
ABSTRACT
Heart failure, a leading driver of global mortality, remains a topic of intense contemporary research interest due to the prevailing unmet need in cardiometabolic therapeutics. Numerous mechanisms with the potential to influence the onset and development of heart failure remain incompletely understood. Firstly, myocardial autophagy, which involves lysosomal degradation of damaged cellular components, confers context-dependent beneficial and detrimental effects. Secondly, sterile inflammation may arise following cardiac stress and exacerbate the progression of heart failure. Inflammation changes in a temporal manner and its onset must be adequately resolved to limit progression of heart failure. Mitochondria are an important factor in contributing to sterile inflammation by releasing damage associated molecular patterns (DAMPs) including mitochondrial DNA (mtDNA). Accordingly, this is one reason why the selective autophagy of mitochondria to maintain optimal function is important in determining cardiac function. In this review, we examine the increasing evidence suggesting crosstalk between autophagy and sterile inflammation together with their role in the development of heart failure. In particular, this is exemplified in the preclinical models of ischaemia/reperfusion injury and pressure overload induced heart failure. We also highlight potential therapeutic approaches focusing on autophagy and addressing sterile inflammation, aiming to enhance outcomes in heart failure.
Introduction
Heart failure is a progressive clinical event with an estimated global prevalence of 64.3 million annual cases [Citation1]. It occurs due to insufficient systemic circulation following structural or functional alterations in the ventricle or vasculature [Citation2]. Associated risk factors of heart failure include obesity, hypertension, diabetes, and atherosclerotic disease [Citation2]. Heart failure may elicit several compensatory mechanisms, including renin-angiotensin system activation, adrenergic nervous system activation, and cytokine production in order to compensate for decreased cardiac function [Citation3]. Ischaemia/reperfusion injury, elicited by coronary artery ligation, and pressure overload-induced damage, induced by aortic constriction, are two well-established preclinical models used to study the development of heart failure. While necessary to resolve cardiac ischaemia, reperfusion can increase production of reactive oxygen species (ROS), subsequent cell death and inflammation [Citation4]. Pressure overload-induced damage is characterised by myocardial remodelling such as hypertrophy and fibrosis [Citation5]. Here we review current knowledge on the alterations in autophagy and sterile inflammation in these models, and their impact on the development of heart failure.
Autophagy is a bulk degradation process involving the clearance of damaged proteins and organelles in response to stressors [Citation6]. Although autophagy is thought to be a beneficial protective response, it also elicits detrimental cardiac effects [Citation7]. As new evidence emerges, the detrimental effects of both increased and reduced autophagy in a context-specific manner are becoming apparent. Several recent studies have indicated that the degree of autophagy changes in a failing heart in direct response to ischaemia/reperfusion injury or pressure overload [Citation8-11]. Additionally, it is important to note that during the course of heart failure progression, temporal changes in autophagy occur [Citation12], which is not yet fully understood. Thus, understanding temporal changes in autophagy using pre-clinical models may aid in identifying the optimal time for therapeutic intervention in cardiac diseases.
The innate immune system, comprised of cell types such as neutrophils, macrophages, mast cells and eosinophils, contributes to myocardial inflammation and metabolic dysfunction [Citation13-16]. Sterile inflammation can be characterised as inflammation in the absence of pathogens and their products, resulting from acute conditions such as ischaemia/reperfusion injury, chemical injury and trauma [Citation17,Citation18]. During the onset of sterile inflammation, damage-associated molecular patterns (DAMPs), endogenous molecules released after tissue injury or cell death, are recognised by pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) and cytoplasmic Nod-like receptors (NLRs) [Citation18,Citation19]. DAMPs are also recognised by non-PRRs, such as the receptor for advanced glycation end products (RAGE), CD44, integrins, and CD91, which activate pro-inflammatory pathways [Citation18,Citation19]. Although the inflammatory response promotes cell debris clearance and wound healing, excessive inflammation may lead to additional tissue damage [Citation20]. Therefore, resolution of inflammation ensures the clearance of dead cells from injury site can occur without adverse consequences of prolonged inflammation [Citation21]. While most experimental studies have focused on mechanisms regulating the onset of sterile inflammation, recent studies have highlighted the resolution of inflammation as an underappreciated component of the innate immune response [Citation22,Citation23]. Therefore, it is imperative to identify both the pathways that initiate sterile inflammation as well as the mechanisms through which the inflammatory response resolves.
The key roles of both autophagy and sterile inflammation in the development of heart failure are well known. However, the interplay between autophagy and inflammation has not yet been extensively and cohesively summarised. In this review, the mechanisms via which autophagy and sterile inflammation crosstalk will be examined. Furthermore, their significance in ischaemia/reperfusion injury and pressure overload-induced damage will be discussed, together with ensuing opportunities for therapeutic intervention.
Autophagy molecular mechanisms
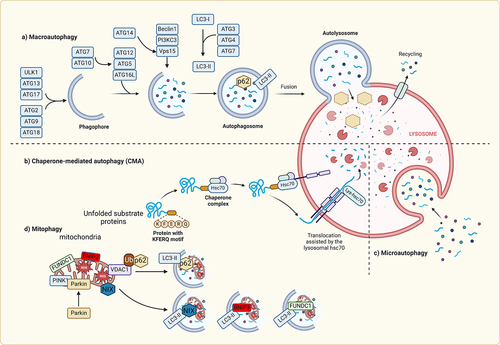
Autophagy primarily functions as a catabolic process aimed at eliminating intracellular components, including dysfunctional organelles and protein aggregates, by proteolytic degradation within lysosomes [Citation24]. To date, at least three forms of autophagy; namely chaperone-mediated autophagy, microautophagy, and macroautophagy have been identified [Citation25,Citation26] They differ in physiological function and mode of cargo delivery to the lysosome [Citation27]. Within the context of chaperone-mediated autophagy, the lysosome-associated membrane protein 2A (LAMP2A) plays a crucial role by facilitating the recruitment of the chaperone HSC70 into lysosomes. HSC70 selectively identifies specific cargoes through the recognition of KFERQ-like motifs [Citation25]. In contrast, microautophagy involves the engulfment of cytosolic components within vesicles that are created through the invagination of lysosomal membranes [Citation28]. Lastly, macroautophagy encompasses a series of intracellular membrane rearrangements to encapsulate cargoes within double-membraned vesicles known as autophagosomes. These autophagosomes then undergo fusion with lysosomes, leading to the degradation of their contents [Citation27]. Extensive research has extensively investigated the significant role of macroautophagy, typically referred to simply as autophagy, in the regulation of cardiac function. Therefore, this review will use the term autophagy to refer to macroautophagy, as it is commonly referred to in the literature [Citation29].
The activation of autophagy is a precisely controlled process that relies on the transcriptional, post-transcriptional, and post-translational regulation of various autophagy related genes (ATGs) and associated proteins [Citation30-32]. The activation of autophagy is a precisely controlled process that relies on the transcriptional, post-transcriptional, and post-translational regulation of various autophagy related genes (ATGs) and associated proteins. Autophagy is initiated by the ULK1 (UNC-51-like kinase 1) complex, which can be activated by several signalling pathways stimulated by nutrient starvation or other stressors [Citation33]. Upon activation, ULK1 binds to and phosphorylates multiple downstream autophagy proteins including multiple components of the VPS34 class III PI3K complex (AMBRA1, ATG14, VPS34, and BECLIN1) [Citation33]. ULK1 activation of the class III PI3K (PI3KC3) complex stimulates phosphatidylinositol-3-phosphate (PI3P) on phagophores, which act to recruit autophagy-related proteins responsible for phagophore expansion. The elongation phase of autophagosome formation depends on the involvement of ATG8 family members, such as LC3 and GABARAP. Initially, these members are cleaved by ATG4 and subsequently activated by ATG7. Finally, they become anchored to the phagophore membranes through conjugation to phosphatidylethanolamine, a process facilitated by ATG3. The maturation of the phagophore to form an autophagosome also requires the participation of ATG8 [Citation34]. Additionally, the process involves the contribution of the small GTPase Rab5 and other components of the endosomal sorting complexes required for transport [Citation35]. After formation, the molecular machinery required for its fusion with lysosomes involves complexes of SNARE proteins [Citation36]. During the autophagosome-lysosome fusion process, various proteins, including small GTPases like Rab7, are involved in the trafficking of autophagosomes to lysosomes [Citation37]. Autolysosomes facilitate digestion of autophagosome cargo via lysosomal hydrolases to yield macromolecules such as amino acids for re-use [Citation38,Citation39].
In the past decade, our understanding of the mechanisms involved in cargo engulfment within phagophores and autophagosomes has significantly advanced [Citation40,Citation41]. Initially viewed as a bulk recycling process, the discovery of selective targeting via chaperone-mediated autophagy prompted further investigation into the mechanisms of cargo selection [Citation25]. Nowadays, all forms of autophagy are recognised to possess selectivity, which is achieved through the binding of ATG8 proteins, such as LC3, to cargo receptors like SQSTM1 (p62), NBR1, and NIX (BNIP3L) [Citation42]. The proximity of these components to the targeted cargo facilitates the elongation of the phagophore around the cargo and its subsequent incorporation into nascent autophagosomes. Cargo receptors interact with conjugated LC3 through a distinctive WXXL motif called the LC3-interacting region which is conserved across evolution [Citation43]. These receptors enable the physical interaction between the cargo and the growing phagophore during elongation by recognising specific self-digestion signals, either dependent or independent of ubiquitination [Citation42].
Mitophagy is the process of selective degradation of dysfunctional mitochondria [Citation44]. While it utilises some of the same cellular machinery, this process can occur somewhat independently of canonical autophagy [Citation45]. Mitophagy can be classified into either PINK1/Parkin-dependent, or receptor mediated. The most well characterised form of mitophagy occurs through the PINK1/Parkin pathway. Under physiological conditions, PINK1 is imported into the mitochondria by the TOM complex for cleavage by PARL, followed by degradation. Under conditions of stress, mitochondria lose their membrane potential, PINK1 stabilises on the outer membrane and signals for the recruitment of the E3 ligase Parkin from the cytosol. Parkin ubiquitinates several proteins, some of which include MFN1, MFN2, and MIRO. These ubiquitinated proteins are then recognised by cargo receptors such as optineurin, NDP52, and SQSTM1 [Citation46].
Alternative mitophagy is mediated via a distinct pathway independent from conventional autophagy or mitophagy and appears to play an important role in various models of heart failure [Citation47]. Indeed, mitophagy mediated by a Ulk1, Rab9 and Drp1 complex conferred cardioprotective effects during ischaemia [Citation48]. Loss of this alternative mechanism for induction of mitophagy through knock in of inactive Rab9 S179A mutant worsened ischaemic injury. An Atg7-independent form of mitophagy has also been shown to be critical in mediating the degradation of damaged cardiac mitochondria in a mouse model of PO [Citation11]. Following transverse aortic constriction (TAC), conventional Atg7-dependent mitophagy was transiently activated but quickly returned to baseline within 1 day. However, 3 days after TAC, there is a more robust induction of mitophagy which was mediated by a Rab9/Ulk/Drp1 axis and sustained longer. In addition, knockout of Ulk1 in the heart abrogated induction of alternative mitophagy, worsened TAC-induced cardiac dysfunction, whilst not affecting conventional autophagy. Thus, whereas the functional significance of conventional Atg5- and Atg7-dependent autophagy has been extensively studied in heart failure models, the studies cited here highlight that alternative mitophagy is perhaps an underappreciated contributor to cardioprotection during the progression of heart failure.
Receptor-mediated mitophagy is another important, though less well-characterised pathway to clear damaged mitochondria. The 3 receptors identified to date include BNIP3, NIX/BNIP3L, and FUNDC1 which contain LCR interaction regions (LIR) to directly bind to and recruit autophagosomes [Citation49]. BNIP3 and NIX have gained attention for studies and their role in promoting cell death. They also promote autophagy by disrupting BCL2 and BECLIN1 interactions, which allows BECLIN1 to form complexes with autophagy proteins to form a phagophore. FUNDC1 is regulated by phosphorylation where its phosphorylation is inhibitory for mitophagy. Dephosphorylation by PGAM5 allows it to bind to LC3 for mitophagy. However, unlike PINK1/PARKIN pathway, the regulatory mechanisms are not well characterised. Other examples of selective autophagy include reticulophagy, lysophagy, nucleophagy, lipophagy, pexophagy [Citation42]. Additionally, while organelles and proteins are commonly targeted, autophagy can also affect RNA by influencing RNA-binding proteins or through direct interaction of LC3 with RNA () [Citation50]. Evidently, processes related to autophagy are strategically positioned to exert a broad impact on cellular function.
Figure 4. Molecular mechanisms of autophagy In macroautophagy, the cargo is sequestered within a unique double membrane cytosolic vesicle, an autophagosome. The autophagosome itself is formed by expansion of the phagophore. The autophagosome undergoes fusion with a late endosome or lysosome to form an autolysosome, in which the sequestered material is degraded. Microautophagy refers to the sequestration of cytosolic components directly by lysosomes through invaginations within their limiting membrane. Chaperone-mediated autophagy involves direct translocation of unfolded substrate proteins (KFERQ-like motif) across the lysosome membrane through the action of a cytosolic and lysosomal chaperone heat shock cognate protein of 70 kDa (HSC70), and the integral membrane receptor lysosome-associated membrane protein type 2A (LAMP-2A). Mitochondria can be removed by receptor mediated mitophagy using NIX, BNIP3, or FUNDC1, which interact with LC3; or via PTEN-induced putative kinase 1 (Pink1) and Parkin-mediated ubiquitination (Ub) of voltage-dependent anion channel 1 VDAC1), recognized by the adapter p62 for removal of stressed.
Role of autophagy in preclinical models and human heart failure
Autophagy is heavily implicated in optimal cardiac function as an important endogenous anti-stress response, yet excessive autophagy can be detrimental and pathological [Citation51-53]. The degradation of misfolded or damaged proteins and organelles via autophagy is especially important for cells with low regenerative capacity, such as cardiomyocytes, to maintain cellular homoeostasis [Citation54,Citation55]. Autophagy is widely associated with different cardiac pathologies [Citation29,Citation56]. Both basal and stress-induced autophagy exhibit cardioprotective and anti-apoptotic effects, particularly in the context of heart failure [Citation7,Citation57]. Specifically, mitophagy has been reported to have beneficial effects on outcome in various preclinical models [Citation58,Citation59]. Consistent with the cardioprotective effects of autophagy, it was reported that the inhibition of autophagy elicits detrimental effects in conditions of acute fasting and nutrient withdrawal where autophagy is normally required [Citation52,Citation60,Citation61]. Numerous studies have demonstrated alterations in autophagy in the context of failing hearts [Citation52,Citation62]. Although basal autophagy levels are important for the maintenance of cardiac function, temporal profile of changes or the magnitude of changes in autophagy has been reported to have detrimental outcomes [Citation9,Citation63]. Therefore, the existing evidence underscores the involvement of disrupted autophagy in cardiac disease, yet further research is necessary to comprehensively elucidate the precise circumstances and mechanisms underlying its significance [Citation52,Citation64].
