
ABSTRACT
Pathogenic protists are a group of organisms responsible for causing a variety of human diseases including malaria, sleeping sickness, Chagas disease, leishmaniasis, and toxoplasmosis, among others. These diseases, which affect more than one billion people globally, mainly the poorest populations, are characterized by severe chronic stages and the lack of effective antiparasitic treatment. Parasitic protists display complex life-cycles and go through different cellular transformations in order to adapt to the different hosts they live in. Autophagy, a highly conserved cellular degradation process, has emerged as a key mechanism required for these differentiation processes, as well as other functions that are crucial to parasite fitness. In contrast to yeasts and mammals, protist autophagy is characterized by a modest number of conserved autophagy-related proteins (ATGs) that, even though, can drive the autophagosome formation and degradation. In addition, during their intracellular cycle, the interaction of these pathogens with the host autophagy system plays a crucial role resulting in a beneficial or harmful effect that is important for the outcome of the infection. In this review, we summarize the current state of knowledge on autophagy and other related mechanisms in pathogenic protists and their hosts. We sought to emphasize when, how, and why this process takes place, and the effects it may have on the parasitic cycle. A better understanding of the significance of autophagy for the protist life-cycle will potentially be helpful to design novel anti-parasitic strategies.
Abbreviations: AAs: amino acids; ATGs: autophagy-related proteins; ADCD; autophagy-dependent cell death; AMPK: 5’ adenosine monophosphate-activated protein kinase; CD40: Cluster of differentiation 40; gHBSS: Hanks’ Balanced Salt Solution; GO: gene ontology; IFN-γ: IFN-gamma; LC3: mammalian microtubule-associated protein light chain 3; LAP; LC3-associated phagocytosis; LECA: last eukaryotic common ancestor; 3-MA: 3-methyladenine; MTOR; Mechanistic target of rapamycin kinase; MDC: monodansylcadaverine; NDP52: nuclear dot protein 52; PAAR: Plasmodium-Associated Autophagy-Related response; PE: phosphatidylethanolamine: PCD: programmed cell death; PND: programmed nuclear death; PtdIns3K: class III phosphatidylinositol 3-kinase; PtdIns3P: phosphatidylinositol 3-phosphate; PV: parasitophorous vacuole; PVM: parasitophorous vacuole membrane; SNARE: soluble N-ethylmaleimide-sensitive-factor attachment receptor; SQSTM1/p62: sequestosome-1; TEM: transmission electron microscopy; TNF-α: tumor necrosis factor-alpha; TVN: tubovesicular network; Ub: ubiquitin; UPS: ubiquitin-proteasome system; Vps: vacuolar protein sorting.
The life-cycle of parasitic protists
Protists are a diverse collection of organisms, mainly unicellular, distributed in the main groups of eukaryotic lineages. Many of them are free-living organisms that are important for the environment because they carry out processes like photosynthesis (plant-like algae) and waste decomposition (slime and water molds), while others (animal-like protists) are medically relevant because they cause a variety of human diseases.
Eukaryotic lineages; that constitute the eukaryotic tree of life (eToL), are currently grouped in a short number of supergroups or clades based almost exclusively on molecular phylogenies, in contrast to earlier models derived from molecular and other biological data [Citation1,Citation2]. Discoba (containing Kinetoplastida) and Metamonads, both belonging to the previous clade Excavate; Apicomplexa (included in the recently named TSAR group: Telonemia, Stramenopila, Alveolata and Rhizaria); and Amoebozoa lineages possess the most important species of protists causing human diseases [Citation3] ().
Table 1. Classification of parasitic protists and associated diseases
The diseases caused by these parasitic protists range from very mild to life-threatening infections. Individuals whose defenses can control but not eliminate a parasitic infection become carriers and constitute a source of infection for others. Many protist infections that are asymptomatic or mild in normal individuals can be life-threatening in immunosuppressed patients, particularly those with acquired immune deficiency syndrome (AIDS). For instance, Toxoplasma gondii, a widely distributed parasite (infects up to a third of the world’s human population), usually causes a rather mild initial illness followed by a long-lasting latent infection [Citation4]. AIDS patients, however, can develop fatal toxoplasmic encephalitis. The lack of effective vaccines, the paucity of reliable drugs, and other problems, including difficulties in vector control, have prompted the World Health Organization to target some specific diseases, collectively named Neglected Tropical Diseases (NTDs), for increased research and training. Four of these NTDs are infections caused by parasitic protists: malaria, American and African trypanosomiasis, and leishmaniasis.
Pathogenic protists display complex life-cycles alternating between several stages that differ in structure and activity. These parasitic forms develop in different types of hosts. The Toxoplasma gondii life-cycle occurs between members of the Felidae family (domestic cats and their relatives), which are the definitive hosts, and humans and other animals that are the intermediate hosts. Plasmodium sp. and trypanosomatids (Leishmania sp., T. brucei, and T. cruzi), on the other hand, alternate between insects and mammals, which are the vectors and the reservoirs of diseases, respectively. Parasitic forms found in insects are extracellular whereas mammalian forms have the ability to enter into host cells and establish their replicative niche there, a strategy to evade the host immune response. Extracellular parasites have developed other strategies to escape the immune system, as exemplified by the bloodstream forms of Trypanosoma brucei. This parasite possesses a large repertoire of genes encoding for Variant Surface Glycoproteins (VSG). During infection, this antigenic variation strategy allows evasion from the mammalian host immune system [Citation5].
In the following paragraphs, we briefly present the life-cycles of protists whose connection with autophagy have been established. shows a graphic representation of these cycles with the developmental stages of the main parasitic protists described therein, their hosts, and the main organs they parasitize. However, it should be noted that there are other pathogenic protists including Metamonada responsible for intestinal (Giardia lamblia) or sexually transmitted (Trichomonas vaginalis) diseases, as well as pathogenic Amoebozoa (like the dysentery-causing Entamoeba histolytica), for which autophagy has been described to some extent, although they are much less studied.
Figure 1. Life-cycle of pathogenic protists: Leishmania spp., T. brucei, T. cruzi, T. gondii, and Plasmodium spp. The figure shows the parasitic forms found in trypanosomatid and apicomplexan protists during their life-cycle. The main host cells targeted by these protists, the organs affected by the infections, and the vector that transmit them are depicted. Created with BioRender.com.
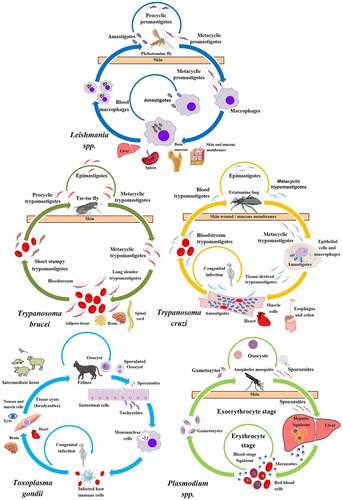
Leishmania species are transmitted by the bite of infected female phlebotomine sandflies during a blood meal. Metacyclic promastigotes, the infective stages, reach the wound and are phagocytized by macrophages and other mononuclear cells. In these cells, they differentiate in the replicative amastigote form inside a vesicular compartment, the Leishmania parasitophorous vacuole. Sandflies become infected by the ingestion of infected cells containing amastigotes, which transform into procyclic promastigotes in the fly gut and migrate to the proboscis as metacyclic promastigotes to start a new cycle of transmission. More than twenty Leishmania species can cause leishmaniasis. There are three main forms of this disease; cutaneous leishmaniasis, the most common form, occurs in the Americas, the Mediterranean basin, the Middle East and Central Asia; mucocutaneous leishmaniasis is present in Bolivia, Brazil, Peru and Ethiopia; and visceral leishmaniasis (kala-azar) is mainly found in Brazil, East-Africa and India.
Trypanosoma brucei metacyclic trypomastigotes are also transmitted by the bite of an insect vector, the tsetse fly, belonging to the Glossina spp. Once transmitted to the blood of mammalian hosts, metacyclic trypomastigotes transform into slender trypomastigotes, with higher replicative capacity. In mammalian hosts, trypanosomes exist in three major niches: early in infection, they populate the blood; later, they breach the blood-brain barrier, and the adipose tissue constitutes a third major reservoir for T. brucei. Bloodstream forms of T. brucei multiply by binary fission in blood and other fluids and then undergo a developmental transition to the non-dividing short stumpy trypomastigotes. The cycle is completed when the tsetse fly ingests blood from an infected organism. In the fly midgut, trypomastigotes become procyclic trypomastigotes, which multiply and then leave the midgut and turn into epimastigotes. Epimastigotes migrate to the salivary glands and replicate until they transform into metacyclic trypomastigotes. Human African trypanosomiasis (sleeping sickness) is located in countries of sub-Saharan Africa. The chronic disease, produced by T. brucei gambiense, is the most frequent form, whereas T. brucei rhodesiense mainly produces acute infections.
Trypanosoma cruzi is the etiologic agent of Chagas disease, a vector-borne and life-threatening illness still highly prevalent in the endemic countries of Latin America and also disseminated to non-endemic countries nowadays. The vectors of T. cruzi are hematophagous insects of the Reduviidae family, also called triatomines. After a blood meal, the infective metacyclic trypomastigotes present in the hindgut of the triatomine bug are released with the feces. Free parasites invade subcutaneous cells at the wound site and, after a short transit in a vacuole, differentiate into amastigotes, which multiply by binary fission in the host cell cytoplasm. Amastigotes then transform into trypomastigotes, which exit cells and can reach the bloodstream (blood trypomastigotes) and disseminate to different tissues. When triatomines ingest blood, trypomastigotes become epimastigotes and replicate in the midgut. In the hindgut, they transform into metacyclic trypomastigotes to restart the cycle.
Plasmodium sp. The causative agents of malaria are unicellular parasites of the genus Plasmodium, which infect different vertebrate hosts, including humans. Malaria is the most prevalent, vector-borne infectious disease worldwide. It threatens approximately half of the human population and poses a major health concern in tropical and sub-tropical regions around the globe. Plasmodium parasites follow a rather complex life-cycle, alternately infecting a vertebrate host and their definitive host, an Anopheles mosquito. A female mosquito infects a vertebrate host during a blood meal, when Plasmodium sporozoites are injected into the dermis. Sporozoites exhibit gliding motility that allows them to move in the skin. Sporozoites actively penetrate blood vessel walls and migrate with the bloodstream to the liver sinusoids. They invade hepatocytes and divide to form multinucleated schizonts. Transition to schizogony depends on Plasmodium species; always occurs in P. falciparum liver stage development, but does not always occur in P. vivax. The P. vivax sporozoite, once it has entered a host hepatocyte, dedifferentiates and can then become a dormant trophozoite, known as the hypnozoite. The hypnozoite can lie dormant for months and even years and then reactivate and fully develop, leading to P. vivax malaria relapses. Late in parasite development, merozoites egress from the liver through the budding of merosomes (merozoite-filled vesicles) to reach the sinusoidal lumen. Merosomes eventually break up inside pulmonary capillaries, resulting in merozoites liberation and red blood cell infection [Citation6]. Within red cells, merozoites mature from ring forms to trophozoites, then to multinucleated schizonts, and finally individual merozoites (completing the intraerythrocytic cycle). Some merozoites differentiate into male or female gametocytes. These cells are ingested by the Anopheles mosquito and mature in the midgut, where sporozoites develop and migrate to the salivary glands of the mosquito. The mosquito completes the transmission cycle by biting another host.
Toxoplasma gondii is the causal agent of toxoplasmosis, a highly disseminated infection throughout the world. The parasite primarily exists in three forms: oocysts, tachyzoites, and bradyzoites. Oocysts are only produced in the definitive host: cats and other felines. Oocysts are discharged within the feces of felines and contaminate water and produce, which may be ingested by humans and other intermediate hosts (warm-blooded vertebrates). Sporozoites developed within oocyst, are released in the intestinal lumen, infect epithelial cells and then transform into tachyzoites, which are the rapidly multiplying form of T. gondii. Consecutive intracellular division cycles cause tissue destruction and the spreading of the infection. Tachyzoites in pregnant women are also capable of infecting the developing fetus. Eventually, under the pressure of the host immune system, tachyzoites differentiate into tissue cysts with bradyzoites localized in muscle tissues and the central nervous system. However, in absence of a fully functional immune system, like in immunosuppressed patients or fetuses, acute toxoplasmosis arises from uncontrolled tachyzoite division. The ingestion of cysts in contaminated meat is the main source of toxoplasmosis transmission, as bradyzoites transform back into tachyzoites upon entering a new host.
Besides the classical forms of transmission described above, because of the presence of infective forms in the blood or organs of infected people, many of these parasites can also be transmitted through blood transfusion or organ transplant. Moreover, not only vertical infection from mother to child during pregnancy is important for toxoplasmosis as described above, but congenital infection is also for instance the most important form of transmission of Chagas disease nowadays and contributes to spreading the infection to non-endemic countries [Citation7]. For Chagas disease, there is also an oral route of infection by ingestion of food and drink contaminated with the feces of triatomines [Citation8].
The autophagy pathway
From yeast to mammalian cells, what we learned about autophagy over time
The autophagic process has been described morphologically in mammalian cells in the late 1950s/early 1960s, with the observation of double-membrane vesicles containing parts of the cytoplasm and organelles in various states of degradation [Citation9]. Yet, the molecular machinery driving the biogenesis of these vesicular compartments, called autophagosomes, has remained unknown for more than 30 years. Then, in the early 1990s, using breakthrough yeast genetic screen experiments, the laboratory of Yoshinori Ohsumi identified a core set of key molecular actors in the autophagic process [Citation10]. This set, now called autophagy-related proteins (ATGs), was then confirmed and further expanded independently by other groups using similar approaches in several yeast models and different experimental setups [Citation11–13]. Strikingly, while there were early reports of autophagy in various animal tissues and organs, fungi, and even plants, it later appeared that the molecular machinery driving the formation of autophagosomes was largely conserved among eukaryotes [Citation14]. In fact, perhaps because of its importance in response to nutrient limitation (which is likely a persistent challenge that many ancient or modern eukaryotes had to face at some point), autophagy is considered an early and fundamental evolutionary adaptation pathway that most likely already existed in the last eukaryotic common ancestor (LECA) [Citation15].
Over the years the autophagic process has been extensively investigated in yeast and mammals, and to a lesser extent in model organisms like the fruit fly Drosophila melanogaster, the nematode Caenorhabditis elegans, and in some plants. However, this does not reflect the whole spectrum of eukaryotic diversity. In this regard, protists are particularly interesting as they represent dozens of major lineages, most of which share both ancestry and overall cellular characteristics with their better-studied multicellular relatives. They also provide a unique window into the diversification of modern eukaryotes [Citation16].
Roughly 43 ATG (Autophagy-related genes) genes have been found in yeast thus far [Citation17–19]. Although initial studies identified several homologs and orthologs across different taxa using BLAST analysis and rudimentary sequencing technology, it was not until the past decades that a clearer understanding of the different factors involved in the autophagy pathway was gained by the new molecular techniques and tools available. Using comparative genomics tools as well as genomic editing tools such as siRNA or CRISPR-Cas-9 genome editing systems, we can now piece together the progression of this system across eukaryotic evolution.
Autophagy can undergo a selective or non-selective process depending on the signaling pathways and components at play. Non-Selective Autophagy includes Macroautophagy and Microautophagy mechanisms where cytoplasmic content is randomly encapsulated and targeted for degradation in response to cellular stress [Citation20]. Selective Autophagy targets the destruction of organelles or cellular components as a result of turnover events, or as a means to recycle damaged or redundant cell parts. Selective autophagy can target organelles such as the mitochondria (mitophagy), Peroxisomes (pexophagy), ribosomes (ribophagy), ER-specific autophagy (ERphagy or reticulophagy), and even portions of the nucleus (Piecemeal micronucleophagy, PMN). Both Macrophagy and Microphagy processes can be selective and non-selective [Citation21,Citation22]. Another major mechanism of the autophagic pathway that is well documented in yeast and filamentous fungi but not well understood in protist parasites is the cytoplasm-to-vacuole (Cvt) pathway. The Cvt pathway involves a series of signaling factors including hydrolases that subsequently undergoes fusion with the phagophore to form a Cvt vesicle with a double membrane. The inner membrane is then degraded, and the digestion/recycling of targeted cargo is released into the cytosol. Chaperone-mediated-autophagy (CMA) is yet another process in autophagy but has only been documented in mammalian systems [Citation23].
While some of the players involved in the autophagic pathways have been lost, some of the core activators and co-regulators are relatively conserved and are essential among the different eukaryotic lineages.
The Macroautophagy Pathway
The overall mechanism of macroautophagy begins with the formation of a phagophore. This process begins with the recruitment of several proteins in response to a range of stimuli from nutrient deprivation or apoptotic signaling. This entire process is tightly regulated and involves a plethora of protein complexes and activators working in coordination. Macroautophagy is best categorized in the yeast model and involves 5 main steps: 1) initiation/induction; 2) nucleation; 3) elongation; 4) fusion and 5) degradation and recycling, which are briefly described below ().
Figure 2. Mechanism of Autophagy. (A) The five steps of the autophagy pathway. 1. Induction/Nucleation begins with the recruitment of ATG induction complex factors (Pink) followed by PtdIns3K complex factors (Purple), recruited to interact with the ATG induction complex. 2. The Elongation step commences by the Ubiq-like complex formation (Green) to conjugate ATG8 proteins to PE through the LC3 Complex factors (Blue) which are recycled by the action of ATG4. This step undergoes elongation of the autophagosome. 3. Completion of autophagosome takes place with insertion of ATG8-PE to vesicle formed and final release with aid of ATG4 to deconjugate ATG8 from PE followed by the 4. Fusion of the autophagosome to the lysosome via several protein factors including Rap7, Ypt7p, and HOPS. 5. Degradation takes place after fusion, the inner membrane of the autophagolysosome is degraded by the action of ATG15 and other proteases. Autophagy is complete with the efflux of degraded proteins (basic molecules and amino acids) into the cytoplasm through the ATG22 efflux pump. (B) Schematic representation of the autophagy complexes and the Atg proteins that compose them.
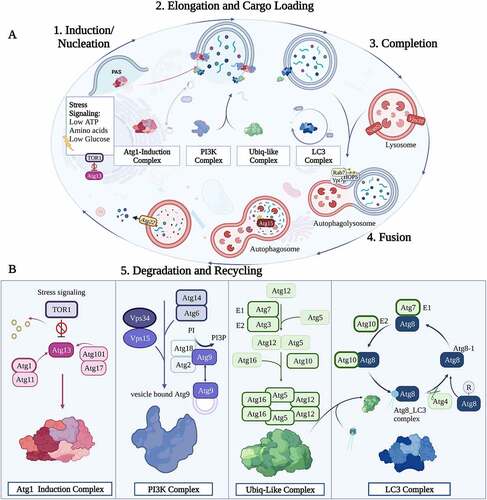
Induction/ initiation
Under regular cellular conditions Mechanistic target of rapamycin kinase (MTOR) inhibits autophagy through phosphorylation of ATG1 (mammalian ULK1). Initiation/induction begins with a series of stress signals such as low ATP, glucose levels, or nutrient deprivation /starvation, which leads to reduced TOR1 serine/threonine kinase levels and dephosphorylation of the Atg1 complex. Dephosphorylated Atg1 dissociates from TOR1 and then initiates the autophagy induction complex. The ATG1 protein forms a complex with ATG13/FIP200 (a Focal Adhesion Kinase [FAK] family-interacting protein), ATG17, ATG101 (mammalian C12orf44) and RB1CC1 (RB1 Inducible Coiled-Coil 1 functioning in protein binding but no evidence is shown of its presence in yeast) to begin the next phase of autophagy [Citation24–26].
Nucleation
Nucleation begins with the formation of the ATG1, ATG101, ATG13, and ATG17 complex. The formation of the membrane begins in peri-vacuolar membrane vesicles termed pre-autophagosomal structures (PAS). The nucleation event consists of a series of protein interactions beginning with the activation of Vps34 (PtdIns3KC3 in mammals) protein, a kinase responsible for the activation of phosphatidylinositol 3-phosphate (PtdIns3P). Several other cofactors are involved in this process including ATG6/BECN1, ATG14 (subunit of the PtdIns3KC3-complex 1 involved in protein binding), Vps15/PtdInsK3R4 (kinase), ATG9, ATG2 (a multifunctional protein involved in membrane tethering and in lipid transfer activity that was recently demonstrated in yeasts and mammals but not found in most protists) and ATG18/WIPI2 (a WD-repeat protein interacting with phosphoinositides [PIP2] binding protein). ATG9 is the only transmembrane ATG identified in both mammalian and yeast organisms. This component is localized in several membrane organelles such as the Golgi apparatus, endosomes, and several trans-Golgi network-derived vesicles called the Atg9/ATG9 vesicles. During starvation, these vesicles are recruited to the assembled ATG1 complex and mainly localize at the site of early autophagosome biogenesis.
Elongation
The Elongation phase of autophagy involves the recruitment of the protein components necessary for vesicle construction through ubiquitin-like conjugation systems. This process involves 3 enzyme-like players. The E1-like enzyme (ATG7) which functions as a homodimer used for activation of ATG8 (mammalian microtubule-associated protein light chain 3 [LC3]) and ATG12, the E2-like enzyme (ATG3), a key protein in the movement of LC3 towards the isolation membrane, and E3-like enzyme (formed by the ATG5-ATG12 complex). The conjugation of ATG12 to ATG5 is facilitated by ATG7 and ATG10. The E3-like enzyme complex then associates with ATG16 to form the ATG16·ATG5–ATG12 complex (ATG16 is found to be conserved in most eukaryotic organisms but is a non-essential component of the autophagy pathway in some unicellular protists). With the assembly of the ubiquitin-like conjugation systems complete, phosphatidylethanolamine (PE) is then conjugated to a glycine (Gly) residue exposed on ATG8/LC3-I (forming ATG8-PE/LC3-II) through processing by ATG4 (a cysteine protease responsible for the post-translational cleavage of the C-terminal residue of ATG8/LC3, to expose a conserved C-terminal glycine) as well as E1 and E2-like enzymes. This lipidation forms a soluble protein needed for the phagophore elongation. In yeast cells, the ATG8 facilitates tethering and hemifusion of liposomes containing ATG8-PE. Autophagosome biogenesis is complete with conjugation of ATG8, and the next phase can commence when the autophagosome undergoes fusion with the lysosome. As the autophagosome is entering completion, the ATG4 is used to deconjugate the ATG8-PE in the outer membrane, releasing ATG8 to be recycled back into the LC3-complex cycle. As discussed below, ATG8 has been a target of interest in the study of the autophagy pathway in protists. It is among the most highly conserved ATGs present in roughly all LCEA (apart from some extreme cases such as Giardia and C. merolae) yet it has been shown to be highly plastic in its roles, with evidence demonstrating roles in organelle development and evasion of host immune responses.
Fusion
The process of fusion to the lysosome is best understood in the yeast model system with the assembly of several protein families that are recruited to the stalk-like curvature of the autophagosome to the lysosome. The autophagosome is transferred toward the lysosome across microtubules. The process of fusion begins with the recruitment of Rab GTPase Ypt7p which functions in the recruitment of HOPS (homotypic fusion and protein sorting) proteins to membranes for fusion. The ATG8 proteins induce autophagy growth and fusion via stalk-mediated fusion events facilitated by the membrane curvature followed by the recruitment of several other protein family complexes such as soluble N-ethylmaleimide-sensitive-factor attachment receptor (SNARE) protein families that play an important role in membrane tethering and fusion of vesicles through the α-helical bundles, and Vam, required for the delivery of alpha-factor receptor-ligand complexes to the vacuole. Several other proteins such as Vam7, Vti, Ykt6, Mon1, and Ccz1 among others are needed to successfully complete the fusion mechanism forming the double membrane autolysosome and the next stage of autophagy commences [Citation27,Citation28].
Degradation and recycling
Post autophagosome fusion and completion, the inner membrane of the autophagic vesicle undergoes degradation by lysosomal enzymes. The low pH content of the lysosome along with the lysosomal hydrolases/lipases and proteases facilitate the process of degradation. In yeast, ATG15 functions as a putative lipase that is likely involved in the intravacuolar lysis of autophagic bodies (ATG15 has only been found in yeast model systems but not in mammalian or protists systems). By comparison, in mammalian cells, the lysosomal proteases (cathepsins B, D, and L) are required for the degradation of autophagosomal contents and the LC3-II of the intra-autophagosomal membrane. Finally, with the degradation of the contents into its simple monomeric units (e.g., amino acids, lipids) the contents are then subject to export into the cytosol through efflux pumps such as ATG22 found in yeast (no identifiable homolog of ATG22 has been found in mammalian cells or protists’ lineages).
Selective Autophagy pathways
Selective autophagy can comprise components of Macroautophagy, Microautophagy, and Cvt pathways. This process involves several of the main actors involved in the macroautophagy pathways but is mediated not by stress signals or nutrient deprivation but instead by specific proteins bound to damaged or redundant organelles/proteins labeled for degradation. Targeted mitophagy for example, is initiated in yeast cells when PINK1 (an inner membrane kinase found in the mitochondria) is transported to the outer membrane when damaged, subsequently recruiting PARK2/Parkin resulting in ubiquitination of mitochondrial substrates. Under healthy mitochondrial conditions, PINK1 accumulates to the inner membrane of the mitochondria through the action of PARL proteins.
Even proteins are subject to degradation through the targeted ubiquitination pathway. In yeast model systems, this process is established through SQSTM1/p62 (sequestosome 1), a ubiquitin- and LC3-binding protein. This protein identifies and targets either damaged, misfolded proteins, protein aggregates, or even bacteria for degradation. SQSTM1/p62 binding acts as an adaptor protein for ATG8-PE/LC3-II interaction which then initiates a form of macroautophagy-specific degradation, forming an autophagosome surrounding the targeted cargo [Citation29].
The Cvt pathway is, yet an additional form of selective autophagy process found in yeast that requires ATP and GTP binding proteins to destroy specifically targeted material. Several Cvt pathway-specific ATG genes have been identified including ATG11, ATG19, ATG20, and ATG2 (among others) but have yet to be identified in mammal systems. The Cvt pathway is activated via four major aminopeptidases including the precursor Aminopeptidase I, (ApeI) that hydrolyzes their substrates. The Pre-Ape1 proteins oligomerize to form a homododecamer. The Ape1 complex interacts with ATG19, a peripheral membrane protein found in yeast, forming the Cvt Complex. Other cofactors are then recruited including the ATG11 and ATG9. The growing complex is transported to the PAS site where phagophore expansion begins. Phagophore expansion and maturation occur (abided by the interaction of ATG19 to ATG8-PE) surrounding the targeted cargo to form a Cvt vesicle. Once formed, the Cvt vesicles are targeted for autophagy through Vps51, Vps53, and Vps54 forming the VFT tethering complex in union with Vps45 and Q-SNARE protein family.
Beyond a conserved core set of ATGs proteins: the autophagy machinery of protists shows losses and diversification
The core autophagic machinery is fairly well conserved between yeasts and mammals (). This is not surprising, as they are all part of the Opisthokonta phylogenetic group of eukaryotes and are thus relatively close from an evolutionary point of view. In the parasitic protists, database-mining studies aiming to establish the ATG repertoire in these eukaryotes highlighted some important differences with the canonical autophagy models [Citation25,Citation27,Citation30,Citation31]. Searching for homologs of yeast ATGs show there is relatively poor conservation of the early components of the machinery in parasitic protists (). In particular, the ATG1 complex is mostly absent (the Atg1 homologs identified show significant domain variation but retain their core functional tasks). It is not currently clear whether this upstream regulating kinase complex has emerged recently in specific eukaryotic lineages, or if it has been lost secondarily in most protists; however, protists have likely evolved alternative ways of regulating the initiation of autophagosome formation.
Figure 3. Conservation of the core autophagy proteins in selected parasitic protists. A Coulson plot [Citation459] was generated to indicate the presence (color) or absence (white) of a homologous protein to the machinery described in yeast. Grey coloring indicates that only a distant homology can be found. Asterisks indicate multiple protein homologs.
![Figure 3. Conservation of the core autophagy proteins in selected parasitic protists. A Coulson plot [Citation459] was generated to indicate the presence (color) or absence (white) of a homologous protein to the machinery described in yeast. Grey coloring indicates that only a distant homology can be found. Asterisks indicate multiple protein homologs.](/cms/asset/0e4960df-410c-4475-aace-4ce8bd9d958a/kauo_a_2149211_f0003_oc.jpg)
Some key components of the membrane/lipid transfer for autophagosome biogenesis, like ATG9 or ATG2, are also seemingly absent from certain protists. The most striking reduction is in Giardia, a pathogen known for displaying some streamlined cellular and metabolic features that possibly only retained components whose function is important for other cellular processes, like the PtdIns3K complex. Trypanosoma and Leishmania also show the highest conservation of Vps34 and Vps15 [Citation32] and the same for Trichomonas and Apicomplexa family members [Citation33].
The conjugation systems, the Ubiquitin-like and LC3 complexes are usually quite well conserved in protists. In some species, ATG10 has been lost or has no obvious ortholog. However, a distant Leishmania ATG10 homolog, despite its weak sequence similarity, was shown by heterologous complementation in yeast to be likely functional [Citation34]. Moreover, as recently shown for Apicomplexa, although ATG10 is absent and ATG12 does not have the C-terminal glycine needed for conjugation, it was shown that ATG12 could be interacting instead with ATG5 via non-covalent bonds and this way allow ATG8 lipidation [Citation35]. These examples show that divergent protist lineages have likely evolved alternative proteins or strategies to perform functions initially described as part of the canonical pathway in yeast and mammals.
While ATG8 is present as a single protein in a few eukaryotes, ATG8 family members have expanded in others, like in the metazoan and plant lineages [Citation36]. This is also the case in trypanosomatids, with up to 25 ATG8 homologs in Leishmania [Citation37], which are categorized into four main families as Atg8, Atg8A, Atg8B and Atg8C. Atg8 and Atg8A are recruited in L. major during starvation and differentiation, but the Atg8A task has been shown to be reserved for puncta formation whereas Atg8 is a component of autophagosome biogenesis. Atg8B and Atg8C show no clear role in autophagy according to a 2009 study by Coombs et al.[Citation37]. This resembles the situation in mammals, where several ATG8 orthologs were identified that were clustered into two subfamilies, LC3 and GATE-16/GABARAP [Citation38]. Both subfamilies were shown to have distinct roles in autophagy with LC3B being the classical ATG8 ortholog with a major role in autophagosome formation, whereas the members of GABARAP family were more involved in the later stages of autophagy, contrary to the situation in yeast with a single ATG8 ortholog. Interestingly, the other members of the Trypanosomatidae family, T. brucei and T. cruzi, preserved three ATG8 homologs (Atg8.1, Atg8.2, and Atg8.3). Atg8.1 is proven to be the functional homolog of the yeast counterpart and remains an active component of the stress-induced autophagic pathway [Citation39]. Atg8.2 does not seem to participate in autophagy and Atg8.3 is shown to be syntenic to that of a protein in L. major which exerts Atg12-like functions[Citation40]. When a phylogenetic analysis was done comparing the protein sequences of the human members of the ATG8 family, yeast Atg8 and the T. cruzi and T. brucei Atg8.1 and Atg8.2, Atg8.1 from both T. cruzi and T. brucei clustered together with the members of the GATE/GABARAP family and yeast ATG8 that was the most distant member of the cluster with amino acid sequence similarity ̴50%. Surprisingly, yeast ATG8 was found to share the lowest amino acid sequence similarity to human LC3 family members, which were grouped into another subfamily with no parasitic members, despite their common function in autophagy. Atg8.2 from T. cruzi and T brucei with no clear role in autophagy were clustered in the third subfamily (Rajkovic and Turk, unpublished). Atg12 appears to have been lost in T. cruzi and T. brucei microorganisms (although some researchers identify Atg5 and Atg10 as orthologs of Atg12) [Citation41].
Consistent with this diversification, these parasites also express several isoforms of the ATG4 cysteine peptidase, whose function is to regulate ATG8 membrane association. It does so by processing ATG8 C-terminal extension to reveal the glycine used for lipid conjugation, and can also cut between the C-terminal carboxyl moiety and the amine group of the lipid to release ATG8 from the membrane[Citation42]. Another unusual feature of some ATG8 homologs of trypanosomatids or Apicomplexa is that they do not have any amino acids extension after the C-terminal glycine residue, which implies ATG4 may only be used for deconjugation/delipidation in that case and suggests different modes of regulation for ATG8 membrane association [Citation25]. While the functional implications of these particular features found in protists ATG8s are not yet fully elucidated, it is possible that this diversification reflects some differences in their physiological roles, some of which may not even be related to canonical autophagy. Interestingly, the T. cruzi Atg4.2, the functional analogue of ATG4, was able to process both natural Atg8 variants from T. cruzi and the human GATE-16, but failed to process all other human homologs (LC3B, GATE-16, GABARAP, and ATGL). In contrast, human ATG4B processed all human and parasite proteins, whereas human ATG4A processed all members of GABARAP family and Atg8.1 from T. cruzi, but not the others (Rajkovic and Turk, unpublished). This somehow suggests that autophagy was developing and specializing during evolution with GATE/GABARAP being perhaps more ancient, but in mammals its role was taken over by another family of proteins.
There is increasing evidence that suggests that many components of the molecular machinery for autophagy also mediates functions that are independent from canonical degradative autophagy [Citation43]. This is for example illustrated by the implication of ATG8 and its related conjugation machinery in the inheritance during cell division of the plastid harbored by several Apicomplexa [Citation44,Citation45]. Hence, the apparent losses or multiplications and specific features highlighted when comparing the ATG repertoire of eukaryotes most probably reflect the functional diversification and specialization that occurred throughout evolution from the ancient function initially present in the LECA [Citation46].
Autophagy in the protist life-cycle
Autophagy and protist metabolism (main physiological modulators of autophagy)
A brief introduction to metabolism and autophagy in eukaryotes
A conserved function of autophagy is to maintain the overall cellular homeostasis in conditions of limiting nutrients (e.g. amino acids, irons [Citation47], or intracellular Acetyl-CoA [Citation48]) and other metabolic perturbations (e.g., hypoxia [Citation49], reduced energy charge [Citation50], or increased ammonia level [Citation51]). In these cases, autophagy enables rapid mobilization of endogenous reserves and a global rewiring of intracellular metabolism, retrieving ATP as well as building blocks to support essential biosynthetic reactions [Citation52]. Depending on the specific type of nutrient shortage or metabolic perturbations, autophagy can be triggered via conserved or distinct metabolic sensors. The MTOR kinase is a protein complex that senses the nutrient/energy state of the cell. Inhibition of MTOR activity is strongly linked to autophagy triggered by amino acid depletion. In the presence of amino acids, MTOR is recruited and activated on the surface of the lysosomes, promoting protein synthesis by phosphorylating eukaryotic translation initiation factor 4E binding protein 1 (EIF4EBP1/4EBP1) and ribosomal protein S6 kinase (RPS6K) [Citation53], and suppressing autophagy by phosphorylating and inhibiting ULK1, autophagy and BECN1/Beclin 1 regulator 1 (AMBRA1), ATG14 and transcription factor EB (TFEB) [Citation54–57]. In the absence of amino acids, particularly glutamine and leucine [Citation58], inactivated MTOR is released from the lysosomal membrane, promoting autophagy and suppressing translation. During glucose starvation, a decrease in cellular ATP boosts the activity of 5’ adenosine monophosphate-activated (AMPK), one of the key energy sensors in the cell [Citation50,Citation59]. As a master regulator of metabolism, AMPK stimulates autophagy by inhibiting MTOR, or by activating ULK1 or components of the PtdIns3K complex directly [Citation54]. Importantly, activated MTOR directly downregulates AMPK signaling suggesting a bidirectional regulation between these two metabolic networks [Citation60].
From an evolutionary perspective, autophagy is thought to be evolved from an ancient mechanism to supply nutrients to a unicellular organism encountering energy or nutrient limitations [Citation61]. In multicellular organisms, autophagy affects not only individual cells but also the maintenance of metabolic balance in surrounding tissues or organs, further impacting the energy homeostasis of the whole body. Given the relatively simple metabolic processes and the diverse living environments, unicellular protists could be an ideal model to study the connection between autophagy and metabolism at the cellular level.
Amino acid transport and sensing during protist autophagy
In addition to protein synthesis, amino acids (AAs) also function as precursors for polyamines and pyrimidines biosynthesis or serve as osmolytes [Citation62–64]. Individual eukaryotic cells can synthesize a subset of AAs, while other AAs must be uptaken from the extracellular environment. Even for AAs that can be synthesized by a cell, supplementary permeases may exist to allow uptake. The AA transporters represent one of the largest families of permeases encoded within the genomes of many eukaryotes [Citation65]. The intracellular levels of AAs are regulated by influx from the extracellular living milieu and recycling of intracellular resources. AA sufficiency, particularly glutamine and leucine sufficiency, is sensed by two signaling pathways controlled by MTOR [Citation66,Citation67] and eIF2a/GCN2 [Citation68], respectively, in higher organisms. Lacking certain AAs in the living milieu or blocking their uptake can affect global protein synthesis and induce autophagy via the inhibition of MTOR activity, through a lysosome centric “inside-out” model [Citation69]. In the “inside-out” model, the intra-lysosomal accumulation of certain amino acids (under fed condition) induces v-ATPase-Ragulator-RagB-Raptor interaction, anchors and activates MTOR on the lysosome surface. Upon amino acids starvation, a specifically lysosome-localized, proton-coupled amino acid transporter PAT1 exports amino acids from lysosome lumen and disrupts the amino acids-sensitive Rag-Ragulator complex, releasing deactivated MTOR from the lysosome membrane [Citation69].
Approximately 30 to 40 genes encoding putative AA permeases/transporters can be identified in each of the L. major, T. brucei and T. cruzi genomes [Citation70]. Some of these transporters have been characterized [Citation71] though most have not. AA-starvation triggers autophagy in many parasitic protists [Citation72–74]. However the AA sensing mechanism remains largely unknown.
