
ABSTRACT
Glaucoma encompasses a spectrum of disorders characterized by the chronic degeneration of retinal ganglion cell (RGC) axons and the progressive loss of RGCs, resulting in visual impairment. In this study, we investigated the effect of autophagy deficiency on two glaucoma hypertensive models, the DBA/2J spontaneous glaucoma model, and the TGFβ2 (transforming growth factor β2) chronic ocular hypertensive model. For this, we used the Atg4b−/− and DBA/2J-Atg4b−/− mice, this latter generated in our laboratory via CRISPR/Cas9 technology, which display impaired autophagy. In contrast to littermate WT controls, mice deficient in Atg4B, did not develop glaucomatous elevation of intraocular pressure (IOP). Moreover, autophagy deficiency protected against RGC death and optic nerve atrophy. Collectively, our data supports a pathogenic role of autophagy in the context of ocular hypertension and glaucoma.
Abbreviations: ATG4B: Autophagy-related 4B; BAX: BCL2-associated X protein; BECN1: Beclin 1; BID: BH3 interacting domain death agonist; CASP8: Caspase 8; IOP: Intraocular Pressure; MAP1LC3B: microtubule-associated proteins 1B; ON: Optic Nerve; RGC: Retinal Ganglion Cells; SQSTSM1: Sequestosome 1; TBK1: TANK-binding kinase 1; TGFβ2: Transforming Growth Factor β2; WT: Wild Type.
Glaucoma is a neurodegenerative disease of the eye, characterized by the progressive loss of retinal ganglion cells (RGCs) and optic nerve (ON) atrophy, ultimately leading to peripheral vision loss and irreversible blindness. Glaucoma’s pathogenesis is closely tied to ocular hypertension, which is the primary modifying risk factor for the condition. However, the molecular mechanisms underlying both ocular hypertension and glaucoma remain largely uncharacterized.
Recent studies have emphasized a significant role of autophagy in the development of ocular hypertension and glaucoma, suggesting that dysfunctional autophagy is closely associated with the disease. Additionally, the findings that OPTN (optineurin) and TBK1 (TANK-binding kinase 1) genes, associated with normal tension glaucoma, are involved in several types of selective autophagy, further connects autophagy to glaucoma pathology. Autophagy is induced in RGCs in response to various insults, including transient elevation in intraocular pressure (IOP) and optic nerve crush. However, its role appears to be context-dependent, with studies reporting both protective and detrimental effects depending on the experimental model.
In our latest work (Citation1), we wanted to gain a deeper understanding of the role of autophagy in the context of glaucoma (). To do so, we investigated the impact of autophagy deficiency in the spontaneous murine glaucoma model DBA/2J. These mice naturally develop elevated IOP around 5-6 months of age, followed by RGC and axon loss. We utilized CRISPR technology to delete the expression of the autophagy gene Atg4b (autophagy related-4b), which resulted in the reduced basal autophagy in the outflow pathway and retina, as evidenced by the absence of lipidated microtubule-associated proteins 1B/Map1Lc3b (Lc3-II) and increased levels of Sqstsm1 (sequestosome 1), evaluated in whole lysates from dissected retina and outflow pathway tissues. Surprisingly, DBA/2J-Atg4b−/− mice did not develop glaucomatous elevation in IOP with aging. Moreover, no signs of RGC death or ON atrophy were observed. Evidently, the absence of neurodegeneration in DBA/2J-Atg4b−/− mice could be easily explained by the fact the autophagy-deficient mice remain normotensive, therefore not triggering glaucomatous damage; however, the findings also highlight a very important remark: ATG4B expression is not necessary for RGC survival during development and normal physiology, possibly because of the presence of the other Atg4 isoforms, i.e., Atg4a, Atg4c and Atg4d.
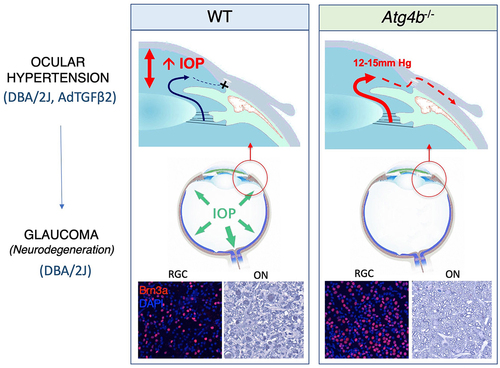
Figure 1. Illustrative summary of the effect of autophagy deficiency in mouse models of ocular hypertension and glaucoma: Elevated IOP leads to glaucoma, characterized by RGC death and ON atrophy. Atg4b deficiency prevented IOP elevation in the DBA/2J and in the TGFβ2-ocular hypertensive mouse models, and rescued RGC survival and ON atrophy, which were evaluated by retinal flat mounts, i.e., Brn3a+ (brain-specific homeobox/POU Domain Protein 3A) cells, and histological cross-sections, respectively.