Autophagy in ischaemia and reperfusion injury
The role of autophagy during ischaemia/reperfusion has been under study for more than a decade. Upon reperfusion, ischaemic tissue is replenished with nutrients and oxygen. However, this restoration process is accompanied by a significant accumulation of reactive oxygen species (ROS), contributing to progressive damage to the myocardium. During ischaemia, autophagy is activated as an adaptive response to nutrient and oxygen deprivation [Citation65]. However, the influence of autophagy during reperfusion varies based on the specific experimental model and the type of autophagy activated. In short, the stimulation of autophagy through BECN1 or AMPK-mTOR pathways can lead to either favourable or unfavourable effects, respectively [Citation66,Citation67]. In the ischaemic phase, the activation of autophagy is instigated by the AMPK pathway’s activation and the inhibition of the Rheb/mTORC1 pathway. Disruption of these mechanisms hampers the activation of autophagy and exacerbates myocardial injury [Citation61,Citation66]. Autophagic activation and cardioprotective effects during ischaemia are also influenced by NOX4 through the activation of ATF4 [Citation68]. The activation of NOX4 and autophagy under conditions of energy deprivation are believed to be negatively regulated by the tyrosine kinase FYN [Citation69]. In the pig heart, it was shown that autophagy can act to mitigate the extent of apoptosis in ischaemic regions [Citation70]. Consequently, it was proposed that interventions aimed at promoting autophagy may confer protection during myocardial ischaemia.
Interestingly, autophagy can be activated during reperfusion independent of the AMPK pathway. Beclin 1, a key regulator of autophagy, is significantly upregulated in the heart during reperfusion in a ROS-dependent manner [Citation66]. Studies using mice with systemic heterozygous deletions of Beclin 1 demonstrated a significant reduction in both autophagy and injury, indicating that the ROS-dependent upregulation of autophagy mediated by Beclin 1 during reperfusion was detrimental [Citation66]. Recent discoveries suggest that the combined administration of PT1, an AMPK-specific activator, and 3HOI-BA-01, a potent mTOR inhibitor, effectively reduces cardiomyocyte death caused by simulated ischaemia/reperfusion. In murine models of ischaemia/reperfusion injury, the introduction of PT1 or 3HOI-BA-01 stimulates autophagy and leads to a decrease in the size of infarct [Citation71]. Likewise, targeted disruption of autophagy in cardiomyocytes, achieved through the conditional knockout of ATG7, leads to myofibrillar disarray, severe contractile dysfunction, and worsens ischaemia/reperfusion injury. This disruption is accompanied by cardiac hypertrophy and extensive cardiac fibrosis [Citation72]. Autophagic inhibitors such as 3-methyladenine and bafilomycin A1 increase myocardial damage in ischaemia/reperfusion [Citation73]. Simvastatin, which is known to be effectively transported into cardiomyocytes, induced autophagy via inhibition of RAC1 GTPase and mTORC1 signalling [Citation74]. This suggests the well-established therapeutic effect of simvastatin may partially be attributed to its ability to induce autophagy. In the context of ischaemia/reperfusion, autosis, a type of cell death triggered by prolonged and excessive autophagic activation, can occur in cardiomyocytes [Citation75]. Autosis is associated with increased RUBCN expression, attenuation of autophagic flux with accumulation of autophagosomes. Moreover, the inhibition of excessive ischaemia/reperfusion induced autophagy by trimetazidine provides protection against ischaemia/reperfusion induced heart failure in rat hearts [Citation76].
The role of autophagy during ischaemia/reperfusion may vary depending on the specific cargo being processed. Mitophagy, unlike general autophagy, seems to have an adverse effect on the heart during ischaemia/reperfusion. Mice with a specific deletion of MFN1 and MFN2 in cardiomyocytes were protected from cardiac injury [Citation77]. Additionally, the use of the inhibitor Mdivi-1, which interferes with mitochondrial fission and can suppress mitophagy, limited the size of myocardial infarction in mice undergoing coronary artery occlusion and reperfusion [Citation78]. Similarly, mice lacking Bnip3 exhibited reduced myocardial damage and maintained cardiac function during ischaemia/reperfusion, primarily due to the role of Bnip3 in apoptotic cell death [Citation79] There is also further evidence suggesting mitophagy can be protective against cardiac ischaemia/reperfusion injury wherein casein kinase 2α (CK2α) was upregulated in a model of cardiac IR injury which contributed to mitochondrial damage and cardiac dysfunction [Citation80]. Cardiac-specific knockout of CK2α yielded protection against cardiac IR injury. Mechanistically, CK2α was found to phosphorylate (and inactivate) FUNDC1 which compromised mitophagy leading to mitochondrial genome instability, cardiolipin oxidation, oxidative stress, and apoptosis. Furthermore, FUNDC1-dependent mitophagy is now known to be a crucial component underlying the protective effects of ischaemic preconditioning [Citation81]. Therefore, FUNDC1 plays a protective role by promoting cell survival via upregulation of mitophagy. Whilst distinct mitophagy receptors may exert different effects, FUNDC1 in particular may play a crucial role in initiating “protective” mitophagy.
Autophagy in pressure overload
Left ventricular pressure overload caused by aortic constriction is a commonly used preclinical model of heart failure [Citation82]. It is now known that the heart undergoes a gradual metabolic remodelling process following cardiac injury induced by pressure overload [Citation83]. This remodelling is characterised by hypertrophy, fibrosis, and insulin resistance with metabolic dysfunction, including reduced GLUT4-mediated glucose uptake and decreased mitochondrial oxidative capacity without a compensatory increase in glycolysis [Citation83]. These changes in substrate utilisation result in inadequate ATP production to meet the heightened metabolic demand, ultimately leading to the development of heart failure [Citation84].
Numerous studies have examined temporal changes in autophagy after pressure overload-induced injury. In some scenarios, inhibition of autophagy can have negative effects during pathological pressure overload. For example, when mice with a cardiac-specific deletion of ATG5 were subjected to transverse aortic constriction (TAC), cardiac dysfunction was exacerbated and this was accompanied by a notable build-up of misfolded proteins, impaired mitochondrial function and damage to sarcomeres [Citation57]. However, some evidence has also implicated the inhibition of autophagy as a protective effect against pressure overload. Heterozygous disruption of Beclin-1 caused a reduction in cardiac hypertrophy and dysfunction [Citation85]. The pharmacological inhibition of autophagy in mice subjected to pressure overload, achieved through the use of trichostatin A (a histone deacetylase inhibitor) and dimethyl α-ketoglutarate (a cytosolic acetyl-CoA precursor), resulted in reduced cardiac hypertrophy and the reversal of cardiac dysfunction [Citation85]. Therefore, autophagy may play a dual role following pressure overload-induced injury and the precise role likely depends on factors such as severity of pressure overload, duration of injury, and timing of therapeutic intervention [Citation86]. Therefore, it is important to investigate the implications of autophagy activation over the course of pressure overload-induced injury.
Numerous studies have focused on the role of mitophagy, with focus on mediators such as such as ULK1-dependent and ALDH2-dependent Nrf1-FUNC1 signalling [Citation87,Citation88]. It was observed that mitophagy is only transiently activated during the acute phase of pressure overload-induced injury and subsequently becomes inactivated [Citation89]. The latter phase with inactivation of mitophagy correlated with mitochondrial and cardiac dysfunction. Notably, the use of an autophagic inducer, TAT-beclin-1, has shown that mitophagic reactivation improves cardiac function following pressure overload [Citation11]. Similarly, activation of mitophagy using the antioxidant alpha-lipoic acid was protective against pressure overload-induced cardiac dysfunction [Citation88]. Mechanistically, alpha lipoic acid activated ALDH2 resulting in upregulation of Nrf1 and FUNDC1, thus promoting mitophagy. These findings suggest that mitophagy serves as an adaptive process preserving mitochondrial function during pathological hypertrophy. However, the mechanism by which mitophagy is deactivated following transient activation during pressure overload remains unknown.
Autophagy in myocardial infarction
The blockage, whether temporary or permanent, of a coronary artery poses a potentially life-threatening shortfall in delivering oxygen and nutrients to heart muscle cells [Citation90]. This ischaemic condition is typically associated with dysfunctional mitochondria and an overproduction of reactive oxygen species [Citation91]. In such circumstances, proficient autophagic responses empower heart muscle cells to cope with nutritional stress effectively
As mentioned earlier, autophagy plays a pivotal role in preserving cellular function and homoeostasis by breaking down long-lived proteins and damaged organelles, thereby averting the build-up of protein aggregates to cytotoxic levels. Baseline autophagy and the inducible enhancement of autophagy serve protective and mitigative functions in the context of ischaemic injury. The adaptively induced autophagy has been discovered to diminish aggregate formation in the heart, thereby alleviating the adverse effects associated with protein aggregation [Citation92]. Additionally, studies have demonstrated that autophagy induction can decrease the infarct size in acute myocardial infarction rat models [Citation73,Citation93]. Conversely, inhibition of autophagy using bafilomycin A1 was shown to significantly increase infarct size in animal models of acute myocardial infarction [Citation73]. G protein-coupled receptor kinase (GRK4) has been reported to increase cardiomyocyte injury during myocardial infarction by inhibiting autophagy and promoting cardiomyocyte apoptosis via HDAC4 phosphorylation and a decrease in Beclin 1 expression [Citation94]. Genetic inhibition of the AMPK signalling pathway resulted in dysfunctional autophagy and increased infarct size in acute myocardial infarction [Citation61]. Furthermore, studies by Wu et al. showed that upregulation of autophagy flux protects cardiomyocytes against ischaemia and mitigates adverse cardiac remodelling after acute myocardial infarction in rat models [Citation95,Citation96]. Conversely, administration of the autophagy inhibitor 3-MA was found to contribute to adverse cardiac remodelling through induction of nuclear factor-κB (NF-κB) activation in animal models of acute myocardial infarction [Citation95]. Collectively, these and other studies highlight autophagy’s cardioprotective effect against ischaemic injury in various rodent models [Citation97-100].
In the course of myocardial infarction, a cascade of multi-component consequences ensues, encompassing intracellular acidification, mitochondrial Ca2+ overload, aberrant metabolism, and the opening of the mitochondrial permeability transition pore complex (mPTP). Collectively, these factors contribute to mitochondrial dysfunction and elevated levels of reactive oxygen species (ROS), ultimately culminating in myocardial cell death and injury. Much of the damage inflicted on the myocardium proves irreversible, presenting challenges in achieving complete or partial recovery, and exacerbating mitochondrial dysfunction and heightened ROS levels, further contributing to myocardial cell death and injury [Citation101,Citation102]. Therefore, effective mitophagy plays a pivotal role in promoting cell survival in the myocardium during ischaemic conditions.
The activation of BNIP3 or BNIP3L/NIX is extensively documented to induce either autophagy or apoptosis, depending on the prevailing stress conditions. The expression of these proteins escalates during hypoxia, cardiac hypertrophy, or ischaemia [Citation103]. Notably, cardiac overexpression of BNIP3L/NIX leads to lethal cardiomyopathy characterised by high levels of apoptosis, while adulthood overexpression exacerbates cardiac dysfunction post-myocardial infarction. Inhibition of both BNIP3 and BNIP3L/NIX under basal conditions worsens both left ventricular and mitochondrial dysfunction, suggesting a synergistic cardioprotective effect [Citation104]. Interestingly, inhibition of BNIP3 or BNIP3L/NIX prior to myocardial infarction is mainly cardioprotective by reducing apoptosis [Citation104]. Permanent coronary artery ligation-induced myocardial infarction results in severe cardiac dysfunction, mitochondrial dysfunction, fibrosis and apoptosis after 4 weeks, all of which were reduced in mice lacking p53, and this was associated with increased BNIP3-mediated mitophagy [Citation105]. A previous study demonstrated that 45 minutes of ischaemia significantly diminishes the inhibitory phosphorylation of FUNDC1 at tyrosine 18 by Src kinase, consequently activating mitophagy [Citation106]. As noted elsewhere in this review, platelet activation plays a crucial role in acute myocardial infarction by releasing platelet-derived mediators that worsen tissue injury [Citation107]. The decrease in oxygen levels caused by ischaemia in platelets leads to excessive FUNDC1-mediated mitophagy by reducing phosphorylated FUNDC1 at tyrosine 18, which enhances the interaction between FUNDC1 and LC3 and activates mitophagy [Citation107]. Genetic deletion of FUNDC1 impairs mitochondrial quality, increases mitochondrial mass, and renders platelets insensitive to ischaemia [Citation107]. It has been reported that myocardial infarction induces mitophagy specifically in the infarct border zone of rats for up to 48 hours post-myocardial infarction, as evidenced by increased autophagosomes containing mitochondria, elevated PINK-1 expression, parkin expression, and its translocation to mitochondria [Citation108]. Activation of OPA-1 with irisin enhances PINK1/parkin-mediated mitophagy and reverses ischaemia-induced cardiac dysfunctions [Citation109]. Animal studies demonstrate that mice lacking parkin (parkin-/-) and subjected to permanent coronary ligation exhibit increased sensitivity to myocardial infarction, with higher mortality rates compared to wild type mice [Citation110]. The observed accumulation of dysfunctional mitochondria in the infarct border zone of parkin-/- mice following myocardial infarction confirms that the deletion of parkin hinders mitophagy and this correlated with enhanced cardiac dysfunction [Citation110].
Autophagy in Diabetic cardiomyopathy
Altered autophagy has been documented in animal models of diabetes [Citation111]. In the hearts of mice with T2DM, increased abundance of lipid droplets and immature autophagosomes were observed to occur over time [Citation111]. Researchers have explored the influence of resveratrol, a recognised enhancer of autophagy, on diastolic dysfunction in the hearts of diabetic mice. Notably, resveratrol exhibited positive effects in alleviating diastolic dysfunction in type 2 diabetic mouse hearts. Conversely, in type 1 diabetic mouse hearts, resveratrol had a contrasting impact, highlighting the context-dependent nature of its potential as a therapeutic target for diabetic cardiomyopathy [Citation111]. Supporting the idea that insulin has an inhibitory effect on autophagy, it was recently discovered that there was an excessive activation of autophagy in a mouse model of insulin-deficient late-stage diabetic cardiomyopathy [Citation112]. In these mice, left ventricular dysfunction, adverse remodelling, fibrosis, and myocyte apoptosis were observed. Diabetes, correlated with hypoadiponectinemia, led to impaired autophagic flux. Wang et al. discovered that the arrest of autophagic flux was linked to exacerbated injury during myocardial ischaemia/reperfusion. The subsequent restoration of autophagic flux through adiponectin receptor activation proved protective against IR-induced cardiac dysfunction in the diabetic state [Citation113].