Taking advantage of the inheritable and inducible RNAi system that is available in T. brucei, it has been performed an RNAi screening of putative AA transporters identified in the T. brucei genome. Upon the depletion of each individual or a group of AA transporters that share near identical DNA sequences, cell viability and autophagy were monitored. While some putative AA transporters are essential for procyclic cell growth in culture, only one transporter (encoded by Tb927.11.15840/Tb927.11.15860) had effects on both cell viability and autophagy. This transporter, named TbAAT16 [Citation75], is a homolog of LdAAT7 and TcAAT7 in L. donovani and T. cruzi, respectively, which have characterized roles in lysine uptake[Citation76]. The specific lysine uptake activity of TbAAT16 was also observed in T. brucei [Citation75]. Upon RNAi silencing of TbAAT16 transcripts, autophagosome number increased in both fed and starvation conditions (). Acidification of the acidocalcisomes (lysosome-related organelles), which has been shown to be required for autophagy induction [Citation21], was also observed (), consistent with autophagy induction in TbAAT16-RNAi cells in both fed and starvation conditions.
Figure 4. Lysine depletion triggers autophagy in T. brucei. (A) The images show the formation of autophagosomes using ectopically expressed YFP::TbATG8.2 as an autophagosome marker in parasites upon RNAi of lysine transporter TbAAT16. (B) Images show acidocalcisome acidification (based on the accumulation of LysoTracker Red) in parasites after RNAi of lysine transporter TbAAT16. Upon RNAi of lysine transporter TbAAT16, autophagosome formation and acidocalcisome acidification were observed in fed conditions (A and B, left panels), and these activities increased further upon starvation (A and B, right panels). (C) Cells ectopically expressing YFP::TbATG8.2 were incubated in gHBSS containing all AAs except for the indicated individual AA for 2 h. The autophagosome number was quantified based on YFP::TbATG8.2-positive puncta. The relative autophagy activity (fold change) was normalized against the negative control (gHBSS with full AAs), where few autophagosomes were formed. gHBSS only was used as a positive control.
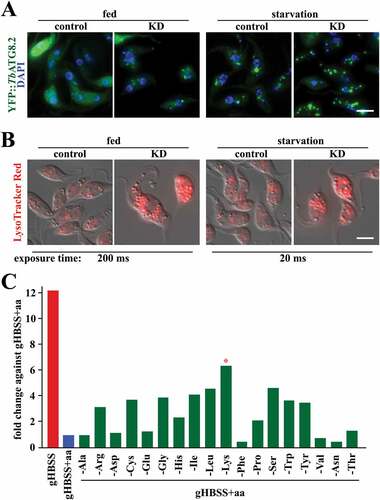
The effect of individual AAs in autophagy induction was also monitored by incubating cells in the gHBSS (Hanks’ Balanced Salt Solution), an amino acid-free buffer that robustly induces autophagy in T. brucei, supplemented with all AAs except for the specified individual AA (). Individual deprivation of several amino acids led to increased autophagy, but deprivation of lysine displayed the highest autophagy level (). Together with the RNAi screening of the AA-transporters, these results suggested that deficiency in lysine or lysine uptake is an efficient trigger for autophagy in procyclic T. brucei.
In a previous study, supplementation of histidine to the gHBSS starvation buffer inhibits autophagosome formation, suggesting that the presence of histidine has an inhibitory role on autophagy [Citation77]. Notably, histidine and lysine are two of three positively-charged AAs (the other one is arginine) stored in the lysosome-related acidocalcisomes [Citation78]. The acidification of acidocalcisomes upon starvation is shown to be crucial for autophagy initiation [Citation21]. Whether the lysine transporter TbAAT16 functions in lysine storage or release in/from the acidocalcisomes and whether the storage/release processes are coupled to proton transport remain unknown. However, TbAAT16 depletion led to acidocalcisome acidification both in fed and starvation conditions (), implying that in T. brucei amino acid sensing may occur via a similar “inside-out” mechanism as shown in mammalian cells [Citation69].
Energy metabolism and autophagy in protists
Glucose and related hexoses, such as fructose, are important carbon sources for many types of cells. While glycolysis using glucose serves as a main ATP production pathway used by parasitic protists [Citation79], ATP can also be produced from amino acids or fatty acids depending on their availability in the different living milieu. In mammal hosts, T. brucei exists in different niches. The bloodstream form (BSF) of T. brucei lives extracellularly in blood, a glucose-rich environment, and as a result, they metabolize high levels of glucose by glycolysis and do not possess a fully functional Krebs cycle or oxidative phosphorylation [Citation80]. In contrast, the adipose tissue forms (ATF) utilize the exogenous fatty acids, such as myristate, to produce ATP by b-oxidation [Citation81]. In the procyclic form (PCF) that lives in the midgut of the tsetse fly, the parasite mitochondrion is fully developed and ATP is generated by oxidative phosphorylation using proline, which is abundant in the tsetse vector and can be converted into glutamate and a-ketoglutarate prior to entering the TCA cycle [Citation82]. Leishmania promastigotes in culture can use either glucose or proline, which are also important nutrients available in the sand fly [Citation83]. When the promastigote enters a macrophage and is internalized into the phagolysosome, energy metabolism shifts to fatty acids and amino acids [Citation84–86]. The proline-glutamate pathway is also conserved in T. cruzi [Citation87]. While T. brucei is auxotrophic for proline [Citation88], T. cruzi is capable of producing proline in its cytosol from glutamate [Citation71]. Different to T. brucei and Leishmania sp., T. cruzi can also convert histidine to glutamate via a four-enzymatic-steps pathway [Citation89].
In mammals, energy shortage usually occurs upon glucose starvation. AMPK senses low energy levels (i.e. high AMP: ATP ratio) and mediates autophagy initiation [Citation54]. In protists, although glucose-starvation/restriction conditions can trigger autophagy in certain parasites such as Trichomonas vaginalis [Citation90–92], the link between AMPK and autophagy is only experimentally confirmed in D. discoideum (see below) [Citation93–95]. In Plasmodium berghei, AMPK homolog PbKIN functions in sporozoite development during the egression to the mosquito’s salivary gland [Citation96], while the P. falciparum homolog PfKIN is involved in transmission from human bloodstream to mosquito midgut [Citation97]. In T. gondii, the AMPKa homolog TOXPK1 responds to glycogen biosynthesis during tachyzoites to bradyzoites differentiation [Citation98]. Similar to Plasmodium, T. brucei AMPK is also involved in cell differentiation from proliferative long slender form to quiescent short stumpy form [Citation99]. Expression of a constitutively active TbAMPKa1 is sufficient to induce differentiation, and inhibiting TbAMPKa1 activity reduces differentiation in mice [Citation99]. Interestingly, depleting TbMCU, a mitochondrial calcium uniporter in T. brucei, induces autophagy accompanied by an increase in cellular AMP: ATP ratio [Citation100]. However, there is no evidence that these two events are directly connected. On the contrary, several lines of evidence suggest that TbAMPK is not involved in autophagy regulation in T. brucei. 1. Glucose starvation rapidly depletes cellular ATP but does not induce autophagy in either procyclic or bloodstream-form cells [Citation101]. 2. Depletion of TbAMPK b or g subunit does not affect amino acids-starvation induced autophagy [Citation101]. 3. TbAMPKa1 activation inhibits TbTOR4 that functions in differentiation [Citation102], but not TbTOR1 that is proposed to function in autophagy regulation [Citation103]. 4. Efficient autophagy induction requires a high level of cellular ATP and autophagic activity is positively correlated with cellular ATP [Citation100]. Last but not least, glucose and proline, the two main nutrients for ATP production in the bloodstream and procyclic trypanosomes respectively, are present at a relatively constant level in the hosts [Citation104,Citation105]. In fact, a systematic study using multiple mammalian cell lines also found that only AA-, but not glucose-depletion induces autophagy [Citation106]. Although glucose starvation activates AMPK, the activation does not significantly increase autophagosome formation [Citation106]. It appears that AMPK is not an absolute requirement for autophagy induction as once thought [Citation107].
Autophagy in organelle turnover and protein degradation (role of autophagy in the protist differentiation)
The most striking feature of parasitic protists is their capacity of surviving in multiple vertebrate and arthropod host environments. To this end, they have evolved complex cellular cycles that entail rigorous morphological changes and circulation between vastly different hosts. These extreme shifts in external conditions throughout the life-cycle have not only imprinted on these obligate parasites the need to undergo vast structural changes but also imply extreme plasticity in their cellular processes and pathways to adjust to these ever-changing circumstances. The mechanism of autophagy lends itself as an example to the adaptive changes undergone in response to organismal adaptive pressures throughout its evolution. This process has evolved and adapted to contribute to diverse cellular functions between different protist lineages including, but not limited to, cell differentiation, protein turnover and the maintenance of the apicoplast (a non-photosynthetic plastid found in several Apicomplexa). By studying this system in parasitic human pathogens, our understanding of the autophagic process has unveiled how these protists not only use the autophagy machinery in a vastly simplified and concise manner to undergo the basic cellular recycling and organelle/protein turnover, but also rely on its components for their development and survival. In the following subsections we will present the different roles of parasite autophagy proteins (or ATGs) in the life-cycle of each pathogenic protist, from the rudimentary process found in Entamoeba or Giardia up to the more developed autophagy of trypanosomatids and Apicomplexa.
Entamoeba sp.
Amoeba are widespread and usually harmless, like the social amoeba Dictyostelium discoideum, but there are pathogenic species like E. histolytica (responsible for amoebic dysentery and amoebic liver abscess), and opportunistic pathogens like Acanthamoeba spp. (that can occasionally cause encephalitis or keratitis in humans). D. discoideum has been used extensively as a model for studying autophagy in amoeba [Citation93,Citation108]. In D. discoideum, MTOR, AMPK and ULK1/ATG1 are conserved with the mammalian orthologs, and their involvement in autophagy is experimentally confirmed. DdAMPKa directly interacts with ULK1/ATG1 via its N-terminal region and promotes the basal autophagy induction [Citation109]. It also displays a fairly well conserved ATG machinery [Citation110] and has been shown to rely on autophagy for differentiation and for its developmental program [Citation111]. In contrast, there is reduced autophagy-related machinery in E. histolytica (), which possesses a rather well-conserved ATG8 conjugation system, but for instance seems to lack the ATG1 complex that is present in D. discoideum. However it should be kept in mind that these two species are quite phylogenetically distant [Citation112]. Moreover, E. histolytica has emerged as a parasite from a mostly nonparasitic clade. Evolutionary history and differences in the way of life may thus have shaped losses and gains within amoeba species. While common autophagy inducers such as nutrient starvation do not lead to autophagosome formation in Entamoaeba, autophagosome-like structures have been observed during encystation of E. invadens [Citation113]. Interestingly, disrupting the autophagy in A. castellanii leads to a reduced cyst formation [Citation114,Citation115]. Altogether, this suggests autophagy could be involved in encystation of pathogenic amoeba, but clearly information obtained so far on the autophagic function is very thin and these models deserve to be studied further.
Giardia sp.
This parasite is seen as an enigmatic, double nucleated eukaryotic microorganism, exhibiting uniquely traits and a relatively small genome. Despite its enveloped nucleus and compartmentalized organelles, Giardia displays traits that are core to all eukaryotes and was believed to be asexual, although recent evidence proves the contrary. It shows no evidence of a fully functional mitochondrion, instead it retains simpler mitochondrial-like remnants called mitosomes and undergoes anaerobic metabolism. It also seems to lack classical lysosomes [Citation116]. Consequently, the autophagy systems in Giardia are not well understood and even suggested to be completely absent in the pathogen. Of the 40 known ATGs across the LECA, there is very little evidence of preserved homologs found to date [Citation117]. Until recently, Vps34 and Vps15 (which also play non-autophagic mechanisms) were thought to be the only ATGs conserved in the Giardia genome. However, there are claims of homology in some ATGs suggesting the existence of a primitive or incomplete form of autophagy at play [Citation74]. In an earlier study aiming to decipher the programmed cell death (PCD) of Giardia, researchers were able to induce stress responses through oxidative stress (H2O2) and incubation with drugs like metronidazole. Through monodansylcadaverine (MDC) staining (an autofluorescent compound labeling autophagic vacuoles) on starved cells, the authors identified the hallmarks of an autophagy-like response. Researchers also performed genome sequencing analysis to identify potential homologs to PCD and autophagy. BLASTP queries compared amino acid and protein sequences to identify hits with high similarity index revealed homologs to some core ATGs such as TOR, ATG1 and ATG16 [Citation73]. However, this remains to be a topic of debate and these genes have yet to be functionally characterized in later studies. In a subsequent study aimed to decipher the pathways of Giardia encystation and the role that MLF (Myeloid leukemia factor protein, an important regulator of cell differentiation)-dependent vesicle formation plays in this mechanism, it was found that ATG8-like proteins (Atg8L) along with a FYVE-domain containing protein (autophagy-linked protein ALFY) colocalized with the MLFVs to form a complex resulting in protein clearance. In this study the MLF, FYVE-HA and Atg8L proteins were detected in exosomes using anti-MLF and anti-HA antibodies through immunofluorescent imaging. It was later discovered that MLFVs belonged to the degradative pathway when analyzed in KO CDK2m3 cell lines. MLF, FYVE, and ATG8L were found to play a positive role in encystation and function in protein clearance pathway giving insights on protein turnover in Giardia [Citation74]. A recent study, however, argues against these claims. This study attempted to identify enzymes involved in the Ub and Ub-Ls conjugation processes using the hidden Markov models (HMMs) from functional Pfam domains [Citation114]. Here, researchers found 118 sequences in the Giardia proteome that were classified into five groups: Ub and Ub-like, E1 and E1-like, E2, E3, and DUB enzymes in G. intestinalis. However, they were unable to find any sequence homology to ATG8 and ATG12 proteins. They did, however, identify an ubiquitin-related protein HUB1 homolog, but it is more closely related to a human from an evolutionary point of view. The study did recognize that a novel non canonical ubiquitin-conjugating enzyme NCUBE (GL50803_8638) is localized in the endoplasmic reticulum (ER) lumen and participates in ER-associated degradation. It is yet still unclear whether or not canonical autophagy occurs in Giardia. Yet, as it contains minimal components needed for complex cellular processes and expresses rudimentary mechanisms, this can reflect its basal evolutionary position or genomic reduction in response to the obligate-parasite’s adaptive needs, and there might be lineage-specific innovations in this organism. This calls for further investigations as the Giardia parasite stands out among most other protist superfamilies because it can potentially bridge the gap of knowledge between the prokaryotic and eukaryotic lineages.
Trichomonads
Trichomonads are a family of anaerobic protists with a drastically modified autophagic repertoire. Trichomonads can be parasitic or thrive in host guts as commensals. Among the trichomonads of particular interest are T. foetus and T. vaginalis, which are flagellated parasites of the urogenital tract of cattle and humans, respectively. A unique trait among these microbes is their lack of mitochondrial organelles. Instead, these organisms meet their energy requirements through hydrogenosomes. Hydrogenosomes are electron-dense organelles, encapsulating enzymes that participate in the metabolism of pyruvate formed during glycolysis. T. vaginalis is one of the most widespread non-viral sexually transmitted infection with approximately 276 million cases reported annually worldwide [Citation72]. Recent bioinformatics analysis showed conservation of the core TvAtg8 genes but most of the core system components of autophagy such as the Initiation complex proteins and Ub-like systems (ATG5-ATG12) were lost or modified [Citation118]. The autophagy system of this organism was studied by Hernandez-Garcıa et al. in a 2018 publication in which indirect immunofluorescence assays were performed with an anti-rTvAtg8 antibody on iron- or glucose-reduced, or rapamycin treated cells. Under these stress conditions parasites presented autophagosome-like vesicles. These TvAtg8-positive vesicles were found in the highest percentage numbers upon glucose reduction conditions compared to the rapamycin or iron reduction conditions. Furthermore, sequence analysis revealed the presence of two ATG8 proteins, Atg8a which showed greater sequence identity to LC3/ATG8 while the second Atg8b more closely related to the GABARAP protein families. Regardless of the sequence similarities, both proteins were found to participate in the biogenesis of autophagosomes [Citation119]. These findings were later expanded on by the same researchers where T. vaginalis was revealed to potentially undergo two different autophagic pathways depending on the stress signals initiated. This study validated the non-canonical autophagic system induced via proteasome inhibition (lactacystin and gliotoxin) or glucose reduction. Yet the ubiquitin-proteasome system (UPS) was shown to be involved in an alternative to degradative autophagy during the stationary phase of high glucose medium cultures, as proteasome inhibition initiated the formation of circular membrane whorls containing organelle fragments. This suggests that Trichomonas not only take part in canonical autophagic processes during instances of environmental stress but also perform basal level autophagy for proteolysis under nutrient-replete conditions [Citation92].
Leishmania sp.
The repertoire of autophagy-related genes is reduced in Leishmania compared to yeast and human, likely a result of the drastic adaptive changes throughout the ancestral lineages of parasitic protists. No genes involved in the initiation of macroautophagy have been identified in Leishmania, except for a possible ATG1 ortholog, which shows significant domain variation but may retain its core function. Genes involved in the process of membrane tethering have been identified bioinformatically, including ATG9 (seeds), VPS34, VPS15, ATG6, ATG18 (PtdIns3K complex) and ATG3, ATG4, ATG5 ATG7, ATG8, ATG10, ATG12 and ATG16 () [Citation25,Citation34]. Leishmania Atg8 has been used as a protein marker for autophagosome formation [Citation120] and functional characterization of all the proteins involved in the ubiquitin-like conjugation, except for ATG16, has been documented. Leishmania ATG5, ATG10 and ATG12 homologs were found to complement their respective S. cerevisiae mutants [Citation121], whilst a functional ATG12-ATG5 conjugation system was shown to be required for ATG8-dependent autophagosome formation [Citation122]. ATG12 is unusual in that it requires C-terminal processing by a peptidase, as yet unidentified. Leishmania mutants lacking ATG5 (Δatg5) were unable to form autophagosomes, had a reduced flagellum and had reduced virulence, likely because of disruption of mitochondrial homeostasis [Citation122]. Leishmania has two ATG4 cysteine peptidases, ATG4.1 and ATG4.2, with differing roles in processing ATG8. Individual ATG4 gene deletions could be generated (Δatg4.1 and Δatg4.2), but not double mutants indicating that ATG4 is essential for parasite viability. There is a level of functional redundancy between the two ATG4 enzymes; ATG4.2 appears to be more important, as Δatg4.2 mutants were less virulent than wild type parasites [Citation123] as they are defective in metacyclogenesis [Citation120]. Interestingly, autophagy and differentiation were also affected in VPS4 mutants. VPS4 is an ATPase involved in the formation of late endosomes and VPS4 mutants were found to be defective in the fusion of autophagosomes to the endosomal/multivesicular tubule-lysosomal compartment [Citation120]. This demonstrates the importance of the function of late endosomes and autophagy in the transformation to the infective metacyclic promastigote. Additionally, lysosomal cysteine peptidases CPA and CPB are important for autophagy, as Leishmania mutants deficient in the peptidases do not effectively degrade autophagosomes and fail to differentiate [Citation34]. Short term starvation or entry into the stationary phase is effective in inducing autophagy. Such quiescent cells have a global reduction in transcription, however, a subset of transcripts did not follow this trend and were relatively upregulated in quiescent populations, including those involved in the autophagy pathway [Citation124]. Not all mutants are defective in autophagy, indeed mutants deficient in the L. major PAS domain-containing phosphoglycerate kinase have an increase in autophagosome formation and cell death [Citation125].
While little is known about the payload of autophagosomes in Leishmania, glycosomes, peroxisome-like organelles that uniquely compartmentalize glycolytic and other metabolic enzymes in Leishmania, have been identified as autophagosome cargo [Citation126]. L. major Δatg5 mutants had significantly greater glycosome numbers in promastigotes and amastigotes, linking autophagy to glycosome homeostasis. Glycosomes were found to be cargo in ~15% of autophagosomes, which were trafficked to the lysosome for degradation. During differentiation from metacyclic promastigote to amastigote the percentage of glycosome-containing autophagosomes remained constant, yet the number of autophagosomes increased 10-fold, indicating that increased turnover of glycosomes was due to an increase in autophagy flux. Mitophagy of the single mitochondrion found in Leishmania was not seen in L. major during growth or differentiation; however, remnants of the mitochondrion formed because of stress-induced fragmentation were found in autophagosomes and lysosomes, indicating that these damaged organelles are recycled by autophagy [Citation126].
T. brucei
In most eukaryotes, a conserved function of autophagy is the maintenance of cellular energy and AA homeostasis in nutrient-limiting conditions. In parasitic protist, the mutual dependence between autophagy and energy metabolism is best characterized in T. brucei [Citation101], It has been well documented that T. brucei participates in complex morphological changes while cycling between its mammalian and the arthropods tsetse fly hosts. This change of hosts requires rapid and drastic cellular remodeling events for the parasite to adapt and thrive between the glucose abundant bloodstreams of their vertebrate hosts to the tsetse fly midgut where conditions may not provide a steady supply of glucose and other necessary nutrients. To adapt to these cycles, the parasite found in vertebrates have to shift from undergoing glycolysis as its major ATP source, carried on the peroxisome-like organelles (glycosomes), to the mitochondrial tricarboxylic acid cycle (TCA) occurred in its procyclic stages within the invertebrate hosts. Here, it develops the mitochondria expressing TCA cycle enzymes, respiratory chain, and oxidative phosphorylation enzymes to ensure its energy requirements are met [Citation127]. The autophagy systems play an important role in the cellular remodeling of its enzymes and organelles during these host cell adaptations. These findings were a result of immunofluorescence assays performed on T. brucei cells using anti-aldolase (ALD) antiserum and an anti-p67 monoclonal antibody targeting glycosomes and lysosomes, (respectively). Observations showed significant increase followed by rapid decrease in glycosomal numbers in the LS (long, slender cells) and I2 (mixture of intermediary and short-stumpy trypanosomes) stages of development. These glycosomes were also seen to be colocalized and enter lysosomes membrane compartment during the differentiation processes from slender to stumpy and from stumpy to procyclic forms of the parasite’s development, expressing the importance of the autophagy system in the parasite’s morphological transitions [Citation128]. T. brucei possesses four MTOR homologs (TbTOR1-4) and at least three MTOR complexes: TbTORC1, TbTORC2 and TbTORC4 [Citation102,Citation129–131]. While TbTORC1 and TbTORC2, respectively, maintains conserved functions in autophagy[Citation129], and protein synthesis and cytokinesis regulation[Citation132], TbTORC4 is unexpectedly connected with the AMPKa1 pathway and functions in T. brucei differentiation from the proliferative slender form to the quiescent stumpy form [Citation99,Citation102]. RNAi depletion of each of the four T. brucei mTOR homologs leads to a mild increase in autophagosomes [Citation133]. However, due to the lack of functional ULK1/ATG1 homologs [Citation134], the mechanism of TbTORs, particularly TbTORC1 in autophagy initiation remains unclear. The degradative activity of autophagy in this parasite also remains poorly understood. To this date, not a single parasite protein has been identified as an autophagic substrate, making autophagosome counting the only method to measure and compare autophagy activity under different conditions. However, a role of autophagy in maintaining new protein synthesis and energy production in starved cells was documented. By using the Click-iT® technique [Citation135–137] it was demonstrated that newly synthesized proteins were significantly reduced in cells starved in gHBSS [Citation39], compared to cells cultivated in fed conditions, A total of 372 proteins were identified as newly synthesized proteins in gHBSS-starved cells, including proteins with unknown functions (28%), and proteins involved in metabolism (24%), translation (13%), proteolysis (8%), protein folding (6%), protein transport (4%), RNA processing (4%) and other pathways (13%). Gene Ontology (GO) enrichment analysis showed that the most enriched GO terms (ranked by Benjamini adjusted p-value) were metabolism, catabolism, translation and protein degradation. KEGG enrichment analysis indicated that the two most enriched pathways were TCA cycle and glycolysis, both involved in cellular energy production. Protein expression in starved WT and autophagy-deficient TbATG8.2−/− cells was also compared. Approximately 2/3 of newly synthesized proteins were found more abundantly expressed in wild type cells, and many of these proteins are involved in ATP production through glucose or proline metabolic pathways. Reduced cellular ATP level was also found in TbATG8.2−/−cells compared with wild type control. Altogether, these results strongly support a role of autophagy in maintaining new protein synthesis and energy production in starved cells, which further facilitates the autophagy process.
T. cruzi
Initial BLAST studies in the genome of trypanosomatids confidently identified genes of PtdIns3K complex and ATG8/LC3 complex [Citation30,Citation127] (). These studies did not find T. cruzi homologs of yeast ATG2, ATG14, and ATG12 proteins. Later individual BLAST searches, found others putative T. cruzi versions of autophagy genes such as ATG5, ATG10, and ATG16 (with low overall homology, but with conserved specific motifs or domains, accession numbers TcCLB.509053.80, TcCLB.509965.280 for ATG5, TcCLB.509911.110 for ATG10 and Tc00.1047053506775.160, Tc00.1047053511167.20 for ATG16, H. Sakamoto, personal communication). A distant T. cruzi ATG1 homolog was also found but its function was still unknown. Despite the low percentage of identity, further experimental data confirmed the participation of several of these genes in T. cruzi autophagy. In 2008, Alvarez and coworkers cloned two ATG8 proteins from T. cruzi, TcAtg8.1, and TcAtg8.2, and also the autophagins TcAtg4.1 and TcAtg4.2 [Citation41]. The authors demonstrated that autophagins have the ability to process the two homologs of ATG8 near the carboxyl terminus, exposing a conserved glycine residue and that both proteins were able to substitute the yeast homologs in functional assays. Later studies showed that TcAtg4.2 has greater proteolytic activity and it is responsible for TcAtg8.1 processing [Citation138]. Other components of the T. cruzi autophagy have also been cloned and characterized. TcVps34 produces PtdIns3P and participates in osmoregulation, acidification, and vesicular trafficking [Citation139]. TcVps15 interacts with TcVps34 to form the PtdIns3K complex that associates with cell membranes. TcVps15 has been shown to be a key regulator of TcVps34 enzymatic activity; both proteins change their subcellular distribution showing a partial colocalization with TcAtg8.1 in autophagosomes under conditions of nutritional stress [Citation140].
Recent studies have shown the signaling pathways that allow T. cruzi to adapt to different stress situations. AMPK, the serine/threonine kinase activated by environmental stress with decreased ATP and increased AMP, has been identified and characterized in this parasite. This enzyme was proposed as a new regulator of nutritional stress in epimastigotes [Citation141]. The presence of a putative T. cruzi MTOR gene was also suggested [Citation142] and further confirmed with the use of rapamycin, that induces autophagy due to MTOR inhibition, similar to mammalian cells [Citation143]. The inositol 1,4,5-triphosphate receptor (TcIP3R) was found in the acidocalcisome, a specialized compartment found in trypanosomatids characterized by acidic pH and large content of Ca2+ and polyphosphates. TcIP3R has been shown to be an IP3-controlled Ca2+ release channel required for Ca2+ uptake by T. cruzi mitochondria, regulating pyruvate dehydrogenase dephosphorylation, mitochondrial O2 consumption, and preventing autophagy [Citation144]. Interestingly, this receptor is localized in the endoplasmic reticulum in animal cells, the main place where the autophagosome formation is initiated. As mentioned above, acidification of acidocalcisomes in T. brucei is a key process that precedes autophagosome formation [Citation145]. In a similar way, inhibition of vacuolar H+-ATPase by bafilomycin prevents acidification of acidocalcisomes and inhibits autophagy in T. cruzi [Citation146], thus evidencing a possible role of acidocalcisomes in the origin of autophagic compartments in this parasite.
The autophagy process is important for parasite survival under conditions of nutritional stress and differentiation [Citation147]. As mentioned in the introduction, T. cruzi displays three different parasitic stages throughout its life-cycle and undergoes different stress conditions. One of the most studied differentiation processes is the metacyclogenesis that occurs in the gut of the insect vector when epimastigotes differentiate into metacyclic trypomastigotes. Autophagy is triggered during spontaneous metacyclogenesis of epimastigotes, which spend long periods of starvation after reaching the stationary phase of growth in vitro. In these experiments, the TcAtg8.1 was found in autophagosomes, only in the intermediate stages, showing that this process is very dynamic [Citation41]. In agreement with this, an increase in the gene expression of TcAtg7 and TcAtg8 was observed during the process of metacyclogenesis [Citation148]. Other stress conditions such as exposition to alkaline or acid media, also induce the autophagic pathway in T. cruzi, contributing to the differentiation process, prior to intense mitochondrial dysfunction (ROS production and overexpression of antioxidant enzymes) [Citation149,Citation150]. These evidences clearly show that autophagy is induced during T. cruzi metacyclogenesis. On the other hand, classic modulators of autophagy that have similar effects on T. cruzi autophagy can modify the metacyclogenesis performance. Starvation, rapamycin, and spermidine stimulate parasitic autophagy and favor metacyclogenesis, while wortmannin inhibits this pathway and thus metacyclogenesis [Citation143]. Interestingly bafilomycin inhibits early steps of autophagy in T cruzi, as evidenced by the low number of Atg8.1-positive vesicles generated under this treatment, in contrast to mammalian cells [Citation143,Citation151]. Spermidine is an autophagy inducer in T. cruzi. Overexpression of heterologous ornithine decarboxylase (ODC) allows T. cruzi to produce polyamines (ornithine, spermidine and spermine) and to increase basal autophagy conferring higher metacyclogenesis capacity than the auxotrophic counterpart [Citation146,Citation152]. In contrast, parasites overexpressing TcIP3R showed decreased metacyclogenesis [Citation153]. Autophagy is also required during the differentiation of trypomastigotes to amastigotes since there is evidence that both starvation and rapamycin can increase this process in a similar degree that low pH, classically used to induce this differentiation [Citation154,Citation155].
One of the organelles involved in the autophagic pathway in T. cruzi is the reservosome, a late endosome-like organelle unique of trypanosomatids that concentrate endocytosed proteins and lipids. Higher content of reservosomes is found in epimastigotes while they practically disappear during differentiation into metacyclic trypomastigotes. The parasite’s main cysteine proteinase, cruzipain, is highly concentrated and active in the reservosomes and is thought to be responsible for the massive proteolysis that accompanies differentiation; since enzyme inhibitors partially block this process and overexpression of proteinases increases its rate [Citation41]. Starvation of epimastigotes and activation of autophagy during metacyclogenesis increases the delivery of cruzipain to reservosomes. The acidity and hydrolytic activity increase in these compartments which results in the enzymatic activation and self-processing of cruzipain. Altogether these results indicate that autophagy-induced activation and self-processing of cruzipain promotes the parasite differentiation [Citation146]. Interestingly, inhibition of cruzipain also inhibits the development and differentiation of parasites in mammalian cells, making this enzyme an interesting target of trypanocidal drugs [Citation149].
Plasmodium sp.
Autophagy is a necessary event for the metamorphosis of Plasmodium sporozoites. Key to the parasite’s successful intracellular development in the liver is the morphologic and metabolic changes necessary for the conversion of the sporozoite (motile form) to a replication-competent (trophozoite) liver form. To invade hepatocytes, the sporozoite contains two types of secretory organelles, e.g., micronemes and rhoptries that discharge their content at the time of host cell contact. Micronemal proteins such as the thrombospondin-related anonymous protein (TRAP) contribute to hepatocyte adhesion [Citation156,Citation157] while the content of the rhoptries is implicated in the biogenesis of the parasitophorous vacuole (PV), a unique niche wherein the parasite multiplies in hepatocytes [Citation158,Citation159]. From 4 h post-invasion (p.i.) onwards, sporozoites undergo spectacular phenotypic changes in liver cells. Scanning EM to decipher the 3-D ultrastructural features of converting sporozoites reveals a bulge in the median region of the parasite that gradually expands while the two distal ends of the sporozoite retract and eventually disappear, leading to parasite sphericalization (). Transmission EM shows that the bulbous region corresponds to the nucleus that protrudes to the cell body (). This shape change is concomitant with the clearance of micronemes and rhoptries, superfluous for parasite replication. Converting parasites discharge the inner membrane complex (IMC), a specialized cortical structure composed of the plasma membrane-associated with flattened membrane cisternae and micronemes. Before release, micronemes are sequestered in double membrane-bound compartments that accumulate at the parasite periphery and are docked on the plasma membrane, suggesting exocytic events. The double membrane structures that sequester micronemes are morphologically similar to autophagosomes Plasmodium spp. possess a rudimentary set of autophagy-related proteins[Citation160–163]. Transcripts of ATG3, ATG7, and ATG8 from the ATG8-complex involved in the expansion of the phagophore are detected in all Plasmodium stages but are upregulated in parasites upon liver infection at day 1[Citation164]. In P. berghei, PbATG8 is solely membrane-associated. Double immunostaining for micronemal TRAP and PbATG8 reveals a significant overlap () that increases as the conversion process progresses up to 12 h p.i., suggesting a role of the parasite autophagic machinery in microneme sequestration. PbATG8 localizes to structures that align along the apicoplast identifiable with the acyl carrier protein (ACP) marker (). The apicoplast is delineated by four membranes and immunogold staining reveals that PbATG8 is located on the two outermost membranes of the apicoplast (), which are enriched with phosphatidylinositol 3-phosphate [Citation165], a lipid that marks mammalian autophagic structures [Citation166,Citation167]. PbATG8 is also found on membranes of vesicles and tubules close to the apicoplast. This suggests that the apicoplast is the source of membranes for phagophore and autophagosome formation. In Plasmodium falciparum, PfAtg8 was revealed to be present and expressed throughout the different sexual and asexual stages of parasite development. Strong evidence of apicoplast colocalization and branching during division was also found by immunofluorescence assays. Additionally, abolishment of apicoplast organelle through Chloramphenicol incubation resulted in disruption of PfAtg8-GFP branching during division [Citation168]. Moreover, PfATG8 knock-down parasites were found to be lacking a functional apicoplast and failed to successfully replicate. As intrahepatic Plasmodium lacks degradative organelles or lysosomes [Citation169], it is unlikely that a process of macroautophagy associated with cargo degradation in autolysosomes occurs. Alternatively, converting parasites may eliminate unwanted organelles sequestered into autophagosomal structures by a process of secretory autophagy (named exophagy), as described in other organisms during differentiation [Citation170]. In exophagy, autophagosomes bypass fusion with lysosomes and instead fuse with the plasma membrane to release their content outside the cell. Exophagy requires the association of the Golgi protein, GRASP with autophagosomes to trigger the fusion of autophagosomes with multivesicular bodies containing VPS4 to form hybrid amphisomes that fuse with the plasma membrane [Citation171,Citation172]. Plasmodium contains genes coding for GRASP and VPS4 homologs [Citation173,Citation174] and GRASP and VPS4 transcripts are expressed in intrahepatic P. berghei 24 h p.i. [Citation169].
Figure 5. Metamorphosis of sporozoites and microneme elimination by autophagy. (A) Scanning EM on converting P. berghei maintained axenically. Bars, 1 µm. (B) Transmission EM of intrahepatic P. yoelii. Bars, 1 µm. (C) Double IFA using anti-TRAP and PbATG8 antibodies on intrahepatic P. berghei 12 h p.i., showing colocalization. (D) Double IFA using anti-PbATG8 and ACP antibodies on intrahepatic P. berghei 22 h p.i., showing colocalization. (E) ImmunoEM on intrahepatic P. berghei using anti-PbATG8 antibody, showing gold particles on the outermost membranes of the apicoplast (api, arrows). Bars, 100 nm [adapted from 160,164,169]. (F) Hypothetical model for microneme exophagy and amphisome detection (see text for description). (G) Triple IFA using anti-ATG8, anti-GRASP and anti-VPS4 antibodies on intrahepatic P. berghei 22 h p.i., showing significant co-association of ATG8, GRASP and VPS4 on same structures [adapted from 169].
![Figure 5. Metamorphosis of sporozoites and microneme elimination by autophagy. (A) Scanning EM on converting P. berghei maintained axenically. Bars, 1 µm. (B) Transmission EM of intrahepatic P. yoelii. Bars, 1 µm. (C) Double IFA using anti-TRAP and PbATG8 antibodies on intrahepatic P. berghei 12 h p.i., showing colocalization. (D) Double IFA using anti-PbATG8 and ACP antibodies on intrahepatic P. berghei 22 h p.i., showing colocalization. (E) ImmunoEM on intrahepatic P. berghei using anti-PbATG8 antibody, showing gold particles on the outermost membranes of the apicoplast (api, arrows). Bars, 100 nm [adapted from 160,164,169]. (F) Hypothetical model for microneme exophagy and amphisome detection (see text for description). (G) Triple IFA using anti-ATG8, anti-GRASP and anti-VPS4 antibodies on intrahepatic P. berghei 22 h p.i., showing significant co-association of ATG8, GRASP and VPS4 on same structures [adapted from 169].](/cms/asset/70a15fac-66df-438b-a6d5-f1aca27940b7/kauo_a_2149211_f0005_oc.jpg)
Based on these data, it has constructed a working model outlining the cellular events leading to the elimination of micronemes by secretory autophagy during sporozoite conversion into liver forms (). In this model, the apicoplast supplies PbATG8-containing membranous structures to generate phagophores (step I); micronemes are sequestered into phagophores and autophagosomes (step II); GRASP is transported from the Golgi to autophagosomes for membrane fusion (step III); autophagosomes fuse with multivesicular bodies (MVB) to form amphisomes (step IV); amphisomes fuse with the plasma membrane for microneme release into the PV (step V) and microneme are degraded within the PV (step VI). To validate step IV of this model and illustrate an exophagic pathway intersecting with Golgi vesicles and multivesicular bodies, liver forms were immunostained for ATG8, GRASP and VPS4. VPS4-and GRASP-positive autophagic compartments were detected in converting parasites (). This result points to a potential cross-talk between the autophagic machinery and exocytic organelles for organelle disposal in liver forms.
To examine whether that PbATG8 plays a key role in microneme elimination by exophagy in intrahepatic Plasmodium, PbATG8 expression has been manipulated by creating a strain that conditionally overexpresses ATG8 (PbATG8-OE parasites [Citation169]) when sporozoites reach the salivary glands and thus in the liver. This strain has an insertion of a 60-bp ‘GC’-rich sequence in the 3′ UTR of ATG8, resulting in the stabilization of ATG8 mRNA and a 2-fold increase of ATG8 expression during liver stage compared to the parental strain. Examination of PbATG8-OE infection in hepatocytes reveals that PbATG8-OE parasites suffer developmental defects; delayed karyokinesis and schizogony, abnormally small PV, and undersized merosomes, the membrane package produced at the end of schizogony that contains hepatic merozoites [Citation169,Citation175].These developmental defects of PbATG8-OE parasites in the liver lead to delays in initiating the first round of blood infection. Other abnormalities found in these parasites were expanded apicoplasts (the PbATG8 signal looks branched into a reticulate network) and delayed microneme evacuation into the PV[Citation169]. Together, these data confirm the participation of Plasmodium autophagy and ATG8 in the development of the parasite infection in hepatocytes.
