
ABSTRACT
The pathological accumulation of the nuclear protein TDP-43 (TAR DNA-binding protein 43 kDa) in the cytoplasm is characteristic of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD-TDP), and its spread through the brain and spinal cord is closely associated with the progression of these two diseases. However, the mechanisms through which the TDP-43 pathology propagates throughout the central nervous system remain unclear. We recently reported the role of (macro)autophagy in the secretion of TDP-43 via extracellular vesicles (EVs). We found that among the autophagy modulators, bafilomycin A1 (Baf) and GRN (granulin precursor) deficiency impair the formation of autolysosomes and promote the secretion of TDP-43 by EVs. TDP-43 loading on EVs involves autophagy-related proteins and the knockdown of TDP-43 augmented Baf-induced EV release. Thus, our results suggest that the loss-of-function of TDP-43 accelerates release of EVs possibly derived from autophagosomes, which may mediate cell-to-cell spread of the TDP-43 pathology.
The nuclear protein TDP-43 regulates exon splicing, gene transcription, and mRNA stability in the nucleus. In the affected brain regions of ALS and FTLD-TDP patients, pathologically aggregated TDP-43 accumulates in the cytoplasm. This transition causes a loss-of-function of TDP-43 in the nucleus and a toxic gain-of-function of TDP-43 in the cytoplasm. Pathological TDP-43 possesses prion-like properties, which can promote conformational changes in the normal form of TDP-43 into its pathological form and thereby accelerate its intracellular accumulation.
The pathological accumulation of TDP-43 initially occurs in specific areas of the brain and spinal cord, and then spreads throughout those tissues leading to a progression of ALS and FTLD-TDP. TDP-43 can be detected in the cerebrospinal fluids (CSFs), and elevated levels have been found in the CSFs from ALS and FTLD-TDP patients. TDP-43 is extracellularly released inside EVs and this secreted form may mediate the cell-to-cell spread of the TDP-43 pathology. However, the mechanisms by which TDP-43 is extracellularly released via EVs have not been elucidated yet.
(Macro)autophagy is a highly conserved intracellular degradative process in which unwanted materials are sequestered within double-membrane vesicles called autophagosomes, which fuse with lysosomes to form autolysosomes and degrade their content. A pathological analysis suggested that autolysosome formation is inhibited in neurons from patients affected by sporadic ALS. Some of causal genes of familial ALS are associated with the autophagy-lysosome system, including OPTN (optineurin) and TBK1 (TANK binding kinase 1). Along this line, we previously demonstrated that GRN (granulin precursor), the insufficiency of which causes FTLD-TDP, positively regulates autophagy and lysosome functions. These observations suggest that the formation of autolysosomes is suppressed in ALS and FTLD-TDP patients. Moreover, aggregated TDP-43 is degraded within the autophagy-lysosome system. Since the prevention of autolysosome formation increases the secretion of EVs containing autophagic components and autophagy cargo receptors, we hypothesized that the inhibition of autolysosome formation may result in the accumulation of TDP-43 within autophagosomes, which leads to their extracellular release via EVs.
To investigate the role of the autophagy-lysosome system in the secretion of TDP-43 via EVs, we examined the effects of autophagic modulators on TDP-43 levels in an EV-enriched fraction prepared by the sequential centrifugation of the conditioned medium from cultured cells [Citation1]. Among autophagic modulators, those affecting autolysosome formation such Baf, an inhibitor of the vacuolar H+-ATPase, and GRN knockdown/knockout increased the levels of TDP-43, the autophagosome marker protein LC3-II (microtubule-associated proteins 1A/1B light chain 3B-II) and the multivesicular body (MVB) marker protein TSG101 (tumor susceptibility 101) in the EV-enriched fraction. In contrast, autophagy inducers such as MG132, rapamycin and serum starvation, slightly decreased TDP-43 levels in the same EV-enriched fraction. However, vacuolin-1, an inhibitor of PIKFYVE (phosphoinositide kinase, FYVE-type zinc finger containing), and the knockdown of STX17 (syntaxin 17), a SNARE (soluble NSF attachment protein receptor) protein regulating autophagosome-lysosome fusion, suppressed the autolysosome formation but failed to induce TDP-43 secretion by EVs, suggesting that the inhibition of autolysosome formation is required but not sufficient for TDP-43 extracellular release via EVs. Treatments that increased TDP-43 in the EV-enriched fraction also promoted the nuclear translocation of TFEB (transcription factor EB), a master transcriptional regulator of lysosomal biogenesis and autophagy. We previously suggested that lysosomal dysfunction resulting from GRN knockout increased the nuclear translocation of TFEB. Thus, lysosomal dysfunction may be necessary in addition to the suppression of autolysosome formation for TDP-43 secretion via EVs.