The majority of energy produced for utilisation in the heart is derived from mitochondrial fatty acid oxidation [Citation114]. In diabetic hearts, there is a decrease in glucose utilisation, leading to a further increased reliance on fatty acids as the primary energy source. This can be associated with elevated levels of oxidative stress and the potential for mitochondrial dysfunction [Citation115]. As a result, damaged mitochondria can release ROS and factors that induce cell death, thereby contributing to cardiac myocyte injury [Citation116,Citation117]. There is substantial evidence suggesting that mitochondrial dysfunction plays a central role in the cell death induced by diabetic cardiomyopathy [Citation118]. In the context of type 1 diabetes mellitus (T1DM) mice, the levels of various crucial components involved in autophagy, such as LC3, ATG5, and ATG12, are reduced in cardiac tissue [Citation119]. Moreover, the levels of both PINK1 and Parkin proteins are diminished. These observations indicate specific alterations in mitophagy signalling in T1DM models [Citation119,Citation120]. These findings suggest that despite the presence of extensive mitochondrial dysfunction, specific mitophagic signalling is reduced in diabetic cardiomyopathy. This leads to the accumulation of dysfunctional mitochondria at the cardiac level, exacerbating tissue damage. Contrasting observations have been documented in the context of type 2 diabetes mellitus. In mice exposed to a high-fat diet, there was an initial increase in autophagic flux in cardiomyocytes, which was followed by a decline after 2 months of treatment [Citation114]. Notably, an increase in mitophagy was observed following high fat diet (HFD) feeding [Citation114]. In response to HFD feeding, impaired mitophagy and increased cardiac diastolic dysfunction were observed when ATG7 or Parkin was deleted, while overexpression of Beclin1 had the opposite effect [Citation114]. These findings suggest a protective role of autophagy and mitophagy in obesity-associated diabetic cardiomyopathy. However, it is crucial to note that despite these seemingly conflicting outcomes, it would be premature to conclude that mitophagy is inhibited in type 1 diabetes mellitus and activated in type 2 diabetes mellitus models of diabetic cardiomyopathy. In a transgenic model of type 1 diabetes mellitus and streptozotocin (STZ)-treated mice, decreased levels of PINK1 and Parkin were observed in cardiac tissue, but the expression of the small GTPase RAB9 was increased [Citation119], which has been linked to an alternative non-canonical autophagy pathway responsible for mitochondrial degradation in the absence of ATG5 [Citation121]. Therefore, it is plausible that when canonical autophagy is inhibited, an alternative autophagy pathway is activated, potentially triggering the activation of mitophagy in the diabetic heart.
Mechanisms of sterile inflammation
Sterile inflammation describes an inflammatory response triggered by harmful substances that are not pathogens nor their by-products, meaning substances other than pathogen-associated molecular patterns (PAMPs). Traditional experimental models for sterile inflammation include the infliction of physical or chemical damage, which subsequently leads to the release of DAMPs. These DAMPs are recognised by various receptors, including TLRs, cytoplasmic NLRs, non-PRRs such as the receptor for advanced glycation end products (RAGE), CD44, integrins, and CD91. PRRs are expressed on immune cells known as sentinels, which include mast cells, macrophages, dendritic cells, innate lymphoid cells, and basophils. However, PRR expression is not exclusive to immune cells, as sentinel cells also include many non-immune cells [Citation122,Citation123]. PRR activation on sentinel cells triggers the production of various proinflammatory cytokines such as TNF-α and IL-1, as well as vasoactive amines like histamine and serotonin, nitric oxide (NO), ROS, neuropeptides and arachidonic acid metabolites including prostaglandins (PGs) and leukotrienes [Citation19].
The role of platelets, which accumulate in the heart following MI, as regulators of inflammation is noteworthy. Antiplatelets have demonstrated a protective effect in mitigating the extent of inflammation and improving post-myocardial infarction (MI) outcomes [Citation124]. Patients with acute coronary syndrome displayed higher expression levels of TLR2 and TLR4 on platelets [Citation125]. Platelets additionally play a role in inflammation by facilitating the release of HMGB1, which in turn promotes the migration of immune cells and activates inflammasomes through signalling via RAGE and TLR2 [Citation125]. This suggests that platelets play a crucial role in mediating a proinflammatory phenotype and can have a detrimental impact on the infarcted heart. A potential mechanism involves platelets exerting harmful effects following ischaemia/reperfusion injury by releasing platelet-activating factor and upregulating P-selectin expression [Citation126]. Augmented platelets with increased P-selectin expression have been demonstrated to intensify cell death in the heart following ischaemia-reperfusion [Citation127]. To validate the functional significance of this concept, a separate study demonstrated that neutralisation of P-selection through the use of monoclonal antibodies exerted cardioprotective effects against ischaemia/reperfusion injury [Citation128].
DAMPs and high mobility group box-1 (HMGB1)
Release of intracellular DAMPs can occur during cell death or other cellular abnormalities leading to altered membrane integrity. DAMPs encompass a broad array of intracellular components, including mtDNA, HMGB1, heat shock proteins, adenosine triphosphate (ATP), and histone. DAMPs may serve as pro-inflammatory stimuli independently of microbial presence or exacerbate pathogenic inflammation () [Citation129].
Figure 1. Signaling pathways activated by DAMPs All immunogenic nucleic acids bind cytosolic DNA sensors or RNA sensors, including retinoid acid-inducible gene I (RIG-I)-like receptors (RLRs), which are required for subsequent recognition by specific pattern recognition receptors to activate innate immune responses. DAMPs such as HMGB1, S100 proteins (S100s), and heat shock proteins (HSPs) recognize the receptor for advanced glycation end products (RAGE), TLR4, or triggering receptor expressed on myeloid cells-1 (TREM-1) and activate the MyD88-MAPK-NF-jB pathway. HMGB1 and RAGE activate the TLR9-MyD88 dependent pathway, which contributes to autoimmune pathogenesis. CD24 is a negative receptor to inhibit the DAMP-induced TLR4 pathway. ATP binding of the P2X7 receptor and uric acid, as well as asbestos and alum, increase activation of caspase-1 by the NLRP3 inflammasome to promote secretion of IL-1β and IL-18.
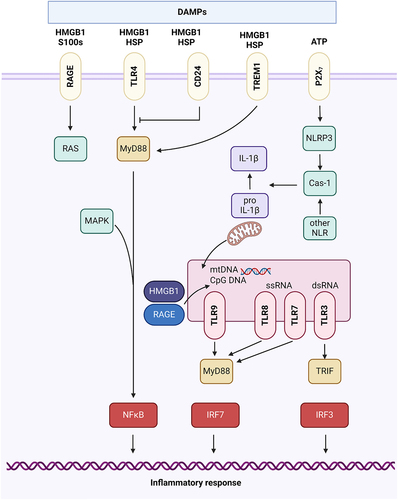
Cytosolic HMGB1 plays a crucial role as a detector of nucleic acid-induced immune responses and can also serve as a natural promoter of autophagy [Citation130,Citation131]. When found in the extracellular space, HMGB1 acts as a potent innate immunity mediator. It functions as a widespread DAMP during tissue injury and cell death [Citation132]. The effects of HMGB1 during sterile inflammation are additionally regulated by posttranslational modification and various compatible receptors. Among these receptors, TLR4 and the advanced glycosylation end product-specific receptor (AGER or RAGE) have been extensively studied. Binding of HMGB1 to these receptors triggers downstream signalling pathways involving NFκB and interferon regulatory factor 3 (IRF3), resulting in the production of immune mediators such as cytokines and chemokines [Citation133,Citation134]. Notably, HMGB1 demonstrates a higher affinity for TLR4 compared to AGER [Citation135,Citation136].
HMGB1, an abundant protein found in almost every tissue, is released during various forms of cell death and sterile tissue injury. Notably, HMGB1 is released earlier than other DAMPs in response to early signals of tissue damage, like ROS [Citation137]. Additionally, HMGB1 has a relatively long half-life due to its ability to bind with other molecules and form large complexes, which enhances extracellular stability. The release of intracellular HMGB1 from tissues through extracellular secretion not only leads to nuclear stress but also triggers the release of other nuclear DAMPs such as histones and genomic DNA. These nuclear DAMPs recruit and activate immune cells that in turn secrete more HMGB1. Moreover, HMGB1 can enhance DNA-sensing pathways, including those involving STING1, TLR3, TLR7, TLR9, and AIM2, which are activated by both host and pathogenic nucleic acids. This effect is achieved through either synergistic interaction or increased affinity between DNA and its sensor [Citation138]. Excessive production of HMGB1 in response to sterile stimuli promotes the release of inflammatory mediators and activation of the complement system, ultimately exacerbating tissue damage and increasing the susceptibility to secondary pathogen infections. However, extracellular HMGB1 can also exhibit growth factor activity by recruiting resident stem cells and initiating healing and tissue regeneration [Citation139]. HMGB1 signalling through TLR4 and AGER can induce both systemic inflammation and wound healing. Consequently, the outcomes of targeting HMGB1 in sterile inflammation may vary depending on the stage of pathology.
mtDNA as an inflammatory mediator
mtDNA is a small double-stranded circular molecule encoding 13 respiratory chain polypeptides, together with associated transfer and ribosomal RNAs that are needed for their translation in the mitochondrial matrix [Citation140]. Mitochondrial proteins are orchestrated by transcriptional co-activators, including the peroxisome proliferator-activated receptor gamma co-activator-1 family, nuclear respiratory factors 1 and 2, and oestrogen-related receptor α [Citation141]. Within the mitochondria, specific proteins may bind to mtDNA, forming a complex known as nucleoids [Citation142]. These nucleoids are regions within the mitochondria that contain DNA along with associated proteins essential for maintaining the integrity of mtDNA. The transcription factors A (TFAM), B1 (TFB1M), and B2 (TFB2M) are examples of mtDNA-binding proteins. They are encoded in the nuclear genome and are subsequently transported into the mitochondria through a protein import machinery after expression. Within the mitochondria, these proteins play a critical role in regulating and maintaining the stability of mtDNA [Citation142].
Dysfunctional mitochondria have been identified as potent triggers of sterile inflammation and we now understand much about the mechanisms through which they initiate inflammatory signalling. One established pathway involves the release of mtDNA from stressed mitochondria. This mtDNA can become oxidised (ox-mtDNA), induce NLRP3 or other inflammasome activation, resulting in the secretion of pro-inflammatory cytokines (such as IL-1β and IL-18) and pyroptotic cell death [Citation143]. mtDNA possesses specific characteristics that contribute to its potential as a potent DAMP. These characteristics include relative hypomethylation, unique structural features, and vulnerability to oxidative damage due to proximity to significant sources of ROS. As a result, mtDNA has the capacity to activate innate immunity, triggering pro-inflammatory pathways and type I interferon (IFN) responses. Current evidence suggests that the inflammation mediated by mtDNA is primarily induced through the activation of TLR9 and the NLRP3 inflammasome [Citation144,Citation145]. Furthermore, regardless of oxidation status, cytosolic mtDNA is recognised by the DNA-sensing cyclic GMP-AMP synthase (cGAS), which in turn activates STING to induce the release of type 1 interferons [Citation146-148].
NLRP3 inflammasome in sterile inflammation
Inflammasomes are a diverse group of large macromolecular complexes that assemble upon the activation of both intra- and extra-cellular receptors designed to detect various harmful stimuli [Citation19]. Inflammasomes are conserved across different species and operate through tightly regulated pathways [Citation149]. As components of the innate immune system, they serve as a connection between the recognition of danger signals, both intra- and extra-cellular, associated with pathogen infections or tissue damage and the subsequent inflammatory response [Citation150]. Inflammasomes can identify a range of danger signals, including microbial and viral molecules, intracellular proteins, nucleic acids, lipids, mitochondrial components, and various organic and inorganic compounds [Citation151,Citation152]. Recognition of DAMPs or PAMPs involves specific extracellular receptors, as well as the involvement of nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), and the AIM2-like receptor (ALR), which collectively contribute to the assembly of the inflammasomes [Citation19]. NLRP3, a pattern recognition receptor (PRR), plays a role in recognising both bacterial and viral PAMPs, as well as various DAMPs involved in tissue and cellular injury. Among the inflammasomes, NLRP3 has been the focus of most extensive research and priming and activation mechanisms are well understood [Citation153]. Thus, NLRP3 is often a critical participant in the inflammatory response triggered by the disturbance of cellular homoeostasis and it contributes significantly to the body’s reaction to tissue damage [Citation153].
Upon activation, NLRP3 undergoes oligomerization and forms a connection (via a PYD-PYD interlinkage) with the adaptor protein apoptosis-associated speck-like protein containing a carboxy-terminal caspase recruitment domain (CARD) also referred to as ASC [Citation153]. Subsequently, ASC polymerises and forms insoluble filamentous structures that serve as a scaffold necessary for recruiting the effector enzyme, pro caspase-1, into the NLRP3 inflammasome through a CARD-CARD interaction [Citation154]. This leads to the proteolytic activation of pro caspase-1into caspase-1, which triggers the release of the pro-inflammatory cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18) [Citation155]. Additionally, caspase-1 is involved in a regulated form of cell death called pyroptosis [Citation156]. Pyroptosis is characterised by disruption of the plasma membrane, resulting in the release of intracellular contents that act as pro-inflammatory mediators, propagating signalling to neighbouring cells [Citation156].
NLRP3 can be expressed by various cell types within the heart. However, each cell type elicits a distinct response. For instance, in endothelial cells, the activation of NLRP3 by leukocytes and fibroblasts leads to the processing of IL-1β [Citation154]. On the other hand, cardiomyocytes may exhibit limited production of IL-1β, instead favouring a form of cell death called pyroptosis. Activation of the NLRP3 pathway in the heart often results in cardiomyocyte loss through pyroptosis [Citation157]. Consequently, a sustained inflammatory condition can progressively diminish the number of cardiomyocytes, ultimately restricting the heart’s contractile capacity.
Toll-like Receptors (TLRs)
DAMPs released by necrotic cells provoke inflammatory responses through engagement of innate immune receptors (such as TLRs) [Citation123]. TLRs have the remarkable ability to detect patterns rather than specific ligands, which allows different members of the TLR family to recognise a wide range of microorganisms. This unique feature extends beyond the recognition of microbe-associated molecular patterns (MAMPs) and encompasses the detection of DAMPs by TLR receptors, with TLRs 2 and 4 being particularly notable in this regard [Citation158].