T. gondii
Like its Plasmodium relative, this protist parasite has a reduced ATG repertoire, but has retained some of the core elements of the autophagy systems. In this Apicomplexa, the single ATG8 homolog is involved in autophagosome formation during starvation [Citation176,Citation177], but like in P. falciparum, it is also recruited to the apicoplast along with its role to apicoplast biogenesis to ensure proper inheritance of the organelle during cell division [Citation178]. The apicoplast is a plastid that originated from a secondary endosymbiosis event. This organelle is surrounded by four membranes, of which the outermost is decorated with TgAtg8. Although nonphotosynthetic, the apicoplast is nevertheless essential for the survival of Apicomplexa, as it is involved in several key metabolic pathways [Citation179]. During the course of parasite replication, the apicoplast divides and must be segregated into each of the daughter cells for proper inheritance across generations. TgAtg8 seems to drive the association between the apicoplast and the centrosomes for correct positioning and segregation of the organelle [Citation44]. The machinery for TgAtg8 membrane conjugation is involved in this original non-degradative function, as shown by defects in apicoplast maintenance and reduced cell survival observed in mutants lacking ATG3 and ATG4 [Citation176,Citation177]. Yet, early players in the autophagy pathway, like TgAtg9 (which is conserved in Toxoplasma, unlike for Plasmodium) is only involved in canonical autophagy, which is not essential for parasite viability in normal growth conditions in culture, but is important for surviving specific stresses [Citation180]. Interestingly, it has been shown that this protein and thus canonical autophagy, plays an essential role in the persistence of the bradyzoite stage (the latent tissue cyst stage of the parasite)[Citation181]. This is consistent with the fact that chemical or genetic ablation of the proteolytic capacity of bradyzoites led to autophagosome accumulation and death in bradyzoites [Citation182]. There is much to be learned about the roles these proteins play in parasite development and can potentially shed light on the host-parasite relationships that keep these organisms thriving. T. gondii has not only managed to make use of its own autophagy system for organelle biogenesis, but also has brilliant ways to manipulate the host’s autophagy systems for its growth and preservation as will be explained below.
Other (non-canonical) functions of autophagy in protists
As shown above, individual ATG proteins of protist organisms can display specialized functions other than canonical autophagy as in the case of apicoplast inheritance of Apicomplexa. That could be one reason for the diversification of some of these proteins observed in these parasites as in trypanosomatids, although the functions of homologs have not been elucidated yet. The whole autophagy process also accomplishes other non-canonical roles as will be described following.
Autophagic cell and nuclear death in protists
In the history of eukaryotic evolution, the concept of cell suicide was assumed to have arisen with multicellularity. However, the discovery of apoptosis in trypanosomes [Citation183,Citation184] and the ciliate Tetrahymena thermophila [Citation185] in the 1990s indicated that a similar process of socially advantageous regulation of cell death also operates in unicellular eukaryotes. By the 2000s, apoptotic markers were described in all major groups of eukaryotes [Citation186]. These findings highlight the utility of the altruistic form of regulated cell death in unicellular organisms for the elimination of deficient mutant or pathological individuals from the community, reminiscent of the selective elimination of unhealthy cells, such as cancer cells, for the maintenance of tissue homeostasis by apoptosis in multicellular organisms.
While most cell death involves apoptosis, studies in multicellular model systems have clearly identified instances of regulated cell death that involve autophagy. Dying cells often display a large-scale accumulation of autophagosomes and autolysosomes in the cytoplasm, which differs considerably from cells undergoing apoptosis or necrosis, and hence adopt a morphology called “autophagic cell death” or type II cell death [Citation187]. Since promoting the use of this term in the 1980s, there have been many reports of autophagic cell death highlighting the significance of autophagy for developmental and pathophysiological cell death. Autophagic cell death has also been reported from a wide group of protists [Citation188], implying an ancient evolutionary origin of autophagy as a suicide program that differs from apoptosis. However, genetic studies suggest that in many cases, autophagy is not directly a cause of cell death but rather accompanies apoptosis or necrosis as a failed effort to mitigate cell damage [Citation189,Citation190]. It is also the case that autophagy contributes to the activation of other death programs [Citation189,Citation190]. The term “autophagic cell death” is now used in at least three different ways based on the roles of autophagy in cell death: (i) autophagy-associated cell death, where the induction of autophagy accompanies apoptosis or other cell death pathways; (ii) autophagy-mediated cell death, where autophagy induction triggers apoptosis or other cell death modalities; and (iii) autophagy-dependent cell death (ADCD), a distinct mechanism of cell death that occurs independently of apoptosis or necrosis [Citation191]. The recent recommendations of the Nomenclature Committee on Cell Death defines ADCD as regulated cell death that depends on the autophagic machinery without involving alternative death pathways [Citation192]. In (i) and (ii), cell death can be mitigated by inhibition and/or genetic manipulation of autophagy mechanisms. Autophagy is however not considered as a genuine apparatus on cell death. The complex relationship between autophagy and other cell death pathways remains to be elucidated.
The strongest evidence for ADCD comes from developmentally programmed cell death in the obsolete larval midgut during Drosophila metamorphosis. Despite the high level of caspase activities, the removal of the midgut depends on only autophagy [Citation193]. Autosis, a type of autophagic cell death due to activation of the Na+/K+-ATPase pump in mammalian cells [Citation194], is also known to meet the criteria of ADCD. An additional clear-cut example of ADCD is found in the amoeba Dictyostelium discoideum that does not encode caspases or the major apoptotic machinery genes found in mammals[Citation195]. Under starvation conditions, about 105 vegetative amoeba cells aggregate into a multicellular organism, which then differentiates to form a fruiting body consisting of viable spores and dead stalk cells in response to cAMP production [Citation110]. The cells forming the stalk undergo cell death with vacuolization in the cytoplasm, which is prevented by genetically blocking autophagy [Citation196,Citation197]. This cell death requires the morphogen differentiation-inducing factor (DIF-1) in addition to starvation[Citation196]. Starvation alone or DIF-1 exposure to non-starved cells is not sufficient to induce cell death in the stalk. This is an intriguing example where combination of two signals converts a survival function of autophagy to a cell death promoting role. The developmental cell death in D. discoideum could provide an important insight into the key questions of ADCD, such as (1) how survival and lethal autophagy differ, (2) what internal/external cues promote a signal to increase the rate of flux or prolong autophagy resulting in the demise of the cell, and (3) whether ADCD involves the degradation of selective cargo or bulk degradation of cytoplasmic components, without the complexity between autophagy and apoptosis.
Autophagy, even if not ADCD, could represent a valuable drug target for parasitic protists, as it may lead to a way of autophagic cell death without side effects to the human body and damages to other animal resources. In the bloodstream-form of T. brucei, rapamycin, the classical inhibitor of MTOR, induces growth arrest leading to autophagic cell death [Citation198]. Note that this effect of rapamycin on T. brucei is only seen at a high concentration, at least 10-fold higher than that required to induce autophagy in yeast. It remains to be determined how the autophagic events observed after treatment with high doses of rapamycin are caused and if these processes are indicative of regulated cell death by autophagy. In the mammalian-infective form of T. brucei, on the other hand, autophagic cell death can be induced by neuropeptides such as vasoactive intestinal peptide, urocortin, and adrenomedullin [Citation199,Citation200]. In this scenario, endocytosed peptides reach the lysosomes whose integrity is subsequently disrupted. The peptides are released from the late endosomes or lysosomes in the cytoplasm, where they interact with glycosomes leading to depletion of cellular ATP. The failure of energy metabolism results in cell death with involvement of autophagy. No signs of apoptosis or necrosis are observed in both the forms of T. brucei, implying predominant or exclusive roles of autophagy in these death processes.
In free-living protists, the freshwater green alga Micrasterias denticulata is an example where the role of autophagy in cell death has been investigated. In this organism, treatment with a high concentration of salt leads to cell death involving autophagosome formation, which is readily observed by transmission electron microscopy [Citation201]. Pigment composition, photosynthesis, and respiration indicate an active metabolism, which supports a regulated form of autophagic cell death rather than necrosis [Citation201]. Cell death in this organism can also be induced by the treatment with H2O2, but it does not display autophagosome formation [Citation202]. The H2O2-induced cell death appears apoptotic as it shows increase in caspase-3-like activity as well as a nucleosome-sized DNA laddering[Citation202], a key feature of nuclear degradation by apoptosis. These results show that different environmental stresses lead to different death pathways in one and the same organism, suggesting a context-specific role of autophagy in the salt-induced cell death. Note that DNA laddering also occurs in the salt-induced cell death [Citation201]. Since neither cytochrome c release from mitochondria nor increase in caspase-3-like activity is detected[Citation201], a caspase-independent apoptosis pathway may participate in the death process. Like the removal of salivary glands during Drosophila metamorphosis which requires both autophagy and apoptosis (hence not ADCD)[Citation203], two programs may function in parallel for the salt-induced cell death, implying an additional mode of autophagic cell death.
Finally, programmed nuclear death (PND) is known to occur in the free-living ciliate Tetrahymena thermophila as a unique and intriguing example of autophagy. Like other ciliates, T. thermophila have evolved remarkable nuclear dualism that involves spatial segregation of the somatic macro- and germline micronucleus in a single cytoplasm [Citation204]. PND is a remarkable macroautophagy process that occurs during sexual reproduction (conjugation), in which only the parental macronucleus is committed to suicide and eliminated from the cytoplasm, but other co-existing nuclei such as developing progeny macro- and micronuclei are unaffected [Citation205]. Once there is commitment to PND, the envelope of the parental macronucleus displays an autophagosome-like property demonstrated by stainability with MDC [Citation206] and localization of ATG5 [Citation207] and ATG8 [Citation208], without sequestration of the nucleus by a double membrane. Subsequently, lysosomes fuse only to the parental macronucleus to form a gigantic autolysosome for the final resorption [Citation206]. During PND, a mitochondrial nuclease-associated protein and apoptosis-inducing factor (AIF) degrade the parental macronuclear DNA into oligonucleosomal size fragments [Citation209,Citation210]. These proteins contribute to the promotion of nuclear degeneration, but PND per se is not blocked in their knockout mutants. In contrast, loss of ATG5, ATG8, and VPS34 results in a block of the autophagy processes, rendering the retention of the parental macronucleus in the cell even after conjugation [Citation207,Citation208,Citation211]. In ciliates with nuclear dualism, the parental cell-derived nucleus is taken over by the progeny macronucleus after conjugation. Therefore, PND is essential to finish the lifespan of soma and reset the developmental time of the cell to zero. The regulatory mechanism of PND appears partly different from another nuclear autophagy that is required for the removal of relic meiotic products, the haploid micronuclei not used for fertilization; loss of VPS34 does not block this autophagy [Citation211], while ATG5 and ATG8 are prerequisite to the autophagy as well as PND [Citation207,Citation208]. Given the context-specific role of autophagy mediating cell death in other organisms, it will be important to investigate the regulatory mechanism of PND that acts specifically to terminate the somatic life equivalent to autophagic cell death in other protists. Each group of protists may use orphan proteins in canonical autophagic pathways and, conversely, may use conserved proteins for more specialized forms of autophagic cell death as well as PND. Understanding the commonalities and differences in autophagic cell death of protists and higher multicellular organisms will illustrate how extant autophagy has acquired its complexities and extended functions, from survival to lethality, during evolution.
Autophagy response during drug treatment
Protist diseases that mostly affect more than 1 billion people worldwide, are mainly vector-borne diseases, have animal reservoirs and are associated with complex life-cycles, factors that make their public-health control challenging [Citation212–216]. Moreover, due to the lack of efficient transmission control strategies and efficient vaccines against most of these diseases; in addition to limited efficacy, high toxicity and increase of drug resistance of the current chemotherapy, the development of new therapeutic interventions is imperative [Citation214,Citation217–221]. To further identify alternative treatments, research groups worldwide are concentrating efforts on designing novel strategies to trigger parasite death in vitro and in vivo. Besides rational drug design, other sources for drug discovery comprise natural-derived compounds and drug repositioning/repurposing [Citation222]. As a result, several novel compounds have been found and their anti-protist activity assessed, but only occasionally further information about their mechanism of action was addressed. In this direction, mechanistic assays must be performed to identify molecular and/or cellular targets, and these findings are pivotal to guaranteeing drug safety as well as to confirm specificity or presence of off-target effects that even can be advantageous (or not) for the course of infection. Additionally, a significant number of active anti-protist compounds presented no predictable biological effect [Citation223,Citation224], reinforcing how crucial is the identification of the molecular targets. Interestingly, several compounds causing parasite death and proposed as potentially antiparasitic therapy seem to activate autophagy regardless of their nature. In this section we are going to summarize the autophagic response of protists to drug treatment. Besides the autophagy cell death presented previously, two other possibilities were observed: autophagy associated cell death and autophagy incidental death. In addition, a new therapeutic strategy based on targeting autophagy for vaccine and drug development will be presented.
Autophagy associated cell death
A couple of years ago, Jeremy Mottram’s team introduced the concept that while autophagy can be considered a regulated cell death process in mammalian cells, this type of death remains controversial for protists [Citation188,Citation225]. Concordantly with this notion, in most of the studies reviewed herein investigating the mechanisms involved in drug-induced parasite death, the authors observed mechanisms other than autophagy contributing to cell death and thus emphasized that this process became activated under drug stress conditions as a compensatory parasite survival mechanism in response to treatment. The autophagic phenotype, or, as introduced above, the autophagy-associated cell death is observed in almost all parasite species, independently of the drug used [Citation226]. Evidence of autophagic activation, mainly include observation of morphological alterations via electron microscopy or the identification of autophagosomal markers [Citation227,Citation228].
Among all approaches to the mechanistic characterization of drugs action, electron microscopy is one of the most frequently employed in almost all cellular models, including parasitic protist [Citation221,Citation229]. Concentric membranous structures or myelin-like figures are one of the most recurrent autophagic phenotypes detected in parasitic protist treated with all classes of compounds [Citation228,Citation230–239] (), followed by the high number of autophagosomes, with different degrees of cargo degradation [Citation91,Citation176,Citation233–235,Citation239–254] (). Another frequent morphological evidence is the appearance of remarkable ER profiles in close contact to cytosolic structures and organelles (). As an example, T. gondii treated with thiolactomycin analogues showed high amounts of expanded concentric membranes, suggestive of ER profiles [Citation255]. In relation to trypanosomatids, elatol and amiodarone led to proeminent ER and mitochondrial swelling in L. amazonensis [Citation251,Citation256], and triazolic naphthoquinone triggered such phenotype surrounding reservosomes in T. cruzi epimastigotes [Citation254].
Figure 6. Ultrastructural autophagic phenotype of Trypanosoma cruzi triggered by drugs. (A-E) Parasites treated with different classes of drugs shared similar autophagic features such as multiple concentric membranous structures (black arrows) distributed in the cytosol, the presence of endoplasmic reticulum surrounding organelles (white arrows) as well as an intense cytosolic vacuolization (black stars) and mitochondrial swelling (black asterisk). (E) Increased formation of autophagosomes (white star), with distinct levels of cargo degradation is also frequently observed after the stress induction. M: mitochondrion; L: lipid; N: nucleus. Bars = 0.5 µm.
To confirm autophagy induction, ultrastructural studies needs to be completed by tracking the autophagosome increase with specific markers. According to Klionsky and colleagues Atg8 expression is the gold standard approach for monitoring autophagy by Western blotting and fluorescence microscopy [Citation257]. The treatment of T. gondii with antimalarial agents, monensin, dithiothreitol, brefeldin A or tunicamycin promoted a time-dependent increase in the number of Atg8 puncta [Citation180,Citation258–260]. Such increase was also observed in T. brucei treated with bacteriocin AS-48 and L-leucine methyl ester [Citation239,Citation261] as well as in cryptolepine-treated L. donovani as described below [Citation253]. On the other hand, several studies still employed the non-specific probe MDC to label autophagic vacuoles. In the literature, there are many reports describing the increased number of MDC puncta in parasites treated with a great variety of compounds [Citation143,Citation247,Citation249,Citation253,Citation262–266]−[Citation46]. This is the case with L. amazonensis promastigotes, whose death was associated with high positivity for MDC, when treated with flavonoids [Citation267]. The association between parasite death and autophagy was also established by enhancing MDC labeling in parasites treated with a combination of a thiosemicarbazone derivative and miltefosine [Citation266]. Other example is antimicrobial peptides (AMPs). These compounds exhibited enhanced antileishmanial activity via a mechanism involving ionic interactions with Leishmania, which is modulated by parasite lipophosphoglycan (LPG). This effect results in membrane potential dissipation and the equilibration of intracellular pH with the extracellular environment. The investigators demonstrated the involvement of autophagy in parasite death, supported by observations using transmission electron microscopy (TEM) revealing cellular damage, including cytoplasmic vacuolization and the degeneration of cellular organization without the disruption of the plasma membrane, as well as the detection of intracellular vacuoles via MDC labeling [Citation227]. Although useful as a first approach to study autophagy, MDC labeling has to be accompanied with other more specific markers to conclude the involvement of autophagy in the response of protists to parasiticidal drugs.
As an alternative approach, an experimental design including the preincubation with autophagic inhibitors, such as wortmannin and 3-methyladenine (3-MA) and subsequent treatment with the compounds has been used to assess the relation between autophagy and the parasite death induced by the drug [Citation257,Citation268]. In T. gondii, 3-MA prevented the mitochondrial effect of monensin [Citation259], but not of different antimalarial drugs, indicating an PI3K-independent mitochondrial injury in this last case [Citation260].
Describing the activation of the autophagic pathway in T. cruzi treated with naphthoimidazoles, Menna-Barreto, and colleagues [Citation228] elegantly associated ultrastructural evidence suggestive of apoptotic-like alterations and autophagic activation with the identification of ATG gene overexpression, along with a marked increase in MDC labeling and the inhibition of death by wortmannin or 3-MA. In the epimastigotes and trypomastigotes stages of T. cruzi, the treatment with either one of three naphthoimidazoles N1, N2, and N3 derived from β-lapachone induces cell death. These antiparasitic compounds are known to affect the organelles of T. cruzi, such as mitochondria, Golgi, and reservosomes [Citation228,Citation269],[Citation270]. Upon treatment, autophagic characteristics in the cytoplasm are observed; the number of concentric membrane structures stainable with MDC, increases significantly [Citation228]. Ultrastructural studies demonstrate that the organelles, especially reservosomes, are surrounded by membrane structures like autophagosomes and lose the integrity of their structures [Citation31,Citation236]. The effect of the naphthoimidazoles on the cell death events is blocked by cysteine protease inhibitors, E64 and calpain I inhibitor [Citation270], suggesting the participation of autophagic machinery in the death process as macroautophagy in mammalian cells requires calpain [Citation271]. The preincubation with the autophagic inhibitors wortmannin and 3-methyladenine completely abolishes the effects of naphthoimidazoles and RT-PCR data indicate the participation of ATG3, ATG4, ATG7 and ATG8. These findings associated with a slight increase in phosphatidylserine exposure suggest that autophagic activation predominates in T. cruzi during naphthoimidazole induced cell death [Citation228]. Eventhough, significant increases in necrosis in treated parasites support the notion that autophagy is insufficient to control drug effects in parasite death [Citation228], indicating that the detailed pathway and proteins involved in the mechanisms of cell death require further investigation.
Cryptolepine, an indoloquinoline alkaloid that presents antimalarial and cytotoxic properties, induces L. donovani promastigotes to produce ROS, ultimately resulting in DNA fragmentation, a hallmark of apoptosis [Citation253]. Alterations suggestive of apoptosis activation, including mitochondrial membrane depolarization, mitochondria swelling, increased ROS production, and phosphatidylserine’s externalization, have been described in L. amazonensis promastigotes treated with similar compounds, i.e., organic salts derived from quinoline[Citation267]. The notion that autophagy functions as a survival mechanism are supported by additional decreases in the number of viable parasites in L. donovani cultures treated with the combination of cryptolepine and 3MA, compared to those treated exclusively with the antileishmanial drug [Citation253]. It is possible that in response to cryptolepine, a stress response is induced by these parasites, thereby initiating an autophagic response as a survival mechanism. By contrast, treatment with quinoline derivatives alone abrogated autophagic vacuole formation, likely preventing the elimination of damaged organelles, which increased the antileishmanial effect of these compounds [Citation267].
Altogether the findings espoused by these studies mainly support the notion that, in protists, the autophagic process is activated when submitted to stress conditions, as has been previously described in mammalian cells [Citation272]. Thus, autophagy functions as a critical process for maintaining parasite survival under the stress produced from treatment with antiparasitic compounds [Citation225].
Autophagy incidental death
In a previous work it was emphasized that until lethal signaling pathways and mechanisms associating autophagy to a death-regulated path are unveiled in protists, it is equivocal to consider cell death as distinctly different from incidental death or unregulated necrosis [Citation225]. In line with this concept, more recently studies have consistently demonstrated that treatment of Leishmania sp. and T. cruzi with antiparasitic compounds caused abnormal activation of the autophagic pathway, subsequently resulting in incidental parasite death.
Using carvedilol, a beta-blocker broadly used to treat hypertension and other cardiovascular diseases and known to modulate autophagy in mammalian cells [Citation273,Citation274], Romano and colleagues [Citation275] reported an intense accumulation of immature autophagosomes in T. cruzi that presented lower acidity and hydrolytic properties. Subsequently, these authors observed an intense impairment in trypomastigote survival in addition to decreased replication of T. cruzi epimastigotes and amastigotes in vitro. Furthermore, carvedilol impaired the peak of parasite burden in infected mice evidencing the effect of this treatment in vivo. Similar to what was observed with other compounds, ultrastructural alterations suggestive of autophagic activation were detected in Leishmania parasites in infected cells treated with the potent anti-cancer Hsp90 inhibitor 17-AAG, as evidenced by intense anti-leishmanial effects both in vitro and in vivo [Citation276,Citation277]. Moreover, transgenic parasites containing GFP-ATG8-labeled autophagosomes treated with 17-AAG exhibited autophagosomes that did not uptake cargo, such as glycosomes, nor fuse with lysosomes. Furthermore, ATG5-knockout (Deltaatg5) parasites, which cannot form autophagosomes, demonstrated lower sensitivity to 17-AAG-induced cell death than wild-type (WT) Leishmania, further indicating the participation of autophagy in 17-AAG-induced cell death. Interestingly, abnormal autophagic activation overloaded the ubiquitin-proteasome system (UPS). Like the UPS system inhibitor, MG132, 17-AAG also provokes a significant accumulation of ubiquitinated proteins in both WT- and Deltaatg5-treated parasites compared to controls. However, the compensative mammalian mechanism of protein aggregate formation, which is characteristic of proteasome overload, was not observed in parasites treated with the Hsp90 inhibitor. In sum, these findings suggest that overload occurs during the formation of immature autophagosomes unable to degrade their cargo, causing, consequently, incidental parasite death [Citation278].
By previous suggestions, the present review reinforces the notion that to convincingly evidence the occurrence of regulated cell death in parasitic protists, it must be demonstrated that the death process can be delayed or abrogated by targeting essential signaling molecules or pathways. This aim could be achieved through genetic manipulation or the chemical inhibition or activation of specific pathways. We believe that such approaches should be actively employed to elucidate whether cell death can be regulated in parasitic protist and, if true, to identify the relevant mechanisms involved.
Targeting protist autophagy as therapeutic intervention: a new strategy of defense against Malaria
As mentioned above (see section #3), overexpression of Plasmodium ATG8 in liver forms result in dysregulation of autophagy leading to aberrant accumulation of micronemes and structural derangement of the apicoplast, These mutant parasites (PbATG8-OE, see above) are expected to be more sensitive to chemoprophylaxis treatment, and hence an ideal antigenic constituent of a Chemoprophylaxis Vaccination regimen (CVac).
CVac is so far the most efficient vaccination strategy using whole organisms developed against malaria and is currently being evaluated in clinical studies [Citation168,Citation279–282]. In contrast to radiation attenuated sporozoite (RAS) vaccination, CVac requires 20-fold fewer sporozoites to induce a sterile protective immune response and provides more robust and long-lasting protection. The immunization of mice with PbATG8-OE parasites then treated with chloroquine for 7 days after each immunization show 100% protection and remain completely blood form-negative at the memory time point of 80 days post-challenge with sporozoite wild-type in comparison with the parental strain subjected to the same protocol that reach only 60% protection [Citation175]. These data point to PbATG8-OE parasites as a prototype of future genetically-attenuated parasites based on autophagy interference and encourage reproducing the ATG8 overexpression phenotype in Plasmodium falciparum to evaluate the protective efficacy of antibodies against natural P. falciparum infection. Additionally, designing autophagy mutants based on alterations of ATG3 or ATG7 expression levels or mutants lacking GASP or VPS4 involved in amphisome formation for microneme clearance () would advantageously be considered as genetically attenuated malaria parasites in case of their arrest during liver stage development.
On the other hand, ATG8 is expressed during all life stages in Plasmodium and is essential for parasite survival as demonstrated in ATG8 gene knockout experiments [Citation164,Citation283,Citation284]. Autophagic machinery of malaria parasite cannot be substituted by other biological systems as mammalian cells or yeasts [Citation285]. This incompatibility could then be exploited for specific interference of Plasmodium ATG proteins. To this point, discerned regions in the parasite ATG3 sequence could be targeted by small molecules to specifically inhibit the Plasmodium ATG8-ATG3 interaction and to further avoid ATG8 lipidation. PfATG3 possesses a short interacting motif of 8 amino acids (PfATG3103-110 region) involved in PfATG8 binding as demonstrated in vitro using recombinant proteins and confirmed by the production of a X-ray co-crystal structure of the PfATG3103-110 peptide and PfATG8 [Citation286]. Importantly, PfATG3103-110 does not bind to mammalian LC3 due to the unique loop present in the Plasmodium ATG8 sequence (the hallmark of ATG8 in all Apicomplexa) necessary for interaction with PfATG3. Among a library of 352 small molecular fragments with an average molecular weight of 150 Da from Zenobia. Therapeutics, inhibition of full length PfAtg8 with PfAtg3 interaction was achieved by pyrogallolic acid (1,2,3-trihydroxybenzene) at low micromolar concentrations (IC50 of 150 μM) [Citation286]. Screening of 200 drug-like and 200 probe-like molecules from the Malaria Medicine Venture Malaria Box for at least 25% inhibition of PfATG8-PfATG3 interaction at the threshold of IC50 of 5 μM identifies 6 molecules [Citation287]. Among them, N-(4-methylphenyl)-4-pyridin-2-yl-1,3-thiazol-2-amine has been further investigated against liver stages of infection in cultured hepatocytes, and shows a 50% decrease in the proportion of cells infected with P. falciparum sporozoites at subtoxic concentration of 30 μM for HC-04 cells. In conclusion, targeting Plasmodium ATG8 lipidation is a rational strategy for drug intervention. In particular, the unique structural motifs required for Plasmodium ATG3-ATG8 interactions (a short interacting motif of 8 amino acids, PfATG3103-110 region [Citation286]) have been to date amenable to chemotherapeutic interference based on in vitro studies. The promising compounds identified from libraries may then be a good starting scaffold for drug optimization, with validation of drug effect on Plasmodium AGT8 in animal models. More broadly, investigating many other autophagy protein interactions that may have drastic effects on normal functions of autophagy complexes could also be a starting avenue for new drug discovery against protist illnesses.
The host autophagy during protist infections
With the exception of the ATG17 homolog that was not present in the Homo sapiens, mammalian cells have conserved all the repertory of autophagy proteins described in yeasts. Mammalian autophagy exhibits higher complexity than protists and displays a conjunct of functions that involve the energy maintenance in conditions of nutrient starvation, protein and organelle turnover, and its participation in cellular functions such as cell growth, cell division, and cell differentiation and cell death. However, autophagy actions in mammalian cells are not limited to these physiological roles, it is also important in pathological processes. Autophagy has a role in different diseases such as cancer, neurodegenerative disorders, and infective processes. In these pathologies autophagy can encompass opposite responses; on one side it can be beneficial for the host but also can be harmful depending on the conditions of the cells. The case of cancer exemplifies this concept: at the initial steps of carcinogenesis, autophagy contributes to eliminating malignant cells by induction of cell death but when the cancer is installed, autophagy contributes to the survival of neoplastic cells in the center of the tumor where nutrients are scarce due to low blood circulation. In the infective processes, autophagy response functions to destroy the intracellular microorganisms integrating the repertory of innate immune responses against pathogens. However, many microorganisms can subvert this host response for their own benefit, to get nutrients, or to generate membrane compartments or vacuoles with favorable environments to live in. The following sections present the current knowledge about the interaction of pathogenic protist with host cell autophagy. Given that each parasite targeted specific types of host cells, autophagy responses from phagocytes and non-professional phagocytic cells will be presented separately. Interestingly, since many of these protists possess models of infection in vivo, mainly displayed in mice; we added a final chapter that describes the effect of autophagy in the development of the infection in the animal model where autophagy responses are participating at different levels by regulation of cellular and even humoral immune responses.
Infection in professional phagocytic cells
Autophagy during Leishmania infection in macrophages
Several intracellular microorganisms uniquely interact with the autophagic pathway by activating or impairing autophagy, ultimately resulting in organism survival or death [Citation288]. This review section focuses on three main subjects approaching the knowledge regarding the role played by autophagy on Leishmania infection: whether Leishmania sp. induce autophagy during infection, how the autophagic pathway interacts with Leishmania-induced intracellular compartments, and how autophagy influences Leishmania infection outcome. During the initial steps of Leishmania-macrophage interaction, these parasites are recognized by a variety of receptors and internalized, leading to the formation of phagosomes containing Leishmania [Citation289–295]. These acidic compartments containing lysosomal enzymes, known as parasitophorous vacuoles (PVs), are surrounded by a membrane enriched with late endosomal/ lysosomal proteins, such as RAB7, macrosialin, LAMP-1, LAMP-2, vacuolar H+-ATPase, and molecules of the antigen presentation machinery. Inside PVs, Leishmania promastigotes transform into amastigotes, which can survive and replicate, leading to infection amplification [Citation296]. The infection outcome depends on different factors, such as parasite species and a balance between the host immune response and the parasite immune-evasion strategies[Citation297,Citation298]. The role played by Leishmania-induced autophagy on the modulation of host cell immune response has been discussed elsewhere [Citation299–301]; therefore, this topic will not be addressed in the present review section.
Although labeling intracellular compartments with markers of autophagy, such as MDC and LC3 by immunofluorescence or detecting LC3 in cell extracts by western-blot indicate the occurrence of autophagy, other techniques, including TEM, the expression of other autophagy-related genes (Atg) than LC3, quantification, and detection of LC3, including accurately measuring autophagic flux, are needed to confirm autophagic activation in eukaryotic cells [Citation191]. Regardless of the methodology employed to evaluate the autophagic activation in host cells during Leishmania spp. infection, data consistently supports the notion that this process occurs during most infections caused by these parasites in mammalian cells [Citation302–309] ().
Figure 7. The host autophagy during Leishmania infection. (A) Leishmania major- and L. amazonensis induced autophagy. Both L. amazonensis and L. major induce autophagy in macrophages. L. major activation of autophagy depends on TLR3, 7, and 9, reducing parasite survival inside the host cell. L. major-infected macrophages present a higher percentage of parasitophorous vacuoles labeled with DQ-BSA and earlier loss of LC3-recruited molecules than L. amazonensis-infected macrophages. (B) Leishmania braziliensis-induced autophagy. L. braziliensis infection induces BECN1–Vps34 complex. This parasite also reduces LC3B expression in cutaneous lesion biopsies and a transient decrease in LC3+ cells in L. braziliensis-infected THP-1 compared to uninfected cells. In addition, L. braziliensis reduces the percentage of LC3+ cells and the number of puncta structures after rapamycin or starvation treatment in macrophages. (C) Leishmania donovani-induced autophagy. L. donovani induces high LC3-II to LC3-I ratios in infected THP1 cell-line, human macrophages, and PMNs. This parasite induces non-canonical autophagy in the early stages after infection and canonical autophagy later in PMNs. In macrophages, this parasite induces non-canonical autophagy at later stages of infection and inhibits classical autophagy at both early and late stages, favoring parasite survival. Created with BioRender.com.
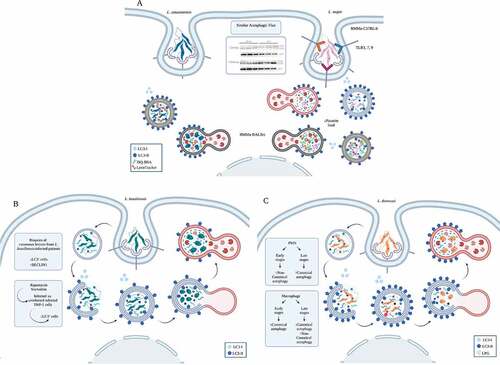
For parasites that cause cutaneous disease, autophagic activation in response to infection seems to be unique to each Leishmania species. LC3 labeling by western-blot was detected in extracts of Leishmania amazonensis-infected macrophages from susceptible and resistant mice and in the RAW 264.7 macrophage cell line infected with this parasite[Citation302]. Morphological alterations suggestive of autophagy activation, along with an increase in the autophagic flux, support the activation of autophagy in Leishmania major-infected bone marrow-derived macrophages (BMDM)[Citation308]. Furthermore, the dependence on Toll-like receptors for the activation of autophagy in the control of L. major infection was proven using TLR3, -7, and -9 knockout (Tlr3/7/9−/−) mouse macrophages, which are incapable of infection control[Citation303]. Interestingly, autophagy-induced after L. donovani engulfment by neutrophils is not dependent on the TLR2/4 receptor since the receptor blockade did not alter parasite lipophosphoglycan (LPG)-induced autophagy [Citation309]. Independent of resistance and susceptibility to infection, L. major- and L. amazonensis-infected CBA macrophages similarly activate the autophagic flux, showing a similar LC3-II/Actin ratio in the extracts of infected cells [Citation305] ().
The activation of autophagy in L. braziliensis infection was suggested by the description of the upregulation of the BECN1/Beclin1 (BECN1) – Vps34 complex, which is known to be associated with phagophore biogenesis and the maturation of autophagosomes [Citation310], in macrophage cultures in vitro and biopsies of cutaneous lesions from L. braziliensis-infected patients. At the same time, LC3B expression was downregulated in cutaneous lesion biopsies. Immunolabeling analysis revealed a slight and transient decrease in LC3+ cells in L. braziliensis-infected THP-1 compared to uninfected cells. L. braziliensis-induced downregulation of autophagy was further supported by the impairment of the percentage of LC3+ cells and the number of puncta structures by rapamycin or starvation treatment of L. braziliensis-infected macrophages compared to uninfected cells. The importance of LC3B reduced expression on infection outcome was highlighted by the infection rate increase in LC3B- but not in BECN1-silenced THP-1 cells [Citation311] ().
One of the primary causative agents of visceral leishmaniasis, Leishmania donovani, induced high LC3-II to LC3-I ratios in THP1 infected cells, indicating parasite-induced activation of the autophagic pathway at later time points [Citation304]. These findings were confirmed by a recent in vitro study demonstrating that L. donovani induces autophagy in macrophages and human polymorphonuclear neutrophils (PMNs)[Citation307]. Consistently, greater LC3-I to LC3-II conversion was found in a human bone marrow sample from a patient with visceral leishmaniasis but not in a sample from a healthy individual [Citation307]. Recently, Pitale et al.[Citation309] comprehensively examined the mechanisms involved in L. donovani-induced autophagy, revealing this process’s complexity. While this parasite induces noncanonical autophagy at early stages after infection, it induces canonical autophagy later in polymorphonuclear cells (PMNs) infection [Citation309]. Furthermore, although L. donovani induces the activation of an alternative autophagic pathway in macrophages at later stages of infection, the classical pathway was found to be inhibited at both early and late infection points in these infected cells to optimize parasite survival [Citation304] ().
Interestingly, autophagic induction, demonstrated by transcriptomic analyses, induced parasite clearance of L. major in infected BMDM in the late phase of infection [Citation308]. One possible explanation for autophagy distinctly influencing infection outcome in vitro relies on the recent description of differences in the parasitophorous vacuoles’ (PVs) autophagic features induced by Leishmania sp. Indeed, compared with mouse macrophages from CBA mice infected with L. amazonensis, L. major-PVs presented a double percentage of vacuoles labeled with the degradative compartment marker DQ-BSA. In addition, LC3-II is lost earlier in L. amazonensis than in L. major PVs, although both compartments were labeled similarly by LysoTracker, an acidic compartment marker [Citation305]. Notably, autophagy inhibition by a specific VPS34 inhibitor, VPS34-IN1, or activation by rapamycin did not alter the degree of LC3 recruitment to L. amazonensis- or L. major-induced PVs. Similarly, rapamycin-induced autophagy did not modify LC3 labeling in L. donovani-infected THP-1 cells compared to untreated cells[Citation304]. In contrast, LC3 positivity and the number of puncta structures were impaired in L. braziliensis-infected THP-1 cells compared to the control, untreated macrophages [Citation311]. These findings also reinforce the notion that during infection, autophagy can be activated via a pathway other than PI3K-Akt-mTOR.
Contradictory results regarding the role played by autophagy in the Leishmania infection outcome probably arise from the type of methodology used to investigate this effect. Thomas et al.[Citation304] showed that the abrogation of autophagic activation by a knockdown of Atg5 and Atg9 in THP-1 cells decreases intracellular L. donovani survival. Furthermore, during L. amazonensis infection, starvation causes differing outcomes depending on the strain of infected mouse macrophages [Citation312]. The intracellular viability of L. amazonensis is increased in macrophages from susceptible BALB/c mice but not in those from resistant mice [Citation312]. This effect was not observed for L. major in BALB/c mouse macrophages. In contrast, using a more specific methodology, autophagy abrogation secondary to Atg5 knockdown in BALB/c[Citation308] and C57BL/6 [Citation303] macrophages increased L. major intracellular viability. Interestingly, autophagic activation before infection profoundly affects macrophage phagocytic capacity [Citation313,Citation314], suggesting that inhibition of autophagy should be made after infection by using a genetic approach or plasmids with inducible promoters. Employing another mouse model, in CBA/j mouse macrophages that were proven resistant to L. major yet susceptible to L. amazonensis, physiological and pharmacological inhibition of autophagy did not alter either L. amazonensis or L. major parasitic load. On the other hand, autophagic induction significantly increased L. major intracellular viability within PVs that notably exhibited a more degradative feature than those induced by L. amazonensis [Citation305].
