
ABSTRACT
Mitophagy is a central component of the mitochondrial quality control machinery, which is necessary for cellular viability and bioenergetics. The E3 ubiquitin ligase CLEC16A (C-type lectin domain containing 16A) forms a tripartite mitophagy regulatory complex together with the E3 ligase RNF41 (ring finger protein 41) and the ubiquitin-specific peptidase USP8 (ubiquitin specific peptidase 8), yet CLEC16A structural/functional domains relevant for mitophagy are unknown. We identify that CLEC16A contains an internal intrinsically disordered region (IDR), which is important for CLEC16A function and stability. IDRs are flexible domains lacking fixed secondary structure and regulate an emerging number of diverse processes, yet they have been largely unstudied in mitophagy. We observe that the internal CLEC16A IDR is essential for CLEC16A degradation and is bound by RNF41 to promote CLEC16A turnover. This IDR also promotes assembly of the CLEC16A-RNF41-USP8 mitophagy regulatory complex. Thus, our study revealed the importance of IDRs in mitophagy via the regulation of CLEC16A abundance by RNF41, opening new structural insights into mitochondrial quality control.
Mitophagy, the selective degradation of dysfunctional or excess mitochondria by autophagy, is crucial to maintain mitochondrial homeostasis . Impairments in mitophagy contribute to numerous diseases, including neurodegenerative disorders, cardiovascular diseases, and diabetes. The diabetes susceptibility gene CLEC16A (C-type lectin domain containing 16A) encodes an E3 ubiquitin ligase, which regulates PRKN (parkin RBR E3 ubiquitin protein ligase)-mediated mitophagy through interactions with the E3 ligase RNF41 (ring finger protein 41) and deubiquitinating enzyme USP8 (ubiquitin specific peptidase 8). We sought to describe the previously unexplored structural/functional domains within CLEC16A. CLEC16A is predicted to have two intrinsically disordered regions (IDRs), which are domains that lack secondary structure but can regulate protein stability, interactions, and enzymatic function through protein binding and posttranslational modification, leading to a fixed structure. We recently described that CLEC16A contains a C-terminal IDR that stabilizes CLEC16A by inhibiting autoubiquitination. However, the function of this putative internal CLEC16A IDR was unknown.
Utilizing several in silico methods, we identified an internal IDR in CLEC16A that is enriched in polar and charged amino acid residues, i.e., lysines and glutamic acids, which are known to promote intrinsic disorderCitation1. We then applied two complementary biophysical methods, i.e., Citation1H-15N heteronuclear single quantum coherence-NMR spectroscopy and far-UV circular dichroism spectroscopy, which demonstrated a lack of secondary structure and confirmed the presence of an IDR. To understand the function of this internal IDR, we initially investigated its effects on CLEC16A stability. Indeed, we observed that a CLEC16A mutant lacking the internal IDR was more stable than wild-type CLEC16A.
We next sought to determine what features of the internal IDPR are necessary for CLEC16A turnover. We hypothesized that the enrichment of lysine residues could be a site for CLEC16A ubiquitination and subsequent degradation. A CLEC16A mutant bearing lysine-to-arginine (K-to-R) mutations within the internal IDR displayed an increase in protein stability. A similar observation was made after generating a CLEC16A variant in which the amino acid sequence within the internal IDR was randomly shuffled. Interestingly, maintaining the position of lysine residues while shuffling the remainder of the internal IDPR sequence had no significant effect on CLEC16A stability. Taken together, these data suggest that both the enrichment of lysine residues and their position within the IDR are vital features to CLEC16A stability. Notably, the K-to-R internal IDR mutant did not impair CLEC16A autoubiquitination, suggesting other proteins may regulate CLEC16A stability.
To evaluate other regulators of CLEC16A turnover, we initially considered the two CLEC16A binding partners USP8 and RNF41. Previous studies have established that USP8 stabilizes RNF41 by removal of destabilizing ubiquitin linkages, while RNF41 reciprocally induces USP8 ubiquitination and its subsequent proteasomal degradation. Similarly, we have previously found that CLEC16A stabilizes RNF41, leading us to hypothesize that RNF41 participates in a similar reciprocal relationship as with USP8 by promoting CLEC16A clearance. Indeed, we observed that RNF41 gain-of-function induced CLEC16A turnover, and CLEC16A stability was enhanced following RNF41 loss-of-function via overexpression of a dominant negative RNF41 mutant. RNF41 was unable to induce turnover of CLEC16A mutants lacking the internal IDR, carrying a shuffled internal IDR, or possessing K-to-R mutation within the IDR, suggesting that RNF41 may act directly on the internal IDR to ubiquitinate and clear CLEC16A. However, RNF41 loss-of-function did not stabilize CLEC16A to the same extent as CLEC16A mutants lacking the internal IDR, suggesting there are additional proteins that target CLEC16A for turnover.