TLRs can activate two major intracellular signalling pathways based on the adaptors they recruit. The first pathway, which is MyD88-dependent, is activated by all TLRs except TLR3. It involves IL-1R-associated kinases (IRAKs), specifically IRAK-1 and IRAK-4, TNF receptor-associated factor 6 (TRAF-6), and mitogen-activated kinases (MAPKs) [Citation159]. This pathway culminates in the activation of the transcription factor NFκB through the IkB kinase (IKK) complex. Subsequently, NFκB regulates the transcription of genes responsible for producing pro-inflammatory cytokines [Citation160]. The second pathway, known as the TRIF pathway, operates independently of MyD88 and can be triggered by the stimulation of TLR3 or TLR4. It involves the recruitment of TRIF and leads to the activation of the interferon-regulated factors (IRF) family of transcription factors. This activation, in turn, induces the synthesis of interferons (IFNs) [Citation161]. TLR signalling triggers the activation of transcription factors that regulate the expression of specific genes, leading to diverse cellular responses [Citation162]. For instance, NFB, AP-1, and IRF5 govern the expression of genes encoding inflammatory cytokines, while IRF3 and IRF7 stimulate the expression of type I interferons (IFNs) and IFN-inducible genes [Citation162]. As a result, a wide array of proteins is synthesised to mediate inflammatory and immune responses. These proteins include inflammatory cytokines like IL-1, IL-6, TNF, IL-12, IFNs, chemokines, adhesion molecules, costimulatory molecules, growth factors, tissue-degrading enzymes such as metalloproteinases, and enzymes involved in generating inflammatory mediators like cyclooxygenase 2 and inducible nitric oxide synthase (iNOS) [Citation163].
Sterile inflammation in preclinical models and human heart failure
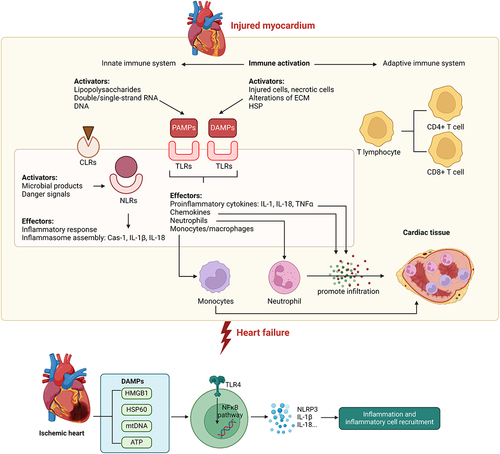
Recurrent sterile inflammation triggered by metabolic dysfunction is a classic example of pathophysiological conditions that occur in patients with cardiometabolic disease [Citation164-166]. Sterile inflammation is initiated by the release of DAMPs during tissue damage, which can initiate an inflammatory response, and migration of leukocytes to and from the site of injury [Citation134,Citation167]. Sterile inflammation and subsequent tissue repair depend on a well-orchestrated migration sequence of leukocytes to and from the site of injury. Increased release of DAMPs has been observed in patients with myocardial infarction with heart failure [Citation168]. Specifically, circulating leukocytic TLR-2 and TLR-4 contributed to myocardium damage in ischaemia/reperfusion injury [Citation169-171]. The HMGB1 protein passively enters the extracellular environment from necrotic cells and serves as a DAMP for leukocytes [Citation172,Citation173]. The rise in HMGB1 levels in response to ischaemic injury of the human heart is well-reported, showing that peak serum levels of HMGB1 were higher in patients with pump failure and cardiac rupture than in those without. Additionally, patients with increased peak HMGB1 levels were more likely to die from cardiac causes [Citation174,Citation175]. Tumour necrosis factor alpha (TNFα) contributes to myocardial dysfunction with increased circulating levels observed in chronic heart failure [Citation176,Citation177]. Moreover, caspase-1 activation, by the NLRP3 inflammasome, is upregulated in murine and human failing hearts [Citation178]. Circulating cytokines from damaged myocardium are increased in patients with chronic heart failure and their levels correlate with the severity [Citation179]. To address the impact of inflammation on heart failure, therapeutic treatments have been developed to improve cardiovascular outcome in patients with heart failure. In mice, genetic ablation of the NLRP3 inflammasome reduced inflammation and proinflammatory cytokine maturation [Citation180]. In humans, the clinical trial revealed that a therapeutic monoclonal antibody targeting IL-1β reduced cardiovascular events in patients with myocardial infarction [Citation181]. However, other clinical trials attempting to modulate inflammation in heart failure were not as effective, emphasising the need for a better understanding of the processes involved [Citation182,Citation183]. Thus, evidence from human and rodent studies reported a critical role of sterile inflammation in heart failure ().
Figure 2. Innate immune activation contributes to the progression of heart failure Upon injury within the myocardium, the innate immune system is activated by various factors such as PAMPs and DAMPs. Activation of the innate immune system is mediated by signaling via NLRs, TLRs, and CLRs to mediate downstream proinflammatory effects such as production of proinflammatory cytokines and chemokines, inflammasome assembly, and promote immune cell infiltration into cardiac tissue, thus contributing to the pathogenesis of heart failure. Within the ischemic heart, DAMPs are known to activate TLR4-NFkB signaling pathway to promote production of specific cytokines leading to sterile inflammation in the heart. Casp-1, caspase-1; CLR, C-type lectin receptor; DAMP, damage-associated molecular pattern; ECM, extracellular matrix; HF, heart failure; HSP, heat-shock protein; IL, interleukin; NLR, NOD-like receptor; PAMP, pathogen-associated molecular pattern; TLR, Toll-like receptor; TNF, tumor necrosis factor.
Myocardial infarction
Permanent left anterior descending (LAD) ligation in mice leads to acute myocardial injury caused by ischaemia, which is commonly referred to as myocardial infarction (MI) [Citation184]. The inflammatory phase that follows MI serves a dual role. On one hand, it plays a crucial role in initiating cardiac repair processes. On the other hand, excessive inflammation during this phase can contribute to the subsequent development of heart failure. Therefore, it is essential to carefully regulate the spatial and temporal aspects of inflammation to ensure sufficient healing of the infarcted area.
MI is pathologically characterised as the death of myocardial cells resulting from prolonged ischaemia [Citation185]. During MI, necrotic cardiomyocytes release various danger signals known as DAMPs. These DAMPs include mtDNA, HMGB1, purine metabolites, sarcomeric protein fragments, and S100 proteins. These molecules are released into the surrounding tissue and play a significant role in triggering inflammatory responses and immune cell activation [Citation186-188]. Upon release, these DAMPs initially activate various sentinel cells, including resident macrophages, endothelial cells, resident cardiac fibroblasts, and surviving cardiomyocytes in the border zone of the infarcted area. Subsequently, these danger signals also attract and activate invading leukocytes, further contributing to the inflammatory response. The activation of these cells and the subsequent inflammatory cascade play a crucial role in the healing process and tissue remodelling following myocardial infarction. The main PRRs of the cardiac sentinel cells include TLRs, RAGE and NLRs [Citation189]. PRRs can be localised in intracellular compartments or on cellular surfaces, leading to distinct roles in phagocytes and non-phagocytes. In the context of MI, the interactions between DAMPs and their corresponding PRRs have been extensively reviewed [Citation189]. Activation of downstream PRR signalling leads to the activation of the NFκB, which upregulates the expression of chemokines, cytokines, and cell adhesion molecules in the infarcted heart wall [Citation189-191]. Chemokines, such as CC-chemokine ligand (CCL)-2, −5, and −7, attract monocytes, lymphocytes, and mast cells, while CXC chemokines, such as CXCL-8, attract neutrophils [Citation190]. Pro-inflammatory cytokines like TNF-α and IL-1β, IL-6, and −18 further amplify the inflammatory response by activating resident and invading effector cells [Citation189]. IL-1β plays a crucial role in myocardial chemokine synthesis and induces pro-inflammatory polarisation in macrophages [Citation190,Citation192]. IL-1 receptor type I deficient mice exhibit reduced inflammation and fibrosis following MI [Citation192]. The mature form of IL-1β is secreted by both leukocytes and activated cardiac fibroblasts and requires Caspase 1-mediated cleavage of its precursor pro-IL-1β within a multiprotein complex called the inflammasome [Citation149]. Immediately following MI, IL-6 upregulates the expression of hyaluronan synthases, which promotes myofibroblast differentiation and cardiac repair. Thus, blocking IL-6 may have detrimental effects after MI [Citation193].
Ischaemia/reperfusion injury
The murine model of transient LAD ligation replicates type 1 MI followed by successful revascularization [Citation194]. Following the diagnosis of ST-elevation myocardial infarction (STEMI) via electrocardiogram, the American Heart Associate Guidelines recommends reperfusion be achieved within 10 minutes. Regardless of the reperfusion strategy chosen, it is imperative to restore blood flow within 60-90 minutes to prevent cell death [Citation195]. Improved awareness of heart attack symptoms and importance of prompt reperfusion have contributed to the declining mortality associated with this acute coronary syndrome [Citation196]. While reperfusion is necessary to rescue ischaemic myocardium from impending damage, it also introduces additional injury that is not always reversible. Under these circumstances, reperfusion will lead to increased infarct size and microvascular dysfunction [Citation197].
Cardiac repair following MI begins with an initial inflammatory phase characterised by the infiltration of immune cells and the release of DAMPs. This inflammatory response is mediated by PRRs that recognise and bind to DAMPs [Citation19,Citation198,Citation199]. Proper coordination and timing of this early inflammatory response is crucial for promoting tissue repair and preventing adverse cardiac remodelling, which will ultimately improve patient outcomes. An excessive or prolonged inflammatory response can lead to ventricular dilatation, systolic dysfunction, and an increased risk of heart failure. On the other hand, insufficient or delayed inflammation may impede the clearance of necrotic cardiac cells and matrix components, hindering proper tissue repair [Citation200]. Thus, a balanced and tightly regulated innate immune activation response is essential for optimal cardiac repair following ischaemia/reperfusion injury.
It is widely acknowledged that non-immune cells, including cardiomyocytes, cardiac fibroblasts and cardiac endothelial cells, express receptors associated with innate immunity [Citation199,Citation201]. Thus, DAMPs can stimulate inflammation via cardiomyocytes and cardiac fibroblasts, leading to the production and release of pro-inflammatory cytokines. Whilst these levels are typically small, they may be sufficient to crosstalk with innate immune cells and induce a cascade of robust inflammation. For instance, pressure overload induced a proinflammatory state which resulted in elevated fibrosis and left ventricular dysfunction. This adverse cardiac remodelling was attributed to altered signalling from cardiomyocytes in the early phase of injury which was ultimately transduced to macrophages [Citation202]. An inflammatory response facilitates the clearance of dead cells and cellular debris from the infarcted site and is normally then counteracted by a resolution phase which suppresses inflammation [Citation189,Citation203]. There is a limited number of cardiac resident immune cells so, following injury, immune cells are recruited and infiltrate infarct areas within approximately 6-8 hours, as observed in human cardiac tissue [Citation204]. The extravasation of immune cells, particularly neutrophils and monocytes, is attributed to the presence of DAMPs, cytokines, and components of the complement system, which play a crucial role in facilitating the recruitment and migration of immune cells to the infarct area [Citation205,Citation206]. The infiltration of specific innate immune response cell types into the infarct area initiates the resolution phase by the phagocytosis of necrotic cells and cellular debris. However, the proliferation of DAMPs may elicit negative cardiac outcomes by promoting increased cell death [Citation186]. The healing process plays a crucial role in preventing cardiac rupture and ultimately involves the production of anti-inflammatory cytokines to dampen the inflammatory response, reducing further cellular damage and promoting cardiac recovery [Citation189,Citation207].
It has been proposed that mtDNA is released intracellularly through permeabilization of both inner and outer mitochondrial membranes in damaged or stressed mitochondria [Citation208]. The early phase following ischaemia/reperfusion injury is associated with upregulation of retinoic acid early transcript 1 (RAE-1) expression in cardiomyocytes, which is believed to be activated through the STING pathway. It is hypothesised that release of mtDNA into the cytosol might indirectly activate RAE-1 expression, which in turn contributes to cardiac fibrosis and remodelling [Citation209,Citation210]. In cardiomyocytes, mtDNA release triggers the NFκB signalling pathway. This activation leads to an increased expression and release of pro-inflammatory cytokines, which play a role in both local and systemic inflammatory responses [Citation186,Citation210].
Several published studies have reported approaches to attenuate sterile inflammation and reduce myocardial ischaemia/reperfusion injury. For example, pharmacological inhibition of the NLRP3 inflammasome using dapansutrile has been shown to reduce infarct size and mitigate ischaemia/reperfusion-induced injury, thus preserving cardiac function [Citation211,Citation212]. Similarly, inhibition of TXNIP by intramyocardial injection of siRNA decreased NLRP3 activation and reduced infarct size in an animal model of myocardial ischaemia/reperfusion injury [Citation213]. Administration of recombinant klotho protein reduced sterile inflammation through inhibition of HMGB1 [Citation214]. Additionally, the bacterial C-type lectin domain family 4 member E (CLEC4E) was strongly associated with robust activation of a PRR during ischaemia/reperfusion and knockout mice lacking CLEC4E demonstrated significant improvements in myocardial metabolism, ROS scavenging, angiogenesis, and fibrosis () [Citation215].
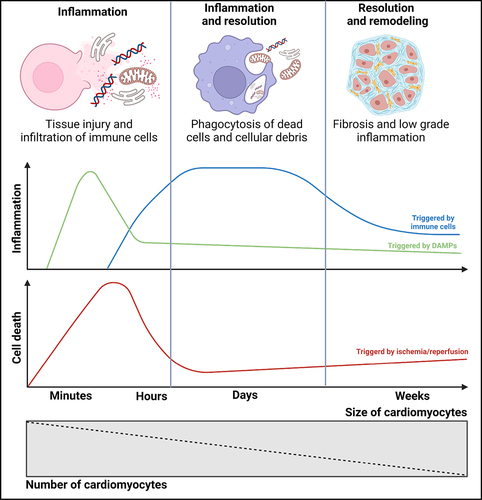
Figure 3. Progression from myocardial infarction to heart failure. Myocardial infarction leads to the death of heart muscle cells, causing the release of DAMPs and the production of pro-inflammatory substances. These substances attract immune cells, which infiltrate the damaged area and clear away cellular debris. As a response to cell death caused by the lack of blood flow and subsequent restoration, the heart initiates remodeling and fibrosis to compensate. In the resolution phase, the inflammation is reduced by anti-inflammatory cytokines, but a low-level chronic inflammation persists. The number of cardiomyocytes decreases due to myocardial necrosis, but their size increases as a result of increased mechanical strain and compensatory hypertrophy.