In sum, the discrepancy involving Leishmania-induced autophagy in host cells should be counteracted by performing studies using complementary comparative methods for expanding the identification of common targets from the autophagic pathway for an appropriate design of new therapeutic strategies in treating leishmaniasis.
LC3-associated phagocytosis during Leishmania infection
As key players of the innate immune system, professional phagocytes recognize, engulf, and destroy pathogens through a process known as phagocytosis. Here, pathogens are internalized in a vacuole, the phagosome, which undergoes a complex maturation program characterized by a series of highly regulated sequential interactions with diverse cellular compartments [Citation315]. These interactions, which are essential for the generation of a microbicidal phagolysosome, entail the accrual of numerous proteins, including the vacuolar proton (H+)-pumping ATPase (V-ATPase) and of an array of degradative hydrolases to these organelles [Citation316,Citation317]. Most microbes are destroyed in such a degradative environment, leading to the generation of peptides through the degradation of microbial antigens. These peptides are loaded on MHC molecules and transported to the cell surface to initiate an adaptive immune response [Citation316,Citation318–320], establishing a link between innate and adaptive immunity [Citation316,Citation321]. Phagolysosome biogenesis thus represents an important means of controlling infections.
A non-canonical autophagic pathway known as LC3-associated phagocytosis (LAP) provides a link between phagocytosis and autophagy [Citation322–325]. The salient feature of LAP is the recruitment of the autophagy-related protein LC3 to phagosomes, which takes place early on during the internalization process [Citation322]. Hence, time-lapse analysis of LC3 association to phagosomes indicated this event occurs rapidly, within 15 min after internalization and is transient, as LC3 starts dissociating from phagosomes after 60 min [Citation322]. Consistent with its role in host defense against pathogens, including bacteria, fungi, and parasites [Citation324], LAP requires the triggering of various pattern recognition receptors including toll-like receptors, Dectin-1, Dectin-2, as well as receptors for phosphatidylserine for the recognition of apoptotic cells [Citation322,Citation326–328]. Although the significance of LC3 recruitment on phagosomal functions remains to be fully elucidated, LAP is characterized by an enhancement of phagosome maturation and microbial killing [Citation322], as well as of MHC class II-mediated antigen presentation [Citation328].
One of the key features of LAP is the essential role of NADPH oxidase (NOX2)-mediated production of reactive oxygen species (ROS) in the recruitment of LC3 to phagosomes [Citation329]. ROS generated by NOX2 play a central role in the biology of phagocytes, including microbial killing and antigen cross-presentation [Citation328,Citation330,Citation331]. Assembly and activation of the NOX2 complex to phagosomes during LAP is a highly regulated process. Upon stimulation of pattern recognition of receptors by pathogens or apoptotic cells, the cytosolic components of NOX2 p40phox, p47phox, and p67phox form a heterotrimer which is then translocated to the phagosome membrane to assemble with the membrane-associated flavocytochrome b558 components gp91phox and p22phox[Citation330]. Although LAP requires a number of components associated with canonical autophagy [Citation324], it remains distinct for a number of characteristics. In particular, LAP necessitates the activity of Rubicon, which controls the phagosomal association of a structure known as the LAPosome [Citation332]. In addition, Rubicon is essential for the phagosomal assembly and activity of NOX2, production of ROS, and recruitment of LC3 to phagosomes [Citation332]. Phagosomal assembly of NOX2 is also regulated by a number of modulators of membrane fusion, including Synaptotagmin XI, VAMP8, and SNAP23 [Citation333–335]. Hence, membrane trafficking mechanisms associated with these SNAREs play an important role during LAP [Citation306,Citation335].
Given the potentially lethal consequences of exposure to toxic ROS, it is not surprising that a number of intracellular pathogens evolved virulence strategies to interfere with phagosomal assembly of NOX2 [Citation336–340]. To establish infection within a hospitable intracellular compartment, both developmental stages of the Leishmania parasite prevent assembly of NOX2 at the phagosome through various strategies. Hence, amastigotes of L. pifanoi induce heme degradation through the activation of heme oxidase-1, preventing maturation of the immature form of gp91phox and ROS production at the phagosome [Citation341]. Amastigotes of L. donovani evade serine phosphorylation of p47phox, impairing the recruitment of both p47phox and p67phox to phagosomes and ROS generation [Citation342]. Promastigotes use at least two distinct strategies to prevent phagosomal assembly of a functional NOX2. One involves inhibiting the recruitment of the cytosolic p47phox/p67/p40phox heterotrimer to the phagosome through the action of the cell surface virulence gycolipid lipophosphoglycan (LPG) [Citation343]. The second strategy consists in the cleavage of the SNARE VAMP8 by the virulence-associated metalloprotease GP63. By cleaving VAMP8, Leishmania prevents NOX2 assembly to the phagosome and the recruitment of LC3 and inhibits LAP [Citation306]. Importantly, inhibition of LAP may require live parasites, as apoptotic L. major promastigotes are readily internalized by macrophages in LC3-positive compartments via LAP [Citation344]. Of note, in contrast to the phagocytosis of other types of particles, Leishmania does not trigger the recruitment of BECN1 to phagosomes [Citation306]. Interestingly, induction of host macrophage autophagy by L. braziliensis is BECN1-independent [Citation311]. Thus, both amastigote and promastigote forms of Leishmania use a panoply of strategies to prevent phagosomal NOX2 assembly/activity which, in addition to protecting the parasite from toxic ROS, leading to the impairment of the microbicidal LAP process in the host phagocytes.
Infection in non-professional phagocytic cells
In contrast to Leishmania sp., which subverts phagocytosis to infect host cells and thus develop only on professional phagocytic cells, T. cruzi and T. gondii exploit many mechanisms to invade cells and therefore can invade both professional and non-professional phagocytes. The effect of host autophagy on the outcome of infection is not always the same for both types of cells and contradictory data can be found in the literature about this matter. Interestingly, although both T. cruzi and T. gondii establish their replicative niches in non-professional phagocytic cells, as myocytes, brain and eyes, they can also infect macrophages, neutrophils and dendritic cells at initial times of infection or to gain access to immunologically privileged tissues such as brain in neurotoxoplasmosis. In contrast, Plasmodium species target hepatocytes and red blood cells establishing its infection only in non-professional phagocytes.
Different phenotypes are displayed by host autophagy during T. cruzi infection
Depending on the stage in the biological cycle, two infective forms of T. cruzi can interact with mammalian host cells, the tissue-derived trypomastigotes (TCT), equivalent to bloodstream trypomastigotes, produced by differentiation of amastigotes in the host cells, and the metacyclic trypomastigotes (MT) present in the insect vector. For TCT, the initial events during the T. cruzi intracellular cycle can be divided in 4 main steps [Citation155,Citation345]: (1) Adhesion of trypomastigotes to the host cell surface; (2) invasion/internalization of trypomastigotes and formation of the T. cruzi parasitophorous vacuole; (3) maturation of PV by fusion with lysosomes; and (4) exit of parasites from PV to cytosol and differentiation into amastigotes (). After that, amastigotes replicate several times until they transform back into trypomastigotes which exit the cell to start a new infective cycle. As T. cruzi can infect all nucleated cells, the host response and the parasite life-cycle in those cells can be peculiar. Features such as different (i) parasitic forms and their source (metacyclic, bloodstream, or tissue culture trypomastigotes (TCT)); (ii) strains; and (iii) host cell type directly influence the parasite-host interaction. In the first description on host autophagy in T. cruzi infection, Romano and colleagues showed LC3 recruitment to the parasitophorous vacuoles (PVs) of TCT from CL Brener strain in a non-phagocytic cell line (CHO: Chinese hamster ovary cell line) [Citation154] (). This recruitment on the PV was also observed in other T. cruzi strains and host cell types such as cardiac-derived cell lines and RAW macrophages [Citation346]. The induction of host autophagy (by starvation or rapamycin treatment) before the infection led to an increase in the number of infected cells at early times after infection but not variation in the amastigote replication occurred when autophagy was induced after the infection, at later times [Citation154]. Inhibitors of canonical autophagy such as wortmannin, 3-methyladenine or vinblastine, and the inhibitor of the spermidine biosynthesis, DFMO (difluoromethylornithine) reverted the effect of starvation and decrease the LC3 recruitment [Citation154,Citation347]. Acquisition of the endosomal/lysosomal markers Lamp-1 and LBPA to PVs increased at starvation conditions suggesting that autophagy induction promoted the infection by favoring the previously described lysosomal dependent entry of T. cruzi in the host cells [Citation348] or by promoting the fusion of lysosomes with the early vacuole, a key step for retention of T. cruzi trypomastigotes inside host cells [Citation349]. A more recent work showed that T. cruzi (Tulahuen strain) increases the number of LC3 dots after infection in a human fibrosarcoma cell line [Citation350]. Increased number of LC3 vesicles was also observed in CHO cells infected with the Y strain of T. cruzi together with the formation of LC3-positive tubules, an interesting feature not found when these cells were treated with rapamycin and whose relevance will need to be explored in the future [Citation155]. Working with metacyclic trypomastigotes of CL strain, Dr. Yoshida laboratory found that starvation of HeLa cells at the same time of T. cruzi infection increases the lysosome formation and scattering required for the invasion of MT to host cells, while TCT enter preferentially by a lysosomal-independent process that is benefited when host cell starvation is made prior infection [Citation351] (). Together, these evidences showed that starvation increases T. cruzi infection in a manner that depends to the mechanism exploited by T. cruzi to infect cells, by the lysosomal-dependent or independent (endocytic-like) route. In the same way interaction of T. cruzi with host cell autophagosomes and maturation of the T. cruzi vacuole by the acquisition of lysosomal markers are differently modulated according to mammalian cell and parasite features. Time of infection is also important. At later times when amastigotes are present in the cytoplasm of host cells, LC3 recruitment to these parasites was very low at basal autophagy conditions [Citation154,Citation350]. Although T. cruzi infection increases the number of LC3-positive vesicles, Onisuka and colleagues showed that these autophagosomes did not acquire syntaxin-17, a SNARE protein associated with autophagosome-lysosome fusion, and lysotracker, suggesting that T. cruzi can inhibit the autophagy flux to evade autophagic capture and degradation, corroborated by non-degraded p62, an autophagy cargo receptor [Citation350] ().
Figure 8. The host autophagy during T. cruzi infection. (A) invasion of tissue cell trypomastigotes (TCT) is characterized by 4 steps, adhesion of parasites to cell surface (step 1), internalización/parasitophorous vacuole formation (step 2), vacuole maturation (step 3) and exit of parasites to cytosol by vacuole membrane disruption (step 4). TCT vacuole is decorated with LC3 and infection increase in pre-starved cells by favoring the vacuole maturation. (B) MT invasion is characterized by plasma membrane wounding and lysosomal exocytosis. Starvation of cells favors infection of MT by increasing lysosomal formation and scattering. (C) T. cruzi infection increases autophagosome number. Amastigotes developed at later times of infection inhibit autophagy flux in non-professional phagocytic cells. (D) In professional phagocytic cells (or in cardiac cells treated with autophagy inducers), amastigotes are targeted by xenophagy and destroyed. Created with BioRender.com.
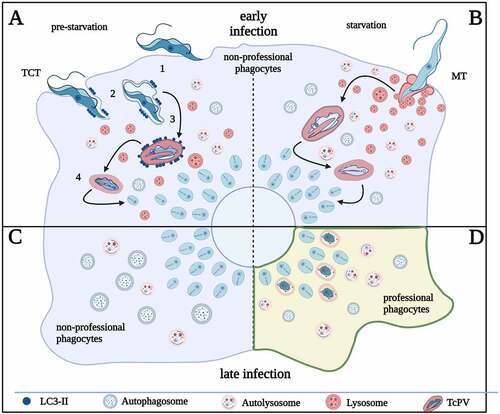
Quite different is the action of autophagy on the T. cruzi infection in phagocytic cells. In this case it was observed that autophagy response results in the T. cruzi elimination by xenophagy of amastigotes at later times of infection [Citation352]. This selective process increases in the presence of autophagy inducers such as ursolic acid [Citation352] and has the opposite effect in the presence of autophagy inhibitors or in the BECN1 heterozygous KO peritoneal cells [Citation353]. Furthermore, other studies corroborate that autophagy displays not only a direct action in the parasite but this process interlinks other host processes to subvert infection in primary culture cell models. In activated C57Bl/6 peritoneal macrophages, infection induced LC3 and p62 expression, and rapamycin decreased the number of infected cells and parasite proliferation in a NLRP3-dependent pathway [Citation354]. Similar data were obtained in peritoneal macrophages and cardiac cells infected with bloodstream trypomastigotes from the T. cruzi Y strain, whereas starvation induced after infection decreased the infection only in cardiac cells, indicating different roles of autophagy regarding cell type [Citation355] (). Also in this study, infected Atg5−/− MEF showed an increased percentage of infection [Citation355]. As it was mentioned above, different host cell and parasite models, as well as distinct experimental designs, could lead to the variety of autophagic phenotypes of T. cruzi infected cell previously reported, emphasizing the huge repertory of molecules involved in the parasite-host cell relationship. This hypothesis is strengthened by the substantial differences in surface protein repertoire between tissue culture, bloodstream, and metacyclic trypomastigotes from the Y strain [Citation351,Citation356]. Those molecules, that modulate several processes during T. cruzi entry such as parasite adhesion, host cytoskeleton remodeling, and lysosome location, could explain the different results obtained in each model. Systematic comparative studies combined to the best technology available, which is another important source of data variation, will need to be conducted in the future to explain the differences found.
Host autophagy during T. gondii infection
Toxoplasma gondii uses an active invasion process to infect many kinds of nucleated mammalian cells, including professional and non-professional phagocytic cells. The process involves the formation of a receptor-ligand mediated tight apposition of the parasite and host plasma membranes known as the moving junction [Citation357]. As the moving junction translocates from the apical to the posterior end of the parasite, it excludes host transmembrane proteins [Citation358], thereby forming a specialized intracellular compartment known as the PV [Citation359]. Excluding host transmembrane proteins renders the PV non-fusogenic [Citation360], thus allowing the parasite to avoid degradation by lysosome fusion, at least initially. Thereafter the parasite secretes lipids and proteins to modify the PV for its replication.
Host cell defense against T. gondii infection involves the autophagy machinery. As early as 2 min post-infection, host LC3 or homologs (GABARAPs) are detected at the surface of T. gondii PV, indicating the detection of the intruder by host autophagy. Lipidated LC3 is directly incorporated in situ in the PV membrane (PVM) and not by the fusion with an LC3-positive autophagosome, indicating the occurrence of a LAP-like process different from canonical autophagy. LC3 lipidation in Toxoplasma infected cells is mediated by the complex ATG5-ATG12-ATG16L1 and the enzymes ATG7 and ATG3. Although the nature of the factor(s) to recruit ATG12-ATG5-ATG16L1 complex to the PV is unknown, it is possible that abnormal membranous structures or a pathogen-associated molecular pattern (PAMP) participate in this recognition. Downstream of LC3, two different mechanisms that mediate parasite killing and control toxoplasmosis have been described. The autophagy-dependent killing of parasites is activated by CD40 and its main ligand CD154[Citation361]. CD40 activation induces recruitment and fusion of host lysosomes to LC3-positive PV followed by parasite destruction [Citation362]. The second process is triggered by interferon-gamma (IFN-γ). IFN-γ is a pleiotropic cytokine that plays a key role in activation of cellular immunity and antitumor immune-response. This protein has a main role against T. gondii in hematopoietic and non-hematopoietic cells [Citation363]. IFN-γ induces the localization of IFNγ-regulated GTPases (IRGs) and guanylate-binding proteins (GBPs) to T. gondii vacuoles [Citation364,Citation365]. These proteins vesiculate the PVM of T. gondii, thereby exposing the denuded parasites to autophagosomes [Citation364,Citation366]. Loading of IRGs on the PVM depends on the prior recruitment of the autophagy proteins ATG3, ATG7 and ATG5-ATG12-ATG16L complex, but not LC3 [Citation366,Citation367]. Also, activation of Arf1 by GABARAPL2 (GATE16) enables GBP to remain in a non-aggregated form, thus promoting GBP loading onto PVM [Citation365]. These mechanisms were observed mainly in mouse-derived cells. In human epithelial cells, ubiquitin, the autophagy receptors NDP52 (nuclear dot protein 52) and SQSTM1/p62, and LC3 are deposited on T. gondii PV by the action of an interferon-stimulated gene 15 (ISG15)-related pathway. This produces the entrapment of the PV by a multilayer structure that restricts parasite growth [Citation368]. In human endothelial cells infected with type-II strains, loading of ubiquitin, SQSTM1/p62 and NDP52 on the PVM is followed by acquisition of Rab7 and fusion of PV with lysosomes leading to parasite destruction [Citation369]. Because this process does not involve ULK1, ULK2, or Atg14L, and classic modulators of autophagy (rapamycin, wortmannin, chloroquine, bafilomycin A1) have no effect, most authors agree that IFNγ functions to limit T. gondii infection in this context via non-canonical autophagy [Citation369–371]. Although both CD40 and IFNγ toxoplasmacidal mechanisms share some molecular components, there are specific differences between them. CD40 activation induced T. gondii killing by autophagy via ULK1, BECN1, PI3KC3, ATG5, ATG7, and lysosomal enzymes [Citation367,Citation372] whereas ULK1 and BECN1 do not participate in IFNγ activated parasite death. In addition, interferon-dependent autophagy is selective for type II parasites, while CD40-induced autophagy kills type I and II parasites [Citation370,Citation371,Citation373,Citation374]. Despite this arsenal of autophagy effectors and IRG at the PV to control infection, infected host cells remain incapable of clearing all parasites. Only a small portion of Toxoplasma PVs (less than 10%) are targeted by LC3. This means that T. gondii has evolved successful strategies to subvert the destructive actions of host autophagy. One strategy Toxoplasma uses involves stimulating the host epidermal growth factor receptor (EGFR)/PI3K/Akt signaling axis. Extracellular T. gondii release proteins (MICs) from apical secretory organelles (micronemes) to support gliding motility and facilitate cell invasion. Several MICs contain multiple domains with homology to EGF [Citation375] that act as ligands of EGFR. MIC/EGFR engagement triggers EGFR autophosphorylation, resulting via several steps in the phosphorylation and activation of the kinase Akt, which in turn activates MTOR to suppress host autophagy [Citation376]. Besides this inhibition of the defense mechanisms of host cells, T. gondii takes advantage of host cell degradation pathways to acquire a sustainable source of nutrients for growth [Citation377]. Microscopic observations showed the particular location of PV containing replicating parasites between the endocytic and secretory pathways. Indeed host endocytic organelles and Rab-positive vesicles are observed in the PV lumen [Citation378]. Non-selective autophagy also benefits the parasite, as another source of nutrients. On one hand, it has been shown that proteins (ROPs) secreted from apical rhoptry organelles can modulate host canonical autophagy. ROP17 interacts with Bcl-2, disrupting Bcl2-BECN1 interaction, and increasing LC3 and p62 expression [Citation379]. On the other hand, ROP16 contributed to Focal Adhesion Kinase/Signal Transducer and Activator of Transcription 3 (FAK/STAT3) activation, impairing autophagy clearance [Citation380]. The induction of host autophagy also impacts T. gondii infection. In human embryonic fibroblasts, rapamycin increased infection [Citation145], nevertheless, in Vero cells and other human fibroblasts, starved cells induced parasite cell death by mitochondrial fragmentation [Citation240]. In primary skeletal muscle cells, autophagy inducers reduced infection [Citation242]. Using human umbilical cord mesenchymal stem cells to mimic congenital toxoplasmosis in vitro, T. gondii downregulated MCL-1, an anti-apoptotic protein, disrupting BECN1 interaction, promoting autophagy and apoptosis by caspase-3-cleavage [Citation381]. Additionally, the parasite uses fatty acids from host lipophagy for its growth, competing with the host mitochondrial network [Citation382].
In conclusion, host cells resort to different innate immunity and cell-autonomous mechanisms to contain the parasitic infection. Some of these mechanisms encompass the activation of the canonical autophagic pathway (), and others that use the ubiquitination machinery (). They turn the PV into a fusogenic or damaged vacuole finally destroyed by the lysosomal degradation pathway. For its part, T. gondii has evolved to limit the activation of autophagy and subsequent lysosomal degradation (). The effect of this pathway is dependent on the host cell type and explains why under certain conditions T. gondii induces autophagy and uses it for its own benefit.
Figure 9. The host autophagic responses during T. gondii infection. (A) activation of host cell autophagy by CD40 stimulus induces the fusion of T. gondii PV with lysosomes followed by parasite destruction. (B) In murine cells, IFN-ɣ induces the recruitment of IRGs and GBPs on the PVM which expose the denuded parasites to destruction by autophagosomes (pathway 1). In human epithelial cells, ISG15 induces the ubiquitination of PVM followed by SQSTM1/p62 and NDP52 mediated LC3 recruitment. Concentric LC3-positive membranes around PV limit parasite replication (pathway 2). In human endothelial cells, ubiquitination and loading of SQSTM1/p62 and NDP52 are followed by Rab7 recruitment and lysosomal fusion leading to parasite destruction (pathway 3). (C) T. gondii resists host cell toxoplasmicidal action by inhibiting selective autophagy via MICs-dependent EGFR activation. Induction of canonical autophagy and lipophagy provide nutrients for T. gondii growth. Created with BioRender.com.
Host cell autophagy-related pathways during Plasmodium liver stage infection
Sporozoites, the infective form of Plasmodium for the vertebrate host, exhibit gliding motility[Citation383], a substrate-dependent locomotion depending on their actin myosin motor located between the inner membrane complex and the parasite plasma membrane [Citation384]. Eventually, they cross the endothelial barrier by either transmigration of Kupffer cells, endothelial cells or paracellularly between two cells [Citation385]. Cell traversal (CT) is not only important for reaching the liver, but also inside the liver parenchyma. Sporozoites first transmigrate several hepatocytes until they finally invade one productively [Citation386]. CT can occur either by disrupting the plasma membrane of the traversed hepatocytes and moving freely in their cytoplasm (, non-productive invasion by cell wounding) or by forming transient vacuoles (TVs). TVs are formed by invagination of the hepatocyte’s plasma membrane when sporozoites transmigrate the cell without forming a moving junction and are subsequently disrupted [Citation387](, non-productive invasion by formation of a transient vacuole). In contrast, productive invasion of Plasmodium sporozoites is characterized by an invagination of the host cell plasma membrane forming a specialized and stable compartment, the parasitophorous vacuole (PV), in which the parasite resides (reviewed in Loubens M, et al 2021 [Citation388]). Proper PV formation requires the establishment of a moving junction complex between the invading parasite and the host cell at the site of entry [Citation357]. The moving junction acts as a molecular sieve, excluding host cell proteins from the emerging PV membrane (PVM)[Citation389,Citation390]. Therefore, a fully functional PVM, which initially does not contain typical host cell plasma membrane proteins, differs from a TV membrane, which contains all host plasma membrane proteins present at the time of parasite entry (, productive invasion). Export of proteins and maintenance of PVM integrity are crucial for successful liver stage development. Interestingly, parasites that exhibit problems with PVM formation or maintenance are commonly subject to elimination, underlining the importance of the PVM in maintaining a reproductive niche for the parasite [Citation391–394]. A functional PVM not only promotes the uptake of nutrients, but also the release of waste products into the host cell and most likely it acts also as a signaling hub between host cytosol and parasite. The PVM contains pores which allow passive diffusion of small molecules up to about 855 Da [Citation395]. PVM modifications include parasite protein export to the PVM and a change in the membrane’s architecture by formation of a tubovesicular network (TVN)[Citation395–398].
Figure 10. Cell invasion by Plasmodium and autophagy associated host responses. (A) Different types of invasion: non-productive invasion by cell wounding, non-productive invasion by formation of a transient vacuole, and productive invasion. (B) Mechanisms of host response against Plasmodium: the Plasmodium-Associated Autophagy-Related (PAAR) response targeting P. berghei parasites, the LAP-like response against P. vivax parasites, and the PI3P-Associated Sporozoite Elimination (PASE) against P. berghei parasites trapped in a transient vacuole. The molecular components involved in each response were depicted (see text for description). (C) Signaling functions of SQSTM1/p62. Middle panel: SQSTM1/p62 is recruited by LC3 and associates with the parasitophorous vacuole membrane of Plasmodium liver stage parasites. Upper panel: p62 interaction with TRAF6 is essential for MTOR activation and suppression of canonical autophagy (pathway 2). Bottom panel: binding of KEAP1 to phosphorylated p62 increases NRF2 activation. NRF2 is stabilized and translocates into the nucleus to induce the expression of cytoprotective genes that benefit Plasmodium (see text for description). INF-γ: interferon-gamma, PLP1: perforin-like protein 1 (also called SPECT2), PV: parasitophorous vacuole, TV: transient vacuole, PVM: parasitophorous vacuole membrane, TVN: tubovesicular network. Created with BioRender.com.
Thus, host cells are confronted with parasites residing in different types of vacuoles and have to react adequately to control infection.
During their development in hepatocytes, Plasmodium parasites face major challenges. On one hand, they require considerable amounts of nutrients and lipids for their extensive growth. On the other hand, parasites must escape host cell defense mechanisms. There is increasing evidence that autophagy-related responses play a role in both aspects. This means that host cell autophagy during the Plasmodium liver stage can have beneficial as well as deleterious effects on the parasite. It is therefore important to distinguish between starvation-induced canonical autophagy, which can supply nutrients and autophagy-related pathways as an intracellular immune response against the parasite. The liver stage Plasmodium parasite is one of the fastest growing eukaryotic organisms [Citation399]. Therefore, it needs a constant supply of nutrients which must be provided by the host cell (reviewed in Nyboer B, et al 2018 [Citation400]). Starvation-induced canonical autophagy represents an important source of nutrients for the growing parasite. Inhibition of canonical autophagy by genetic manipulation of FIP200 and ATG5 lead to a reduction in parasite growth [Citation401–403]. This growth defect could be partly compensated by amino acid supplementation of infected ATG5−/− cells [Citation401]. These data, also supported by in vivo experiments (see below), depict the positive role of host canonical autophagy on parasite growth. Defense action of autophagy-related pathways against the parasite will be explained in the following paragraphs.
All Plasmodium species analyzed until now are recognized via non-canonical autophagy mechanisms that are independent of the ULK complex and the nutritional status of the cell. These mechanisms are: the Plasmodium-Associated Autophagy-Related (PAAR) response; the LAP-like response; and the PI3P-associated sporozoite elimination (PASE), ().
The most important autophagy-related process targeting P. berghei and similarly P. yoelii liver stages is the Plasmodium-Associated Autophagy-Related (PAAR) response. During the PAAR response LC3 is directly incorporated into the parasite’s PVM immediately after invasion. LC3 labeling of the PVM is long-lasting and occurs independent of both the ULK and PI3K complex [Citation401–404]. New insights suggested LC3 lipidation at the PVM involves V-ATPase assembly and non-canonical recruitment of ATG16L1 (unpublished results). If this can be confirmed, the PAAR response closely resembles a process termed V-ATPase-ATG16L1-induced LC3B lipidation (VAIL)[Citation405] and a process called Conjugation of ATG8 proteins to Single Membranes (CASM)[Citation406]. Around 90% of P. berghei liver stage parasites are targeted by the PAAR response [Citation401–403]. However, parasites provoking the PAAR response are not necessarily being eliminated, around 50% of P. berghei parasites persist and successfully develop (, PAAR response) [Citation401]. Interestingly, infection of FIP200−/− cells that are deficient in canonical autophagy but still can execute PAAR response, resulted in a decreased parasite size. However, in comparison to wildtype cells, FIP200−/− cells could eliminate the same number of parasites suggesting that canonical autophagy is not required for parasite elimination but rather supports parasite growth by supplying additional nutrients. On the other hand, in ATG5−/− cells which are deficient in canonical autophagy and PAAR response, parasite survival is enhanced [Citation401–403]. This suggests the PAAR response is responsible for the elimination of a significant proportion of parasites during the liver stage.
Autophagic responses of host cells vary considerably between different Plasmodium species investigated so far. Although some features are shared in different species, the molecular mechanisms seem to differ substantially. LC3 labeling of the PVM appears to be a common denominator during hepatocyte infection as it was observed during P. berghei, P. yoelii and P. vivax liver stage infection [Citation401,Citation402,Citation407]. However, P. vivax parasites are targeted by a process, which rather resembles LC3-associated phagocytosis (LAP) and only targets around 30% of parasites. Similar to LAP, LC3 labeling of the PVM follows PI3P formation by the PI3K complex and leads to efficient parasite elimination (, LAP-like response)[Citation407]. In contrast to the PAAR response, LC3 labeling of the P. vivax PVM depends on IFN-γ and does not occur spontaneously. Thus, there appears to be substantial differences in autophagic targeting between the rodent and human Plasmodium species. These differences might stem from their different lifestyles: P. vivax parasites are able to form dormant stages in the liver called hypnozoites, which can reactivate months or even years after initial infection [Citation408]. P. vivax thus might have evolved a different evasion strategy that prevents immediate detection by host cell autophagy and allows dormancy. Low metabolic activity might also be a reason why at least dormant P. vivax parasites are not spontaneously recognized.
Interestingly, a small fraction of P. berghei parasites is also eliminated in a pathway resembling LAP. In the first hours of hepatocyte infection a maximum of 5 % of parasites are transiently labeled with PI3P followed by weak LC3 labeling and acidification through lysosomal fusion. This is in stark contrast to the long-lasting association of LC3 with the PVM during the PAAR response which is not preceded by PtdIns3P labeling. Surprisingly, elimination of PtdIns3P-positive parasites partly occurs also in ATG5−/− cells and is therefore not dependent on autophagy. The parasites targeted by this PtdIns3P-associated sporozoite elimination (PASE) were identified as transmigrating sporozoites which are contained within a TV (, PASE) [Citation404]. To traverse cells, parasites engage perforin-like protein 1 (PLP1 also known as SPECT2) [Citation409,Citation410]. In vitro infection of hepatocytes with CT-deficient, SPECT2-negative parasites resulted in a higher percentage of parasites targeted by PASE, as they are not able to exit their TV. Sporozoites captured in their TV are successfully acidified by lysosomal fusion [Citation404].
Although no autophagosome membrane is formed around the parasite and classical selective autophagy can be excluded as a mechanism of parasite recognition, ubiquitin and the selective autophagy receptors (SARs) SQSTM1/p62, NBR1 (neighbor of BRCA1 gene 1) and to a lesser extent NDP52 associate with the PVM of P. berghei parasites [Citation401,Citation411]. Interestingly, in P. berghei infected cells, labeling with SARs does not follow the selective autophagy pathway. The order of membrane labeling appears to be inverted from LC3 recruiting SARs which in turn recruit ubiquitin [Citation411]. This raises the question, how LC3 itself is recruited to the PVM and what the function of SARs is at the PVM if not recruiting LC3.
The answer to the last question can be found in the signaling function of SQSTM1/p62. This protein is also a central hub in cell signaling as it is involved in many pathways including NF-kB signaling, nutrient sensing and NRF2-driven oxidative stress response. None of these functions depend on the LC3 interacting region (LIR) motif or the ubiquitin-binding domain that are needed for successful function of SQSTM1/p62 in selective autophagy. Therefore, SQSTM1/p62 represents a link between autophagy and signaling (reviewed in Katsuragi Y el al 2015 [Citation412]). Interfering with host cell signaling is a popular strategy of intracellular pathogens to secure survival and establish a perfect niche for growth [Citation413]. NRF2 is a transcription factor involved in oxidative stress response and pro-survival mechanisms. Under basal conditions, NRF2 is constantly bound by its negative regulator KEAP1, leading to ubiquitination and subsequent proteasomal degradation of the complex [Citation414]. Sequestration of KEAP1 by SQSTM1/p62 allows stabilization and therefore activation of NRF2. Phosphorylation of SQSTM1/p62 at Ser349 drastically enhances affinity of KEAP1 for p62 (; bottom panel) [Citation415]. It was recently shown that in P. berghei-infected hepatocytes, host cell p62 is recruited to the PVM and binds KEAP1. This KEAP1 recruitment to the Plasmodium PVM-associated SQSTM1/p62 appears to activate NRF2 signaling in infected hepatocytes, thereby ensuring host cell and thus parasite survival (; 1) [Citation416]. Several NRF2 target genes were shown to be upregulated upon Plasmodium infection in a recent in vivo single cell transcriptomics screen. Among these were proteins involved in glutathione and iron metabolism such as glutathione S-transferases (Gstm1 and Gstm3) and Hmox1, Ftl1, Fth and Slc40a1 respectively. In addition, SQSTM1/p62 expression itself, which is also controlled by Nrf2, was found to be upregulated in infected host cells [Citation417]. Additionally, SQSTM1/p62 is involved in other signaling pathways. It has been shown that it interacts with TRAF6 and other players in the nutrient sensing machinery of the cell [Citation418,Citation419]. Interaction between SQSTM1/p62 and TRAF6 is needed for activation of MTOR [Citation419], a master regulator of nutrient sensing, promoting anabolic processes when nutrients are available and activating autophagy when nutrients are scarce [Citation420]. Thus, SQSTM1/p62 might function as an adaptor for assembly of the nutrient sensing machinery on the lysosomal membrane and mTORC1 activation thereby regulating autophagy inhibition in response to AAs (; upper panel). However, it remains to be shown how the parasite interferes with the cell’s nutrient sensing and metabolic signaling to induce canonical autophagy that could provide additional nutrients for the parasite (; 2).
Related to the LC3 interaction to the PVM, it has been speculated whether this is beneficial or deleterious for the parasite. The answer is not straightforward as it turned out that it can be both. The PVM-resident protein UIS3 was reported to be crucial for autophagy evasion [Citation421]. This protein interacts with LC3 and likely competes with the binding of LIR-motif containing proteins to LC3, therefore allowing evasion from autophagic degradation[Citation421]. Under normal conditions UIS3(-) parasites are efficiently eliminated by the host cell as they have problems maintaining an intact PVM [Citation422]. Importantly, UIS3(-) parasites can successfully develop in autophagy-deficient ATG5−/−, ATG7−/− or ATG3−/− cells, confirming the importance of autophagy and related pathways for parasite elimination. In the opposite side, it was shown that a small molecule, C4 (4-{[4-(4-{5-[3-(trifluoromethyl) phenyl]-1,2,4-oxadiazol-3-yl}benzyl)piperazino]carbonyl}benzonitrile), that inhibits interaction between the parasite-derived protein called Upregulated in infective sporozoites 3 (UIS3) and LC3 is capable of impairing infection [Citation423] arguing that LC3 labeling of the PVM is beneficial for parasite survival.
Clearly, liver stage Plasmodium parasites need additional strategies to evade degradation by lysosomal fusion. A very efficient way to avoid autophagic responses in general is enhanced membrane shedding towards the TVN [Citation397]. By shedding autophagy proteins and lysosomes from the PVM, the parasite evades harmful consequences of PVM targeting by the host cell. This way the parasite does not only evade elimination but might even benefit from lysosomal fusion at the TVN as an additional source of nutrients. This suggests that there is an ideal amount of lysosomal material at the PVM allowing the parasite to benefit from nutrients and lipids while avoiding lysosomal elimination. The equilibrium is maintained by constant PVM shedding. Arrested parasites that are impaired in membrane shedding exhibit a strong PVM labeling with the lysosomal marker LAMP1 followed by acidification and elimination of the parasite [Citation424]. Interestingly, successfully developing parasites are not acidified even though lysosomes fuse with the PVM and material of the endo-lysosomal network is found in the cytoplasm of the parasite [Citation424,Citation425]. One possible explanation for that might be that the PVM contains pores which allow diffusion of molecules below 855 Da [Citation395] and would thus most likely allow protons to freely diffuse into the host cell cytoplasm avoiding a drop of the pH in the PV.
In summary, autophagy and related responses can have beneficial as well as deleterious aspects for intracellular development of Plasmodium parasites. To benefit from the capability of providing nutrients and membrane material parasites must balance activation of autophagy and preventing elimination by autophagy-related pathways. The PVM plays a central role in both the host cell’s ability to target of the intruder and the parasite’s evasion strategy. Interestingly, a highly dynamic vesicle exchange between the host cell and the parasite appears to take place and is not only restricted to lysosomes and late endosomes but also concerns Golgi-derived vesicles [Citation426]. The P. berghei PVM is able to fuse with host late endocytic vesicles in a PtdIns(3,5)P2 -dependent manner, allowing the exchange of material between the host and the parasite, which is essential for successful infection [Citation427]. However, the precise molecular mechanism that drives the interaction between host cell vesicles and the PVM and how parasites can access the potential nutrients in the vesicles remains elusive.
Downstream molecules involved in efficient vesicle fusion are usually proteins of the SNARE family and small GTPases of the Ras-related in brain (RAB) protein family, which should be subject of future investigations. If we finally understand better the fine balance of beneficial and deleterious autophagy responses in infected hepatocytes, it might be possible to generate drugs that shift this balance to complete elimination of parasites.