Baf-induced TDP-43 secretion via EVs was suppressed in ATG16L1 (autophagy-related 16 like 1)-deficient autophagy-null cells, indicating that an intact autophagy machinery is required for this extracellular release of TDP-43. Moreover, the knockdown of GRN increased TDP-43 and TSG101 levels in the EV-enriched fraction from the culture medium of wild-type cells, but not of ATG16L1-deficient cells (TSG101 levels were unaltered), suggesting that autophagy is a critical mechanism for loading TDP-43 on EV. LC3-dependent extracellular vesicle loading and secretion (LDELS), secretory autophagosomes, and amphisome could be mechanism mediate TDP-43 secretion via EVs. Since TDP-43 secretion is accompanied with increase of TSG101 and LC3-II, amphisome, a hybrid organelle formed by the fusion between autophagosome and MVBs, may mediate the TDP-43 secretion via EVs.
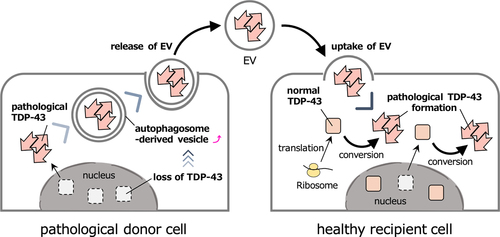
Our study suggested that the suppression of autophagosome-lysosome fusion accompanied by lysosomal dysfunction increases the release of TDP-43 via EVs, possibly derived from autophagosomes. The knockdown of TDP-43 accelerated this process when induced by Baf, suggesting that a loss-of-function of TDP-43 promoted this EV transport system. Pathological TDP-43 converts newly translated or nuclear-exported wild-type TDP-43 into its pathological form in the cytoplasm, in a prion-like manner. This conversion leads to a decrease in functional TDP-43 and the loss-of-function of TDP-43 in the nucleus induces the extracellular release of the pathological TDP-43 via EVs. These pathological TDP-43-containing EVs are then taken up by healthy cells and the pathological TDP-43 released into the cytoplasm of the recipient cells triggering the de novo pathogenesis and spreading of the disease (). The mechanisms by which TDP-43 is loaded on the EVs or the molecules that mediate this EV transport system currently remain unknown. Further studies are required to unveil the mechanism of extracellular TDP-43 release, which may provide new therapeutic targets for the treatment of ALS and FTLD-TDP.
Figure 1. Hypothetical model of the cell-to-cell spread of pathological TDP-43 via EV.
Abbreviations
| ATG16L1: | = | autophagy-related 16 like 1; |
| Baf: | = | bafilomycin A1; |
| CSFs: | = | cerebrospinal fluids; |
| EVs: | = | extracellular vesicles; |
| LC3-II: | = | microtubule-associated proteins 1A/1B light chain 3B-II; |
| MVB: | = | multivesicular body; |
| GRN: | = | granulin precursor; |
| LDELS: | = | LC3-dependent extracellular vesicle loading and secretion; |
| PIKfyve: | = | phosphoinositide kinase, FYVE-type zinc finger containing; |
| OPTN: | = | optineurin; |
| SNARE: | = | soluble NSF attachment protein receptor; |
| TBK1: | = | TANK binding kinase 1; |
| TFEB: | = | transcription factor EB; |
| TDP-43: | = | TAR DNA-binding protein 43 kDa; |
| TSG101: | = | tumor susceptibility 101 |
Disclosure statement
There are no potential conflicts of interest with the content of this article
Additional information
Funding
Reference
- Tanaka Y, Ito SI, Honma Y, Hasegawa M, Kametani F, Suzuki G, Kozuma L, Takeya K, Eto M. Dysregulation of the Progranulin-driven Autophagy-lysosomal Pathway Mediates Secretion of the Nuclear Protein TDP-43. J Biol Chem. 2023. 105272.