Pressure overload
Pathological cardiac hypertrophy and fibrosis in models of pressure overload induced by transverse aortic constriction (TAC), a commonly used approach to mimic the increased left ventricular load in patients, are preceded by significant alterations in energy metabolism, leading to significantly increased risk of heart failure [Citation216]. Pressure overload-induced cardiac hypertrophy is an adaptive response and is believed to be a compensatory response to physiological stimuli or pathological insults in the heart by diminishing wall stress and oxygen consumption [Citation217]. Although inflammation has been implicated in these processes, the mechanisms via which inflammation regulates metabolic dysfunction, hypertrophy, and fibrosis are not fully understood [Citation218].
Many reports have now studied the role of sterile inflammation in adverse cardiac remodelling upon pressure overload [Citation219,Citation220]. For example, CaMKIIδ, a calcium/calmodulin-dependent protein kinase II isoform, plays a crucial role in the pathogenesis of pressure overload and increased expression and activation of CaMKIIδ have been observed in both rabbit and human heart failure [Citation221]. Treatment with CaMKII inhibitors has shown beneficial effects on contractility and cardiac function in patients with failing myocardium [Citation222]. Activation of the NLRP3 inflammasome, mediated by CaMKIIδ, has been implicated in fibrosis associated with pressure overload. In a separate study, inflammasome activation occurs early during angiotensin II (Ang II) infusion, preceding macrophage recruitment. Inhibition of NLRP3 inflammasome through knockout or inhibition significantly reduces inflammation, macrophage accumulation, and fibrosis during Ang II infusion, similar to the effects observed with CaMKIIδ deletion [Citation223,Citation224]. It’s also demonstrated that deletion of CaMKIIδ specifically in cardiomyocytes reduced the accumulation of CD68+ macrophages and the production of pro-inflammatory cytokines such as IL-1β and IL-18 in response to pressure overload. Cardiomyocytes isolated from mice with cardiomyocyte-specific CaMKIIδ deletion and subjected to TAC exhibited decreased levels of NLRP3, reduced caspase-1 activity, and inhibited IL-18 activation. These findings suggest that CaMKIIδ activation in cardiomyocytes in response to pressure overload stimulates the activation of the NLRP3 inflammasome [Citation225].
It is also of interest to note that right ventricular failure is one of the leading causes of death in cardiac failure associated with pulmonary hypertension or in congenital heart diseases. In a pulmonary artery banding-induced pressure overload rat model, inhibition of TLR9-NFκB signalling by TLR9 inhibitor E6446 and NFκB inhibitor pyrrolidine dithiocarbonate improved right ventricular function [Citation226]. Transcriptional control of proinflammatory cytokines plays an important role in the inflammation process. Regnase-1 is an RNase degrades certain proinflammatory cytokine mRNA in immune cells [Citation227]. Cardiomyocyte-specific Regnase-1 deficiency in mice showed exacerbated inflammation (upregulated IL-6 mRNA) and cardiomyopathy following pressure overload. Thus, transcriptional control of proinflammatory cytokine mRNA by Regnase-1 in cardiomyocytes can play a critical role in limiting sterile inflammation during pressure overload [Citation227].
Diabetic cardiomyopathy
Diabetic cardiomyopathy is a distinct type of chronic disease characterised by myocardial insulin resistance [Citation228]. It is further exacerbated by compensatory hyperinsulinemia and the advancement of hyperglycaemia. Importantly, this condition develops independently of other cardiovascular risk factors like coronary artery disease and hypertension. The compromised cardiac function in diabetic cardiomyopathy may manifest as both systolic and diastolic dysfunction, and the progression of diabetic cardiomyopathy increases the risk of developing heart failure [Citation228]. Dysregulation of both the innate and adaptive immune systems can contribute to the development of diabetic cardiomyopathy [Citation229,Citation230]. Additionally, diabetic cardiomyopathy is frequently associated with obesity, and it is well known that obese visceral adipose tissue exhibits an inflammatory phenotype [Citation231]. This chronic pro-inflammatory milieu is characterised by macrophage M1 polarisation that often occurs in obesity and insulin resistant conditions. Macrophage M1 polarisation is associated with increased release of pro inflammatory cytokines that contribute to myocardial inflammation and the progression of heart failure [Citation232].
Multiple mechanisms appear to be relevant in the pathogenesis of heart failure in diabetic models. Activation of myocardial innate immunity in diabetic cardiomyopathy involves PRRs, including TLRs, leading to activation of NLRP3 inflammasome and NFκB and expression of pro-inflammatory cytokines such as TNF-α, IL-6, IL-1β, and IL-18. In the streptozotocin (STZ)-induced diabetic rat model, there is an increase in expression of TNF-α and it has been observed that inhibiting TNF-α can reduce the development of diabetic cardiomyopathy [Citation233]. Furthermore, in diabetic rat models with a combination of a high-fat diet and STZ injection, reducing the expression of NLRP3 has been shown to improve cardiac function and decrease the expression of mature IL-1β [Citation234]. Also, in animal models of type 2 diabetes, cardiac dysfunction can be ameliorated by dapagliflozin and rosuvastatin, and these protective effects are likely mediated through the inhibition of NLRP3 inflammasome [Citation235,Citation236]. Moreover, the suppression of NOD2 through knockdown has been shown to reduce myocardial fibrosis and cell apoptosis in diabetic mice [Citation237]. Additionally, there is an upregulation of NOD1 expression in both heart tissues and cardiomyocytes of mice with T2DM, suggesting a potential involvement of NOD1 in the development of diabetic cardiomyopathy [Citation238]. It was also shown that there was a significant increase in the expression of miRNA-30d in both STZ-induced diabetic rats and cardiomyocytes treated with high glucose, which contributed to the progression of cardiomyocyte pyroptosis [Citation239]. Conversely, this process was attenuated by miRNA-30d knockdown [Citation239]. Finally, it was reported that chemerin and its G-protein-coupled receptor CMKLR1 strongly contributed to cardiac dysfunction in the model of high-fat diet combined with STZ injection. By silencing CMKLR1, cardiac function was partially restored and suppressed NLRP3 formation [Citation240].
Interplay between sterile inflammation and autophagy in heart failure
DAMPs and autophagy
We will begin by exploring how inflammation induced by various DAMPs can impact autophagy. The intricate relationship between autophagy and HMGB1 is intricate. Autophagy, instead of apoptosis, significantly contributes to the regulation of the localisation and release of HMGB1 in response to ROS during early cellular stress events [Citation131,Citation241]. HMGB1 competes with Bcl-2 for interaction with Beclin 1, directing Beclin 1 towards autophagosomes. The interaction between HMGB1 and Beclin 1 depends on the autophagic complex ULK1-mAtg13-FIP200. Additionally, HMGB1 can be involved in the regulation of Bcl-2 phosphorylation through the extracellular signal-regulated kinase (ERK)/MAPK pathway [Citation242]. The intramolecular disulphide bridge (C23/45) of HMGB1 is crucial for its binding to Beclin 1 and for sustaining autophagy [Citation131]. Along with its downstream mediator HSPB1/HSP27, HMGB1 modulates mitochondrial respiration and morphology by sustaining autophagy/mitophagy, indicating its essential role in mitochondrial quality control. Furthermore, HMGB1 forms highly inflammatory complexes with ssDNA, LPS, IL-1β, and nucleosomes, interacting with TLR9, TLR4, IL-1R, and TLR2 receptors, respectively [Citation243]. Thus, current evidence suggests that HMGB1 serves as a crucial node in the interaction between inflammation and autophagy.
TLRs, NLRs and autophagy
The induction of autophagy by TLR signalling seems to be dependent on both MyD88 and TRIF. TLR signalling has been demonstrated to strengthen the interaction between MyD88 and TRIF with Beclin 1, concurrently diminishing the binding of Beclin 1 to Bcl-2 in macrophages [Citation244]. Notably, ubiquitination of Beclin 1 facilitated by tumour necrosis factor receptor (TNFR)-associated factor 6 (TRAF6), amplifies TLR4-induced autophagy [Citation245]. Conversely, the deubiquitinating enzyme A20 decreases ubiquitination of Beclin 1, thereby limiting the induction of autophagy [Citation245]. Additionally, phosphorylation of Beclin 1 on Thr 119 in the BH3 domain by death-associated protein kinase (DAPK) promotes autophagy [Citation246]. Several protein modifications have been identified in the assembly of the Beclin 1-PI3KC3 complex, indicating that Beclin 1 likely plays a pivotal role in TLR-mediated autophagy through post-translational modification [Citation247].
In mammalian cells, NLR family members, such as NOD1 and NOD2, induce autophagy as a response to control bacterial infection and facilitate antigen presentation [Citation248]. Ablation of autophagy regulators ATG16L1 or ATG7, with resultant inhibition of autophagy, exacerbates LPS-induced inflammasome activation, leading to enhanced processing of pro IL-1β into IL-1β [Citation249]. Macrophages demonstrate autophagy in response to various inflammasome stimuli, achieved through the activation of nucleotide exchange on RalB [Citation250]. Blocking mitophagy/autophagy results in the accumulation of damaged mitochondria that generate ROS, subsequently activating the NLRP3 inflammasome. In contrast, autophagic proteins play a regulatory role in NLRP3-dependent inflammation by preserving mitochondrial integrity [Citation251]. These findings suggest that autophagy contributes to maintaining inflammasome homoeostasis by clearing dysfunctional mitochondria and reducing ROS production. Additionally, NLR members may negatively regulate autophagosome maturation by interacting with Beclin1 [Citation252]. Notably, autophagy plays a dual role in IL-1β signalling and inflammasome activation, as it inhibits IL-1β release by targeting pro IL-1β for p62-mediated lysosomal degradation, while also promoting IL-1b release through an unconventional secretory pathway [Citation253].
Autophagy orchestrates inflammatory response in cardiac injury
Sterile inflammation and autophagy interact through various potential mechanisms. Inflammatory mediators can engage with autophagy proteins, promoting an increase in autophagic flux and conferring cardioprotective benefits. One pivotal participant in this interplay is HMGB1, a nuclear binding protein with roles in regulating chromosome structure and contributing significantly to heart repair [Citation254]. HMGB1 exerts its influence on the inflammatory response in individuals with heart failure through a process involving its translocation from the nucleus to the cytoplasm. This relocation of HMGB1 contributes to the modulation of inflammatory reactions associated with heart failure [Citation255]. The translocation of HMGB1 from the nucleus to the cytoplasm is facilitated by molecules such as angiotensin II (Ang II) and endothelin-1. This phenomenon has been observed in neonatal rat cardiomyocytes in vitro [Citation256]. HMGB1 interacts with the receptor for advanced glycation end products (RAGE), triggering an inflammatory response. Additionally, HMGB1 directly enhances autophagic flux by interacting with Beclin1 and displacing Beclin2. Mutations affecting cysteine 106 in HMGB1 and the intramolecular disulphide bridge (Cysteine 23/45) promote cytosolic localisation, leading to binding with Beclin1 and sustaining autophagy. Furthermore, ROS facilitates the cytosolic translocation of HMGB1, inducing autophagy and disrupting the formation of the Beclin1-Bcl-2 complex. Therefore, targeting HMGB1 may offer therapeutic potential for heart failure by mitigating its cytokine-like activity and its ability to promote autophagy [Citation257,Citation258]. Although AngII can mediate HMGB1 translocation, Ang II itself acts as an inflammatory mediator through the AT1 receptor. Ang II exerts pro-inflammatory effects and increases the ATG5 mRNA expression level in macrophages. Ang II treatment significantly stimulates the expression of LC3, an autophagy marker, in cultured macrophages in vitro. Haplo-deficiency of ATG5, a key autophagy-related protein, substantially reduces the formation of cytosolic autophagic vacuoles induced by Ang II. This reduction in autophagy contributes to increased NFκB activity and mitochondrial ROS production in macrophages, subsequently worsening hypertension-induced cardiac inflammation and injury in mice [Citation259].
Considering the crucial role of neutrophils in acute myocardial ischaemia/reperfusion injury, chronic ischaemia, and remodelling, it is plausible that autophagic flux in neutrophils plays a critical role in regulating cardiac injury and remodelling [Citation260,Citation261]. It has been demonstrated in a mouse model of permanent infarction that neutrophil-secreted factors, including neutrophil gelatinase-associated lipocalin (NGAL), play a role in regulating macrophage polarisation during the healing phase following ischaemic myocardial injury [Citation260]. A more recent study shed light on the significance of endothelial autophagy in the regulation of neutrophil infiltration at sites of inflammation [Citation262]. In an experimental model, inflamed venular endothelial cells selectively upregulated autophagy at endothelial cell junctions during the peak of neutrophil trafficking. The deficiency of the ATG5 protein in endothelial cells resulted in excessive transendothelial migration of neutrophils and uncontrolled leukocyte movement in murine models of inflammation. Conversely, pharmacologically induced autophagy suppressed the infiltration of neutrophils into tissues. However, the specific mechanisms underlying neutrophil extravasation in the ischaemic myocardium have not been extensively investigated, primarily due to the challenges in imaging leukocyte trafficking in the working heart. The molecular processes governing neutrophil diapedesis in remote areas of the infarcted heart or the reperfused infarction zone may be speculated to resemble those studied in venules of other inflammatory sites [Citation263]. Overexpression of transcription factor EB (TFEB), a key regulator of lysosome biogenesis, in macrophages resulted in the mitigation of post-ischaemia/reperfusion cardiac remodelling and reduced inflammation, and a similar observation was made in or in Tfeb-transgenic mice. Surprisingly, these beneficial effects were observed independently of ATG5 expression in myeloid cells. This suggests TFEB signalling at the lysosome can regulate inflammation to attenuate the post MI remodelling independently from autophagy. However, as this study focused only on a single time point after 4 weeks of ischaemia/reperfusion injury, further in-depth investigation is necessary to elucidate the precise role of autophagy in monocyte-macrophage responses to cardiac injury [Citation264].
The interplay between autophagy and sterile inflammation has been highlighted in an in vitro model of cardiomyocytes treated with hypoxia/reoxygenation. Many studies have sought to regulate autophagy to mitigate hypoxia/reoxygenation injury in cardiomyocytes. Various microRNAs have been identified as modulators of autophagy, serving as potential therapeutic targets for alleviating hypoxia/reoxygenation injury through diverse mechanisms [Citation86,Citation265-273]. As described above, HMGB1 plays a crucial role in regulating cardiomyocyte autophagy and can be targeted to alleviate hypoxia/reoxygenation injury [Citation273,Citation274]. Moreover, mitochondrial autophagy is now established as a potential therapeutic target for attenuating hypoxia/reoxygenation injury in cardiomyocytes [Citation275,Citation276]. The induction of PINK1/Parkin-dependent mitophagy has been shown to sustain mitochondrial ATP, maintain membrane potential, and reduce mitochondrial damage. This process helps prevent downstream inflammatory signalling. Additionally, the activation of AMPK inhibits the inflammatory response by modulating the JNK-NFκB signalling pathway [Citation277]. Additionally, upregulation of C1q/TNF-related protein 12 inhibits inflammation via Nrf2-dependent upregulation of anti-inflammatory genes HO-1 and NQO-1 [Citation278,Citation279]. Therefore, regulating interplay between autophagy and sterile inflammation in cardiomyocytes can effectively alleviate hypoxia/reoxygenation injury.