Role of autophagy on in vivo models of infection
Autophagy during trypanosomatids infection in mice
Despite the high number of in vitro studies showing the interplay of autophagy and parasites in different scenarios of the host cell, protist species and the types of infective forms, there is a relatively modest quantity of works describing the effect of autophagy on trypanosomatid infections in animal models. This difference is based in the increased level of complexity that means to pass from in vitro to in vivo models of infection. Similar to cell culture experiments, data obtained from mice infection have to be interpreted in the light of the type of intervention, genetic or pharmacological, used to interrupt or activate autophagy in the whole organism or in a specific tissue or organ. In the case of T. cruzi, first studies showed that autophagy played a protective role during the infection. Acute T. cruzi infection in the autophagy deficient BECN1 heterozygous knock-out mice (Becn1+/-), generated in the C57BL6/J background in the Beth Levine laboratory [Citation428], was associated with higher parasitemia, cardiac parasitism and earlier death compared to their WT counterpart. Peritoneal cells derived from Becn1+/- animals and RAW macrophages treated with autophagy inhibitors displayed higher levels of infection compared to controls. Interestingly, in RAW cells free cytosolic parasites are surrounded by LC3 protein and other markers of selective autophagy such as Ubiquitin, SQSTM1/p62 and NDP52, features that were observed in lesser extension in autophagy-deficient cells, indicating that this process contributed to control the infection by elimination of intracellular amastigotes [Citation353]. Strikingly, expression of inducible nitric oxide synthase (iNOS) in peritoneal cells and NO2− plasma levels of Becn1+/- mice measured 5 days after T. cruzi infection showed significantly lower increase than Becn1+/+compared to uninfected animals (Casassa and Romano, unpublished data). Additionally, Duque and colleagues showed that induction of autophagy by treatment of mice with the autophagy inducer Rapamycin protected the cardiac function by reducing myocarditis, cardiac damage and the production of pro-inflammatory cytokines by the heart during acute T. cruzi infection without modulation of cardiac infection and parasitemia [Citation355]. Therefore, beneficial actions of autophagy in mice infected with T. cruzi are displayed at different levels, from a cell-autonomous immune defense process through the engulfment of cytoplasmic amastigotes [Citation352,Citation353], to the modulation of inflammation and immune responses [Citation429,Citation430] It is known that C57BL6/J mice, that preferentially develop Th1 responses, are able to resist T. cruzi infection by activation of iNOS and the increase of NO level, a potent anti-T. cruzi agent [Citation431]. In contrast, BALB/c mice, which are more susceptible to T. cruzi infection, displayed a predominance of Th2 responses and polyamine synthesis that benefit the parasite growth [Citation432]. In this sense it was recently shown that chronic Rapamycin pretreatment contributed to improve the balance between Th1 and Th2 effectors in elderly C57BL6/J mice, which exhibited a better parasitological control, reduced heart inflammation and microstructural damage [Citation433].
Balance between Th1 and Th2 responses is also important during the experimental infection of mice with Leishmania amazonensis. Il4−/− BALB/c mice displayed increased Th1 response and developed smaller lesions when infected with moderate parasite inoculum compared to Il4+/+ mice. Similar results were obtained in Il4+/+ mice treated with indomethacin, an inhibitor of PGE(2) synthesis [Citation434]. Interestingly, induction of autophagy in macrophages from BALB/c mice displayed higher parasitic load compared to macrophages from C57BL6/J mice. Increased parasite load was associated with a reduction in NO levels and increased number of lipid bodies and amounts of PGE(2) thus indicating that in macrophages from these mice strain can induce autophagy and generate products that favor L. amazonensis replication and reduces the production of toxic compounds at the same time [Citation312]. Moreover in skin lesions of BALB/c mice infected with L. amazonensis, a positive reaction was observed by immunohistochemistry with anti-LC3 antibody indicating autophagy induction [Citation302]. In contrast, action of autophagy in the control of Leishmania infection was demonstrated, as presented in the previous section, in the case of L. major, in which autophagy was shown to function downstream of endosomal TLRs signaling of macrophages from C57BL6/J mice [Citation303]. In addition, rapamycin treatment reduced inflammatory lesions formed in the ears of Leishmania-infected C57BL/6 and Tlr3/7/9−/− mice.
In conclusion, up to the moment the effect of autophagy on in vivo infections with trypanosomatids seems to be related to the type of immune response predominant in the mice strain; favoring the replication of parasites in the susceptible BALB/c mice, that preferentially develops a Th2 response, or contributing to the parasite elimination in the resistant C57BL6/J mice which preferentially activate Th1 responses.
Autophagy during in vivo infections of Plasmodium
As mentioned above autophagy inhibition by genetic manipulation impaired the liver Plasmodium growth mainly by the reduction of nutrients required for the fastest growing of this eukaryotic organism [Citation399]. Therefore, it needs a constant supply of nutrients which must be provided by the host cell (reviewed in Nyboer B, et al 2018 [Citation400]). In vivo experiments support these findings as in Atg5-deficient mice; parasite liver loads were significantly reduced [Citation402]. However, the exact mechanism behind the reduced liver load is not known. While parasites in autophagy-deficient cells are clearly smaller, they can still successfully develop into detached cells harboring infective merozoites [Citation402]. Hence, while canonical autophagy contributes to parasite growth, it is not essential for parasite development. One reason might be that the intracellular parasites also control other potential nutrient sources in that it participates in the host cell Golgi vesicular traffic [Citation423]. However, it is not known what kind of nutrients they gain from associating with the host cell Golgi and how they are eventually processed.
Interestingly, activation of both starvation-induced and drug-mediated canonical autophagy by treatment with the MTOR inhibitor rapamycin results in an extraordinary 25-fold increase in parasite load in vivo, that arises from significantly increased parasite size but mainly from enhanced parasite survival [Citation401]. The mechanism underlying enhanced parasite survival upon activation of canonical autophagy induction is still unknown, but it is remarkable that the in vivo effect of autophagy activation is much stronger than in in vitro cultivated cells including primary hepatocytes. A reason could be that in vivo, autophagy responses are more pronounced because they underlie physiological conditions, whereas in vitro, medium conditions including supplements and fetal calf serum downregulate such autophagy responses. It is also important to note that increased growth of parasites in rapamycin-treated mice could be due to the immunosuppressive effect of this compound in vivo [Citation435,Citation436] resulting in the impairment of the immunity against Plasmodium. An interesting hypothesis concerning the extraordinary growth and survival of liver stage parasites in vivo upon rapamycin treatment or starvation is that different autophagy pathways might compete for limited concentration of autophagy proteins. In normal cells most autophagy-related proteins (i.e. LC3) are free in the cytosol. Upon parasite invasion, autophagy proteins are sufficient to target the parasite and about 50% are eliminated by the PAAR response. In contrast, when canonical autophagy is induced by starvation or rapamycin treatment, autophagy-related proteins are required for the generation of autophagic vesicles and less parasites are attacked by the PAAR response. Together, the autophagic status of the host cell appears to drastically influence the outcome of infection in vivo and to a lesser extent also in vitro. The beneficial effect of autophagy can be modulated by the same parasite. It is showed that Plasmodium yoelii rodent malaria parasites perturb hepatocyte regulatory pathways involved in cell survival, proliferation, and autophagy. The pro-death protein p53 was substantially decreased in infected hepatocytes suggesting that it could be targeted by the parasite to foster survival [Citation437]. In contrast, other host pathways are hijacked by Plasmodium as was shown by the detrimental parasite infection observed during the knockdown of genes involved in protein trafficking pathways [Citation438]. Targeting of host proteins might be a smart strategy to expand the repertoire of prophylactic drugs against malaria as in the case of p53 agonists that were effective when administered to humanized mice infected with Plasmodium falciparum [Citation439].
Effect of host autophagy on T. gondii infection in vivo
As discussed above, the most important mechanism of T. gondii to avoid killing by the host cell is the formation of a specialized intracellular compartment (the PV) that does not fuse with compartments from the endolysosomal system. The two main host mechanisms to induce parasite killing, and control toxoplasmosis, involving either autophagy or autophagy proteins, have been recently reviewed focusing on the importance of these processes in the outcome of in vivo infections [Citation440]. The autophagy-dependent killing of parasites is activated by CD40 and its main ligand CD154 [Citation361]. Existence of this mechanism was confirmed in vivo in CD40–/– and CD154–/– mice, which displayed increased susceptibility to cerebral and ocular toxoplasmosis [Citation373,Citation441] despite seemingly unimpaired expression of IFN-γ, the main cytokine that mediates protection against T. gondii in mice [Citation442,Citation443]. Similar increased susceptibility to cerebral and ocular toxoplasmosis was demonstrated in Becn1+/- mice, mice lacking Atg7 in myeloid cells (Atg7flox/flox-Lyz-M Cre mice), and mice deficient in the pro-autophagy protein PKR (PKR–/–) [Citation373,Citation444]. This confirms that CD40-induced death of T. gondii (type I strain) is independent of IFN-γ and effector responses downstream of this cytokine and depends on autophagy. Besides macrophages and microglia, CD40 restriction of infection is also functional in brain endothelial cells, an important port of entry of T. gondii to the Central Nervous System (CNS), mainly the brain and retina [Citation445]. The toxoplasmicidal actions of endothelial cells is acquired upon interaction of these cells with T. gondii-infected dendritic cells and macrophages, and depends on the expression of CD40 by endothelial cells and the expression of BECN1 and the inducible heat shock protein 70 in dendritic cells [Citation445]. Other actions of CD40 pathway include reduced anti-T. gondii antibody production [Citation446] and protection against CD8+ T cell exhaustion[Citation447], which have implications in the establishment of the chronic infection.
The second mechanism to control T. gondii infection is displayed by cell-autonomous actions elicited by INF-γ in which autophagy proteins are involved. In this case, parasite killing is independent of endosomal/lysosomal fusion with the PV and depends on the recruitment of certain autophagy proteins Atg3, Atg7, and the complex Atg5-Atg12-Atg16L [Citation366,Citation448]. In mouse cells these proteins trigger the recruitment of GKS IRGs to the PVM, followed by deposition of ubiquitin, SQSTM1/p62, and the E3 ubiquitin ligases TRAF6 and TRIM21, which promotes sustained decoration of PVM with ubiquitin [Citation449]. This process leads to delivery of guanylate-binding proteins (GBPs) to PVM followed by vesiculation and rupture of the vacuole, and finally death of susceptible strains [Citation448,Citation450], type II and III (, left panel). Virulent T. gondii strains (type I) avoid this action by the production of ROP kinases that acts synergistically to impair the recruitment of IRGs to the PVM, and control acute infection in mice [Citation451]. Control of T. gondii infection by INF-γ in human cells is quite different than in mouse cells since it does not rely on IRGs and has notable cell-type specificity. In human epithelial cells infected with type II and III strains, ubiquitin deposition on PVM leads to recruitment of SQSTM1/p62, NDP52, and optineurin receptors followed by the formation of a LC3-positve multilayer that restricts the parasite replication [Citation370] (, central panel). In contrast, in human endothelial cells infected with type-II strain, ubiquitin and SQSTM1/p62 deposition is followed by recruitment of Rab7, fusion with lysosomes, and parasite killing [Citation452] (, right panel). In this case LC3 is not recruited around PVM, and this process is not dependent on autophagy [Citation452].
IFN-γ-mediated immunity against T. gondii seems to be more critical in mice than humans. Patients with an autosomal dominant defect in IFN-γR1 resulting in deletion of the signal transducer and activator of transcription 1 (STAT1) binding site are not susceptible to toxoplasmosis, although their macrophages are unable to display anti-T. gondii activity in response to IFN-γ [Citation453]. Treatment with tumor necrosis factor-alpha (TNF-α) partially restored antimicrobial activity of macrophages from these patients, indicating that this factor contributes to the control of infection [Citation453]. CD40 may also contribute to IFN-γ-independent mechanisms of protection in humans [Citation454], likely in part by inducing TNF-α production [Citation455,Citation456].
To investigate T. gondii evasion of host autophagic clearance in vivo, the role of EGF and EGFR was demonstrated in transgenic mice that express a dominant-negative (DN) mutant of EGFR that impairs EGFR autophosphorylation and EGFR transactivation. Expression of DN EGFR exclusively in endothelial cells was accompanied by a reduction in parasite load and histopathology in the brain and retina after T. gondii infection [Citation457].
In summary, the main host mechanisms that operate in vitro against T. gondii have been investigated in mouse models of infection. Remarkably, in contrast to toxoplasmicidal actions triggered by INF-γ that are not operative across species, parasite strains, or host cell types, autophagic killing functions in all scenarios. This feature points to autophagy modulation (or impairment of T. gondii evasion of autophagy) as a potential strategy for drug development against toxoplasmosis.
Effect of parasite autophagy on apicomplexan infections in vivo
Few in vivo infection studies have been performed on parasite autophagy in apicomplexans for two main reasons. First, much of the work on autophagy in malaria parasites has been done with P. falciparum, for which animal models are limited. Second, as noted above, many ATG proteins in apicomplexans are essential for parasite replication because of their non-canonical role in apicoplast inheritance. Since conditional disruption of ATG proteins involved in apicoplast inheritance severely affects parasite replication, the impact on in vivo infection is assumed to be equally pronounced.
As noted above, overexpressing ATG8 by 2-fold significantly delayed the liver stage development of rodent infective P. berghei parasites in infected mice [Citation175], likely due to disrupting both the canonical and non-canonical roles of ATG8.
In the case of T. gondii, two studies have examined the canonical role of the parasite ATG9 in autophagy during experimental infection of mice [Citation180,Citation458]. The first study showed that complete ablation or conditional knockdown of TgATG9 markedly reduced parasite burden during acute infection and significantly attenuated parasite virulence, resulting in >70% mouse survival with the knockout strain and or 100% mouse survival from the conditional knockdown [Citation180]. This is particularly notable given that the mutants were created in a highly virulent strain of T. gondii that normally causes 100% mortality. Disruption of TgATG9 also reduced parasite levels in mouse macrophages. Since macrophages are known to restrict T. gondii by limiting certain essential amino acids and by generating reactive oxygen species, the authors suggested that parasite autophagy supports parasite survival by recycling amino acids or turning over oxidatively damaged parasite proteins, structures, or organelles. The second study knocked out TgATG9 in an avirulent T. gondii strain that is competent for differentiating from the acute to chronic stage [Citation458]. Consistent with the earlier study, mice infected with parasites lacking TgATG9 showed 100% survival compared to 70-80% survival of mice infected with parental or genetically complemented strains. Also, ~5-fold fewer TgATG9 parasites were detected upon dissemination to the brain on day 7 post-infection, further supporting a role for canonical autophagy during acute infection. However, TgATG9 deficient parasites showed a >200-fold loss of parasite burden in the brain at 5 weeks post-infection during the chronic stage. Death of chronic stage TgATG9 deficient parasites in vitro was associated with markedly abnormal mitochondria, implying a critical role for autophagy in support of mitochondrial homeostasis. Whether the canonical autophagy pathway in T. gondii has additional roles during persistence warrants further investigation.
Concluding remarks
In this work, we have reviewed the biological cycle of pathogenic protists and the participation of the autophagic pathway in the different stages of their cycles. The particular lifestyles of these parasites, which alternate between vertebrate and invertebrate hosts, in addition to the necessity to adapt to different environments, allow the conservation of cellular processes like autophagy. We have seen that autophagy displayed different levels of complexity through this early evolutionary branch of the eukaryote domain, from a rudimentary process observed in the Giardia genus up to the more complex pathway displayed in Trypanosomatids and Apicomplexa families with the diversification of autophagic proteins such as Atg4 and Atg8 in a similar way to what is observed in humans, a feature whose presence is still not completely understood. Parasite autophagy has been shown to participate in many processes, including survival under nutrient deprivation, formation and turnover of organelles, and cell and nuclear death during specific situations. Contrary to these physiological functions, autophagy is activated in the presence of antiprotist drugs as a quick survival response to the stress generated by the treatment. Since this is a phenomenon observed with most drugs, the use of inhibitors of parasite autophagy as adjuvants of anti-parasitic drugs could be interesting to explore in the future.
The interaction of pathogenic parasites with the host autophagy has also been reviewed. In most cases, autophagy is activated in the host cell after infection, being a protective response for the host, which can be subverted by the pathogen to favor growth and survival. Therefore, depending on parameters like type of the host cell-targeted, time of infection, and life-styles and virulence of pathogenic protists, host autophagy plays two opposite roles, promoting parasite survival or parasite death. More complex is the scenario when the function of autophagy is studied on in vivo models of infection, where the immune system can modulate the autophagy response and vice versa, modifying the fate of infection. Finally, we have to consider that parasitic autophagy can also play an important role in the intracellular protist life, another aspect that should be considered for future treatments. In summary, protist autophagy is needed at different stages of the protist life-cycle, in axenic environments, or inside host cells. On the other way, host cells have developed different mechanisms to destroy parasites during infection. They are carried out by canonical or selective autophagy pathways, autophagy proteins, and/or autophagy-related processes to finally kill the pathogen. However, virulent pathogens can take advantage of host autophagy responses to resist destruction. The application of this knowledge in the search and design of new drugs that interrupt the mechanisms of resistance of protists in addition to the improvement of immune responses and cell-autonomous processes of hosts will allow the discovery of efficient therapies against these still successful pathogens in the future.
Acknowledgments
We thank Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina and Gonçalo Moniz Institute, Fiocruz Bahia, Brazil for financial and institutional support.P.S.R. holds a research grant from Agencia Nacional de Promoción Científica y Tecnológica, Argentina (PICT 2017-1456 and PICT 2019-00640); P.S.T.V. holds a grant for productivity in research from CNPq (305235/2019-2). This work was supported by JSPS KAKENHI Grant Number 21K20643 to T.A. V.B.C is supported by US National Institutes of Health grants R01 AI120607 and R21 AI160610). Work by L.F.J. and C.Y.H. are supported by Singapore Ministry of Education Research Grants (MOE-T2EP30221-0002 and R-154-000-C60-114). We also wish to thank Mr. Ng Jun Jie, Angus for technical assistance in the experiments shown in . The authors would like to thank Andris K. Walter for English language revision and manuscript copyediting assistance. AD is supported by the Canadian Institutes of Health Research (grant PJT-156416) and is the holder of the Canada Research Chair on the Biology of intracellular parasitism.
Additional information
Funding
References
- Desmond E, Brochier-Armanet C, Forterre P, et al. On the last common ancestor and early evolution of eukaryotes: reconstructing the history of mitochondrial ribosomes. Research in microbiology [Internet]. 2011 [cited 2022 Nov 12];162:53–108. Available from: https://pubmed.ncbi.nlm.nih.gov/21034815/.
- Kazamia E, Helliwell KE, Purton S, et al. How mutualisms arise in phytoplankton communities: building eco-evolutionary principles for aquatic microbes. Ecology letters [Internet]. 2016 [cited 2022 Nov 12];19:810–822. Available from: https://pubmed.ncbi.nlm.nih.gov/27282316/.
- Burki F, Roger AJ, Brown MW, et al. The New Tree of Eukaryotes. Trends in ecology & evolution [Internet]. 2020 [cited 2022 Nov 12];35:43–55. Available from: https://pubmed.ncbi.nlm.nih.gov/31606140/.
- Sanchez SG, Besteiro S. The pathogenicity and virulence of Toxoplasma gondii. Virulence [Internet]. 2021 [cited 2022 Jul 18];12:3095–3114. Available from: https://pubmed.ncbi.nlm.nih.gov/34895084/.
- Mugnier MR, Stebbins CE, Papavasiliou FN. Masters of Disguise: Antigenic Variation and the VSG Coat in Trypanosoma brucei. PLoS pathogens [Internet]. 2016 [cited 2022 Jul 18];12. Available from: https://pubmed.ncbi.nlm.nih.gov/27583379/.
- Vaughan AM, Kappe SHI. Malaria Parasite Liver Infection and Exoerythrocytic Biology. Cold Spring Harbor perspectives in medicine [Internet]. 2017 [cited 2022 Nov 12];7. Available from: https://pubmed.ncbi.nlm.nih.gov/28242785/.
- Kemmerling U, Osuna A, Schijman AG, et al. Congenital Transmission of Trypanosoma cruzi: A Review About the Interactions Between the Parasite, the Placenta, the Maternal and the Fetal/Neonatal Immune Responses. Frontiers in microbiology [Internet]. 2019 [cited 2022 Jul 18];10. Available from: https://pubmed.ncbi.nlm.nih.gov/31474955/.
- Klotz SA, Schmidt JO. Autochthonous Chagas Disease: How Are These Infections Happening? The American journal of medicine [Internet]. 2020 [cited 2022 Jul 18];133:e683–e686. Available from: https://pubmed.ncbi.nlm.nih.gov/32682868/.
- Mizushima N. A brief history of autophagy from cell biology to physiology and disease. Nature cell biology [Internet]. 2018 [cited 2022 Jul 19];20:521–527. Available from: https://pubmed.ncbi.nlm.nih.gov/29686264/.
- Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Letters [Internet]. 1993 [cited 2022 Jul 19];333:169–174. Available from: https://onlinelibrary.wiley.com/doi/full/10.1016/0014-5793%2893%2980398-E.
- Thumm M, Egner R, Koch B, et al. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS letters [Internet]. 1994 [cited 2022 Jul 19];349:275–280. Available from: https://pubmed.ncbi.nlm.nih.gov/8050581/.
- Harding TM, Morano KA, Scott S V., et al. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. The Journal of cell biology [Internet]. 1995 [cited 2022 Jul 19];131:591–602. Available from: https://pubmed.ncbi.nlm.nih.gov/7593182/.
- Titorenko VI, Keizer I, Harder W, et al. Isolation and characterization of mutants impaired in the selective degradation of peroxisomes in the yeast Hansenula polymorpha. Journal of bacteriology [Internet]. 1995 [cited 2022 Jul 19];177:357–363. Available from: https://pubmed.ncbi.nlm.nih.gov/7814324/.
- King JS. Autophagy across the eukaryotes: is S. cerevisiae the odd one out? Autophagy [Internet]. 2012 [cited 2022 Jul 19];8:1159–1162. Available from: https://pubmed.ncbi.nlm.nih.gov/22722653/.
- Hughes T, Rusten TE. Origin and evolution of self-consumption: autophagy. Advances in experimental medicine and biology [Internet]. 2007 [cited 2022 Jul 13];607:111–118. Available from: https://pubmed.ncbi.nlm.nih.gov/17977463/.
- Keeling PJ, Burki F. Progress towards the Tree of Eukaryotes. Current biology : CB [Internet]. 2019 [cited 2022 Jul 19];29:R808–R817. Available from: https://pubmed.ncbi.nlm.nih.gov/31430481/.
- Nakatogawa H, Suzuki K, Kamada Y, et al. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nature reviews. Molecular cell biology [Internet]. 2009 [cited 2022 Jul 13];10:458–467. Available from: https://pubmed.ncbi.nlm.nih.gov/19491929/.
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nature cell biology [Internet]. 2010 [cited 2022 Jul 13];12:814–822. Available from: https://pubmed.ncbi.nlm.nih.gov/20811353/.
- Fukuda T, Ebi Y, Saigusa T, et al. Atg43 tethers isolation membranes to mitochondria to promote starvation-induced mitophagy in fission yeast. eLife [Internet]. 2020 [cited 2022 Jul 13];9:1–29. Available from: https://pubmed.ncbi.nlm.nih.gov/33138913/.
- Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxidants & redox signaling [Internet]. 2014 [cited 2022 Jul 13];20:460–473. Available from: https://pubmed.ncbi.nlm.nih.gov/23725295/.
- Li FJ, He CY. Acidocalcisome is required for autophagy in Trypanosoma brucei. Autophagy [Internet]. 2014 [cited 2022 Jul 12];10:1978–1988. Available from: https://pubmed.ncbi.nlm.nih.gov/25484093/.
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nature reviews. Molecular cell biology [Internet]. 2001 [cited 2022 Jul 13];2:211–216. Available from: https://pubmed.ncbi.nlm.nih.gov/11265251/.
- Matsuura A, Tsukada M, Wada Y, et al. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene [Internet]. 1997 [cited 2022 Jul 13];192:245–250. Available from: https://pubmed.ncbi.nlm.nih.gov/9224897/.
- Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxidants & redox signaling [Internet]. 2011 [cited 2022 Jul 13];14:2201–2214. Available from: https://pubmed.ncbi.nlm.nih.gov/20712405/.
- Sakamoto H, Nakada‐tsukui K, Besteiro S. The Autophagy Machinery in Human-Parasitic Protists; Diverse Functions for Universally Conserved Proteins. Cells [Internet]. 2021 [cited 2022 Jul 13];10. Available from: https://pubmed.ncbi.nlm.nih.gov/34069694/.
- Cernikova L, Faso C, Hehl AB. Roles of Phosphoinositides and Their binding Proteins in Parasitic Protozoa. Trends in parasitology [Internet]. 2019 [cited 2022 Jul 13];35:996–1008. Available from: https://pubmed.ncbi.nlm.nih.gov/31615721/.
- Brennand A, Gualdrón-López M, Coppens I, et al. Autophagy in parasitic protists: unique features and drug targets. Molecular and biochemical parasitology [Internet]. 2011 [cited 2022 Jul 12];177:83–99. Available from: https://pubmed.ncbi.nlm.nih.gov/21315770/.
- Jiang W, Chen X, Ji C, et al. Key Regulators of Autophagosome Closure. Cells [Internet]. 2021 [cited 2022 Jul 13];10. Available from: https://pubmed.ncbi.nlm.nih.gov/34831036/.
- Faruk MO, Ichimura Y, Komatsu M. Selective autophagy. Cancer science [Internet]. 2021 [cited 2022 Jul 13];112:3972–3978. Available from: https://pubmed.ncbi.nlm.nih.gov/34407274/.
- Rigden DJ, Herman M, Gillies S, et al. Implications of a genomic search for autophagy-related genes in trypanosomatids. Biochemical Society transactions [Internet]. 2005 [cited 2022 Aug 16];33:972–974. Available from: https://pubmed.ncbi.nlm.nih.gov/16246023/.
- Duszenko M, Ginger ML, Brennand A, et al. Autophagy in protists. Autophagy [Internet]. 2011 [cited 2022 Jul 19];7:127–158. Available from: https://pubmed.ncbi.nlm.nih.gov/20962583/.
- Brennand A, Rico E, Michels PAM. Autophagy in Trypanosomatids. Cells. 2012;1:346–371.
- Hassett MR, Sternberg AR, Riegel BE, et al. Heterologous Expression, Purification, and Functional Analysis of Plasmodium falciparum Phosphatidylinositol 3’-Kinase. Biochemistry [Internet]. 2017 [cited 2022 Aug 25];56:4335–4345. Available from: https://pubmed.ncbi.nlm.nih.gov/28719180/.
- Williams RA, Tetley L, Mottram JC, et al. Cysteine peptidases CPA and CPB are vital for autophagy and differentiation in Leishmania mexicana. Molecular microbiology [Internet]. 2006 [cited 2022 Jul 19];61:655–674. Available from: https://pubmed.ncbi.nlm.nih.gov/16803590/.
- Pang Y, Yamamoto H, Sakamoto H, et al. Evolution from covalent conjugation to non-covalent interaction in the ubiquitin-like ATG12 system. Nature structural & molecular biology [Internet]. 2019 [cited 2022 Jul 19];26:289–296. Available from: https://pubmed.ncbi.nlm.nih.gov/30911187/.
- Kellner R, De la Concepcion JC, Maqbool A, et al. ATG8 Expansion: A Driver of Selective Autophagy Diversification? Trends in plant science [Internet]. 2017 [cited 2022 Jul 19];22:204–214. Available from: https://pubmed.ncbi.nlm.nih.gov/28038982/.
- Williams RAM, Woods KL, Juliano L, et al. Characterization of unusual families of ATG8-like proteins and ATG12 in the protozoan parasite Leishmania major. Autophagy [Internet]. 2009 [cited 2022 Jul 19];5:159–172. Available from: https://pubmed.ncbi.nlm.nih.gov/19066473/.
- Weidberg H, Shvets E, Shpilka T, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. The EMBO journal [Internet]. 2010 [cited 2022 Aug 16];29:1792–1802. Available from: https://pubmed.ncbi.nlm.nih.gov/20418806/.
- Li FJ, Shen Q, Wang C, et al. A role of autophagy in Trypanosoma brucei cell death. Cellular microbiology [Internet]. 2012 [cited 2022 Jul 12];14:1242–1256. Available from: https://pubmed.ncbi.nlm.nih.gov/22463696/.
- Proto WR, Jones NG, Coombs GH, et al. Tracking autophagy during proliferation and differentiation of Trypanosoma brucei. Microbial cell (Graz, Austria) [Internet]. 2014 [cited 2022 Jul 13];1:9–20. Available from: https://pubmed.ncbi.nlm.nih.gov/28357206/.
- Alvarez VE, Kosec G, Sant’Anna C, et al. Autophagy Is Involved in Nutritional Stress Response and Differentiation in Trypanosoma cruzi. Journal of Biological Chemistry. 2008;283:3454–3464.
- Maruyama T, Noda NN. Autophagy-regulating protease Atg4: structure, function, regulation and inhibition. The Journal of antibiotics [Internet]. 2017 [cited 2022 Jul 19];71:72–78. Available from: https://pubmed.ncbi.nlm.nih.gov/28901328/.
- Galluzzi L, Green DR. Autophagy-Independent Functions of the Autophagy Machinery. Cell [Internet]. 2019 [cited 2022 Jul 19];177:1682–1699. Available from: https://pubmed.ncbi.nlm.nih.gov/31199916/.
- Lévêque MF, Berry L, Cipriano MJ, et al. Autophagy-Related Protein ATG8 Has a Noncanonical Function for Apicoplast Inheritance in Toxoplasma gondii. mBio [Internet]. 2015 [cited 2022 Jul 13];6. Available from: https://pubmed.ncbi.nlm.nih.gov/26507233/.
- Walczak M, Ganesan SM, Niles JC, et al. ATG8 Is Essential Specifically for an Autophagy-Independent Function in Apicoplast Biogenesis in Blood-Stage Malaria Parasites. mBio [Internet]. 2018 [cited 2022 Jul 19];9. Available from: https://pubmed.ncbi.nlm.nih.gov/29295911/.
- Zhang S, Hama Y, Mizushima N. The evolution of autophagy proteins - diversification in eukaryotes and potential ancestors in prokaryotes. Journal of cell science [Internet]. 2021 [cited 2022 Jul 19];134. Available from: https://pubmed.ncbi.nlm.nih.gov/34228793/.
- Mancias JD, Wang X, Gygi SP, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature [Internet]. 2014 [cited 2022 Aug 17];509:105–109. Available from: https://pubmed.ncbi.nlm.nih.gov/24695223/.
- Mariño G, Pietrocola F, Eisenberg T, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Molecular cell [Internet]. 2014 [cited 2022 Jul 12];53:710–725. Available from: https://pubmed.ncbi.nlm.nih.gov/24560926/.
- Bohensky J, Shapiro IM, Leshinsky S, et al. HIF-1 regulation of chondrocyte apoptosis: induction of the autophagic pathway. Autophagy [Internet]. 2007 [cited 2022 Jul 12];3:207–214. Available from: https://pubmed.ncbi.nlm.nih.gov/17224629/.
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature reviews. Molecular cell biology [Internet]. 2012 [cited 2022 Jul 12];13:251–262. Available from: https://pubmed.ncbi.nlm.nih.gov/22436748/.
- Eng CH, Yu K, Lucas J, et al. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Science signaling [Internet]. 2010 [cited 2022 Jul 12];3. Available from: https://pubmed.ncbi.nlm.nih.gov/20424262/.
- Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell metabolism [Internet]. 2011 [cited 2022 Jul 12];13:495–504. Available from: https://pubmed.ncbi.nlm.nih.gov/21531332/.
- Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nature reviews. Molecular cell biology [Internet]. 2014 [cited 2022 Jul 12];15:155–162. Available from: https://pubmed.ncbi.nlm.nih.gov/24556838/.
- Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology [Internet]. 2011 [cited 2022 Jul 12];13:132–141. Available from: https://pubmed.ncbi.nlm.nih.gov/21258367/.
- Nazio F, Strappazzon F, Antonioli M, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nature cell biology [Internet]. 2013 [cited 2022 Jul 12];15:406–416. Available from: https://pubmed.ncbi.nlm.nih.gov/23524951/.
- Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nature Cell Biology [Internet]. 2013 [cited 2018 Aug 29];15:741–750. Available from: http://www.nature.com/articles/ncb2757.
- Settembre C, Fraldi A, Medina DL, et al. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nature reviews. Molecular cell biology [Internet]. 2013 [cited 2022 Jul 12];14:283–296. Available from: https://pubmed.ncbi.nlm.nih.gov/23609508/.
- Jewell JL, Kim YC, Russell RC, et al. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science (New York, N.Y.) [Internet]. 2015 [cited 2022 Jul 12];347:194–198. Available from: https://pubmed.ncbi.nlm.nih.gov/25567907/.
- Lin SC, Hardie DG. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell metabolism [Internet]. 2018 [cited 2022 Jul 12];27:299–313. Available from: https://pubmed.ncbi.nlm.nih.gov/29153408/.
- Ling NXY, Kaczmarek A, Hoque A, et al. mTORC1 directly inhibits AMPK to promote cell proliferation under nutrient stress. Nature metabolism [Internet]. 2020 [cited 2022 Jul 12];2:41–49. Available from: https://pubmed.ncbi.nlm.nih.gov/31993556/.
- Yang J, Zhou R, Ma Z. Autophagy and Energy Metabolism. Advances in experimental medicine and biology [Internet]. 2019 [cited 2022 Jul 12];1206:329–357. Available from: https://pubmed.ncbi.nlm.nih.gov/31776993/.
- Dall’Asta V, Bussolati O, Sala R, et al. Amino acids are compatible osmolytes for volume recovery after hypertonic shrinkage in vascular endothelial cells. The American journal of physiology [Internet]. 1999 [cited 2022 Jul 12];276. Available from: https://pubmed.ncbi.nlm.nih.gov/10199817/.
- Tabor CW, Tabor H. Polyamines. Annual review of biochemistry [Internet]. 1984 [cited 2022 Jul 12];53:749–790. Available from: https://pubmed.ncbi.nlm.nih.gov/6206782/.
- Reeds PJ, Biolo G. Non-protein roles of amino acids: an emerging aspect of nutrient requirements. Current opinion in clinical nutrition and metabolic care [Internet]. 2002 [cited 2022 Jul 12];5:43–45. Available from: https://pubmed.ncbi.nlm.nih.gov/11790948/.
- Zuzarte-Luís V, Mota MM. Parasite Sensing of Host Nutrients and Environmental Cues. Cell host & microbe [Internet]. 2018 [cited 2022 Jul 12];23:749–758. Available from: https://pubmed.ncbi.nlm.nih.gov/29902440/.
- Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science (New York, N.Y.) [Internet]. 2008 [cited 2022 Jul 12];320:1496–1501. Available from: https://pubmed.ncbi.nlm.nih.gov/18497260/.
- Kim E, Goraksha-Hicks P, Li L, et al. Regulation of TORC1 by Rag GTPases in nutrient response. Nature cell biology [Internet]. 2008 [cited 2022 Jul 12];10:935–945. Available from: https://pubmed.ncbi.nlm.nih.gov/18604198/.
- Tallóczy Z, Jiang W, Virgin IV HW, et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proceedings of the National Academy of Sciences of the United States of America [Internet]. 2002 [cited 2022 Jul 12];99:190–195. Available from: https://pubmed.ncbi.nlm.nih.gov/11756670/.
- Zoncu R, Bar-Peled L, Efeyan A, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science (New York, N.Y.) [Internet]. 2011 [cited 2022 Jul 12];334:678–683. Available from: https://pubmed.ncbi.nlm.nih.gov/22053050/.
- Aslett M, Aurrecoechea C, Berriman M, et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic acids research [Internet]. 2010 [cited 2022 Jul 12];38. Available from: https://pubmed.ncbi.nlm.nih.gov/19843604/.
- Marchese L, Nascimento JDF, Damasceno FS, et al. The Uptake and Metabolism of Amino Acids, and Their Unique Role in the Biology of Pathogenic Trypanosomatids. Pathogens (Basel, Switzerland) [Internet]. 2018 [cited 2022 Jul 12];7. Available from: https://pubmed.ncbi.nlm.nih.gov/29614775/.
- Global prevalence and incidence of selected curable sexually transmitted infections : overview and estimates [Internet]. [cited 2022 Jul 13]. Available from: https://apps.who.int/iris/handle/10665/66818.
- Bagchi S, Oniku AE, Topping K, et al. Programmed cell death in Giardia. Parasitology [Internet]. 2012 [cited 2022 Jul 13];139:894–903. Available from: https://pubmed.ncbi.nlm.nih.gov/22405231/.
- Wu JH, Tung SY, Ho CC, et al. A myeloid leukemia factor homolog involved in encystation-induced protein metabolism in Giardia lamblia. Biochimica et biophysica acta. General subjects [Internet]. 2021 [cited 2022 Jul 13];1865. Available from: https://pubmed.ncbi.nlm.nih.gov/33581251/.
- Mathieu C, Macêdo JP, Hürlimann D, et al. Arginine and Lysine Transporters Are Essential for Trypanosoma brucei. PloS one [Internet]. 2017 [cited 2022 Jul 12];12. Available from: https://pubmed.ncbi.nlm.nih.gov/28045943/.
- Inbar E, Canepa GE, Carrillo C, et al. Lysine transporters in human trypanosomatid pathogens. Amino acids [Internet]. 2012 [cited 2022 Jul 12];42:347–360. Available from: https://pubmed.ncbi.nlm.nih.gov/21170560/.
- Schmidt RS, Buẗikofer P. Autophagy in Trypanosoma brucei: amino acid requirement and regulation during different growth phases. PloS one [Internet]. 2014 [cited 2022 Jul 12];9. Available from: https://pubmed.ncbi.nlm.nih.gov/24699810/.
- Docampo R. The origin and evolution of the acidocalcisome and its interactions with other organelles. Molecular and biochemical parasitology [Internet]. 2016 [cited 2022 Jul 12];209:3–9. Available from: https://pubmed.ncbi.nlm.nih.gov/26523947/.
- Santos HJ, Makiuchi T, Nozaki T. Reinventing an Organelle: The Reduced Mitochondrion in Parasitic Protists. Trends in parasitology [Internet]. 2018 [cited 2022 Jul 12];34:1038–1055. Available from: https://pubmed.ncbi.nlm.nih.gov/30201278/.
- Glucose uptake mechanisms as potential targets for drugs against trypanosomatids. 1991 [cited 2022 Aug 17];379–386. Available from: https://www.taylorfrancis.com/chapters/edit/10.4324/9780203221259-35/glucose-uptake-mechanisms-potential-targets-drugs-trypanosomatids.
- Trindade S, Rijo-Ferreira F, Carvalho T, et al. Trypanosoma brucei Parasites Occupy and Functionally Adapt to the Adipose Tissue in Mice. Cell host & microbe [Internet]. 2016 [cited 2022 Jul 12];19:837–848. Available from: https://pubmed.ncbi.nlm.nih.gov/27237364/.
- Bringaud F, Barrett MP, Zilberstein D. Multiple roles of proline transport and metabolism in trypanosomatids. Frontiers in bioscience (Landmark edition) [Internet]. 2012 [cited 2022 Jul 12];17:349–374. Available from: https://pubmed.ncbi.nlm.nih.gov/22201748/.
