light-flavour Baijiu during spontaneous fermentation. Scientific Reports, 8(1), 3396. https://doi.org/10.1038/s41598-018-21814-y
- Qu, G., Tang, J., Jiang, J., Yang, Q., Liu, C., & Wu, Q. (2020). Characteristics of higher alcohols synthesized by microbiota in the fermentation process of light aroma type Baijiu started by Xiaoqu. Food and Fermentation Industries, 47, 32–37. https://doi.org/10.13995/j.cnki.11-1802/ts.025697
- Rauter, M., Prokoph, A., Kasprzak, J., Becker, K., Baronian, K., Bode, R., Kunze, G., & Vorbrodt, H.- M. (2015). Coexpression of Lactobacillus brevis ADH with GDH or G6PDH in Arxula adeninivorans for the synthesis of 1-(R)-phenylethanol. Applied Microbiology and Biotechnology, 99(11), 4723–4733. https://doi.org/10.1007/s00253-014-6297-z
- Robinson, G., Bailey, C., & Dowds, B. (1994). Gene structure and amino acid sequences of alcohol dehydrogenases of Bacillus stearothermophilus. Biochimica et Biophysica Acta, 1218(3), 432–434. https://doi.org/10.1016/0167-4781(94)90199-6
- Shen, M., Zhang, C., Wang, Y., Wang, J., Cai, X., & Xing, L. (2018). Research progress on microbial enzyme system in Baijiu Daqu. China Brewing, 37, 7–11. https://doi.org/10.11882/j.issn.0254-5071.2018.04.002
- Sheng, M., Lu, Z., Zhang, X., Chai, L., Yu, Y., & Shi, J. (2019). Species-level diversity of lactate-producing bacteria in acetic acid fermentation of Zhenjiang aromatic vinegar. Chin J Appl Environ Biol, 25, 1434–1440. https://doi.org/10.19675/j.cnki.1006-687x.2019.02005
- Sun, H., Liu, S., Mao, J., Yu, Z., Lin, Z., & Mao, J. (2020). New insights into the impacts of Huangjiu components on intoxication. Food Chemistry, 317, 126420. https://doi.org/10.1016/j.foodchem.2020.126420
- Sun, L., Zhang, W., Xu, J., Liu, J., Xin, X., & Ren, Q. (2020). Changes of microbial community structure and effects on higher alcohols in oat Huangjiu fermentation process. Journal of Food Science and Technology. 38, 43–52. https://doi.org/10.3969/j.issn.2095-6002.2020.01.006
- Thiour-Mauprivez, C., Devers-Lamrani, M., Mounier, A., Beguet, J., Spor, A., Calvayrac, C., Barthelmebs, L., & Martin-Laurent, F. (2020). Design of a degenerate primer pair to target a bacterial functional community: The hppd bacterial gene coding for the enzyme targeted by herbicides, a study case. Journal of Microbiological Methods, 170, 105839. https://doi.org/10.1016/j.mimet.2020.105839
- Thompson, C., Freitas, L., & Salzano, F. (2018). Molecular evolution and functional divergence of alcohol dehydrogenases in animals, fungi and plants. Genetics and Molecular Biology, 41(1 suppl 1), 341–354. https://doi.org/10.1590/1678-4685-GMB-2017-0047
- UniProt Consortium. (2021). UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Research, 49(D1), D480–D489. https://doi.org/10.1093/nar/gkaa1100
- Wang, X., Du, H., & Xu, Y. (2018). Spatial distribution of microbial community during Chinese light-flavor liquor fermentation. Food and Fermentation Industries, 44, 1–8. https://doi.org/10.13995/j.cnki.11-1802/ts.017334
- Wang, S., Miao, L., Zhang, M., Liu, P., & Liu, J. (2017). Analysis of the flavor compounds fermented by sauce-flavor-producing Bacillus strain isolated from fermented grains of mixed strong-sauce-flavor Baijiu. China Brewing, 36, 61–65. https://doi.org/10.11882/j.issn.0254-5071.2017.10.014
- Wang, P., Wu, Q., & Xu, Y. (2018). Core microbiota in Chinese liquor fermentation and associations with environmental factors. Acta Microbiol Sin, 58, 142–153. https://doi.org/10.13343/j.cnki.wsxb.20170060
- Wang, B., Wu, Q., Xu, Y., & Sun, B. (2021). Recent advances and perspectives in study of microbiome in Chinese Jiuqu starter. Microbiol Chi, 48, 1737–1746. https://doi.org/10.13344/j.microbiol.china.200650
- Whitham, J., Pawlak, J., & Grunden, A. (2016). Clostridium ljungdahlii: A Review of the Development of an Industrial Bio-catalyst. Curr Biotechnol, 5, 54–70. https://doi.org/10.2174/2211550105666151208211335
- Woloshuk, C., & Payne, G. A. (1994). The alcohol dehydrogenase gene adh1 is induced in Aspergillus flavus grown on medium conducive to aflatoxin biosynthesis. Applied and Environmental Microbiology, 60(2), 670–676. https://doi.org/10.1002/bit.260430310
- Xiu, L., Kunliang, G., & Hongxun, Z. (2012). Determination of microbial diversity in Daqu, a fermentation starter culture of Maotai liquor, using nested PCR-denaturing gradient gel electrophoresis. World Journal of Microbiology & Biotechnology, 28(6), 2375–2381. https://doi.org/10.1007/s11274-012-1045-y
- Xu, Y., Fan, W., Wu, Q., & Wang, H. (2012). Research advance in flavor-oriented analytic technology for liquor-making. Liquor-Making Sci Technol, 1, 17–23. https://doi.org/10.13746/j.njkj.2014533
- Yilmaz, P., Kottmann, R., Field, D., Knight, R., Cole, J. R., Amaral-Zettler, L., Gilbert, J. A., Karsch-Mizrachi, I., Johnston, A., Cochrane, G., Vaughan, R., Hunter, C., Park, J., Morrison, N., Rocca-Serra, P., Sterk, P., Arumugam, M., Bailey, M., Baumgartner, L., … Glöckner, F. O. (2011). Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nature Biotechnology, 29(5), 415–420. https://doi.org/10.1038/nbt.1823
- Zhang, L., Wu, C., Ding, X., Zheng, J., & Zhou, R. (2014). Characterisation of microbial communities in Chinese liquor fermentation starters Daqu using nested PCR-DGGE. World Journal of Microbiology & Biotechnology, 30(12), 3055–3063. https://doi.org/10.1007/s11274-014-1732-y
- Zhao, C., Yan, X., Yang, S., & Chen, F. (2017). Screening of Bacillus strains from Luzhou-flavor liquor making for high-yield ethyl hexanoate and low-yield propanol. LWT, 77, 60–66. https://doi.org/10.1007/s11274-014-1732-y
- Zhu, J., Shi, J., Lu, Y., Liu, L., & Liu, Y. (2016). Application of strains of Geotrichum spp. to decrease higher alcohols and to increase esters. Journal of the Institute of Brewing, 122(1), 147–155. https://doi.org/10.1002/jib.287
- Zou, W., Zhao, C., & Luo, H. (2018). Diversity and function of microbial community in Chinese strong-flavor Baijiu ecosystem: A review. Frontiers in Microbiology, 9, 671. https://doi.org/10.3389/fmicb.2018.00671