The role of mtDNA in the pathogenesis of heart failure
The relationship between autophagy, mtDNA, and inflammation in the heart has gained prominence and been a fruitful area of research in recent years. The Otsu group demonstrated that when mtDNA escapes autophagy, it can lead to the activation of TLR9. This was evidenced by accelerated myocarditis and heart failure observed after the induction of a pressure overload model in mice with a cardiac-specific deletion of lysosomal DNase II (DNase2a-/-) [Citation280]. In DNase2a−/− mice treated with a transverse aortic constriction, cardiomyocytes exhibited heightened expression of pro-inflammatory cytokines IL-1β and IL-6, leading to a subsequent infiltration of CD68+ macrophages and Ly6G+ cells into affected cardiac tissue. mtDNA in heart tissue of DNase2a−/− mice were only observed within autolysosomes. This indicated that the mitochondria were engulfed in autophagosomes which then fused with lysosomes. However, due to the absence of DNase II, mtDNA could not be properly degraded. It was hypothesised that mtDNA could activate Toll-like receptor 9 (TLR9) present in endolysosomes. This interaction between undigested mtDNA and TLR9 was speculated to trigger inflammatory signalling. DNase II abolishes cardiac inflammation and dysfunction caused by pressure overload [Citation281]. Therefore, TLR9 signalling is essential for recognising undegraded mtDNA and regulating inflammation in stressed cardiomyocytes. Mice lacking TLR9 subjected to pressure overload exhibited reduced macrophage infiltration and improved recovery from heart failure [Citation280]. Overall, excess production and release or inadequate degradation of mtDNA promotes inflammation via TLR9 signalling ultimately contributing to the pathogenesis of heart failure.
It has been proposed that mtDNA is released intracellularly through permeabilization of both the inner and outer mitochondrial membrane in damaged or stressed mitochondria [Citation208]. During the early phase following ischaemia/reperfusion injury, there is an upregulation of retinoic acid early transcript 1 (RAE-1) expression in cardiomyocytes. This increased expression is believed to be activated through the STING pathway. It is hypothesised that mtDNA might indirectly activate RAE-1 expression, which in turn contributes to cardiac fibrosis and remodelling during the remodelling phase [Citation209,Citation210]. In cardiomyocytes, mitochondrial mtDNA activation can also trigger activation of the nuclear factor NFκB signalling pathway. This activation leads to the expression and release of pro-inflammatory cytokines which play a role in both local and systemic inflammatory responses [Citation186,Citation210].
When mitochondria become depolarised or damaged, they are recognised and targeted for removal through a process involving PINK1/Parkin and p62. The PINK1/Parkin pathway leads to the recruitment of the forming autophagosome to the ubiquitin-decorated mitochondrial fragment with the assistance of p62 [Citation282]. Ubiquitination of mitofusin 2 (MFN2) prevents fusion of damaged mitochondria with the remaining mitochondrial network. Proteins such as DRP1 and MFN2 are involved in mitophagy. Nix and Bnip3 stimulate mitophagy, likely by triggering depolarisation and subsequent recruitment of Parkin. However, it has been demonstrated that Nix and Bnip3 can have a maladaptive role, suggesting that autophagy/mitophagy may have deleterious effects in certain contexts.
Clinical perspectives on therapeutic targeting of autophagy and inflammation in heart failure
Given the preceding text, it is abundantly clear that there are numerous scenarios were targeting autophagy, inflammation, and the crosstalk between them could have important therapeutic implications.
Numerous hormones and cytokines play a crucial role in the regulation of autophagy. Adiponectin, for instance, has been demonstrated to induce autophagy in a variety of cell types [Citation283]. Aged mice lacking adiponectin demonstrate reduced autophagy induction versus wild-type mice, which contributed to pressure overload-induced cardiac dysfunction [Citation8]. Adiponectin is also known for its anti-inflammatory effects. The anti-inflammatory effects of adiponectin are due in part to its ability to inhibit TLR-NFkB signalling, stimulate production of anti-inflammatory cytokines, and promote polarisation of macrophages towards an anti-inflammatory phenotype [Citation284-287]. Indeed, mice lacking adiponectin display elevated expression of proinflammatory cytokines and decreased levels of anti-inflammatory mediators in various models of cardiometabolic disease [Citation288,Citation289]. Additionally, adiponectin has been shown to inhibit neutrophil activation in response to pressure overload, and attenuate LPS-stimulated NLRP3 activation [Citation290,Citation291]. Recent research has also indicated that the exacerbated cardiac dysfunction in adiponectin knockout mice after LPS injection is associated with reduced autophagy through an AMPK-mTOR-ULK1-dependent mechanism [Citation292]. Therefore, adiponectin-based therapeutics [Citation290] could modulate both autophagy and inflammation which may improve outcomes following cardiovascular events such as pressure overload.
IL-1 signalling has long been known to contribute to inflammation and the development of heart failure [Citation293]. As a result, there is growing interest in targeting IL-1 signalling as a therapeutic for heart failure. There are currently three approved strategies for inhibiting IL-1 signalling that are clinically available. In the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) trial, inhibition of IL-1ß using a monoclonal antibody (canakinumab) was shown to demonstrate effectiveness against heart failure [Citation293].In the CANTOS randomised and double-blind clinical trial, 10,061 patients with prior history of myocardial infarction were assigned placebo or canakinumab administration every 3 months. In a 3.7-year follow-up, canakinumab reduced rates of inflammation and adverse cardiovascular events including myocardial infarction, stroke, and death. Rilonacept is a receptor antagonist which acts as a decoy to prevent il-1α and il-1ß from binding to the IL-1 receptor [Citation294]. In a phase 3 clinical trial, rilonacept was shown to prevent subsequent pericarditis and alleviated pericardial inflammation [Citation294]. Anakinra is another IL-1 receptor antagonist which functions through competitive inhibition. The use of anakinra in patients who suffered a prior myocardial infarction resulted in lowering of systemic inflammation, reduced progression of heart failure, and reduced all-cause death [Citation295,Citation296]. Therefore, IL-1ß/IL-1R targeted therapies remain a promising therapeutic strategy for providing cardioprotection through regulation of inflammation.
Trehalose, a disaccharide, displays cardioprotective effects by inducing autophagy. In a mouse model of myocardial infarction, trehalose boosted autophagy, leading to decreased apoptosis, fibrosis, and adverse cardiac remodelling. The positive impacts of trehalose were diminished in heterozygous beclin 1 knockout mice, underscoring its specificity for autophagy [Citation64]. These cardioprotective effects of trehalose were echoed in a rat model of myocardial infarction [Citation297]. Howeveer, a clinical study examined the impact of intravenous trehalose administration on atherogenesis in humans, revealing a safe profile but no notable reduction in arterial wall inflammation [Citation298]. The limited sample size suggests larger studies are necessary to comprehensively evaluate the compound’s efficacy in a clinical setting.
Statins have become the most widely prescribed medication globally as a preventative strategy against dyslipidemia and subsequent coronary heart diseases [Citation299]. Statins work by lowering serum cholesterol by inhibiting synthesis and promoting clearance. However, it is now known that statins can provide pleiotropic cardioprotective effects beyond lowering cholesterol [Citation299]. For instance, mitophagy has been demonstrated to play a crucial role in mediating the cardioprotective effects of the statin, simvastatin, against ischaemia/reperfusion injury [Citation300]. Specifically, upregulation of PINK1/Parkin-dependent mitophagy by simvastatin reduced infarct size. It was also demonstrated that the protective effect of statins against ischaemia/reperfusion (I/R) injury occurs in the early/acute phase of injury, suggesting that the timing of intervention will be a key determinant of success. Thus, modulation of mitophagy underlies the cardioprotective effects of statins in I/R injury.
AMPK could be a suitable therapeutic target for heart failure due to its implication in regulating autophagy, inflammation, and conferring cardioprotective effects. In addition to autophagy [Citation301], AMPK signalling is also known to facilitate the induction of mitophagy, which has also been shown to protect against heart failure [Citation302]. In failing hearts from heart failure patients or in vivo model of pressure overload, there was an observed decrease in expression and activity of AMPKα2 [Citation302]. Mechanistically, AMPKα2 interacted with PINK1 to increase cardiac mitophagy, decrease ROS, reduce cardiomyocyte apoptosis, and ultimately prevent the development of heart failure [Citation302]. Furthermore, AMPK has been implicated in providing anti-inflammatory properties. This is exemplified through the use of sodium glucose co-transporter 2 (SGLT2) inhibitors. Although SGLT2 inhibitors treats primarily diabetes, they have gained increasing attention for their off target cardioprotective properties [Citation303]. In fact, the SGLT2 inhibitor empagliflozin has demonstrated anti-inflammatory properties in cardiomyocytes and macrophages [Citation303]. In LPS-treated mice, empagliflozin prevented upregulation of proinflammatory cytokines, promoted M2 macrophage polarisation, and preserved cardiac contractility [Citation303]. The beneficial effects of empagliflozin were independent of its glucose lowering property and dependent on AMPK activation [Citation303].The cardioprotective effects of AMPK is further highlighted using PXL770, the first allosteric AMPK activator to demonstrate clinical safety and efficacy[Citation304]. Although it was originally developed for treatment of nonalcoholic steatohepatitis and metabolic diseases, it is now known to exert cardioprotective effects [Citation305]. In ZSF-1 rats, an in vivo model of heart failure with preserved ejection fraction (HFpEF), long term treatment with PXL770 preserved left ventricular function, reduced signs of cardiac fibrosis and hypertrophy [Citation305]. This suggests that enhancement of AMPK activation is a promising pharmacological target for preventing the development of heart failure ().
Figure 5. Targeting autophagy and inflammation in heart failure (A) Adiponectin is a cytokine that can affect both autophagy and inflammation. Adiponectin is positively correlated with autophagy upregulation. Adiponectin exhibits anti-inflammatory effects by inhibiting TLR-NF-kB signaling and promoting alternative macrophage polarization. (B) Activation of AMPK, such as via PXL770, upregulates autophagy, mitophagy, and exerts anti-inflammatory effects. These processes collectively provide protection against the development of heart failure.
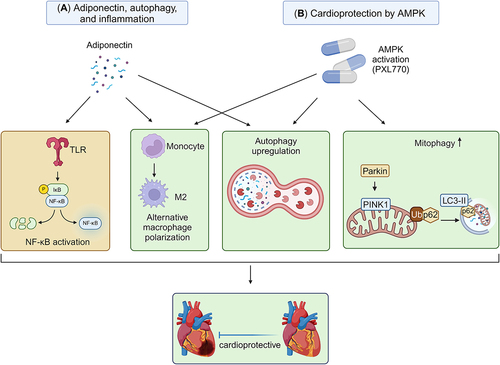
Conclusions and future perspectives
The interplay between sterile inflammation and autophagy has emerged as a crucial factor in heart failure development. Specifically, in models of ischaemia/reperfusion and pressure overload-induced injuries, the interaction between autophagy and sterile inflammation is mediated by DAMPs, TLRs, and NLRs. DAMPs, including mtDNA, can trigger a proinflammatory signal, a response typically countered by autophagy. TLR and NLR signalling, in turn, enhance autophagy as an adaptive measure to alleviate stress, such as that arising from mitochondrial damage. Additionally, the regulation of autophagy is context-specific, as both the deletion and upregulation of autophagy can offer protection depending on the stage of heart failure progression. The emerging role of mitochondrial autophagy, mitophagy, has been investigated, and it can have both detrimental and beneficial effects in cardiac pathologies (). However, evidence suggests that FUNDC1 may act as a potential mediator, switching mitophagy into a cardioprotective process. Therefore, further clarification is needed to fully understand the temporal changes in autophagy and sterile inflammation during heart failure. The clinical applications for targeting the interface of autophagy and inflammation in heart failure has been demonstrated. Therapeutics based on adiponectin show promise as a strategy due to its ability to promote autophagy, exhibit anti-inflammatory effects, and enhance cardiac outcomes in models of pressure overload. Similarly, AMPK activation mediates anti-inflammatory effects, autophagy upregulation, and confers protection against heart failure. In conclusion, autophagy and sterile inflammation are intricately linked in heart failure, and therapeutic strategies that simultaneously regulate these two processes may improve outcomes in patients with heart failure.
Figure 6. Interaction between autophagy and inflammation in heart failure The intricate interplay between sterile inflammation and autophagy is a central aspect of heart failure pathogenesis. This association encompasses the involvement of DAMPs (exemplified by mtDNA and HMGB1) and activation of inflammasomes which can be modulated by autophagy. Furthermore, the activation of inflammasomes results in amplification of autophagic activity to mitigate cellular damage from various stressors. Collectively, various forms of autophagy at the appropriate time and magnitude can be cardioprotective, whilst too much or too little are associated with worsening of adverse events.
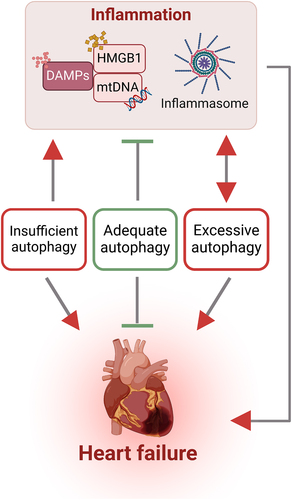
Acknowledgments
Related work in the authors laboratories is funded via research funding from Heart & Stroke Foundation and Canadian Institutes of Health Research (GS) and the Hong Kong Research Grants Council/Area of Excellence (AoE/M-707/18) (AX). We also thank Dana Al Rijjal for helpful text changes and Michael Sun for careful proof reading of the text.
Additional information
Funding
References
- Spencer LJ, Degu A, Kalkidan HA, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018 Nov 10;392(10159):1789–52. doi: 10.1016/S0140-6736(18)32279-7
- Yancy CW, Jessup M, Bozkurt B, et al. ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013 Oct 15;128(16):e240–327.
- Hartupee J, Mann DL. Neurohormonal activation in heart failure with reduced ejection fraction. Nat Rev Cardiol. 2017 Jan;14(1):30–38.
- Wu MY, Yiang GT, Liao WT, et al. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol Biochem. 2018;46(4):1650–1667. doi: 10.1159/000489241
- Nagalingam RS, Chattopadhyaya S, Al-Hattab DS, et al. Scleraxis and fibrosis in the pressure-overloaded heart. Eur Heart J. 2022 Dec 1;43(45):4739–4750.