- ter Kulle BH. Glucose and proline transport in kinetoplastids. Parasitology today (Personal ed.) [Internet]. 1993 [cited 2022 Jul 12];9:206–210. Available from: https://pubmed.ncbi.nlm.nih.gov/15463755/.
- Burchmore RJS, Barrett MP. Life in vacuoles–nutrient acquisition by Leishmania amastigotes. International journal for parasitology [Internet]. 2001 [cited 2022 Jul 12];31:1311–1320. Available from: https://pubmed.ncbi.nlm.nih.gov/11566299/.
- McConville MJ, de Souza D, Saunders E, et al. Living in a phagolysosome; metabolism of Leishmania amastigotes. Trends in parasitology [Internet]. 2007 [cited 2022 Jul 12];23:368–375. Available from: https://pubmed.ncbi.nlm.nih.gov/17606406/.
- Naderer T, McConville MJ. The Leishmania-macrophage interaction: a metabolic perspective. Cellular microbiology [Internet]. 2008 [cited 2022 Jul 12];10:301–308. Available from: https://pubmed.ncbi.nlm.nih.gov/18070117/.
- Paes LS, Suárez Mantilla B, Zimbres FM, et al. Proline dehydrogenase regulates redox state and respiratory metabolism in Trypanosoma cruzi. PloS one [Internet]. 2013 [cited 2022 Jul 12];8. Available from: https://pubmed.ncbi.nlm.nih.gov/23894476/.
- Mantilla BS, Marchese L, Casas-Sánchez A, et al. Proline Metabolism is Essential for Trypanosoma brucei brucei Survival in the Tsetse Vector. PLoS pathogens [Internet]. 2017 [cited 2022 Jul 12];13. Available from: https://pubmed.ncbi.nlm.nih.gov/28114403/.
- Barisón MJ, Damasceno FS, Mantilla BS, et al. The active transport of histidine and its role in ATP production in Trypanosoma cruzi. Journal of bioenergetics and biomembranes [Internet]. 2016 [cited 2022 Jul 12];48:437–449. Available from: https://pubmed.ncbi.nlm.nih.gov/27222029/.
- Huang KY, Chen YYM, Fang YK, et al. Adaptive responses to glucose restriction enhance cell survival, antioxidant capability, and autophagy of the protozoan parasite Trichomonas vaginalis. Biochimica et biophysica acta [Internet]. 2014 [cited 2022 Jul 13];1840:53–64. Available from: https://pubmed.ncbi.nlm.nih.gov/23958562/.
- Hernández-García MS, Miranda-Ozuna JFT, Salazar-Villatoro L, et al. Biogenesis of Autophagosome in Trichomonas vaginalis during Macroautophagy Induced by Rapamycin-treatment and Iron or Glucose Starvation Conditions. The Journal of eukaryotic microbiology [Internet]. 2019 [cited 2022 Jul 13];66:654–669. Available from: https://pubmed.ncbi.nlm.nih.gov/30620421/.
- Huang KY, Chen RM, Lin HC, et al. Potential role of autophagy in proteolysis in Trichomonas vaginalis. Journal of microbiology, immunology, and infection = Wei mian yu gan ran za zhi [Internet]. 2019 [cited 2022 Jul 13];52:336–344. Available from: https://pubmed.ncbi.nlm.nih.gov/30503389/.
- Mesquita A, Cardenal-Muñoz E, Dominguez E, et al. Autophagy in Dictyostelium: Mechanisms, regulation and disease in a simple biomedical model. Autophagy [Internet]. 2017 [cited 2022 Jul 19];13:24–40. Available from: https://pubmed.ncbi.nlm.nih.gov/27715405/.
- Fischer S, Eichinger L. Dictyostelium discoideum and autophagy - a perfect pair. The International journal of developmental biology [Internet]. 2019 [cited 2022 Jul 12];63:485–495. Available from: https://pubmed.ncbi.nlm.nih.gov/31840786/.
- Gross JD, Pears CJ. Possible Involvement of the Nutrient and Energy Sensors mTORC1 and AMPK in Cell Fate Diversification in a Non-Metazoan Organism. Frontiers in cell and developmental biology [Internet]. 2021 [cited 2022 Jul 12];9. Available from: https://pubmed.ncbi.nlm.nih.gov/34820379/.
- Tewari R, Straschil U, Bateman A, et al. The systematic functional analysis of Plasmodium protein kinases identifies essential regulators of mosquito transmission. Cell host & microbe [Internet]. 2010 [cited 2022 Jul 12];8:377–387. Available from: https://pubmed.ncbi.nlm.nih.gov/20951971/.
- Bracchi V, Langsley G, Thélu J, et al. PfKIN, an SNF1 type protein kinase of Plasmodium falciparum predominantly expressed in gametocytes. Molecular and biochemical parasitology [Internet]. 1996 [cited 2022 Jul 12];76:299–303. Available from: https://pubmed.ncbi.nlm.nih.gov/8920016/.
- Identification of two protein serine/threonine kinase genes and molecular cloning of a SNF1 type protein kinase gene from Toxoplasma gondii - PubMed [Internet]. [cited 2022 Jul 12]. Available from: https://pubmed.ncbi.nlm.nih.gov/7735130/.
- Saldivia M, Ceballos-Pérez G, Bart JM, et al. The AMPKα1 Pathway Positively Regulates the Developmental Transition from Proliferation to Quiescence in Trypanosoma brucei. Cell reports [Internet]. 2016 [cited 2022 Jul 12];17:660–670. Available from: https://pubmed.ncbi.nlm.nih.gov/27732844/.
- Huang G, Vercesi AE, Docampo R. Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nature communications [Internet]. 2013 [cited 2022 Jul 12];4. Available from: https://pubmed.ncbi.nlm.nih.gov/24305511/.
- Li FJ, Xu ZS, Soo ADS, et al. ATP-driven and AMPK-independent autophagy in an early branching eukaryotic parasite. Autophagy [Internet]. 2017 [cited 2022 Jul 12];13:715–729. Available from: https://pubmed.ncbi.nlm.nih.gov/28121493/.
- Barquilla A, Saldivia M, Diaz R, et al. Third target of rapamycin complex negatively regulates development of quiescence in Trypanosoma brucei. Proceedings of the National Academy of Sciences of the United States of America [Internet]. 2012 [cited 2022 Jul 12];109:14399–14404. Available from: https://pubmed.ncbi.nlm.nih.gov/22908264/.
- Barquilla A, Crespo JL, Navarro M. Rapamycin inhibits trypanosome cell growth by preventing TOR complex 2 formation. Proceedings of the National Academy of Sciences of the United States of America [Internet]. 2008 [cited 2022 Jul 12];105:14579–14584. Available from: https://pubmed.ncbi.nlm.nih.gov/18796613/.
- Synthesis of proline by fat body of the tsetse fly (Glossina morsitans): metabolic pathways. [Internet]. [cited 2022 Jul 13]. Available from: https://agris.fao.org/agris-search/search.do?recordID=GB19780304219.
- Wang Y, Utzinger J, Saric J, et al. Global metabolic responses of mice to Trypanosoma brucei brucei infection. Proceedings of the National Academy of Sciences of the United States of America [Internet]. 2008 [cited 2022 Jul 13];105:6127–6132. Available from: https://pubmed.ncbi.nlm.nih.gov/18413599/.
- Nwadike C, Williamson LE, Gallagher LE, et al. AMPK Inhibits ULK1-Dependent Autophagosome Formation and Lysosomal Acidification via Distinct Mechanisms. Molecular and cellular biology [Internet]. 2018 [cited 2022 Jul 12];38. Available from: https://pubmed.ncbi.nlm.nih.gov/29507183/.
- Corona Velazquez AF, Jackson WT. So Many Roads: the Multifaceted Regulation of Autophagy Induction. Molecular and cellular biology [Internet]. 2018 [cited 2022 Jul 12];38. Available from: https://pubmed.ncbi.nlm.nih.gov/30126896/.
- Domínguez-Martín E, Cardenal-Muñoz E, King JS, et al. Methods to Monitor and Quantify Autophagy in the Social Amoeba Dictyostelium discoideum. Cells 2017, Vol. 6, Page 18 [Internet]. 2017 [cited 2022 Jul 19];6:18. Available from: https://www.mdpi.com/2073-4409/6/3/18/htm.
- Maurya R, Kumar R, Saran S. AMPKα promotes basal autophagy induction in Dictyostelium discoideum. Journal of cellular physiology [Internet]. 2020 [cited 2022 Jul 12];235:4941–4953. Available from: https://pubmed.ncbi.nlm.nih.gov/31680241/.
- Calvo-Garrido J, Carilla-Latorre S, Kubohara Y, et al. Autophagy in Dictyostelium: genes and pathways, cell death and infection. Autophagy [Internet]. 2010 [cited 2022 Jul 19];6:686–701. Available from: https://pubmed.ncbi.nlm.nih.gov/20603609/.
- Otto GP, Wu MY, Kazgan N, et al. Macroautophagy is required for multicellular development of the social amoeba Dictyostelium discoideum. The Journal of biological chemistry [Internet]. 2003 [cited 2022 Jul 19];278:17636–17645. Available from: https://pubmed.ncbi.nlm.nih.gov/12626495/.
- Song J, Xu Q, Olsen R, et al. Comparing the Dictyostelium and Entamoeba genomes reveals an ancient split in the Conosa lineage. PLoS computational biology [Internet]. 2005 [cited 2022 Jul 19];1:e71. Available from: https://pubmed.ncbi.nlm.nih.gov/16362072/.
- Picazarri K, Nakada-Tsukui K, Nozaki T. Autophagy during proliferation and encystation in the protozoan parasite Entamoeba invadens. Infection and Immunity [Internet]. 2008 [cited 2022 Jul 19];76:278–288. Available from: https://journals.asm.org/doi/10.1128/IAI.00636-07.
- Song SM, Han BI, Moon EK, et al. Autophagy protein 16-mediated autophagy is required for the encystation of Acanthamoeba castellanii. Molecular and biochemical parasitology [Internet]. 2012 [cited 2022 Jul 13];183:158–165. Available from: https://pubmed.ncbi.nlm.nih.gov/22426571/.
- Kim SH, Moon EK, Hong Y, et al. Autophagy protein 12 plays an essential role in Acanthamoeba encystation. Experimental parasitology [Internet]. 2015 [cited 2022 Jul 13];159:46–52. Available from: https://pubmed.ncbi.nlm.nih.gov/26297678/.
- Vanacova S, Liston DR, Tachezy J, et al. Molecular biology of the amitochondriate parasites, Giardia intestinalis, Entamoeba histolytica and Trichomonas vaginalis. International journal for parasitology [Internet]. 2003 [cited 2022 Jul 13];33:235–255. Available from: https://pubmed.ncbi.nlm.nih.gov/12670510/.
- Castellanos IC, Calvo EP, Wasserman M. A new gene inventory of the ubiquitin and ubiquitin-like conjugation pathways in Giardia intestinalis. Memorias do Instituto Oswaldo Cruz [Internet]. 2020 [cited 2022 Jul 13];115. Available from: https://pubmed.ncbi.nlm.nih.gov/32130365/.
- Shao Q, Liu T, Wang W, et al. The Chinese herbal prescription JZ-1 induces autophagy to protect against herpes simplex Virus-2 in human vaginal epithelial cells by inhibiting the PI3K/Akt/mTOR pathway. Journal of ethnopharmacology [Internet]. 2020 [cited 2022 Jul 13];254. Available from: https://pubmed.ncbi.nlm.nih.gov/32088246/.
- Hernández-García MS, Miranda-Ozuna JFT, Salazar-Villatoro L, et al. Biogenesis of Autophagosome in Trichomonas vaginalis during Macroautophagy Induced by Rapamycin-treatment and Iron or Glucose Starvation Conditions. The Journal of eukaryotic microbiology [Internet]. 2019 [cited 2022 Jul 19];66:654–669. Available from: https://pubmed.ncbi.nlm.nih.gov/30620421/.
- Besteiro S, Williams RAM, Morrison LS, et al. Endosome Sorting and Autophagy Are Essential for Differentiation and Virulence of Leishmania major. Journal of Biological Chemistry. 2006;281:11384–11396.
- Williams RAM, Woods KL, Juliano L, et al. Characterization of unusual families of ATG8-like proteins and ATG12 in the protozoan parasite Leishmania major. Autophagy [Internet]. 2009 [cited 2022 Jul 13];5:159–172. Available from: https://pubmed.ncbi.nlm.nih.gov/19066473/.
- Williams RAM, Smith TK, Cull B, et al. ATG5 is essential for ATG8-dependent autophagy and mitochondrial homeostasis in Leishmania major. PLoS pathogens [Internet]. 2012 [cited 2022 Aug 18];8. Available from: https://pubmed.ncbi.nlm.nih.gov/22615560/.
- Williams RAM, Mottram JC, Coombs GH. Distinct roles in autophagy and importance in infectivity of the two ATG4 cysteine peptidases of Leishmania major. The Journal of biological chemistry [Internet]. 2013 [cited 2022 Aug 18];288:3678–3690. Available from: https://pubmed.ncbi.nlm.nih.gov/23166325/.
- Jara M, Barrett M, Maes I, et al. Transcriptional Shift and Metabolic Adaptations during Leishmania Quiescence Using Stationary Phase and Drug Pressure as Models. Microorganisms [Internet]. 2022 [cited 2022 Aug 18];10. Available from: https://pubmed.ncbi.nlm.nih.gov/35056546/.
- Adhikari A, Biswas S, Mukherjee A, et al. PAS domain-containing phosphoglycerate kinase deficiency in Leishmania major results in increased autophagosome formation and cell death. The Biochemical journal [Internet]. 2019 [cited 2022 Aug 18];476:1303–1321. Available from: https://pubmed.ncbi.nlm.nih.gov/30988012/.
- Cull B, Godinho JLP, Rodrigues JCF, et al. Glycosome turnover in Leishmania major is mediated by autophagy. Autophagy [Internet]. 2014 [cited 2022 Jul 13];10:2143–2157. Available from: https://pubmed.ncbi.nlm.nih.gov/25484087/.
- Herman M, Gillies S, Michels PA, et al. Autophagy and related processes in trypanosomatids: insights from genomic and bioinformatic analyses. Autophagy [Internet]. 2006 [cited 2022 Jul 13];2:107–118. Available from: https://pubmed.ncbi.nlm.nih.gov/16874069/.
- Herman M, Pérez-Morga D, Schtickzelle N, et al. Turnover of glycosomes during life-cycle differentiation of Trypanosoma brucei. Autophagy. 2008;4:294–308.
- Crespo L, Navarro M, Barquilla A. Rapamycin inhibits trypanosome cell growth by preventing TOR complex 2 formation. 2008;105:14579–14584.
- De Jesus TCL, Tonelli RR, Nardelli SC, et al. Target of rapamycin (TOR)-like 1 kinase is involved in the control of polyphosphate levels and acidocalcisome maintenance in Trypanosoma brucei. The Journal of biological chemistry [Internet]. 2010 [cited 2022 Jul 12];285:24131–24140. Available from: https://pubmed.ncbi.nlm.nih.gov/20495004/.
- Saldivia M, Barquilla A, Bart JM, et al. Target of rapamycin (TOR) kinase in Trypanosoma brucei: an extended family. Biochemical Society transactions [Internet]. 2013 [cited 2022 Jul 12];41:934–938. Available from: https://pubmed.ncbi.nlm.nih.gov/23863159/.
- Barquilla A, Navarro M. Trypanosome TOR complex 2 functions in cytokinesis. Cell cycle (Georgetown, Tex.) [Internet]. 2009 [cited 2022 Jul 12];8:697–699. Available from: https://pubmed.ncbi.nlm.nih.gov/19221474/.
- Proto WR, Jones NG, Coombs GH, et al. Tracking autophagy during proliferation and differentiation of Trypanosoma brucei. Microbial cell (Graz, Austria) [Internet]. 2014 [cited 2022 Jul 12];1:9–20. Available from: https://pubmed.ncbi.nlm.nih.gov/28357206/.
- Földvári-Nagy L, Ari E, Csermely P, et al. Starvation-response may not involve Atg1-dependent autophagy induction in non-unikont parasites. Scientific reports [Internet]. 2014 [cited 2022 Jul 12];4. Available from: https://pubmed.ncbi.nlm.nih.gov/25059978/.
- Wang J, Zhang J, Lee YM, et al. Quantitative chemical proteomics profiling of de novo protein synthesis during starvation-mediated autophagy. Autophagy [Internet]. 2016 [cited 2022 Jul 12];12:1931–1944. Available from: https://pubmed.ncbi.nlm.nih.gov/27463841/.
- Dieterich DC, Lee JJ, Link AJ, et al. Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nature protocols [Internet]. 2007 [cited 2022 Jul 12];2:532–540. Available from: https://pubmed.ncbi.nlm.nih.gov/17406607/.
- Kiick KL, Saxon E, Tirrell DA, et al. Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proceedings of the National Academy of Sciences of the United States of America [Internet]. 2002 [cited 2022 Jul 12];99:19–24. Available from: https://pubmed.ncbi.nlm.nih.gov/11752401/.
- Rajković J, Poreba M, Caglič D, et al. Biochemical Characterization and Substrate Specificity of Autophagin-2 from the Parasite Trypanosoma cruzi. Journal of Biological Chemistry. 2015;290:28231–28244.
- Schoijet AC, Miranda K, Girard-dias W, et al. A Trypanosoma cruzi Phosphatidylinositol 3-Kinase (TcVps34) Is Involved in Osmoregulation and. 2008;283:31541–31550.
- Gimenez AM, Gesumaría MC, Schoijet AC, et al. Phosphatidylinositol kinase activities in Trypanosoma cruzi epimastigotes. Molecular and biochemical parasitology. 2015;203:14–24.
- Sternlieb T, Schoijet AC, Genta PD, et al. An AMP-activated protein kinase complex with two distinctive alpha subunits is involved in nutritional stress responses in Trypanosoma cruzi. PLoS neglected tropical diseases [Internet]. 2021 [cited 2022 Aug 16];15. Available from: https://pubmed.ncbi.nlm.nih.gov/34029334/.
- Digirolamo FA, Miranda MR, Bouvier LA, et al. [The mammalian TOR pathway is present in Trypanosoma cruzi. In silico reconstruction and possible functions]. Medicina. 2012;72:221–226.
- Vanrell MC, Losinno AD, Cueto JA, et al. The regulation of autophagy differentially affects Trypanosoma cruzi metacyclogenesis. PLoS Neglected Tropical Diseases [Internet]. 2017 [cited 2020 Dec 29];11. Available from: https://pubmed.ncbi.nlm.nih.gov/29091711/.
- Chiurillo MA, Lander N, Vercesi AE, et al. IP 3 receptor-mediated Ca 2+ release from acidocalcisomes regulates mitochondrial bioenergetics and prevents autophagy in Trypanosoma cruzi. Cell calcium [Internet]. 2020 [cited 2022 Aug 16];92. Available from: https://pubmed.ncbi.nlm.nih.gov/32947181/.
- Gao D, Zhang J, Zhao J, et al. Autophagy activated by Toxoplasma gondii infection in turn facilitates Toxoplasma gondii proliferation. Parasitology research [Internet]. 2014 [cited 2022 Jul 26];113:2053–2058. Available from: https://pubmed.ncbi.nlm.nih.gov/24696274/.
- Losinno AD, Martínez SJ, Labriola CA, et al. Induction of autophagy increases the proteolytic activity of reservosomes during Trypanosoma cruzi metacyclogenesis. Autophagy [Internet]. 2021 [cited 2022 Jul 13];17:439–456. Available from: https://pubmed.ncbi.nlm.nih.gov/31983275/.
- VE A, GT N, JJ C. Metacaspases, autophagins and metallocarboxypeptidases: potential new targets for chemotherapy of the trypanosomiases. Current medicinal chemistry [Internet]. 2013 [cited 2022 Aug 16];20:3069–3077. Available from: https://pubmed.ncbi.nlm.nih.gov/23514417/.
- Cruz-Saavedra L, Vallejo GA, Guhl F, et al. Transcriptional remodeling during metacyclogenesis in Trypanosoma cruzi I. Virulence [Internet]. 2020 [cited 2022 Aug 16];11:969–980. Available from: https://pubmed.ncbi.nlm.nih.gov/32715914/.
- Alvarez VE, Niemirowicz GT, Cazzulo JJ. The peptidases of Trypanosoma cruzi: digestive enzymes, virulence factors, and mediators of autophagy and programmed cell death. Biochimica et biophysica acta [Internet]. 2012 [cited 2022 Aug 16];1824:195–206. Available from: https://pubmed.ncbi.nlm.nih.gov/21621652/.
- Pedra-Rezende Y, Fernandes MC, Mesquita-Rodrigues C, et al. Starvation and pH stress conditions induced mitochondrial dysfunction, ROS production and autophagy in Trypanosoma cruzi epimastigotes. Biochimica et biophysica acta. Molecular basis of disease [Internet]. 2021 [cited 2022 Aug 16];1867. Available from: https://pubmed.ncbi.nlm.nih.gov/33248274/.
- Losinno AD, Martínez SJ, Labriola CA, et al. Induction of autophagy increases the proteolytic activity of reservosomes during Trypanosoma cruzi metacyclogenesis. Autophagy [Internet]. 2020 [cited 2020 Dec 29]; Available from: https://pubmed.ncbi.nlm.nih.gov/31983275/.
- Barclay JJ, Morosi LG, Vanrell MC, et al. Trypanosoma cruzi coexpressing ornithine decarboxylase and green fluorescence proteins as a tool to study the role of polyamines in chagas disease pathology. Enzyme Research [Internet]. 2011 [cited 2021 Apr 28];2011. Available from: https://pubmed.ncbi.nlm.nih.gov/21687606/.
- Hashimoto M, Nara T, Enomoto M, et al. A dominant negative form of inositol 1,4,5-trisphosphate receptor induces metacyclogenesis and increases mitochondrial density in Trypanosoma cruzi. Biochemical and biophysical research communications [Internet]. 2015 [cited 2022 May 10];466:475–480. Available from: https://pubmed.ncbi.nlm.nih.gov/26367178/.
- Romano PS, Arboit MA, Vázquez CL, et al. The autophagic pathway is a key component in the lysosomal dependent entry of Trypanosoma cruzi into the host cell. Autophagy. 2009;5:6–18.
- Salassa BN, Romano PS. Autophagy: A necessary process during the Trypanosoma cruzi life-cycle [Internet]. Virulence. Taylor and Francis Inc.; 2019 [cited 2020 Dec 29]. p. 460–469. Available from: https://pubmed.ncbi.nlm.nih.gov/30489206/.
- Robson KJH, Frevert U, Reckmann I, et al. Thrombospondin-related adhesive protein (TRAP) of Plasmodium falciparum: expression during sporozoite ontogeny and binding to human hepatocytes. The EMBO journal [Internet]. 1995 [cited 2022 Aug 26];14:3883–3894. Available from: https://pubmed.ncbi.nlm.nih.gov/7664729/.
- Sinnis P, Sim BKL. Cell invasion by the vertebrate stages of Plasmodium. Trends in Microbiology [Internet]. 1997 [cited 2022 Aug 26];5:52–58. Available from: https://pubmed.ncbi.nlm.nih.gov/9108930/.
- Robson KJH, Hall JRS, Jennings MW, et al. A highly conserved amino-acid sequence in thrombospondin, properdin and in proteins from sporozoites and blood stages of a human malaria parasite. Nature [Internet]. 1988 [cited 2022 Aug 26];335:79–82. Available from: https://pubmed.ncbi.nlm.nih.gov/3045563/.
- Tufet-Bayona M, Janse CJ, Khan SM, et al. Localisation and timing of expression of putative Plasmodium berghei rhoptry proteins in merozoites and sporozoites. Molecular and biochemical parasitology [Internet]. 2009 [cited 2022 Aug 26];166:22–31. Available from: https://pubmed.ncbi.nlm.nih.gov/19428669/.
- Jayabalasingham B, Bano N, Coppens I. Metamorphosis of the malaria parasite in the liver is associated with organelle clearance. Cell research [Internet]. 2010 [cited 2022 Jul 13];20:1043–1059. Available from: https://pubmed.ncbi.nlm.nih.gov/20567259/.
- Coppens I. Metamorphoses of malaria: the role of autophagy in parasite differentiation. Essays in biochemistry [Internet]. 2011 [cited 2022 Aug 26];51:127–136. Available from: https://pubmed.ncbi.nlm.nih.gov/22023446/.
- Brennand A, Gualdrón-López M, Coppens I, et al. Autophagy in parasitic protists: unique features and drug targets. Molecular and biochemical parasitology [Internet]. 2011 [cited 2022 Jul 13];177:83–99. Available from: https://pubmed.ncbi.nlm.nih.gov/21315770/.
- Duszenko M, Ginger ML, Brennand A, et al. Autophagy in protists. 2011;1–32.
- Jayabalasingham B, Voss C, Ehrenman K, et al. Characterization of the ATG8-conjugation system in 2 Plasmodium species with special focus on the liver stage: possible linkage between the apicoplastic and autophagic systems? Autophagy [Internet]. 2014 [cited 2022 Jul 13];10:269–284. Available from: https://pubmed.ncbi.nlm.nih.gov/24342964/.
- Tawk L, Chicanne G, Dubremetz JF, et al. Phosphatidylinositol 3-phosphate, an essential lipid in Plasmodium, localizes to the food vacuole membrane and the apicoplast. Eukaryotic cell [Internet]. 2010 [cited 2022 Aug 26];9:1519–1530. Available from: https://pubmed.ncbi.nlm.nih.gov/20709789/.
- Dall’Armi C, Devereaux KA, Di Paolo G. The role of lipids in the control of autophagy. Current biology : CB [Internet]. 2013 [cited 2022 Aug 26];23. Available from: https://pubmed.ncbi.nlm.nih.gov/23305670/.
- Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. The Journal of cell biology [Internet]. 2008 [cited 2022 Aug 26];182:685–701. Available from: https://pubmed.ncbi.nlm.nih.gov/18725538/.
- Roestenberg M, Teirlinck AC, McCall MBB, et al. Long-term protection against malaria after experimental sporozoite inoculation: an open-label follow-up study. Lancet (London, England) [Internet]. 2011 [cited 2022 Aug 26];377:1770–1776. Available from: https://pubmed.ncbi.nlm.nih.gov/21514658/.
- Voss C, Ehrenman K, Mlambo G, et al. Overexpression of Plasmodium berghei ATG8 by Liver Forms Leads to Cumulative Defects in Organelle Dynamics and to Generation of Noninfectious Merozoites. mBio [Internet]. 2016 [cited 2022 Aug 26];7. Available from: https://pubmed.ncbi.nlm.nih.gov/27353755/.
- Giuliani F, Grieve A, Rabouille C. Unconventional secretion: a stress on GRASP. Current opinion in cell biology [Internet]. 2011 [cited 2022 Aug 26];23:498–504. Available from: https://pubmed.ncbi.nlm.nih.gov/21571519/.
- Vinke FP, Grieve AG, Rabouille C. The multiple facets of the Golgi reassembly stacking proteins. The Biochemical journal [Internet]. 2011 [cited 2022 Aug 26];433:423–433. Available from: https://pubmed.ncbi.nlm.nih.gov/21235525/.
- Hanson PI, Cashikar A. Multivesicular body morphogenesis. Annual review of cell and developmental biology [Internet]. 2012 [cited 2022 Aug 26];28:337–362. Available from: https://pubmed.ncbi.nlm.nih.gov/22831642/.
- Yang M, Coppens I, Wormsley S, et al. The Plasmodium falciparum Vps4 homolog mediates multivesicular body formation. Journal of cell science [Internet]. 2004 [cited 2022 Aug 26];117:3831–3838. Available from: https://pubmed.ncbi.nlm.nih.gov/15252121/.
- Struck NS, de Souza Dias S, Langer C, et al. Re-defining the Golgi complex in Plasmodium falciparum using the novel Golgi marker PfGRASP. Journal of cell science [Internet]. 2005 [cited 2022 Aug 26];118:5603–5613. Available from: https://pubmed.ncbi.nlm.nih.gov/16306223/.
- Sahu T, Gehrke EJ, Flores-Garcia Y, et al. Chemoprophylaxis vaccination with a Plasmodium liver stage autophagy mutant affords enhanced and long-lasting protection. NPJ vaccines [Internet]. 2021 [cited 2022 Jul 26];6. Available from: https://pubmed.ncbi.nlm.nih.gov/34376691/.
- Besteiro S, Brooks CF, Striepen B, et al. Autophagy protein Atg3 is essential for maintaining mitochondrial integrity and for normal intracellular development of Toxoplasma gondii tachyzoites. PLoS pathogens [Internet]. 2011 [cited 2022 Jul 23];7. Available from: https://pubmed.ncbi.nlm.nih.gov/22144900/.
- Kong-Hap MA, Mouammine A, Daher W, et al. Regulation of ATG8 membrane association by ATG4 in the parasitic protist Toxoplasma gondii. Autophagy [Internet]. 2013 [cited 2022 Jul 19];9:1334–1348. Available from: https://pubmed.ncbi.nlm.nih.gov/23748741/.
- Lévêque MF, Nguyen HM, Besteiro S. Repurposing of conserved autophagy-related protein ATG8 in a divergent eukaryote. Communicative & integrative biology [Internet]. 2016 [cited 2022 Jul 19];9. Available from: https://pubmed.ncbi.nlm.nih.gov/27574540/.
- Sheiner L, Vaidya AB, McFadden GI. The metabolic roles of the endosymbiotic organelles of Toxoplasma and Plasmodium spp. Current opinion in microbiology [Internet]. 2013 [cited 2022 Aug 20];16:452–458. Available from: https://pubmed.ncbi.nlm.nih.gov/23927894/.
- Nguyen HM, El Hajj H, El Hajj R, et al. Toxoplasma gondii autophagy-related protein ATG9 is crucial for the survival of parasites in their host. Cellular microbiology [Internet]. 2017 [cited 2022 Jul 26];19. Available from: https://pubmed.ncbi.nlm.nih.gov/27992947/.
- Smith D, Kannan G, Coppens I, et al. Toxoplasma TgATG9 is critical for autophagy and long-term persistence in tissue cysts. eLife [Internet]. 2021 [cited 2022 Jul 26];10. Available from: https://pubmed.ncbi.nlm.nih.gov/33904393/.
- Di Cristina M, Dou Z, Lunghi M, et al. Toxoplasma depends on lysosomal consumption of autophagosomes for persistent infection. Nature microbiology [Internet]. 2017 [cited 2022 Jul 19];2. Available from: https://pubmed.ncbi.nlm.nih.gov/28628099/.
- Ameisen JC, Idziorek T, Billaut-Mulot O, et al. Apoptosis in a unicellular eukaryote (Trypanosoma cruzi): implications for the evolutionary origin and role of programmed cell death in the control of cell proliferation, differentiation and survival. Cell death and differentiation [Internet]. 1995 [cited 2022 Jul 19];2:285–300. Available from: https://pubmed.ncbi.nlm.nih.gov/17180034/.
- Welburn SC, Dale C, Ellis D, et al. Apoptosis in procyclic Trypanosoma brucei rhodesiense in vitro. Cell death and differentiation [Internet]. 1996 [cited 2022 Jul 19];3:229–236. Available from: https://pubmed.ncbi.nlm.nih.gov/17180087/.
- Christensen ST, Wheatley DN, Rasmussen MI, et al. Mechanisms controlling death, survival and proliferation in a model unicellular eukaryote Tetrahymena thermophila. Cell death and differentiation [Internet]. 1995 [cited 2022 Jul 19];2:301–308. Available from: https://pubmed.ncbi.nlm.nih.gov/17180035/.
- Deponte M. Programmed cell death in protists. Biochimica et biophysica acta [Internet]. 2008 [cited 2022 Jul 19];1783:1396–1405. Available from: https://pubmed.ncbi.nlm.nih.gov/18291111/.
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nature reviews. Molecular cell biology [Internet]. 2008 [cited 2022 Jul 19];9:1004. Available from: /pmc/articles/PMC2727358/.
- Duszenko M, Ginger ML, Brennand A, et al. Autophagy in protists. Autophagy [Internet]. 2011 [cited 2022 Jul 13];7:127–158. Available from: https://pubmed.ncbi.nlm.nih.gov/20962583/.
- Bialik S, Dasari SK, Kimchi A. Autophagy-dependent cell death - where, how and why a cell eats itself to death. Journal of cell science [Internet]. 2018 [cited 2022 Jul 19];131. Available from: https://pubmed.ncbi.nlm.nih.gov/30237248/.
- Denton D, Kumar S. Autophagy-dependent cell death. Cell death and differentiation [Internet]. 2019 [cited 2022 Jul 19];26:605–616. Available from: https://pubmed.ncbi.nlm.nih.gov/30568239/.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) [Internet]. Autophagy. Taylor and Francis Inc.; 2016 [cited 2020 Dec 29]. p. 1–222. Available from: https://pubmed.ncbi.nlm.nih.gov/26799652/.
- Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell death and differentiation [Internet]. 2018 [cited 2022 Jul 19];25:486–541. Available from: https://pubmed.ncbi.nlm.nih.gov/29362479/.
- Denton D, Shravage B, Simin R, et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Current biology : CB [Internet]. 2009 [cited 2022 Jul 19];19:1741–1746. Available from: https://pubmed.ncbi.nlm.nih.gov/19818615/.
- Liu Y, Shoji-Kawata S, Sumpter RM, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proceedings of the National Academy of Sciences of the United States of America [Internet]. 2013 [cited 2022 Jul 19];110:20364–20371. Available from: https://pubmed.ncbi.nlm.nih.gov/24277826/.
- Cornillon S, Foa C, Davoust J, et al. Programmed cell death in Dictyostelium. Journal of cell science [Internet]. 1994 [cited 2022 Jul 19];107 (Pt 10):2691–2704. Available from: https://pubmed.ncbi.nlm.nih.gov/7876338/.
- Giusti C, Tresse E, Luciani MF, et al. Autophagic cell death: analysis in Dictyostelium. Biochimica et biophysica acta [Internet]. 2009 [cited 2022 Jul 19];1793:1422–1431. Available from: https://pubmed.ncbi.nlm.nih.gov/19133302/.
- Luciani MF, Giusti C, Harms B, et al. Atg1 allows second-signaled autophagic cell death in Dictyostelium. Autophagy [Internet]. 2011 [cited 2022 Jul 19];7:501–508. Available from: https://pubmed.ncbi.nlm.nih.gov/21301205/.
- Denninger V, Koopmann R, Muhammad K, et al. Chapter Twenty‐Five Kinetoplastida: Model Organisms for Simple Autophagic Pathways? 2008;451:373–408.
- Delgado M, Anderson P, Garcia-Salcedo JA, et al. Neuropeptides kill African trypanosomes by targeting intracellular compartments and inducing autophagic-like cell death. Cell death and differentiation [Internet]. 2009 [cited 2022 Jul 19];16:406–416. Available from: https://pubmed.ncbi.nlm.nih.gov/19057622/.
- Campos-Salinas J, Gonzalez-Rey E. Autophagy and neuropeptides at the crossroad for parasites: to survive or to die? Autophagy [Internet]. 2009 [cited 2022 Jul 19];5:551–554. Available from: https://pubmed.ncbi.nlm.nih.gov/19337024/.
- Affenzeller MJ, Darehshouri A, Andosch A, et al. Salt stress-induced cell death in the unicellular green alga Micrasterias denticulata. Journal of experimental botany [Internet]. 2009 [cited 2022 Jul 19];60:939–954. Available from: https://pubmed.ncbi.nlm.nih.gov/19213813/.
- Darehshouri A, Affenzeller M, Lütz-Meindl U. Cell death upon H(2)O(2) induction in the unicellular green alga Micrasterias. Plant biology (Stuttgart, Germany) [Internet]. 2008 [cited 2022 Jul 19];10:732–745. Available from: https://pubmed.ncbi.nlm.nih.gov/18950431/.
- Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell [Internet]. 2007 [cited 2022 Jul 19];131:1137–1148. Available from: https://pubmed.ncbi.nlm.nih.gov/18083103/.
- Orias E, Cervantes MD, Hamilton EP. Tetrahymena thermophila, a unicellular eukaryote with separate germline and somatic genomes. Research in microbiology [Internet]. 2011 [cited 2022 Jul 19];162:578–586. Available from: https://pubmed.ncbi.nlm.nih.gov/21624459/.
- (PDF) Programmed nuclear death and its relation to apoptosis and autophagy during sexual reproduction in Tetrahymena thermophila [Internet]. [cited 2022 Aug 16]. Available from: https://www.researchgate.net/publication/284485546_Programmed_nuclear_death_and_its_relation_to_apoptosis_and_autophagy_during_sexual_reproduction_in_Tetrahymena_thermophila.
- Akematsu T, Pearlman RE, Endoh H. Gigantic macroautophagy in programmed nuclear death of Tetrahymena thermophila. Autophagy [Internet]. 2010 [cited 2022 Jul 19];6:901–911. Available from: https://pubmed.ncbi.nlm.nih.gov/20798592/.
- Bo T, Kang Y, Liu Y, et al. Atg5 Regulates Selective Autophagy of the Parental Macronucleus during Tetrahymena Sexual Reproduction. Cells [Internet]. 2021 [cited 2022 Jul 19];10. Available from: https://pubmed.ncbi.nlm.nih.gov/34831293/.
- Liu ML, Yao MC. Role of ATG8 and autophagy in programmed nuclear degradation in Tetrahymena thermophila. Eukaryotic cell [Internet]. 2012 [cited 2022 Jul 19];11:494–506. Available from: https://pubmed.ncbi.nlm.nih.gov/22366125/.
- Osada E, Akematsu T, Asano T, et al. A novel mitochondrial nuclease-associated protein: a major executor of the programmed nuclear death in Tetrahymena thermophila. Biology of the cell [Internet]. 2014 [cited 2022 Jul 19];106:97–109. Available from: https://pubmed.ncbi.nlm.nih.gov/24392831/.
- Akematsu T, Endoh H. Role of apoptosis-inducing factor (AIF) in programmed nuclear death during conjugation in Tetrahymena thermophila. BMC cell biology [Internet]. 2010 [cited 2022 Jul 19];11. Available from: https://pubmed.ncbi.nlm.nih.gov/20146827/.