Further insights into the role of autophagy deficiency in glaucomatous neurodegeneration were gained from DBA/2J-Atg4b+/- mice. In contrast to knockout mice, DBA/2J mice heterozygous for Atg4b did indeed develop ocular hypertension at levels similar to their wild-type (WT) littermates. Interestingly, despite the elevation in IOP, DBA/2J-Atg4b+/- mice exhibited partial survival of RGC and full protection against ON atrophy, compared to WT mice. Some ON enlargement was still present, indicating a degree of stress, but no signs of gliosis were observed. Together, these provide clear evidence supporting a protective role of autophagy deficiency against glaucomatous RGC death.
Our results contrast with some previous studies that reported a protective role of autophagy in RGC cell survival. The discrepancies could be attributed to differences in experimental models, as previous studies often focused on acute models of ocular hypertension or axonal injury that do not fully replicate glaucoma pathogenesis. Additionally, our study uniquely employed genetic manipulation of autophagy rather than pharmacological approaches, which tend to be less specific. In this regard, the findings in this study perfectly align with previous work from our laboratory, obtained with the transgenic DBA/2J:GFP-LC3 mice. In this model, overexpression of the GFP-LC3 transgene further elevated IOP. Moreover, DBA/2J::GFP-LC3 mice showed exacerbated RGC death and ON atrophy. ON atrophy was accompanied by the presence of large autophagic structures within degenerating axons, suggesting a detrimental role of autophagy in glaucomatous neurodegeneration.
The debate continues on whether autophagy directly promotes cell death, referred to as autophagy-dependent cell death, or if cell death is merely associated with or mediated by autophagy. Although experimental validation is still pending, the current prevailing view suggests that the latter is the most plausible mechanism. Evidence from studies on DBA/2J mice with a deficiency of BAX (BCL2-associated X protein) indicates a protective effect against glaucomatous IOP elevation and RGC death, reinforcing the significant role of apoptosis in RGC death associated with glaucoma. While BAX is primarily recognized for its involvement in apoptosis, emerging evidence suggests its potential influence on autophagy under specific circumstances. Cells undergoing BAX-mediated apoptosis also exhibit signs of autophagy, whereas BAX-deficient cells display resistance to autophagic cell death. The precise mechanisms by which BAX can induce autophagy and autophagic cell death remain incompletely understood, but they appear to involve lysosome permeability. Additionally, a cross-regulation between autophagy and apoptosis may involve several other interconnected processes. For instance, caspase-mediated cleavage of BECN1 (Beclin 1) leads to autophagy inhibition. In contrast, autophagic degradation of CASP8 (caspase 8) or the activation of BID (BH3 interacting domain death agonist) by BECN1 prevents apoptosis.
Finally, the discovery that DBA/2J-Atg4b−/− mice did not develop glaucomatous IOP elevation with aging raised the question of whether this phenomenon was specific to DBA/2J or could be applicable to other ocular hypertensive models. To address this, we examined the effect of autophagy deficiency in the TGFβ2 (transforming growth factor β2) ocular hypertensive model. Lentiviral particles expressing constitutively active TGFβ2 were intravitreally injected into the anterior pole of Atg4b−/− mice. The delivery of active TGFβ2 gradually increased IOP, starting at approximately 12 days post-injection and maintaining elevated levels throughout the study. Interestingly, the TGFβ2-induced elevation in IOP was significantly reduced in Atg4b−/− mice compared to their wild-type littermates.
In summary, our findings collectively provide valuable insights into the complex relationship between autophagy and glaucoma, highlighting the context-specific nature of autophagy’s impact on RGC survival and offering potential avenues for future research and therapeutic interventions in glaucoma. As lipidation of LC3 plays a role in various cellular processes, including LC3-associated phagocytosis and LC3-associated endocytosis, it is important to recognize that our findings may also reflect disruptions in these other cellular mechanisms. This consideration is particularly relevant when studying the DBA/2J model, where the release of pigment particles and their uptake by outflow cells is a pivotal aspect of the pathogenic mechanisms.
Disclosure statement
The authors declare that they have no conflict of interest.
Additional information
Funding
Reference
- Dixon A, Shim MS et al. Autophagy deficiency protects against ocular hypertension and neurodegeneration in experimental and spontaneous glaucoma mouse models. Cell Death Dis. 2023; 14(8):554.