- Mizushima N, Levine B, Longo DL. Autophagy in Human Diseases. N Engl J Med. 2020 Oct 15;383(16):1564–1576
- Delbridge LMD, Mellor KM, Taylor DJ, et al. Myocardial stress and autophagy: mechanisms and potential therapies. Nat Rev Cardiol. 2017 Jul;14(7):412–425. doi: 10.1038/nrcardio.2017.35
- Jahng JW, Turdi S, Kovacevic V, et al. Pressure Overload-Induced Cardiac Dysfunction in Aged Male Adiponectin Knockout Mice Is Associated With Autophagy Deficiency. Endocrinology. 2015 Jul;156(7):2667–2677. doi: 10.1210/en.2015-1162
- Ma X, Liu H, Foyil SR, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012 Jun 26;125(25):3170–3181.
- Mariño G, Pietrocola F, Kong Y, et al. Dimethyl α-ketoglutarate inhibits maladaptive autophagy in pressure overload-induced cardiomyopathy. Autophagy. 2014 May;10(5):930–932. doi: 10.4161/auto.28235
- Shirakabe A, Zhai P, Ikeda Y, et al. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation. 2016 Mar 29;133(13):1249–1263.
- Wellnitz K, Taegtmeyer H. Mechanical unloading of the failing heart exposes the dynamic nature of autophagy. Autophagy. 2010 Jan;6(1):155–156.
- Meng X, Yang J, Dong M, et al. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol. 2016 Mar;13(3):167–179. doi: 10.1038/nrcardio.2015.169
- Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. Cardiovasc Res. 2017 Mar 15;113(4):389–398.
- Wolf D, Ley K. Immunity and Inflammation in Atherosclerosis. Circ Res. 2019 Jan 18;124(2):315–327.
- Zhang Y, Bauersachs J, Langer HF. Immune mechanisms in heart failure. Eur J Heart Fail. 2017 Nov;19(11):1379–1389.
- Epelman S, Liu PP, Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. 2015 Feb;15(2):117–129.
- Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010 Dec;10(12):826–837.
- Gong T, Liu L, Jiang W, et al. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020 Feb;20(2):95–112. doi: 10.1038/s41577-019-0215-7
- Kologrivova I, Shtatolkina M, Suslova T, et al. Cells of the Immune System in Cardiac Remodeling: Main Players in Resolution of Inflammation and Repair After Myocardial Infarction. Front Immunol. 2021;12:664457.
- Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. 2016 Aug;15(8):551–567.
- Liu J, Wang H, Li J. Inflammation and Inflammatory Cells in Myocardial Infarction and Reperfusion Injury: A Double-Edged Sword. Clin Med Insights Cardiol. 2016;10:79–84.
- Saparov A, Ogay V, Nurgozhin T, et al. Role of the immune system in cardiac tissue damage and repair following myocardial infarction. Inflamm Res. 2017 Sep;66(9):739–751. doi: 10.1007/s00011-017-1060-4
- Cuervo AM, Bergamini E, Brunk UT, et al. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005 Oct-Dec;1(3):131–140. doi: 10.4161/auto.1.3.2017
- Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018 Jun;19(6):365–381.
- Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012 Apr;69(7):1125–1136.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008 Jan 11;132(1):27–42.
- Wang L, Klionsky DJ, Shen HM. The emerging mechanisms and functions of microautophagy. Nat Rev Mol Cell Biol. 2023 Mar;24(3):186–203.
- Sciarretta S, Maejima Y, Zablocki D, et al. The Role of Autophagy in the Heart. Annu Rev Physiol. 2018 Feb 10;80(1):1–26.
- Wesselborg S, Stork B. Autophagy signal transduction by ATG proteins: from hierarchies to networks. Cell Mol Life Sci. 2015 Dec;72(24):4721–4757.
- Diaz-Troya S, Perez-Perez ME, Florencio FJ, et al. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy. 2008 Oct;4(7):851–865. doi: 10.4161/auto.6555
- Banreti A, Sass M, Graba Y. The emerging role of acetylation in the regulation of autophagy. Autophagy. 2013 Jun 1;9(6):819–829.
- Lin MG, Hurley JH. Structure and function of the ULK1 complex in autophagy. Curr Opin Cell Biol. 2016 Apr;39:61–68.
- Martinez-Lopez N, Singh R. ATGs: Scaffolds for MAPK/ERK signaling. Autophagy. 2014 Mar;10(3):535–537.
- Zhou F, Wu Z, Zhao M, et al. Rab5-dependent autophagosome closure by ESCRT. J Cell Biol. 2019 Jun 3;218(6):1908–1927.
- Tian X, Teng J, Chen J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy. 2021 Oct;17(10):2680–2688.
- Langemeyer L, Frohlich F, Ungermann C. Rab GTPase Function in Endosome and Lysosome Biogenesis. Trends Cell Biol. 2018 Nov;28(11):957–970.
- Yang Z, Huang J, Geng J, et al. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol Biol Cell. 2006 Dec;17(12):5094–5104. doi: 10.1091/mbc.e06-06-0479
- Diao J, Liu R, Rong Y, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015 Apr 23;520(7548):563–566.
- Qiao L, Hu J, Qiu X, et al. LAMP2A, LAMP2B and LAMP2C: similar structures, divergent roles. Autophagy. 2023 Nov;19(11):2837–2852. doi: 10.1080/15548627.2023.2235196
- Szatmari Z, Sass M. The autophagic roles of Rab small GTPases and their upstream regulators: a review. Autophagy. 2014 Jul;10(7):1154–1166.
- Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018 Mar;20(3):233–242.
- Ichimura Y, Kumanomidou T, Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008 Aug 15;283(33):22847–22857.
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8(1): 3–5. Spring. doi: 10.1089/rej.2005.8.3.
- Hirota Y, Yamashita S, Kurihara Y, et al. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy. 2015;11(2):332–343. doi: 10.1080/15548627.2015.1023047
- Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010 Feb;12(2):119–131. doi: 10.1038/ncb2012
- Sadoshima J. Alternative mitophagy is a major form of mitophagy in the chronically stressed heart. Autophagy. 2022 Sep;18(9):2252–2253.
- Saito T, Nah J, Oka SI, et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Invest. 2019 Feb 1;129(2):802–819.
- Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012 Jan 22;14(2):177–185.
- Gibbings D, Mostowy S, Jay F, et al. Selective autophagy degrades DICER and AGO2 and regulates miRNA activity. Nat Cell Biol. 2012 Dec;14(12):1314–1321. doi: 10.1038/ncb2611
- Gatica D, Chiong M, Lavandero S, et al. Molecular mechanisms of autophagy in the cardiovascular system. Circ Res. 2015 Jan 30;116(3):456–467.
- Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and Mitophagy in Cardiovascular Disease. Circ Res. 2017 May 26;120(11):1812–1824.
- Lavandero S, Troncoso R, Rothermel BA, et al. Cardiovascular autophagy: concepts, controversies, and perspectives. Autophagy. 2013 Oct;9(10):1455–1466. doi: 10.4161/auto.25969
- Morales PE, Arias-Duran C, Avalos-Guajardo Y, et al. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol Aspects Med. 2020 Feb;71:100822.
- Bergmann O, Zdunek S, Felker A, et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell. 2015 Jun 18;161(7):1566–1575.
- Shirakabe A, Ikeda Y, Sciarretta S, et al. Aging and Autophagy in the Heart. Circ Res. 2016 May 13;118(10):1563–1576.
- Nakai A, Yamaguchi O, Takeda T, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007 May 13(5):619–624. doi: 10.1038/nm1574
- Onishi M, Yamano K, Sato M, et al. Molecular mechanisms and physiological functions of mitophagy. Embo j. 2021 Feb 1;40(3):e104705.
- Luan Y, Luan Y, Feng Q, et al. Emerging Role of Mitophagy in the Heart: Therapeutic Potentials to Modulate Mitophagy in Cardiac Diseases. Oxid Med Cell Longev. 2021;2021:3259963.
- Gurkar AU, Chu K, Raj L, et al. Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nat Commun. 2013;4(1):2189. doi: 10.1038/ncomms3189
- Sciarretta S, Zhai P, Shao D, et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012 Mar 6;125(9):1134–1146.
- Abdellatif M, Sedej S, Carmona-Gutierrez D, et al. Autophagy in Cardiovascular Aging. Circ Res. 2018 Sep 14;123(7):803–824.
- Zhai P, Sadoshima J. Glycogen synthase kinase-3β controls autophagy during myocardial ischemia and reperfusion. Autophagy. 2012 Jan;8(1):138–139.
- Sciarretta S, Yee D, Nagarajan N, et al. Trehalose-Induced Activation of Autophagy Improves Cardiac Remodeling After Myocardial Infarction. J Am Coll Cardiol. 2018 May 8;71(18):1999–2010.
- Valentim L, Laurence KM, Townsend PA, et al. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol. 2006 Jun;40(6):846–852. doi: 10.1016/j.yjmcc.2006.03.428
- Matsui Y, Takagi H, Qu X, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007 Mar 30;100(6):914–922.
- Ma X, Liu H, Foyil SR, et al. Autophagy is impaired in cardiac ischemia-reperfusion injury. Autophagy. 2012 Sep;8(9):1394–1396. doi: 10.4161/auto.21036
- Sciarretta S, Zhai P, Shao D, et al. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ Res. 2013 Nov 8;113(11):1253–1264.
- Matsushima S, Kuroda J, Zhai P, et al. Tyrosine kinase FYN negatively regulates NOX4 in cardiac remodeling. J Clin Invest. 2016 Sep 1;126(9):3403–3416.
- Yan L, Vatner DE, Kim SJ, et al. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005 Sep 27;102(39):13807–13812.
- Huang L, Dai K, Chen M, et al. The AMPK Agonist PT1 and mTOR Inhibitor 3HOI-BA-01 Protect Cardiomyocytes After Ischemia Through Induction of Autophagy. J Cardiovasc Pharmacol Ther. 2016 Jan;21(1):70–81. doi: 10.1177/1074248415581177
- Li S, Liu C, Gu L, et al. Autophagy protects cardiomyocytes from the myocardial ischaemia-reperfusion injury through the clearance of CLP36. Open Biol. 2016 Aug;6(8):160177. doi: 10.1098/rsob.160177
- Kanamori H, Takemura G, Goto K, et al. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am J Physiol Heart Circ Physiol. 2011 Jun;300(6):H2261–H2271. doi: 10.1152/ajpheart.01056.2010
- Wei YM, Li X, Xu M, et al. Enhancement of autophagy by simvastatin through inhibition of Rac1-mTOR signaling pathway in coronary arterial myocytes. Cell Physiol Biochem. 2013;31(6):925–937. doi: 10.1159/000350111
- Nah J, Zhai P, Huang CY, et al. Upregulation of Rubicon promotes autosis during myocardial ischemia/reperfusion injury. J Clin Invest. 2020 Jun 1;130(6):2978–2991.
- Wu S, Chang G, Gao L, et al. Trimetazidine protects against myocardial ischemia/reperfusion injury by inhibiting excessive autophagy. J Mol Med (Berl). 2018 Aug 96(8):791–806. doi: 10.1007/s00109-018-1664-3
- Hall AR, Burke N, Dongworth RK, et al. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016 May 26;7(5):e2238–e2238.
- Ong SB, Subrayan S, Lim SY, et al. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010 May 11;121(18):2012–2022.
- Diwan A, Krenz M, Syed FM, et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007 Oct;117(10):2825–2833. doi: 10.1172/JCI32490
- Zhou H, Zhu P, Wang J, et al. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018 Jun;25(6):1080–1093. doi: 10.1038/s41418-018-0086-7
- Wang J, Zhu P, Li R, et al. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020 Feb;30:101415.
- Sanz J, Sánchez-Quintana D, Bossone E, et al. Anatomy, Function, and Dysfunction of the Right Ventricle: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019 Apr 2;73(12):1463–1482.
- Zhang L, Jaswal JS, Ussher JR, et al. Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ Heart Fail. 2013 Sep 1;6(5):1039–1048.
- Huo S, Shi W, Ma H, et al. Alleviation of Inflammation and Oxidative Stress in Pressure Overload-Induced Cardiac Remodeling and Heart Failure via IL-6/STAT3 Inhibition by Raloxifene. Oxid Med Cell Longev. 2021;2021:6699054.
- Zhu H, Tannous P, Johnstone JL, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007 Jul;117(7):1782–1793. doi: 10.1172/JCI27523
- Cao Z, Liu J, Zhao Z, et al. miR-16-5p Regulates PTPN4 and Affects Cardiomyocyte Apoptosis and Autophagy Induced by Hypoxia/Reoxygenation. Evid Based Complement Alternat Med. 2021;2021:5599031.
- Nah J, Shirakabe A, Mukai R, et al. Ulk1-dependent alternative mitophagy plays a protective role during pressure overload in the heart. Cardiovasc Res. 2022 Sep 20;118(12):2638–2651.
- Li W, Yin L, Sun X, et al. Alpha-lipoic acid protects against pressure overload-induced heart failure via ALDH2-dependent Nrf1-FUNDC1 signaling. Cell Death Dis. 2020 Jul 30;11(7):599.
- Saito T, Hamano K, Sadoshima J. Molecular mechanisms and clinical implications of multiple forms of mitophagy in the heart. Cardiovasc Res. 2021 Dec 17;117(14):2730–2741.
- Wang X, Guo Z, Ding Z, et al. Inflammation, Autophagy, and Apoptosis After Myocardial Infarction. J Am Heart Assoc. 2018 Apr 21;7(9). doi: 10.1161/JAHA.117.008024
- Lopez-Crisosto C, Pennanen C, Vasquez-Trincado C, et al. Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat Rev Cardiol. 2017 Jun 14(6):342–360. doi: 10.1038/nrcardio.2017.23
- Rothermel BA, Hill JA. Autophagy in load-induced heart disease. Circ Res. 2008 Dec 5;103(12):1363–1369
- Wu D, Zhang K, Hu P. The Role of Autophagy in Acute Myocardial Infarction. Front Pharmacol. 2019;10:551.
- Li L, Fu W, Gong X, et al. The role of G protein-coupled receptor kinase 4 in cardiomyocyte injury after myocardial infarction. Eur Heart J. 2021 Apr 7;42(14):1415–1430.
- Wu X, He L, Chen F, et al. Impaired autophagy contributes to adverse cardiac remodeling in acute myocardial infarction. PLoS One. 2014;9(11):e112891. doi: 10.1371/journal.pone.0112891
- Wu X, Zheng D, Qin Y, et al. Nobiletin attenuates adverse cardiac remodeling after acute myocardial infarction in rats via restoring autophagy flux. Biochem Biophys Res Commun. 2017 Oct 14;492(2):262–268.