- Akematsu T, Fukuda Y, Attiq R, et al. Role of class III phosphatidylinositol 3-kinase during programmed nuclear death of Tetrahymena thermophila. Autophagy [Internet]. 2014 [cited 2022 Jul 19];10:209–225. Available from: https://pubmed.ncbi.nlm.nih.gov/24280724/.
- Chagas disease (also known as American trypanosomiasis) [Internet]. [cited 2022 Aug 16]. Available from: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis).
- Van Vugt M, Van Beest A, Sicuri E, et al. Malaria treatment and prophylaxis in endemic and nonendemic countries: evidence on strategies and their cost-effectiveness. Future microbiology [Internet]. 2011 [cited 2022 Aug 16];6:1485–1500. Available from: https://pubmed.ncbi.nlm.nih.gov/22122444/.
- De Menezes JPB, Guedes CES, De Oliveira Almeida Petersen AL, et al. Advances in Development of New Treatment for Leishmaniasis. BioMed research international [Internet]. 2015 [cited 2022 Aug 16];2015. Available from: https://pubmed.ncbi.nlm.nih.gov/26078965/.
- MA V-S, SL DC. Electron microscopy in antiparasitic chemotherapy: a (close) view to a kill. Current drug targets [Internet]. 2009 [cited 2022 Aug 16];10:246–260. Available from: https://pubmed.ncbi.nlm.nih.gov/19275561/.
- CDC - Parasites [Internet]. [cited 2022 Aug 21]. Available from: https://www.cdc.gov/parasites/index.html.
- Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clinical microbiology reviews [Internet]. 2006 [cited 2022 Jul 19];19:111–126. Available from: https://pubmed.ncbi.nlm.nih.gov/16418526/.
- Chappuis F, Sundar S, Hailu A, et al. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nature reviews. Microbiology [Internet]. 2007 [cited 2022 Jul 19];5:873–882. Available from: https://pubmed.ncbi.nlm.nih.gov/17938629/.
- Jain K, Jain NK. Novel therapeutic strategies for treatment of visceral leishmaniasis. Drug discovery today [Internet]. 2013 [cited 2022 Jul 19];18:1272–1281. Available from: https://pubmed.ncbi.nlm.nih.gov/23973338/.
- Frézard F, Demicheli C, Ribeiro RR. Pentavalent antimonials: new perspectives for old drugs. Molecules (Basel, Switzerland) [Internet]. 2009 [cited 2022 Jul 19];14:2317–2336. Available from: https://pubmed.ncbi.nlm.nih.gov/19633606/.
- Salomao K, Figueiredo Sadok Menna-Barreto R, Lisboa de Castro S. Stairway to Heaven or Hell? Perspectives and Limitations of Chagas Disease Chemotherapy. Current topics in medicinal chemistry [Internet]. 2016 [cited 2022 Jul 19];16:2266–2289. Available from: https://pubmed.ncbi.nlm.nih.gov/27072716/.
- Laura Sbaraglini M, Cristina Vanrell M, Leticia Bellera C, et al. Neglected Tropical Protozoan Diseases: Drug Repositioning as a Rational Option. Current topics in medicinal chemistry [Internet]. 2016 [cited 2022 Jul 19];16:2201–2222. Available from: https://pubmed.ncbi.nlm.nih.gov/26881713/.
- Chatelain E, Ioset JR. Drug discovery and development for neglected diseases: the DNDi model. Drug design, development and therapy [Internet]. 2011 [cited 2022 Jul 19];5:175–181. Available from: https://pubmed.ncbi.nlm.nih.gov/21552487/.
- Gaspar MM, Calado S, Pereira J, et al. Targeted delivery of paromomycin in murine infectious diseases through association to nano lipid systems. Nanomedicine: Nanotechnology, Biology and Medicine. 2015;11:1851–1860.
- Proto WR, Coombs GH, Mottram JC. Cell death in parasitic protozoa: regulated or incidental? Nature reviews. Microbiology [Internet]. 2013 [cited 2022 Jul 19];11:58–66. Available from: https://pubmed.ncbi.nlm.nih.gov/23202528/.
- Menna-Barreto RFS. Cell death pathways in pathogenic trypanosomatids: lessons of (over)kill. Cell death & disease [Internet]. 2019 [cited 2022 Jul 19];10. Available from: https://pubmed.ncbi.nlm.nih.gov/30700697/.
- Bera A, Singh S, Nagaraj R, et al. Induction of autophagic cell death in Leishmania donovani by antimicrobial peptides. Molecular and Biochemical Parasitology. 2003;127:23–35.
- Menna-Barreto RFS, Corrêa JR, Cascabulho CM, et al. Naphthoimidazoles promote different death phenotypes in Trypanosoma cruzi. Parasitology [Internet]. 2009 [cited 2022 Jul 23];136:499–510. Available from: https://pubmed.ncbi.nlm.nih.gov/19281638/.
- Corrêa G, Vilela R, Menna-Barreto RFS, et al. Cell death induction in Giardia lamblia: effect of beta-lapachone and starvation. Parasitology international [Internet]. 2009 [cited 2022 Jul 19];58:424–437. Available from: https://pubmed.ncbi.nlm.nih.gov/19703583/.
- VANNIER‐SANTOS MA, URBINA JA, MARTINY A, et al. Alterations induced by the antifungal compounds ketoconazole and terbinafine in Leishmania. The Journal of eukaryotic microbiology [Internet]. 1995 [cited 2022 Jul 19];42:337–346. Available from: https://pubmed.ncbi.nlm.nih.gov/7620457/.
- Araujo-Silva CA, De Souza W, Martins-Duarte ES, et al. HDAC inhibitors Tubastatin A and SAHA affect parasite cell division and are potential anti-Toxoplasma gondii chemotherapeutics. International journal for parasitology. Drugs and drug resistance [Internet]. 2021 [cited 2022 Jul 19];15:25–35. Available from: https://pubmed.ncbi.nlm.nih.gov/33360687/.
- Crouch ML, Benchimol M, Alderete JF. Binding of fibronectin by Trichomonas vaginalis is influenced by iron and calcium. Microbial pathogenesis [Internet]. 2001 [cited 2022 Jul 19];31:131–144. Available from: https://pubmed.ncbi.nlm.nih.gov/11500098/.
- Braga M V., Magaraci F, Lorente SO, et al. Effects of inhibitors of Delta24(25)-sterol methyl transferase on the ultrastructure of epimastigotes of Trypanosoma cruzi. Microscopy and microanalysis : the official journal of Microscopy Society of America, Microbeam Analysis Society, Microscopical Society of Canada [Internet]. 2005 [cited 2022 Jul 19];11:506–515. Available from: https://pubmed.ncbi.nlm.nih.gov/17481329/.
- Granthon AC, Braga M V., Rodrigues JCF, et al. Alterations on the growth and ultrastructure of Leishmania chagasi induced by squalene synthase inhibitors. Veterinary parasitology [Internet]. 2007 [cited 2022 Jul 19];146:25–34. Available from: https://pubmed.ncbi.nlm.nih.gov/17367936/.
- Uzcátegui NL, Denninger V, Merkel P, et al. Dihydroxyacetone induced autophagy in African trypanosomes. Autophagy [Internet]. 2007 [cited 2022 Jul 19];3:626–629. Available from: https://pubmed.ncbi.nlm.nih.gov/17786028/.
- Menna-Barreto RFS, Salomão K, Dantas AP, et al. Different cell death pathways induced by drugs in Trypanosoma cruzi: an ultrastructural study. Micron (Oxford, England : 1993) [Internet]. 2009 [cited 2022 Jul 19];40:157–168. Available from: https://pubmed.ncbi.nlm.nih.gov/18849169/.
- Carvalho CS, De Melo EJT, Tenório RP, et al. Anti-parasitic action and elimination of intracellular Toxoplasma gondii in the presence of novel thiosemicarbazone and its 4-thiazolidinone derivatives. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas [Internet]. 2010 [cited 2022 Jul 19];43:139–149. Available from: https://pubmed.ncbi.nlm.nih.gov/19893994/.
- Bombaça ACS, Von Dossow D, Barbosa JMC, et al. TrypanocidalActivity of Natural Sesquiterpenoids Involves Mitochondrial Dysfunction, ROS Production and Autophagic Phenotype in Trypanosomacruzi. Molecules (Basel, Switzerland) [Internet]. 2018 [cited 2022 Jul 19];23. Available from: https://pubmed.ncbi.nlm.nih.gov/30373326/.
- Martínez-García M, Bart JM, Campos-Salinas J, et al. Autophagic-related cell death of Trypanosoma brucei induced by bacteriocin AS-48. International journal for parasitology. Drugs and drug resistance [Internet]. 2018 [cited 2022 Jul 19];8:203–212. Available from: https://pubmed.ncbi.nlm.nih.gov/29649664/.
- Ghosh D, Walton JL, Roepe PD, et al. Autophagy is a cell death mechanism in Toxoplasma gondii. Cellular microbiology [Internet]. 2012 [cited 2022 Jul 26];14:589–607. Available from: https://pubmed.ncbi.nlm.nih.gov/22212386/.
- Veiga-Santos P, Desoti VC, Miranda N, et al. The natural compounds piperovatine and piperlonguminine induce autophagic cell death on Trypanosoma cruzi. Acta tropica [Internet]. 2013 [cited 2022 Jul 23];125:349–356. Available from: https://pubmed.ncbi.nlm.nih.gov/23228524/.
- Souto XM, Barbosa HS, Menna-Barreto RFS. The morphological analysis of autophagy in primary skeletal muscle cells infected with Toxoplasma gondii. Parasitology research [Internet]. 2016 [cited 2022 Jul 26];115:2853–2861. Available from: https://pubmed.ncbi.nlm.nih.gov/27075305/.
- Nguyen HM, Berry L, Sullivan WJ, et al. Autophagy participates in the unfolded protein response in Toxoplasma gondii. FEMS microbiology letters [Internet]. 2017 [cited 2022 Jul 23];364. Available from: https://pubmed.ncbi.nlm.nih.gov/28859319/.
- Scariot DB, Britta EA, Moreira AL, et al. Induction of Early Autophagic Process on Leishmania amazonensis by Synergistic Effect of Miltefosine and Innovative Semi-synthetic Thiosemicarbazone. Frontiers in microbiology [Internet]. 2017 [cited 2022 Jul 19];8. Available from: https://pubmed.ncbi.nlm.nih.gov/28270805/.
- Wold MS, Lim J, Lachance V, et al. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Molecular Neurodegeneration. 2016;11:76.
- Huang KY, Chen RM, Lin HC, et al. Potential role of autophagy in proteolysis in Trichomonas vaginalis. Journal of microbiology, immunology, and infection = Wei mian yu gan ran za zhi [Internet]. 2019 [cited 2022 Jul 12];52:336–344. Available from: https://pubmed.ncbi.nlm.nih.gov/30503389/.
- de Paula JC, Bakoshi ABK, Lazarin-Bidóia D, et al. Antiproliferative activity of the dibenzylideneacetone derivate (E)-3-ethyl-4-(4-nitrophenyl)but‑3-en-2-one in Trypanosoma cruzi. Acta tropica [Internet]. 2020 [cited 2022 Jul 23];211. Available from: https://pubmed.ncbi.nlm.nih.gov/32777226/.
- Silva RCMC, Fox EGP, Gomes FM, et al. Venom alkaloids against Chagas disease parasite: search for effective therapies. Scientific reports [Internet]. 2020 [cited 2022 Jul 19];10. Available from: https://pubmed.ncbi.nlm.nih.gov/32606423/.
- Nishi L, Sanfelice RADS, Bortoleti BTDS, et al. Moringa oleifera extract promotes apoptosis-like death in Toxoplasma gondii tachyzoites in vitro. Parasitology [Internet]. 2021 [cited 2022 Jul 23];148:1447–1457. Available from: https://www.cambridge.org/core/journals/parasitology/article/abs/moringa-oleifera-extract-promotes-apoptosislike-death-in-toxoplasma-gondii-tachyzoites-in-vitro/69BDB8D9B38ABA2FB904E504F37CA864.
- Zhang J, Si H, Lv K, et al. Licarin-B Exhibits Activity Against the Toxoplasma gondii RH Strain by Damaging Mitochondria and Activating Autophagy. Frontiers in cell and developmental biology [Internet]. 2021 [cited 2022 Jul 23];9. Available from: https://pubmed.ncbi.nlm.nih.gov/34179016/.
- Dos Santos AO, Veiga-Santos P, Ueda-Nakamura T, et al. Effect of elatol, isolated from red seaweed Laurencia dendroidea, on Leishmania amazonensis. Marine drugs [Internet]. 2010 [cited 2022 Jul 19];8:2733–2743. Available from: https://pubmed.ncbi.nlm.nih.gov/21139841/.
- Schurigt U, Schad C, Glowa C, et al. Aziridine-2,3-dicarboxylate-based cysteine cathepsin inhibitors induce cell death in Leishmania major associated with accumulation of debris in autophagy-related lysosome-like vacuoles. Antimicrobial agents and chemotherapy [Internet]. 2010 [cited 2022 Jul 19];54:5028–5041. Available from: https://pubmed.ncbi.nlm.nih.gov/20855728/.
- Sengupta S, Chowdhury S, BoseDasgupta S, et al. Cryptolepine-Induced Cell Death of Leishmania donovani Promastigotes Is Augmented by Inhibition of Autophagy. Molecular biology international [Internet]. 2011 [cited 2022 Jul 23];2011:1–12. Available from: https://pubmed.ncbi.nlm.nih.gov/22091398/.
- Fernandes MC, Da Silva EN, Pinto A V., et al. A novel triazolic naphthofuranquinone induces autophagy in reservosomes and impairment of mitosis in Trypanosoma cruzi. Parasitology [Internet]. 2012 [cited 2022 Jul 23];139:26–36. Available from: https://pubmed.ncbi.nlm.nih.gov/21939585/.
- Martins-Duarte ES, Jones SM, Gilbert IH, et al. Thiolactomycin analogues as potential anti-Toxoplasma gondii agents. Parasitology international [Internet]. 2009 [cited 2022 Jul 19];58:411–415. Available from: https://pubmed.ncbi.nlm.nih.gov/19698800/.
- Macedo-Silva ST de, Oliveira Silva TLA de, Urbina JA, et al. Antiproliferative, Ultrastructural, and Physiological Effects of Amiodarone on Promastigote and Amastigote Forms of Leishmania amazonensis. Molecular biology international [Internet]. 2011 [cited 2022 Jul 19];2011:1–12. Available from: https://pubmed.ncbi.nlm.nih.gov/22091415/.
- Klionsky DJ, Petroni G, Amaravadi RK, et al. Autophagy in major human diseases. The EMBO journal [Internet]. 2021 [cited 2022 Jul 23];40. Available from: https://pubmed.ncbi.nlm.nih.gov/34459017/.
- Besteiro S, Brooks CF, Striepen B, et al. Autophagy protein Atg3 is essential for maintaining mitochondrial integrity and for normal intracellular development of Toxoplasma gondii tachyzoites. PLoS pathogens [Internet]. 2011 [cited 2022 Aug 20];7. Available from: https://pubmed.ncbi.nlm.nih.gov/22144900/.
- Lavine MD, Arrizabalaga G. Analysis of monensin sensitivity in Toxoplasma gondii reveals autophagy as a mechanism for drug induced death. PloS one [Internet]. 2012 [cited 2022 Jul 23];7. Available from: https://pubmed.ncbi.nlm.nih.gov/22848721/.
- Varberg JM, LaFavers KA, Arrizabalaga G, et al. Characterization of Plasmodium Atg3-Atg8 Interaction Inhibitors Identifies Novel Alternative Mechanisms of Action in Toxoplasma gondii. Antimicrobial agents and chemotherapy [Internet]. 2018 [cited 2022 Jul 23];62. Available from: https://pubmed.ncbi.nlm.nih.gov/29158278/.
- Koh HX, Aye HM, Tan KSW, et al. The lysosomotropic drug LeuLeu-OMe induces lysosome disruption and autophagy-independent cell death in Trypanosoma brucei. Microbial cell (Graz, Austria) [Internet]. 2015 [cited 2022 Jul 23];2:288–298. Available from: https://pubmed.ncbi.nlm.nih.gov/28357304/.
- Fernandes MC, Da Silva EN, Pinto A V., et al. A novel triazolic naphthofuranquinone induces autophagy in reservosomes and impairment of mitosis in Trypanosoma cruzi. Parasitology [Internet]. 2012 [cited 2022 Jul 19];139:26–36. Available from: https://pubmed.ncbi.nlm.nih.gov/21939585/.
- Lazarin-Bidóia D, Desoti VC, Ueda-Nakamura T, et al. Further evidence of the trypanocidal action of eupomatenoid-5: confirmation of involvement of reactive oxygen species and mitochondria owing to a reduction in trypanothione reductase activity. Free radical biology & medicine [Internet]. 2013 [cited 2022 Jul 23];60:17–28. Available from: https://pubmed.ncbi.nlm.nih.gov/23376033/.
- Veiga-Santos P, Desoti VC, Miranda N, et al. The natural compounds piperovatine and piperlonguminine induce autophagic cell death on Trypanosoma cruzi. Acta tropica [Internet]. 2013 [cited 2022 Jul 19];125:349–356. Available from: https://pubmed.ncbi.nlm.nih.gov/23228524/.
- Cristina Desoti V, Lazarin-Bidóia D, Bueno Sudatti D, et al. Additional evidence of the trypanocidal action of (-)-elatol on amastigote forms through the involvement of reactive oxygen species. Marine drugs [Internet]. 2014 [cited 2022 Jul 23];12:4973–4983. Available from: https://pubmed.ncbi.nlm.nih.gov/25257785/.
- Scariot DB, Britta EA, Moreira AL, et al. Induction of Early Autophagic Process on Leishmania amazonensis by Synergistic Effect of Miltefosine and Innovative Semi-synthetic Thiosemicarbazone. Frontiers in microbiology [Internet]. 2017 [cited 2022 Jul 23];8. Available from: https://pubmed.ncbi.nlm.nih.gov/28270805/.
- Calixto SL, Glanzmann N, Xavier Silveira MM, et al. Novel organic salts based on quinoline derivatives: The in vitro activity trigger apoptosis inhibiting autophagy in Leishmania spp. Chemico-biological interactions [Internet]. 2018 [cited 2022 Jul 23];293:141–151. Available from: https://pubmed.ncbi.nlm.nih.gov/30098941/.
- Pasquier B. Autophagy inhibitors. Cellular and molecular life sciences : CMLS [Internet]. 2016 [cited 2022 Jul 23];73:985–1001. Available from: https://pubmed.ncbi.nlm.nih.gov/26658914/.
- Menna-Barreto RFS, Henriques-Pons A, Pinto A V., et al. Effect of a beta-lapachone-derived naphthoimidazole on Trypanosoma cruzi: identification of target organelles. The Journal of antimicrobial chemotherapy [Internet]. 2005 [cited 2022 Jul 23];56:1034–1041. Available from: https://pubmed.ncbi.nlm.nih.gov/16269551/.
- Menna-Barreto RFS, Corrêa JR, Pinto A V., et al. Mitochondrial disruption and DNA fragmentation in Trypanosoma cruzi induced by naphthoimidazoles synthesized from beta-lapachone. Parasitology research [Internet]. 2007 [cited 2022 Jul 23];101:895–905. Available from: https://pubmed.ncbi.nlm.nih.gov/17546464/.
- Demarchi F, Bertoli C, Copetti T, et al. Calpain is required for macroautophagy in mammalian cells. The Journal of cell biology [Internet]. 2006 [cited 2022 Jul 23];175:595–605. Available from: https://pubmed.ncbi.nlm.nih.gov/17101693/.
- Yu L, Chen Y, Tooze SA. Autophagy pathway: Cellular and molecular mechanisms [Internet]. Autophagy. Taylor and Francis Inc.; 2018 [cited 2020 Dec 29]. p. 207–215. Available from: https://pubmed.ncbi.nlm.nih.gov/28933638/.
- Meng D, Li Z, Wang G, et al. Carvedilol attenuates liver fibrosis by suppressing autophagy and promoting apoptosis in hepatic stellate cells. Biomedicine & Pharmacotherapy. 2018;108:1617–1627.
- Wong WT, Li LH, Rao YK, et al. Repositioning of the β-Blocker Carvedilol as a Novel Autophagy Inducer That Inhibits the NLRP3 Inflammasome. Frontiers in immunology [Internet]. 2018 [cited 2022 Jul 23];9. Available from: https://pubmed.ncbi.nlm.nih.gov/30186288/.
- Rivero CV, Martínez SJ, Novick P, et al. Repurposing Carvedilol as a Novel Inhibitor of the Trypanosoma cruzi Autophagy Flux That Affects Parasite Replication and Survival. Frontiers in cellular and infection microbiology [Internet]. 2021 [cited 2022 Jul 23];11. Available from: https://pubmed.ncbi.nlm.nih.gov/34476220/.
- Petersen AL de OA, Guedes CES, Versoza CL, et al. 17-AAG Kills Intracellular Leishmania amazonensis while Reducing Inflammatory Responses in Infected Macrophages. Ojcius DM, editor. PLoS ONE. 2012;7:e49496.
- Santos DM, Petersen ALOA, Celes FS, et al. Chemotherapeutic Potential of 17-AAG against Cutaneous Leishmaniasis Caused by Leishmania (Viannia) braziliensis. Kelly BL, editor. PLoS Neglected Tropical Diseases. 2014;8:e3275.
- Petersen AL de OA, Cull B, Dias BRS, et al. 17-AAG-Induced Activation of the Autophagic Pathway in Leishmania Is Associated with Parasite Death. Microorganisms [Internet]. 2021 [cited 2022 Jul 23];9. Available from: https://pubmed.ncbi.nlm.nih.gov/34069389/.
- Murphy SC, Deye GA, B. Kim Lee Sim, et al. PfSPZ-CVac efficacy against malaria increases from 0% to 75% when administered in the absence of erythrocyte stage parasitemia: A randomized, placebo-controlled trial with controlled human malaria infection. PLoS pathogens [Internet]. 2021 [cited 2022 Aug 26];17. Available from: https://pubmed.ncbi.nlm.nih.gov/34048504/.
- Nussenzweig RS, Vanderberg J, Most H, et al. Protective immunity produced by the injection of x-irradiated sporozoites of plasmodium berghei. Nature [Internet]. 1967 [cited 2022 Aug 26];216:160–162. Available from: https://pubmed.ncbi.nlm.nih.gov/6057225/.
- Clyde DF, Most H, McCarthy VC, et al. Immunization of man against sporozite-induced falciparum malaria. The American journal of the medical sciences [Internet]. 1973 [cited 2022 Aug 26];266:169–177. Available from: https://pubmed.ncbi.nlm.nih.gov/4583408/.
- Belnoue E, Costa FTM, Frankenberg T, et al. Protective T cell immunity against malaria liver stage after vaccination with live sporozoites under chloroquine treatment. Journal of immunology (Baltimore, Md. : 1950) [Internet]. 2004 [cited 2022 Aug 26];172:2487–2495. Available from: https://pubmed.ncbi.nlm.nih.gov/14764721/.
- Le Roch KG, Zhou Y, Blair PL, et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science (New York, N.Y.) [Internet]. 2003 [cited 2022 Aug 26];301:1503–1508. Available from: https://pubmed.ncbi.nlm.nih.gov/12893887/.
- Walczak M, Ganesan SM, Niles JC, et al. ATG8 Is Essential Specifically for an Autophagy-Independent Function in Apicoplast Biogenesis in Blood-Stage Malaria Parasites. mBio [Internet]. 2018 [cited 2022 Jul 13];9. Available from: https://pubmed.ncbi.nlm.nih.gov/29295911/.
- Jayabalasingham B, Voss C, Ehrenman K, et al. Characterization of the ATG8-conjugation system in 2 Plasmodium species with special focus on the liver stage: possible linkage between the apicoplastic and autophagic systems? Autophagy [Internet]. 2014 [cited 2022 Aug 26];10:269–284. Available from: https://pubmed.ncbi.nlm.nih.gov/24342964/.
- Hain AUP, Weltzer RR, Hammond H, et al. Structural characterization and inhibition of the Plasmodium Atg8-Atg3 interaction. Journal of structural biology [Internet]. 2012 [cited 2022 Aug 26];180:551–562. Available from: https://pubmed.ncbi.nlm.nih.gov/22982544/.
- Hain AUP, Bartee D, Sanders NG, et al. Identification of an Atg8-Atg3 protein-protein interaction inhibitor from the medicines for Malaria Venture Malaria Box active in blood and liver stage Plasmodium falciparum parasites. Journal of medicinal chemistry [Internet]. 2014 [cited 2022 Aug 26];57:4521–4531. Available from: https://pubmed.ncbi.nlm.nih.gov/24786226/.
- Colombo MI. Pathogens and autophagy: subverting to survive. Cell death and differentiation [Internet]. 2005 [cited 2022 Aug 22];12 Suppl 2:1481–1483. Available from: https://pubmed.ncbi.nlm.nih.gov/16247495/.
- Mosser DM, Edelson PJ. Activation of the alternative complement pathway by Leishmania promastigotes: parasite lysis and attachment to macrophages. Journal of immunology (Baltimore, Md. : 1950) [Internet]. 1984 [cited 2022 Jul 23];132:1501–1505. Available from: https://pubmed.ncbi.nlm.nih.gov/6363545/.
- Blackwell JM, Ezekowitz RAB, Roberts MB, et al. Macrophage complement and lectin-like receptors bind Leishmania in the absence of serum. The Journal of experimental medicine [Internet]. 1985 [cited 2022 Jul 23];162:324–331. Available from: https://pubmed.ncbi.nlm.nih.gov/3891904/.
- Wilson ME, Pearson RD. Roles of CR3 and mannose receptors in the attachment and ingestion of Leishmania donovani by human mononuclear phagocytes. Infection and immunity [Internet]. 1988 [cited 2022 Jul 23];56:363–369. Available from: https://pubmed.ncbi.nlm.nih.gov/2962944/.
- Guy RA, Belosevic M. Comparison of receptors required for entry of Leishmania major amastigotes into macrophages. Infection and immunity [Internet]. 1993 [cited 2022 Jul 23];61:1553–1558. Available from: https://pubmed.ncbi.nlm.nih.gov/8454363/.
- Brittingham A, Mosser D. Exploitation of the complement system by Leishmania promastigotes. Parasitology Today. 1996;12:444–447.
- Brittingham A, Chen G, Mcgwire BS, et al. Interaction of Leishmania gp63 with cellular receptors for fibronectin. Infection and immunity [Internet]. 1999 [cited 2022 Jul 23];67:4477–4484. Available from: https://pubmed.ncbi.nlm.nih.gov/10456889/.
- Ueno N, Wilson ME. Receptor-mediated phagocytosis of Leishmania: implications for intracellular survival. Trends in parasitology [Internet]. 2012 [cited 2022 Jul 23];28:335–344. Available from: https://pubmed.ncbi.nlm.nih.gov/22726697/.
- Courret N, Fréhel C, Gouhier N, et al. Biogenesis of Leishmania-harbouring parasitophorous vacuoles following phagocytosis of the metacyclic promastigote or amastigote stages of the parasites. Journal of cell science [Internet]. 2002 [cited 2022 Jul 23];115:2303–2316. Available from: https://pubmed.ncbi.nlm.nih.gov/12006615/.
- Pérez-Cabezas B, Cecílio P, Gaspar TB, et al. Understanding Resistance vs. Susceptibility in Visceral Leishmaniasis Using Mouse Models of Leishmania infantum Infection. Frontiers in cellular and infection microbiology [Internet]. 2019 [cited 2022 Jul 23];9. Available from: https://pubmed.ncbi.nlm.nih.gov/30881923/.
- Veras PST, Castro-Gomes T, Menezes JPB de. Elucidating the Complex Interrelationship on Early Interactions between Leishmania and Macrophages. Macrophages -140 Years of Their Discovery [Working Title] [Internet]. 2022 [cited 2022 Jul 23]; Available from: undefined/state.item.id.
- Ghartey-Kwansah G, Adu-Nti F, Aboagye B, et al. Autophagy in the control and pathogenesis of parasitic infections. Cell & bioscience [Internet]. 2020 [cited 2022 Aug 23];10. Available from: https://pubmed.ncbi.nlm.nih.gov/32944216/.
- Veras PST, de Menezes JPB, Dias BRS. Deciphering the Role Played by Autophagy in Leishmania Infection. Frontiers in immunology [Internet]. 2019 [cited 2022 Jul 23];10. Available from: https://pubmed.ncbi.nlm.nih.gov/31736955/.
- Evans RJ, Sundaramurthy V, Frickel EM. The Interplay of Host Autophagy and Eukaryotic Pathogens. Frontiers in cell and developmental biology [Internet]. 2018 [cited 2022 Jul 13];6. Available from: https://pubmed.ncbi.nlm.nih.gov/30271774/.
- Cyrino LT, Araújo AP, Joazeiro PP, et al. In vivo and in vitro Leishmania amazonensis infection induces autophagy in macrophages. Tissue & cell [Internet]. 2012 [cited 2022 Jul 23];44:401–408. Available from: https://pubmed.ncbi.nlm.nih.gov/22939777/.
- Franco LH, Fleuri AKA, Pellison NC, et al. Autophagy downstream of endosomal Toll-like receptor signaling in macrophages is a key mechanism for resistance to Leishmania major infection. The Journal of biological chemistry [Internet]. 2017 [cited 2022 Jul 23];292:13087–13096. Available from: https://pubmed.ncbi.nlm.nih.gov/28607148/.
- Thomas SA, Nandan D, Kass J, et al. Countervailing, time-dependent effects on host autophagy promotes intracellular survival of Leishmania. The Journal of biological chemistry [Internet]. 2018 [cited 2022 Jul 23];293:2617–2630. Available from: https://pubmed.ncbi.nlm.nih.gov/29269416/.
- Dias BRS, de Souza CS, Almeida N de J, et al. Autophagic Induction Greatly Enhances Leishmania major Intracellular Survival Compared to Leishmania amazonensis in CBA/j-Infected Macrophages. Frontiers in microbiology [Internet]. 2018 [cited 2022 Jul 23];9. Available from: https://pubmed.ncbi.nlm.nih.gov/30158914/.
- Matte C, Casgrain PA, Séguin O, et al. Leishmania major Promastigotes Evade LC3-Associated Phagocytosis through the Action of GP63. PLoS pathogens [Internet]. 2016 [cited 2022 Jul 23];12. Available from: https://pubmed.ncbi.nlm.nih.gov/27280768/.
- Mitroulis I, Kourtzelis I, Papadopoulos VP, et al. In vivo induction of the autophagic machinery in human bone marrow cells during Leishmania donovani complex infection. Parasitology international [Internet]. 2009 [cited 2022 Jul 23];58:475–477. Available from: https://pubmed.ncbi.nlm.nih.gov/19591960/.
- Frank B, Marcu A, De Oliveira Almeida Petersen AL, et al. Autophagic digestion of Leishmania major by host macrophages is associated with differential expression of BNIP3, CTSE, and the miRNAs miR-101c, miR-129, and miR-210. Parasites & vectors [Internet]. 2015 [cited 2022 Jul 23];8. Available from: https://pubmed.ncbi.nlm.nih.gov/26226952/.
- Pitale DM, Gendalur NS, Descoteaux A, et al. Leishmania donovani Induces Autophagy in Human Blood-Derived Neutrophils. Journal of immunology (Baltimore, Md. : 1950) [Internet]. 2019 [cited 2022 Jul 23];202:1163–1175. Available from: https://pubmed.ncbi.nlm.nih.gov/30635391/.
- Morris DH, Yip CK, Shi Y, et al. BECLIN 1-VPS34 COMPLEX ARCHITECTURE: UNDERSTANDING THE NUTS AND BOLTS OF THERAPEUTIC TARGETS. Frontiers in biology [Internet]. 2015 [cited 2022 Jul 23];10:398–426. Available from: https://pubmed.ncbi.nlm.nih.gov/26692106/.
- Duque TLA, Serrão TC de SLC, Gonçalves AJ da S, et al. Leishmania (V.) braziliensis infection promotes macrophage autophagy by a LC3B-dependent and BECLIN1-independent mechanism. Acta tropica [Internet]. 2021 [cited 2022 Jul 26];218. Available from: https://pubmed.ncbi.nlm.nih.gov/33744245/.
- Pinheiro RO, Nunes MP, Pinheiro CS, et al. Induction of autophagy correlates with increased parasite load of Leishmania amazonensis in BALB/c but not C57BL/6 macrophages. Microbes and infection/Institut Pasteur. 2009;11:181–190.
- Lima JGB, de Freitas Vinhas C, Gomes IN, et al. Phagocytosis is inhibited by autophagic induction in murine macrophages. Biochemical and biophysical research communications [Internet]. 2011 [cited 2022 Jul 23];405:604–609. Available from: https://pubmed.ncbi.nlm.nih.gov/21272565/.
- Bonilla DL, Bhattacharya A, Sha Y, et al. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity [Internet]. 2013 [cited 2022 Jul 23];39:537–547. Available from: https://pubmed.ncbi.nlm.nih.gov/24035364/.
- Levin R, Grinstein S, Canton J. The life cycle of phagosomes: formation, maturation, and resolution. Immunological reviews [Internet]. 2016 [cited 2022 Jul 26];273:156–179. Available from: https://pubmed.ncbi.nlm.nih.gov/27558334/.
- Jutras I, Desjardins M. Phagocytosis: at the crossroads of innate and adaptive immunity. Annual review of cell and developmental biology [Internet]. 2005 [cited 2022 Jul 26];21:511–527. Available from: https://pubmed.ncbi.nlm.nih.gov/16212505/.
- Goyette G, Boulais J, Carruthers NJ, et al. Proteomic characterization of phagosomal membrane microdomains during phagolysosome biogenesis and evolution. Molecular & cellular proteomics : MCP [Internet]. 2012 [cited 2022 Jul 26];11:1365–1377. Available from: https://pubmed.ncbi.nlm.nih.gov/22915823/.
- Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nature immunology [Internet]. 2006 [cited 2022 Jul 26];7:1029–1035. Available from: https://pubmed.ncbi.nlm.nih.gov/16985500/.
- Ramachandra L, Song R, Harding C V. Phagosomes are fully competent antigen-processing organelles that mediate the formation of peptide:class II MHC complexes. Journal of immunology (Baltimore, Md. : 1950) [Internet]. 1999 [cited 2022 Jul 26];162:3263–3272. Available from: https://pubmed.ncbi.nlm.nih.gov/10092778/.
- Houde M, Bertholet S, Gagnon E, et al. Phagosomes are competent organelles for antigen cross-presentation. Nature [Internet]. 2003 [cited 2022 Jul 26];425:402–406. Available from: https://pubmed.ncbi.nlm.nih.gov/14508490/.
- Botelho RJ, Grinstein S. Phagocytosis. Current biology : CB [Internet]. 2011 [cited 2022 Jul 26];21. Available from: https://pubmed.ncbi.nlm.nih.gov/21783028/.
- Sanjuan MA, Dillon CP, Tait SWG, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature [Internet]. 2007 [cited 2022 Jul 26];450:1253–1257. Available from: https://pubmed.ncbi.nlm.nih.gov/18097414/.
- Richmond JM, Frisoli ML, Harris JE. Innate immune mechanisms in vitiligo: danger from within. Current opinion in immunology [Internet]. 2013 [cited 2022 Jul 26];25:676–682. Available from: https://pubmed.ncbi.nlm.nih.gov/24238922/.
- Upadhyay S, Philips JA. LC3-associated phagocytosis: host defense and microbial response. Current opinion in immunology [Internet]. 2019 [cited 2022 Jul 26];60:81–90. Available from: https://pubmed.ncbi.nlm.nih.gov/31247378/.
- Herb M, Gluschko A, Schramm M. LC3-associated phagocytosis - The highway to hell for phagocytosed microbes. Seminars in cell & developmental biology [Internet]. 2020 [cited 2022 Jul 26];101:68–76. Available from: https://pubmed.ncbi.nlm.nih.gov/31029766/.
- Martinez J, Almendinger J, Oberst A, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proceedings of the National Academy of Sciences of the United States of America [Internet]. 2011 [cited 2022 Jul 26];108:17396–17401. Available from: https://pubmed.ncbi.nlm.nih.gov/21969579/.
- Lamprinaki D, Beasy G, Zhekova A, et al. LC3-Associated Phagocytosis Is Required for Dendritic Cell Inflammatory Cytokine Response to Gut Commensal Yeast Saccharomyces cerevisiae. Frontiers in immunology [Internet]. 2017 [cited 2022 Jul 26];8. Available from: https://pubmed.ncbi.nlm.nih.gov/29118762/.
- Ma J, Becker C, Lowell CA, et al. Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. The Journal of biological chemistry [Internet]. 2012 [cited 2022 Jul 26];287:34149–34156. Available from: https://pubmed.ncbi.nlm.nih.gov/22902620/.
- Huang J, Canadien V, Lam GY, et al. Activation of antibacterial autophagy by NADPH oxidases. Proceedings of the National Academy of Sciences of the United States of America [Internet]. 2009 [cited 2022 Jul 26];106:6226–6231. Available from: https://pubmed.ncbi.nlm.nih.gov/19339495/.
- Moghadam ZM, Henneke P, Kolter J. From Flies to Men: ROS and the NADPH Oxidase in Phagocytes. Frontiers in cell and developmental biology [Internet]. 2021 [cited 2022 Jul 26];9. Available from: https://pubmed.ncbi.nlm.nih.gov/33842458/.
- Mantegazza AR, Savina A, Vermeulen M, et al. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood [Internet]. 2008 [cited 2022 Jul 26];112:4712–4722. Available from: https://pubmed.ncbi.nlm.nih.gov/18682599/.
- Martinez J, Malireddi RKS, Lu Q, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nature cell biology [Internet]. 2015 [cited 2022 Jul 26];17:893–906. Available from: https://pubmed.ncbi.nlm.nih.gov/26098576/.
- Matheoud D, Moradin N, Bellemare-Pelletier A, et al. Leishmania evades host immunity by inhibiting antigen cross-presentation through direct cleavage of the SNARE VAMP8. Cell host & microbe [Internet]. 2013 [cited 2022 Jul 26];14:15–25. Available from: https://pubmed.ncbi.nlm.nih.gov/23870310/.