Assembly of the CLEC16A-RNF41-USP8 mitophagy regulatory complex is vital for optimal mitochondrial quality control. Thus, we next tested the importance for the CLEC16A internal IDR in the formation of this complex. Following deletion or shuffling of the internal IDR, we observed a reduction in CLEC16A binding and ubiquitination of RNF41. Moreover, mutation of the CLEC16A internal IDR reduced binding between RNF41 and USP8. We interrogated whether the loss of RNF41-CLEC16A binding was due to changes in CLEC16A structure or loss of a key RNF41 binding site by individually overexpressing the internal IDR alone. Indeed, we found that the individual IDR was sufficient to bind RNF41. Together, these data suggest that the internal IDR promotes the assembly of the CLEC16A-RNF41-USP8 complex and is a binding site important for the reciprocal control of RNF41 stability.
We identified an internal IDPR within CLEC16A that regulates CLEC16A interaction with RNF41 (). Given the reciprocal control of both CLEC16A and USP8 by RNF41, as well as the known role for RNF41 on PRKN proteasomal degradation, our findings potentially position RNF41 as a central hub for the orchestration of mitophagy. Future studies will be essential to determine the direct impact of RNF41 on the mitochondrial quality control pathway. Further, our work implicates IDRs as crucial domains in the regulation of CLEC16A-mediated mitophagy and adds to previous observations on the importance of IDRs within several proteins in the macroautophagy machinery. The presence of a putative IDR within USP8 adds yet another unstudied domain that may be central in the regulation of the mitochondrial quality control pathway. Several questions related to CLEC16A remain, including how the opposing functions of the internal IDR (important for CLEC16A turnover) and the C-terminal IDR (important for CLEC16A stability) are regulated to not only control CLEC16A protein levels but its ubiquitin ligase activity and protein-protein interactions. These may also beget a closer look at degron motifs within the CLEC16A internal IDR, which may be sites for turnover by RNF41 and other unknown regulators of CLEC16A stability. These questions will be of high interest in the future to determine how CLEC16A stability and function could be harnessed for therapeutic approaches to treat the human diseases associated with CLEC16A.
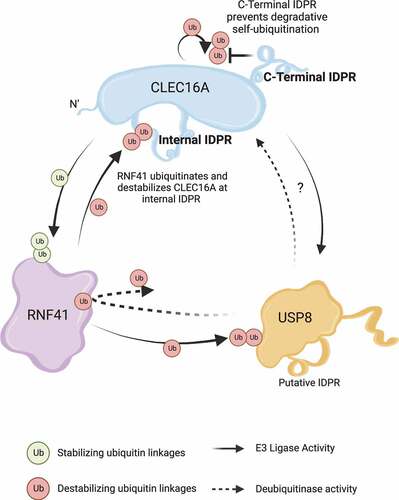
Figure 1. Schema of the contributions of IDRs in the control of CLEC16A-RNF41-USP8 complex assembly. Stability of the CLEC16A-RNF41-USP8 complex is essential for optimal mitophagy. The CLEC16A C-terminal IDR increases CLEC16A stability by preventing degradative self-ubiquitination. CLEC16A stabilizes RNF41 via non-degradative ubiquitination, while RNF41 ubiquitinates and destabilizes CLEC16A by modifying the CLEC16A internal IDR. USP8 and RNF41 also participate in reciprocal cross-regulation. Overexpression of CLEC16A increases USP8 expression, yet the underlying mechanisms of this and the potential importance of the putative USP8 IDR in mitophagy remain unknown.
Abbreviations
ATG3 autophagy-related protein 3
CLEC16A C-type lectin domain containing 16A
IDR intrinsically disordered region
PRKN parkin RBR E3 ubiquitin protein ligase
RNF41 ring finger protein 41
USP8 ubiquitin specific peptidase 8
Disclosures
S.A.S. has received grant funding from Ono Pharmaceutical Co., Ltd. and is a consultant for Novo Nordisk.
Acknowledgements
S.M. acknowledges support from the National Institutes of Health (T32-AI007413). S.A.S. acknowledges funding support from JDRF (COE-2019-861 and SRA-2023-1392), the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (R01-DK108921, R01 DK135268, R01 DK135032, R01 DK136671, U01 DK127747), the Department of Veterans Affairs (I01 BX004444), the Brehm family, the Anthony family, and a Brehm T1D Pilot and Feasibility Grant from the Michigan Diabetes Research Center (P30-DK020572).
Additional information
Funding
Reference
- Gingerich MA, Zhu J, Chai B, Vincent MP, Xie N, Sidarala V, Kotov NA, Sahu D, Klionsky DJ, Schnell S, Soleimanpour SA: Reciprocal regulatory balance within the CLEC16A-RNF41 mitophagy complex depends on an intrinsically disordered protein region. Journal of Biological Chemistry, 2023 Apr;299(4):103057. doi: 10.1016/j.jbc.2023.103057. PMID: 36822331.