- Arango Duque G, Fukuda M, Descoteaux A. Synaptotagmin XI regulates phagocytosis and cytokine secretion in macrophages. Journal of immunology (Baltimore, Md. : 1950) [Internet]. 2013 [cited 2022 Jul 26];190:1737–1745. Available from: https://pubmed.ncbi.nlm.nih.gov/23303671/.
- Hatsuzawa K, Sakurai C. Regulatory Mechanism of SNAP23 in Phagosome Formation and Maturation. Yonago acta medica [Internet]. 2020 [cited 2022 Jul 26];63:135–145. Available from: https://pubmed.ncbi.nlm.nih.gov/32884432/.
- Gallois A, Klein JR, Allen L-AH, et al. Salmonella pathogenicity island 2-encoded type III secretion system mediates exclusion of NADPH oxidase assembly from the phagosomal membrane. Journal of immunology (Baltimore, Md. : 1950) [Internet]. 2001 [cited 2022 Jul 26];166:5741–5748. Available from: https://pubmed.ncbi.nlm.nih.gov/11313417/.
- Carlyon JA, Latif DA, Pypaert M, et al. Anaplasma phagocytophilum utilizes multiple host evasion mechanisms to thwart NADPH oxidase-mediated killing during neutrophil infection. Infection and immunity [Internet]. 2004 [cited 2022 Jul 26];72:4772–4783. Available from: https://pubmed.ncbi.nlm.nih.gov/15271939/.
- Allen L-AH, Beecher BR, Lynch JT, et al. Helicobacter pylori disrupts NADPH oxidase targeting in human neutrophils to induce extracellular superoxide release. Journal of immunology (Baltimore, Md. : 1950) [Internet]. 2005 [cited 2022 Jul 26];174:3658–3667. Available from: https://pubmed.ncbi.nlm.nih.gov/15749904/.
- Harada T, Miyake M, Imai Y. Evasion of Legionella pneumophila from the bactericidal system by reactive oxygen species (ROS) in macrophages. Microbiology and immunology [Internet]. 2007 [cited 2022 Jul 26];51:1161–1170. Available from: https://pubmed.ncbi.nlm.nih.gov/18094534/.
- Rikihisa Y. Molecular events involved in cellular invasion by Ehrlichia chaffeensis and Anaplasma phagocytophilum. Veterinary parasitology [Internet]. 2010 [cited 2022 Jul 26];167:155–166. Available from: https://pubmed.ncbi.nlm.nih.gov/19836896/.
- Pham NK, Mouriz J, Kima PE. Leishmania pifanoi amastigotes avoid macrophage production of superoxide by inducing heme degradation. Infection and immunity [Internet]. 2005 [cited 2022 Jul 26];73:8322–8333. Available from: https://pubmed.ncbi.nlm.nih.gov/16299330/.
- Lodge R, Descoteaux A. Phagocytosis of Leishmania donovani amastigotes is Rac1 dependent and occurs in the absence of NADPH oxidase activation. European journal of immunology [Internet]. 2006 [cited 2022 Jul 26];36:2735–2744. Available from: https://pubmed.ncbi.nlm.nih.gov/16955522/.
- Lodge R, Diallo TO, Descoteaux A. Leishmania donovani lipophosphoglycan blocks NADPH oxidase assembly at the phagosome membrane. Cellular microbiology [Internet]. 2006 [cited 2022 Jul 26];8:1922–1931. Available from: https://pubmed.ncbi.nlm.nih.gov/16848789/.
- Crauwels P, Bohn R, Thomas M, et al. Apoptotic-like Leishmania exploit the host’s autophagy machinery to reduce T-cell-mediated parasite elimination. Autophagy [Internet]. 2015 [cited 2022 Jul 26];11:285–297. Available from: https://pubmed.ncbi.nlm.nih.gov/25801301/.
- Salassa BN, Cueto JA, Gambarte Tudela J, et al. Endocytic Rabs Are Recruited to the Trypanosoma cruzi Parasitophorous Vacuole and Contribute to the Process of Infection in Non-professional Phagocytic Cells. Frontiers in cellular and infection microbiology [Internet]. 2020 [cited 2022 Jul 26];10. Available from: https://pubmed.ncbi.nlm.nih.gov/33194787/.
- Romano PS, Cueto JA, Casassa AF, et al. Molecular and cellular mechanisms involved in the Trypanosoma cruzi/host cell interplay. IUBMB life. 2012;64:387–396.
- Vanrell MC, Cueto JA, Barclay JJ, et al. Polyamine depletion inhibits the autophagic response modulating Trypanosoma cruzi infectivity. Autophagy [Internet]. 2013 [cited 2018 Aug 30];9:1080–1093. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23697944.
- Tardieux I, Webster P, Ravesloot J, et al. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell [Internet]. 1992 [cited 2022 Aug 23];71:1117–1130. Available from: https://pubmed.ncbi.nlm.nih.gov/1473148/.
- Andrade LO, Andrews NW. The Trypanosoma cruzi–host-cell interplay: location, invasion, retention. Nature Reviews Microbiology. 2005;3:819–823.
- Onizuka Y, Takahashi C, Uematsu A, et al. Inhibition of autolysosome formation in host autophagy by Trypanosoma cruzi infection. Acta Tropica. 2017;170:57–62.
- Cortez C, Real F, Yoshida N. Lysosome biogenesis/scattering increases host cell susceptibility to invasion by Trypanosoma cruzi metacyclic forms and resistance to tissue culture trypomastigotes. Cellular microbiology [Internet]. 2016 [cited 2022 Aug 23];18:748–760. Available from: https://pubmed.ncbi.nlm.nih.gov/26572924/.
- Vanrell MC, Martinez SJ, Muñoz LI, et al. Induction of Autophagy by Ursolic Acid Promotes the Elimination of Trypanosoma cruzi Amastigotes From Macrophages and Cardiac Cells. Frontiers in Cellular and Infection Microbiology [Internet]. 2022 [cited 2022 Aug 23];0:951. Available from: https://www.frontiersin.org/articles/10.3389/fcimb.2022.919096/full.
- Casassa AF, Vanrell MC, Colombo MI, et al. Autophagy plays a protective role against Trypanosoma cruzi infection in mice. Virulence [Internet]. 2019 [cited 2019 Nov 25];10:151–165. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30829115.
- Matteucci KC, Pereira GJS, Weinlich R, et al. Frontline Science: Autophagy is a cell autonomous effector mechanism mediated by NLRP3 to control Trypanosoma cruzi infection. Journal of leukocyte biology [Internet]. 2019 [cited 2022 Apr 12];106:531–540. Available from: https://pubmed.ncbi.nlm.nih.gov/31071239/.
- Duque TLA, Cascabulho CM, Oliveira GM, et al. Rapamycin Treatment Reduces Acute Myocarditis Induced by Trypanosoma cruzi Infection. Journal of Innate Immunity [Internet]. 2020 [cited 2021 Mar 13];12:321–332. Available from: /pmc/articles/PMC7383291/.
- Brunoro GVF, Caminha MA, Ferreira AT da S, et al. Reevaluating the Trypanosoma cruzi proteomic map: The shotgun description of bloodstream trypomastigotes. Journal of proteomics [Internet]. 2015 [cited 2022 Jul 26];115:58–65. Available from: https://pubmed.ncbi.nlm.nih.gov/25534883/.
- Shen B, Sibley LD. The moving junction, a key portal to host cell invasion by apicomplexan parasites. Current opinion in microbiology [Internet]. 2012 [cited 2022 Jul 14];15:449–455. Available from: https://pubmed.ncbi.nlm.nih.gov/22445360/.
- Charron AJ, Sibley LD. Molecular partitioning during host cell penetration by Toxoplasma gondii. Traffic (Copenhagen, Denmark) [Internet]. 2004 [cited 2022 Aug 24];5:855–867. Available from: https://pubmed.ncbi.nlm.nih.gov/15479451/.
- David Sibley L. Invasion and intracellular survival by protozoan parasites. Immunological reviews [Internet]. 2011 [cited 2022 Jul 26];240:72–91. Available from: https://pubmed.ncbi.nlm.nih.gov/21349087/.
- Clough B, Frickel EM. The Toxoplasma Parasitophorous Vacuole: An Evolving Host–Parasite Frontier. Trends in Parasitology. 2017;33:473–488.
- Andrade RM, Wessendarp M, Gubbels MJ, et al. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. The Journal of clinical investigation [Internet]. 2006 [cited 2022 Jul 26];116:2366–2377. Available from: https://pubmed.ncbi.nlm.nih.gov/16955139/.
- Van Grol J, Muniz-Feliciano L, Portillo JAC, et al. CD40 induces anti-toxoplasma gondii activity in nonhematopoietic cells dependent on autophagy proteins. Infection and Immunity [Internet]. 2013 [cited 2022 Jul 26];81:2002–2011. Available from: https://journals.asm.org/doi/10.1128/IAI.01145-12.
- Subauste CS, Remington JS. Role of gamma interferon in Toxoplasma gondii infection. European journal of clinical microbiology & infectious diseases : official publication of the European Society of Clinical Microbiology [Internet]. 1991 [cited 2022 Jul 26];10:58–67. Available from: https://pubmed.ncbi.nlm.nih.gov/1907542/.
- Park S, Choi J, Biering SB, et al. Targeting by AutophaGy proteins (TAG): Targeting of IFNG-inducible GTPases to membranes by the LC3 conjugation system of autophagy. Autophagy [Internet]. 2016 [cited 2022 Jul 26];12:1153–1167. Available from: https://pubmed.ncbi.nlm.nih.gov/27172324/.
- Sasai M, Sakaguchi N, Ma JS, et al. Essential role for GABARAP autophagy proteins in interferon-inducible GTPase-mediated host defense. Nature immunology [Internet]. 2017 [cited 2022 Jul 26];18:899–910. Available from: https://pubmed.ncbi.nlm.nih.gov/28604719/.
- Choi J, Park S, Biering SB, et al. The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin-like conjugation systems of autophagy. Immunity [Internet]. 2014 [cited 2022 Aug 24];40:924–935. Available from: https://pubmed.ncbi.nlm.nih.gov/24931121/.
- Zhao Z, Fux B, Goodwin M, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell host & microbe [Internet]. 2008 [cited 2022 Jul 26];4:458–469. Available from: https://pubmed.ncbi.nlm.nih.gov/18996346/.
- Bhushan J, Radke JB, Perng YC, et al. ISG15 Connects Autophagy and IFN-γ-Dependent Control of Toxoplasma gondii Infection in Human Cells. mBio [Internet]. 2020 [cited 2022 Jul 26];11:1–19. Available from: https://pubmed.ncbi.nlm.nih.gov/33024031/.
- Subauste CS. Recent Advances in the Roles of Autophagy and Autophagy Proteins in Host Cells During Toxoplasma gondii Infection and Potential Therapeutic Implications. Frontiers in Cell and Developmental Biology. 2021;9:1335.
- Selleck EM, Orchard RC, Lassen KG, et al. A noncanonical autophagy pathway restricts toxoplasma gondii growth in a strain-specific manner in IFN-γ-activated human cells. mBio [Internet]. 2015 [cited 2022 Jul 26];6. Available from: https://journals.asm.org/doi/10.1128/mBio.01157-15.
- Clough B, Wright JD, Pereira PM, et al. K63-Linked Ubiquitination Targets Toxoplasma gondii for Endo-lysosomal Destruction in IFNγ-Stimulated Human Cells. PLoS pathogens [Internet]. 2016 [cited 2022 Jul 26];12. Available from: https://pubmed.ncbi.nlm.nih.gov/27875583/.
- Liu E, Corcino YL, Portillo JAC, et al. Identification of Signaling Pathways by Which CD40 Stimulates Autophagy and Antimicrobial Activity against Toxoplasma gondii in Macrophages. Infection and immunity [Internet]. 2016 [cited 2022 Jul 26];84:2616–2626. Available from: https://pubmed.ncbi.nlm.nih.gov/27354443/.
- Portillo JAC, Okenka G, Reed E, et al. The CD40-Autophagy Pathway Is Needed for Host Protection Despite IFN-Γ-Dependent Immunity and CD40 Induces Autophagy via Control of P21 Levels. PLOS ONE [Internet]. 2010 [cited 2022 Jul 26];5:e14472. Available from: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0014472.
- D’Avila H, Freire-de-Lima C, Roque N, et al. Host cell lipid bodies triggered by Trypanosoma cruzi infection and enhanced by the uptake of apoptotic cells are associated with prostaglandin E₂ generation and increased parasite growth. The Journal of infectious diseases [Internet]. 2011 [cited 2021 Oct 25];204:951–961. Available from: https://pubmed.ncbi.nlm.nih.gov/21849292/.
- Meissner M, Reiss M, Viebig N, et al. A family of transmembrane microneme proteins of Toxoplasma gondii contain EGF-like domains and function as escorters. Journal of cell science [Internet]. 2002 [cited 2022 Aug 24];115:563–574. Available from: https://pubmed.ncbi.nlm.nih.gov/11861763/.
- Muniz-Feliciano L, Van Grol J, Portillo JAC, et al. Toxoplasma gondii-induced activation of EGFR prevents autophagy protein-mediated killing of the parasite. PLoS pathogens [Internet]. 2013 [cited 2022 Jul 26];9:1–15. Available from: https://pubmed.ncbi.nlm.nih.gov/24367261/.
- Coppens I. How Toxoplasma and malaria parasites defy first, then exploit host autophagic and endocytic pathways for growth. Current opinion in microbiology [Internet]. 2017 [cited 2022 Jul 27];40:32–39. Available from: https://pubmed.ncbi.nlm.nih.gov/29102900/.
- Coppens I, Dunn JD, Romano JD, et al. Toxoplasma gondii sequesters lysosomes from mammalian hosts in the vacuolar space. Cell [Internet]. 2006 [cited 2022 Aug 24];125:261–274. Available from: https://pubmed.ncbi.nlm.nih.gov/16630815/.
- Guo M, Sun J, Wang W tao, et al. Toxoplasma gondii ROP17 promotes autophagy via the Bcl-2-Beclin 1 pathway. Folia parasitologica [Internet]. 2021 [cited 2022 Jul 26];68:1–10. Available from: https://pubmed.ncbi.nlm.nih.gov/34180401/.
- Portillo JAC, Muniz-Feliciano L, Lopez Corcino Y, et al. Toxoplasma gondii induces FAK-Src-STAT3 signaling during infection of host cells that prevents parasite targeting by autophagy. PLoS pathogens [Internet]. 2017 [cited 2022 Jul 26];13. Available from: https://pubmed.ncbi.nlm.nih.gov/29036202/.
- Chu JQ, Jing KP, Gao X, et al. Toxoplasma gondii induces autophagy and apoptosis in human umbilical cord mesenchymal stem cells via downregulation of Mcl-1. Cell cycle (Georgetown, Tex.) [Internet]. 2017 [cited 2022 Jul 26];16:477–486. Available from: https://pubmed.ncbi.nlm.nih.gov/28112581/.
- Pernas L, Bean C, Boothroyd JC, et al. Mitochondria Restrict Growth of the Intracellular Parasite Toxoplasma gondii by Limiting Its Uptake of Fatty Acids. Cell metabolism [Internet]. 2018 [cited 2022 Jul 26];27:886–897.e4. Available from: https://pubmed.ncbi.nlm.nih.gov/29617646/.
- VANDERBERG JP. Studies on the motility of Plasmodium sporozoites. The Journal of protozoology [Internet]. 1974 [cited 2022 Jul 14];21:527–537. Available from: https://pubmed.ncbi.nlm.nih.gov/4138523/.
- Kappe SHI, Buscaglia CA, Bergman LW, et al. Apicomplexan gliding motility and host cell invasion: overhauling the motor model. Trends in parasitology [Internet]. 2004 [cited 2022 Jul 14];20:13–16. Available from: https://pubmed.ncbi.nlm.nih.gov/14700584/.
- Tavares J, Formaglio P, Thiberge S, et al. Role of host cell traversal by the malaria sporozoite during liver infection. The Journal of experimental medicine [Internet]. 2013 [cited 2022 Jul 14];210:905–915. Available from: https://pubmed.ncbi.nlm.nih.gov/23610126/.
- Mota MM, Pradel G, Vanderberg JP, et al. Migration of Plasmodium sporozoites through cells before infection. Science (New York, N.Y.) [Internet]. 2001 [cited 2022 Jul 14];291:141–144. Available from: https://pubmed.ncbi.nlm.nih.gov/11141568/.
- Risco-Castillo V, Topçu S, Marinach C, et al. Malaria Sporozoites Traverse Host Cells within Transient Vacuoles. Cell host & microbe [Internet]. 2015 [cited 2022 Jul 14];18:593–603. Available from: https://pubmed.ncbi.nlm.nih.gov/26607162/.
- Loubens M, Vincensini L, Fernandes P, et al. Plasmodium sporozoites on the move: Switching from cell traversal to productive invasion of hepatocytes. Molecular microbiology [Internet]. 2021 [cited 2022 Jul 14];115:870–881. Available from: https://pubmed.ncbi.nlm.nih.gov/33191548/.
- Amino R, Giovannini D, Thiberge S, et al. Host cell traversal is important for progression of the malaria parasite through the dermis to the liver. Cell host & microbe [Internet]. 2008 [cited 2022 Jul 14];3:88–96. Available from: https://pubmed.ncbi.nlm.nih.gov/18312843/.
- Spielmann T, Montagna GN, Hecht L, et al. Molecular make-up of the Plasmodium parasitophorous vacuolar membrane. International journal of medical microbiology : IJMM [Internet]. 2012 [cited 2022 Jul 14];302:179–186. Available from: https://pubmed.ncbi.nlm.nih.gov/22898489/.
- Annoura T, Van Schaijk BCL, Ploemen IHJ, et al. Two Plasmodium 6-Cys family-related proteins have distinct and critical roles in liver-stage development. FASEB journal : official publication of the Federation of American Societies for Experimental Biology [Internet]. 2014 [cited 2022 Jul 14];28:2158–2170. Available from: https://pubmed.ncbi.nlm.nih.gov/24509910/.
- Labaied M, Harupa A, Dumpit RF, et al. Plasmodium yoelii sporozoites with simultaneous deletion of P52 and P36 are completely attenuated and confer sterile immunity against infection. Infection and immunity [Internet]. 2007 [cited 2022 Jul 14];75:3758–3768. Available from: https://pubmed.ncbi.nlm.nih.gov/17517871/.
- Ploemen IHJ, Croes HJ, van Gemert GJJ, et al. Plasmodium berghei Δp52&p36 parasites develop independent of a parasitophorous vacuole membrane in Huh-7 liver cells. PloS one [Internet]. 2012 [cited 2022 Jul 14];7. Available from: https://pubmed.ncbi.nlm.nih.gov/23227206/.
- Hanson KK, Ressurreicao AS, Buchholz K, et al. Torins are potent antimalarials that block replenishment of Plasmodium liver stage parasitophorous vacuole membrane proteins. Proceedings of the National Academy of Sciences of the United States of America [Internet]. 2013 [cited 2022 Jul 14];110. Available from: https://pubmed.ncbi.nlm.nih.gov/23836641/.
- Bano N, Romano JD, Jayabalasingham B, et al. Cellular interactions of Plasmodium liver stage with its host mammalian cell. International journal for parasitology [Internet]. 2007 [cited 2022 Jul 14];37:1329–1341. Available from: https://pubmed.ncbi.nlm.nih.gov/17537443/.
- Sturm A, Graewe S, Franke-Fayard B, et al. Alteration of the parasite plasma membrane and the parasitophorous vacuole membrane during exo-erythrocytic development of malaria parasites. Protist [Internet]. 2009 [cited 2022 Jul 14];160:51–63. Available from: https://pubmed.ncbi.nlm.nih.gov/19026596/.
- Agop-Nersesian C, De Niz M, Niklaus L, et al. Shedding of host autophagic proteins from the parasitophorous vacuolar membrane of Plasmodium berghei. Scientific reports [Internet]. 2017 [cited 2022 Jul 14];7. Available from: https://pubmed.ncbi.nlm.nih.gov/28526861/.
- Grützke J, Rindte K, Goosmann C, et al. The spatiotemporal dynamics and membranous features of the Plasmodium liver stage tubovesicular network. Traffic (Copenhagen, Denmark) [Internet]. 2014 [cited 2022 Jul 14];15:362–382. Available from: https://pubmed.ncbi.nlm.nih.gov/24423236/.
- Prudêncio M, Rodriguez A, Mota MM. The silent path to thousands of merozoites: the Plasmodium liver stage. Nature reviews. Microbiology [Internet]. 2006 [cited 2022 Jul 14];4:849–856. Available from: https://pubmed.ncbi.nlm.nih.gov/17041632/.
- Nyboer B, Heiss K, Mueller AK, et al. The Plasmodium liver-stage parasitophorous vacuole: A front-line of communication between parasite and host. International journal of medical microbiology : IJMM [Internet]. 2018 [cited 2022 Jul 14];308:107–117. Available from: https://pubmed.ncbi.nlm.nih.gov/28964681/.
- Prado M, Eickel N, De Niz M, et al. Long-term live imaging reveals cytosolic immune responses of host hepatocytes against Plasmodium infection and parasite escape mechanisms. Autophagy [Internet]. 2015 [cited 2022 Jul 14];11:1561–1579. Available from: https://pubmed.ncbi.nlm.nih.gov/26208778/.
- Thieleke-Matos C, Lopes da Silva M, Cabrita-Santos L, et al. Host cell autophagy contributes to Plasmodium liver development. Cellular microbiology [Internet]. 2016 [cited 2022 Jul 14];18:437–450. Available from: https://pubmed.ncbi.nlm.nih.gov/26399761/.
- Wacker R, Eickel N, Schmuckli-Maurer J, et al. LC3-association with the parasitophorous vacuole membrane of Plasmodium berghei liver stages follows a noncanonical autophagy pathway. Cellular microbiology [Internet]. 2017 [cited 2022 Jul 14];19. Available from: https://pubmed.ncbi.nlm.nih.gov/28573684/.
- Bindschedler A, Wacker R, Egli J, et al. Plasmodium berghei sporozoites in nonreplicative vacuole are eliminated by a PI3P-mediated autophagy-independent pathway. Cellular microbiology [Internet]. 2021 [cited 2022 Jul 14];23. Available from: https://pubmed.ncbi.nlm.nih.gov/32979009/.
- Fischer TD, Wang C, Padman BS, et al. STING induces LC3B lipidation onto single-membrane vesicles via the V-ATPase and ATG16L1-WD40 domain. The Journal of cell biology [Internet]. 2020 [cited 2022 Jul 14];219. Available from: https://pubmed.ncbi.nlm.nih.gov/33201170/.
- Durgan J, Lystad AH, Sloan K, et al. Non-canonical autophagy drives alternative ATG8 conjugation to phosphatidylserine. Molecular cell [Internet]. 2021 [cited 2022 Jul 14];81:2031–2040.e8. Available from: https://pubmed.ncbi.nlm.nih.gov/33909989/.
- Boonhok R, Rachaphaew N, Duangmanee A, et al. LAP-like process as an immune mechanism downstream of IFN-γ in control of the human malaria Plasmodium vivax liver stage. Proceedings of the National Academy of Sciences of the United States of America [Internet]. 2016 [cited 2022 Jul 14];113:E3519–E3528. Available from: https://pubmed.ncbi.nlm.nih.gov/27185909/.
- Cogswell FB. The hypnozoite and relapse in primate malaria. Clinical microbiology reviews [Internet]. 1992 [cited 2022 Jul 14];5:26–35. Available from: https://pubmed.ncbi.nlm.nih.gov/1735093/.
- Ishino T, Chinzei Y, Yuda M. A Plasmodium sporozoite protein with a membrane attack complex domain is required for breaching the liver sinusoidal cell layer prior to hepatocyte infection. Cellular microbiology [Internet]. 2005 [cited 2022 Jul 14];7:199–208. Available from: https://pubmed.ncbi.nlm.nih.gov/15659064/.
- Ishino T, Yano K, Chinzei Y, et al. Cell-passage activity is required for the malarial parasite to cross the liver sinusoidal cell layer. PLoS biology [Internet]. 2004 [cited 2022 Jul 14];2. Available from: https://pubmed.ncbi.nlm.nih.gov/14737184/.
- Schmuckli-Maurer J, Reber V, Wacker R, et al. Inverted recruitment of autophagy proteins to the Plasmodium berghei parasitophorous vacuole membrane. PloS one [Internet]. 2017 [cited 2022 Jul 14];12. Available from: https://pubmed.ncbi.nlm.nih.gov/28841718/.
- Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. The FEBS journal [Internet]. 2015 [cited 2022 Jul 14];282:4672–4678. Available from: https://pubmed.ncbi.nlm.nih.gov/26432171/.
- Heussler VT, Küenzi P, Rottenberg S. Inhibition of apoptosis by intracellular protozoan parasites. International journal for parasitology [Internet]. 2001 [cited 2022 Jul 14];31:1166–1176. Available from: https://pubmed.ncbi.nlm.nih.gov/11563357/.
- Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes to cells : devoted to molecular & cellular mechanisms [Internet]. 2011 [cited 2022 Jul 14];16:123–140. Available from: https://pubmed.ncbi.nlm.nih.gov/21251164/.
- Ichimura Y, Waguri S, Sou Y shin, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Molecular cell [Internet]. 2013 [cited 2022 Jul 14];51:618–631. Available from: https://pubmed.ncbi.nlm.nih.gov/24011591/.
- Bindschedler A, Schmuckli-Maurer J, Wacker R, et al. Plasmodium berghei-Mediated NRF2 Activation in Infected Hepatocytes Enhances Parasite Survival. Botelho R, editor. Cellular Microbiology [Internet]. 2022 [cited 2022 Jul 14];2022:1–17. Available from: https://www.hindawi.com/journals/cmi/2022/7647976/.
- Afriat A, Zuzarte-Luís V, Halpern KB, et al. A spatiotemporally resolved single cell atlas of the Plasmodium liver stage. bioRxiv [Internet]. 2021 [cited 2022 Jul 14];2021.12.03.471111. Available from: https://www.biorxiv.org/content/10.1101/2021.12.03.471111v1.
- Duran A, Amanchy R, Linares JF, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Molecular cell [Internet]. 2011 [cited 2022 Jul 14];44:134–146. Available from: https://pubmed.ncbi.nlm.nih.gov/21981924/.
- Linares JF, Duran A, Yajima T, et al. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Molecular cell [Internet]. 2013 [cited 2022 Jul 14];51:283–296. Available from: https://pubmed.ncbi.nlm.nih.gov/23911927/.
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nature reviews. Cancer [Internet]. 2006 [cited 2022 Jul 14];6:729–734. Available from: https://pubmed.ncbi.nlm.nih.gov/16915295/.
- Real E, Rodrigues L, Cabal GG, et al. Plasmodium UIS3 sequesters host LC3 to avoid elimination by autophagy in hepatocytes. Nature microbiology [Internet]. 2018 [cited 2022 Jul 14];3:17–25. Available from: https://pubmed.ncbi.nlm.nih.gov/29109477/.
- Mueller AK, Labaled M, Kappe SHI, et al. Genetically modified Plasmodium parasites as a protective experimental malaria vaccine. Nature [Internet]. 2005 [cited 2022 Jul 14];433:164–167. Available from: https://pubmed.ncbi.nlm.nih.gov/15580261/.
- Setua S, Enguita FJ, Chora ÂF, et al. Disrupting Plasmodium UIS3-host LC3 interaction with a small molecule causes parasite elimination from host cells. Communications biology [Internet]. 2020 [cited 2022 Jul 14];3. Available from: https://pubmed.ncbi.nlm.nih.gov/33214643/.
- Niklaus L, Agop-Nersesian C, Schmuckli-Maurer J, et al. Deciphering host lysosome-mediated elimination of Plasmodium berghei liver stage parasites. Scientific reports [Internet]. 2019 [cited 2022 Jul 14];9. Available from: https://pubmed.ncbi.nlm.nih.gov/31138850/.
- Lopes da Silva M, Thieleke-Matos C, Cabrita-Santos L, et al. The host endocytic pathway is essential for Plasmodium berghei late liver stage development. Traffic (Copenhagen, Denmark) [Internet]. 2012 [cited 2022 Jul 14];13:1351–1363. Available from: https://pubmed.ncbi.nlm.nih.gov/22780869/.
- de Niz M, Caldelari R, Kaiser G, et al. Hijacking of the host cell Golgi by Plasmodium berghei liver stage parasites. Journal of cell science [Internet]. 2021 [cited 2022 Jul 14];134. Available from: https://pubmed.ncbi.nlm.nih.gov/34013963/.
- Thieleke-Matos C, da Silva ML, Cabrita-Santos L, et al. Host PI(3,5)P2 activity is required for Plasmodium berghei growth during liver stage infection. Traffic (Copenhagen, Denmark) [Internet]. 2014 [cited 2022 Nov 12];15:1066–1082. Available from: https://pubmed.ncbi.nlm.nih.gov/24992508/.
- Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. Journal of Clinical Investigation. 2003;112:1809–1820.
- Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nature Reviews Immunology. 2007;7:767–777.
- Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell host & microbe. 2009;5:527–549.
- Hoft DF, Lynch RG, Kirchhoff L V. Kinetic analysis of antigen-specific immune responses in resistant and susceptible mice during infection with Trypanosoma cruzi. Journal of immunology (Baltimore, Md. : 1950) [Internet]. 1993 [cited 2022 Aug 25];151:7038–7047. Available from: https://pubmed.ncbi.nlm.nih.gov/8258708/.
- Peluffo G, Piacenza L, Irigoín F, et al. L-arginine metabolism during interaction of Trypanosoma cruzi with host cells. Trends in parasitology [Internet]. 2004 [cited 2022 Mar 30];20:363–369. Available from: https://pubmed.ncbi.nlm.nih.gov/15246319/.
- Pereira-Santos M, Gonçalves-Santos E, Souza MA, et al. Chronic rapamycin pretreatment modulates arginase/inducible nitric oxide synthase balance attenuating aging-dependent susceptibility to Trypanosoma cruzi infection and acute myocarditis. Experimental gerontology [Internet]. 2022 [cited 2022 Aug 25];159. Available from: https://pubmed.ncbi.nlm.nih.gov/34968674/.
- Guimarães ET, Santos LA, Ribeiro dos Santos R, et al. Role of interleukin-4 and prostaglandin E2 in Leishmania amazonensis infection of BALB/c mice. Microbes and infection [Internet]. 2006 [cited 2022 Aug 25];8:1219–1226. Available from: https://pubmed.ncbi.nlm.nih.gov/16531090/.
- Suthanthiran M, Morris RE, Strom TB. Immunosuppressants: Cellular and molecular mechanisms of action. American Journal of Kidney Diseases. 1996;28:159–172.
- Maciolek JA, Alex Pasternak J, Wilson HL. Metabolism of activated T lymphocytes. Current opinion in immunology [Internet]. 2014 [cited 2022 Aug 25];27:60–74. Available from: https://pubmed.ncbi.nlm.nih.gov/24556090/.
- Kaushansky A, Ye AS, Austin LS, et al. Suppression of host p53 is critical for Plasmodium liver-stage infection. Cell reports [Internet]. 2013 [cited 2022 Nov 12];3:630–637. Available from: https://pubmed.ncbi.nlm.nih.gov/23478020/.
- Raphemot R, Toro-Moreno M, Lu KY, et al. Discovery of Druggable Host Factors Critical to Plasmodium Liver-Stage Infection. Cell chemical biology [Internet]. 2019 [cited 2022 Nov 12];26:1253–1262.e5. Available from: https://pubmed.ncbi.nlm.nih.gov/31257182/.
- Douglass AN, Kain HS, Abdullahi M, et al. Host-based Prophylaxis Successfully Targets Liver Stage Malaria Parasites. Molecular therapy : the journal of the American Society of Gene Therapy [Internet]. 2015 [cited 2022 Nov 12];23:857–865. Available from: https://pubmed.ncbi.nlm.nih.gov/25648263/.
- Subauste CS. Recent Advances in the Roles of Autophagy and Autophagy Proteins in Host Cells During Toxoplasma gondii Infection and Potential Therapeutic Implications. Frontiers in cell and developmental biology [Internet]. 2021 [cited 2022 Jul 13];9. Available from: https://pubmed.ncbi.nlm.nih.gov/34179003/.
- Reichmann G, Walker W, Villegas EN, et al. The CD40/CD40 ligand interaction is required for resistance to toxoplasmic encephalitis. Infection and immunity [Internet]. 2000 [cited 2022 Aug 25];68:1312–1318. Available from: https://pubmed.ncbi.nlm.nih.gov/10678943/.
- Gavrilescu LC, Butcher BA, Del Rio L, et al. STAT1 is essential for antimicrobial effector function but dispensable for gamma interferon production during Toxoplasma gondii infection. Infection and immunity [Internet]. 2004 [cited 2022 Aug 25];72:1257–1264. Available from: https://pubmed.ncbi.nlm.nih.gov/14977926/.
- Lieberman LA, Banica M, Reiner SL, et al. STAT1 plays a critical role in the regulation of antimicrobial effector mechanisms, but not in the development of Th1-type responses during toxoplasmosis. Journal of immunology (Baltimore, Md. : 1950) [Internet]. 2004 [cited 2022 Aug 25];172:457–463. Available from: https://pubmed.ncbi.nlm.nih.gov/14688355/.
- Ogolla PS, Portillo JAC, White CL, et al. The protein kinase double-stranded RNA-dependent (PKR) enhances protection against disease cause by a non-viral pathogen. PLoS pathogens [Internet]. 2013 [cited 2022 Aug 25];9. Available from: https://pubmed.ncbi.nlm.nih.gov/23990781/.
- Portillo JAC, Grol J Van, Saffo S, et al. CD40 in Endothelial Cells Restricts Neural Tissue Invasion by Toxoplasma gondii. Infection and immunity [Internet]. 2019 [cited 2022 Aug 25];87. Available from: https://pubmed.ncbi.nlm.nih.gov/31109947/.
- Portillo JAC, Yu JS, Hansen S, et al. A cell-penetrating CD40-TRAF2,3 blocking peptide diminishes inflammation and neuronal loss after ischemia/reperfusion. FASEB journal : official publication of the Federation of American Societies for Experimental Biology [Internet]. 2021 [cited 2022 Aug 25];35. Available from: https://pubmed.ncbi.nlm.nih.gov/33675257/.
- Bhadra R, Gigley JP, Khan IA. Cutting edge: CD40-CD40 ligand pathway plays a critical CD8-intrinsic and -extrinsic role during rescue of exhausted CD8 T cells. Journal of immunology (Baltimore, Md. : 1950) [Internet]. 2011 [cited 2022 Aug 25];187:4421–4425. Available from: https://pubmed.ncbi.nlm.nih.gov/21949017/.
- Zhao Z, Fux B, Goodwin M, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell host & microbe [Internet]. 2008 [cited 2022 Aug 24];4:458–469. Available from: https://pubmed.ncbi.nlm.nih.gov/18996346/.
- Haldar AK, Foltz C, Finethy R, et al. Ubiquitin systems mark pathogen-containing vacuoles as targets for host defense by guanylate binding proteins. Proceedings of the National Academy of Sciences of the United States of America [Internet]. 2015 [cited 2022 Aug 25];112:E5628–E5637. Available from: https://pubmed.ncbi.nlm.nih.gov/26417105/.
- Martens S, Parvanova I, Zerrahn J, et al. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS pathogens [Internet]. 2005 [cited 2022 Aug 25];1:0187–0201. Available from: https://pubmed.ncbi.nlm.nih.gov/16304607/.
- Etheridge RD, Alaganan A, Tang K, et al. The Toxoplasma pseudokinase ROP5 forms complexes with ROP18 and ROP17 kinases that synergize to control acute virulence in mice. Cell host & microbe [Internet]. 2014 [cited 2022 Aug 25];15:537–550. Available from: https://pubmed.ncbi.nlm.nih.gov/24832449/.
- Clough B, Wright JD, Pereira PM, et al. K63-Linked Ubiquitination Targets Toxoplasma gondii for Endo-lysosomal Destruction in IFNγ-Stimulated Human Cells. PLOS Pathogens [Internet]. 2016 [cited 2022 Jul 26];12:e1006027. Available from: https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1006027.
- Janssen R, van Wengen A, Verhard E, et al. Divergent role for TNF-alpha in IFN-gamma-induced killing of Toxoplasma gondii and Salmonella typhimurium contributes to selective susceptibility of patients with partial IFN-gamma receptor 1 deficiency. Journal of immunology (Baltimore, Md. : 1950) [Internet]. 2002 [cited 2022 Aug 25];169:3900–3907. Available from: https://pubmed.ncbi.nlm.nih.gov/12244188/.
- Subauste CS. Autophagy in immunity against Toxoplasma gondii. Current topics in microbiology and immunology [Internet]. 2009 [cited 2022 Aug 25];335:251–265. Available from: https://pubmed.ncbi.nlm.nih.gov/19802569/.
- Andrade RM, Portillo JAC, Wessendarp M, et al. CD40 signaling in macrophages induces activity against an intracellular pathogen independently of gamma interferon and reactive nitrogen intermediates. Infection and Immunity [Internet]. 2005 [cited 2022 Jul 26];73:3115–3123. Available from: https://journals.asm.org/doi/10.1128/IAI.73.5.3115-3123.2005.
- Subauste CS, Wessendarp M. CD40 restrains in vivo growth of Toxoplasma gondii independently of gamma interferon. Infection and Immunity [Internet]. 2006 [cited 2022 Jul 26];74:1573–1579. Available from: https://journals.asm.org/doi/10.1128/IAI.74.3.1573-1579.2006.
- Lopez Corcino Y, Gonzalez Ferrer S, Mantilla LE, et al. Toxoplasma gondii induces prolonged host epidermal growth factor receptor signalling to prevent parasite elimination by autophagy: Perspectives for in vivo control of the parasite. Cellular microbiology [Internet]. 2019 [cited 2022 Aug 25];21. Available from: https://pubmed.ncbi.nlm.nih.gov/31290228/.
- Smith D, Kannan G, Coppens I, et al. Toxoplasma TgATG9 is critical for autophagy and long-term persistence in tissue cysts. eLife [Internet]. 2021 [cited 2022 Jul 13];10. Available from: https://pubmed.ncbi.nlm.nih.gov/33904393/.
- Field HI, Coulson RMR, Field MC. An automated graphics tool for comparative genomics: the Coulson plot generator. BMC bioinformatics [Internet]. 2013 [cited 2022 Jul 19];14. Available from: https://pubmed.ncbi.nlm.nih.gov/23621955/.