- Zhang X, Wang Q, Wang X, et al. Tanshinone IIA protects against heart failure post-myocardial infarction via AMPKs/mTOR-dependent autophagy pathway. Biomed Pharmacother. 2019 Apr;112:108599.
- Wang D, Lv L, Xu Y, et al. Cardioprotection of Panax Notoginseng saponins against acute myocardial infarction and heart failure through inducing autophagy. Biomed Pharmacother. 2021 Apr 136:111287.
- Foglio E, Puddighinu G, Germani A, et al. HMGB1 Inhibits Apoptosis Following MI and Induces Autophagy via mTORC1 Inhibition. J Cell Physiol. 2017 May;232(5):1135–1143. doi: 10.1002/jcp.25576
- Buss SJ, Riffel JH, Katus HA, et al. Augmentation of autophagy by mTOR-inhibition in myocardial infarction: When size matters. Autophagy. 2010 Feb;6(2):304–306. doi: 10.4161/auto.6.2.11135
- Ramachandra CJA, Hernandez-Resendiz S, Crespo-Avilan GE, et al. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine. 2020 Jul;57:102884.
- Ajoolabady A, Chiong M, Lavandero S, et al. Mitophagy in cardiovascular diseases: molecular mechanisms, pathogenesis, and treatment. Trends Mol Med. 2022 Oct;28(10):836–849. doi: 10.1016/j.molmed.2022.06.007
- Marinkovic M, Sprung M, Novak I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy. 2021 May;17(5):1232–1243.
- Lampert MA, Orogo AM, Najor RH, et al. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy. 2019 Jul;15(7):1182–1198. doi: 10.1080/15548627.2019.1580095
- Hoshino A, Matoba S, Iwai-Kanai E, et al. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J Mol Cell Cardiol. 2012 Jan;52(1):175–184. doi: 10.1016/j.yjmcc.2011.10.008
- Chen G, Han Z, Feng D, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014 May 8;54(3):362–377.
- Zhang W, Ren H, Xu C, et al. Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife. 2016 Dec 20;5. 5. doi: 10.7554/eLife.21407
- Ji W, Wei S, Hao P, et al. Aldehyde Dehydrogenase 2 Has Cardioprotective Effects on Myocardial Ischaemia/Reperfusion Injury via Suppressing Mitophagy. Front Pharmacol. 2016;7:101.
- Xin T, Lu C. Irisin activates Opa1-induced mitophagy to protect cardiomyocytes against apoptosis following myocardial infarction. Aging (Albany NY). 2020 Mar 10;12(5):4474–4488.
- Kubli DA, Zhang X, Lee Y, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013 Jan 11;288(2):915–926.
- Kanamori H, Takemura G, Goto K, et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy. 2015;11(7):1146–1160. doi: 10.1080/15548627.2015.1051295
- Madonna R, Moscato S, Cufaro MC, et al. Empagliflozin inhibits excessive autophagy through the AMPK/GSK3beta signalling pathway in diabetic cardiomyopathy. Cardiovasc Res. 2023 May 22;119(5):1175–1189.
- Wang Y, Liang B, Lau WB, et al. Restoring diabetes-induced autophagic flux arrest in ischemic/reperfused heart by ADIPOR (adiponectin receptor) activation involves both AMPK-dependent and AMPK-independent signaling. Autophagy. 2017;13(11):1855–1869. doi: 10.1080/15548627.2017.1358848
- Tong M, Saito T, Zhai P, et al. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ Res. 2019 Apr 26;124(9):1360–1371.
- Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008 Feb 29;102(4):401–414.
- Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, et al. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018 Jan 25;9(2):119.
- Wilson AJ, Gill EK, Abudalo RA, et al. Reactive oxygen species signalling in the diabetic heart: emerging prospect for therapeutic targeting. Heart. 2018 Feb;104(4):293–299. doi: 10.1136/heartjnl-2017-311448
- Sung MM, Hamza SM, Dyck JR. Myocardial metabolism in diabetic cardiomyopathy: potential therapeutic targets. Antioxid Redox Signal. 2015 Jun 10;22(17):1606–1630.
- Xu X, Hua Y, Nair S, et al. Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. J Mol Cell Biol. 2013 Feb;5(1):61–63. doi: 10.1093/jmcb/mjs055
- Tang Y, Liu J, Long J. Phosphatase and tensin homolog-induced putative kinase 1 and Parkin in diabetic heart: Role of mitophagy. J Diabetes Investig. 2015 May;6(3):250–255.
- Nishida Y, Arakawa S, Fujitani K, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009 Oct 1;461(7264):6548.
- Andonegui G, Zhou H, Bullard D, et al. Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J Clin Invest. 2009 Jul;119(7):1921–1930. doi: 10.1172/jci36411
- Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008 Jul;48(1):322–335.
- Liu Y, Gao XM, Fang L, et al. Novel role of platelets in mediating inflammatory responses and ventricular rupture or remodeling following myocardial infarction. Arterioscler Thromb Vasc Biol. 2011 Apr;31(4):834–841. doi: 10.1161/ATVBAHA.110.220467
- Gurses KM, Kocyigit D, Yalcin MU, et al. Enhanced Platelet Toll-like Receptor 2 and 4 Expression in Acute Coronary Syndrome and Stable Angina Pectoris. Am J Cardiol. 2015 Dec 1;116(11):1666–1671.
- Schanze N, Bode C, Duerschmied D. Platelet Contributions to Myocardial Ischemia/Reperfusion Injury. Front Immunol. 2019;10:1260.
- Mirabet M, Garcia-Dorado D, Inserte J, et al. Platelets activated by transient coronary occlusion exacerbate ischemia-reperfusion injury in rat hearts. Am J Physiol Heart Circ Physiol. 2002 Sep;283(3):H1134–H1141. doi: 10.1152/ajpheart.00065.2002
- Weyrich AS, Ma XY, Lefer DJ, et al. In vivo neutralization of P-selectin protects feline heart and endothelium in myocardial ischemia and reperfusion injury. J Clin Invest. 1993 Jun;91(6):2620–2629. doi: 10.1172/JCI116501
- Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012 Mar 1;4(3):a006049–a006049.
- Yanai H, Ban T, Wang Z, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009 Nov 5;462(7269):99–103.
- Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010 Sep 6;190(5):881–892.
- Tsung A, Sahai R, Tanaka H, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005 Apr 4;201(7):1135–1143.
- Kim S, Kim SY, Pribis JP, et al. Signaling of high mobility group box 1 (HMGB1) through toll-like receptor 4 in macrophages requires CD14. Mol Med. 2013 May 20;19(1):88–98.
- Sims GP, Rowe DC, Rietdijk ST, et al. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28(1):367–388. doi: 10.1146/annurev.immunol.021908.132603
- Ling Y, Yang ZY, Yin T, et al. Heparin changes the conformation of high-mobility group protein 1 and decreases its affinity toward receptor for advanced glycation endproducts in vitro. Int Immunopharmacol. 2011 Feb;11(2):187–193. doi: 10.1016/j.intimp.2010.11.014
- He M, Bianchi ME, Coleman TR, et al. Exploring the biological functional mechanism of the HMGB1/TLR4/MD-2 complex by surface plasmon resonance. Mol Med. 2018 May 10;24(1):21.
- Tang D, Shi Y, Kang R, et al. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J Leukoc Biol. 2007 Mar;81(3):741–747. doi: 10.1189/jlb.0806540
- Andreeva L, Hiller B, Kostrewa D, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature. 2017 Sep 21;549(7672):394–398.
- Tirone M, Tran NL, Ceriotti C, et al. High mobility group box 1 orchestrates tissue regeneration via CXCR4. J Exp Med. 2018 Jan 2;215(1):303–318.
- Yan C, Duanmu X, Zeng L, et al. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells. 2019 Apr 25;8(4):379.
- Picca A, Lezza AM. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: Useful insights from aging and calorie restriction studies. Mitochondrion. 2015 Nov;25:67–75.
- Bonawitz ND, Clayton DA, Shadel GS. Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol Cell. 2006 Dec 28;24(6):813–825.
- Shimada K, Crother TR, Karlin J, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012 Mar 23;36(3):401–414.
- Ohto U, Shibata T, Tanji H, et al. Structural basis of CpG and inhibitory DNA recognition by Toll-like receptor 9. Nature. 2015 Apr 30;520(7549):702–705.
- Zhong Z, Liang S, Sanchez-Lopez E, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018 Aug;560(7717):198–203. doi: 10.1038/s41586-018-0372-z
- Xian H, Watari K, Sanchez-Lopez E, et al. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity. 2022 Aug 9;55(8):1370–1385.e8.
- Aarreberg LD, Esser-Nobis K, Driscoll C, et al. Interleukin-1β Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol Cell. 2019 May 16;74(4):801–815.e6.
- Marchi S, Guilbaud E, Tait SWG, et al. Mitochondrial control of inflammation. Nat Rev Immunol. 2023 Mar;23(3):159–173. doi: 10.1038/nrmicro2070
- Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016 Jul;16(7):407–420.
- Mauro AG, Bonaventura A, Mezzaroma E, et al. NLRP3 Inflammasome in Acute Myocardial Infarction. J Cardiovasc Pharmacol. 2019 Sep;74(3):175–187. doi: 10.1097/FJC.0000000000000717
- Mishra SR, Mahapatra KK, Behera BP, et al. Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int J Biochem Cell Biol. 2021 Jul;136:106013.
- Jo EK, Kim JK, Shin DM, et al. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016 Mar;13(2):148–159. doi: 10.1038/cmi.2015.95
- Rock KL, Latz E, Ontiveros F, et al. The sterile inflammatory response. Annu Rev Immunol. 2010;28(1):321–342. doi: 10.1146/annurev-immunol-030409-101311.
- Kawaguchi M, Takahashi M, Hata T, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011 Feb 15;123(6):594–604.
- Toldo S, Mezzaroma E, Buckley LF, et al. Targeting the NLRP3 inflammasome in cardiovascular diseases. Pharmacol Ther. 2022 Aug;236:108053.
- Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009 Feb;7(2):99–109.
- Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018 Apr;15(4):203–214.
- Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med. 2010 Nov;14(11):2592–2603.
- Walsh MC, Lee J, Choi Y. Tumor necrosis factor receptor-associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol Rev. 2015 Jul;266(1):72–92.
- Mitchell S, Vargas J, Hoffmann A. Signaling via the NFkappaB system. Wiley Interdiscip Rev Syst Biol Med. 2016 May;8(3):227–241.
- Feldman N, Rotter-Maskowitz A, Okun E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res Rev. 2015 Nov;24(Pt A):29–39.
- Ullah MO, Sweet MJ, Mansell A, et al. TRIF-dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J Leukoc Biol. 2016 Jul;100(1):27–45. doi: 10.1189/jlb.2RI1115-531R
- Chen JQ, Szodoray P, Zeher M. Toll-Like Receptor Pathways in Autoimmune Diseases. Clin Rev Allergy Immunol. 2016 Feb;50(1):1–17.
- Castiglione V, Aimo A, Vergaro G, et al. Biomarkers for the diagnosis and management of heart failure. Heart Fail Rev. 2022 Mar;27(2):625–643. doi: 10.1007/s10741-021-10105-w
- Coggins M, Rosenzweig A. The fire within: cardiac inflammatory signaling in health and disease. Circ Res. 2012 Jan 6;110(1):116–125.
- Marchant DJ, Boyd JH, Lin DC, et al. Inflammation in myocardial diseases. Circ Res. 2012 Jan 6;110(1):126–144.
- Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008 Jul 24;454(7203):428–435.
- Maekawa Y, Anzai T, Yoshikawa T, et al. Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction:a possible role for left ventricular remodeling. J Am Coll Cardiol. 2002 Jan 16;39(2):241–246.
- Arslan F, Smeets MB, O’Neill LA, et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation. 2010 Jan 5;121(1):80–90.
- Sakata Y, Dong JW, Vallejo JG, et al. Toll-like receptor 2 modulates left ventricular function following ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007 Jan;292(1):H503–H509. doi: 10.1152/ajpheart.00642.2006
- Fairweather D, Yusung S, Frisancho S, et al. IL-12 receptor beta 1 and Toll-like receptor 4 increase IL-1 beta- and IL-18-associated myocarditis and coxsackievirus replication. J Immunol. 2003 May 1;170(9):4731–4737.
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002 Jul 11;418(6894):191–195.
- Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005 Apr;5(4):331–342.
- Kohno T, Anzai T, Naito K, et al. Role of high-mobility group box 1 protein in post-infarction healing process and left ventricular remodelling. Cardiovasc Res. 2009 Feb 15;81(3):565–573.
- Goldstein RS, Gallowitsch-Puerta M, Yang L, et al. Elevated high-mobility group box 1 levels in patients with cerebral and myocardial ischemia. Shock. 2006 Jun;25(6):571–574. doi: 10.1097/01.shk.0000209540.99176.72
- Bradham WS, Bozkurt B, Gunasinghe H, et al. Tumor necrosis factor-alpha and myocardial remodeling in progression of heart failure: a current perspective. Cardiovasc Res. 2002 Mar;53(4):822–830. doi: 10.1016/S0008-6363(01)00503-X
- Levine B, Kalman J, Mayer L, et al. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990 Jul 26;323(4):236–241.
- Merkle S, Frantz S, Schön MP, et al. A role for caspase-1 in heart failure. Circ Res. 2007 Mar 16;100(5):645–653.
- Deswal A, Petersen NJ, Feldman AM, et al. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation. 2001 Apr 24;103(16):2055–2059.
- Bracey NA, Beck PL, Muruve DA, et al. The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Exp Physiol. 2013 Feb;98(2):462–472. doi: 10.1113/expphysiol.2012.068338
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017 Sep 21;377(12):1119–1131.
- Chung ES, Packer M, Lo KH, et al. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003 Jul 1;107(25):3133–3140.
- Mann DL, McMurray JJ, Packer M, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation. 2004 Apr 6;109(13):1594–1602.
- Lutgens E, Daemen MJAP, de Muinck ED, et al. Chronic myocardial infarction in the mouse: cardiac structural and functional change1. Cardiovasc Res. 1999;41(3):586–593. doi: 10.1016/S0008-6363(98)00216-8
- Thygesen K, Alpert JS, Jaffe AS, et al. Fourth Universal Definition of Myocardial Infarction (2018). Circulation. 2018 Nov 13;138(20):e618–e651.
- Bliksøen M, Mariero LH, Torp MK, et al. Extracellular mtDNA activates NF-κB via toll-like receptor 9 and induces cell death in cardiomyocytes. Basic Res Cardiol. 2016 Jul;111(4):42. doi: 10.1007/s00395-016-0553-6
- Krysko DV, Agostinis P, Krysko O, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011 Apr;32(4):157–164. doi: 10.1016/j.it.2011.01.005
- Lipps C, Nguyen JH, Pyttel L, et al. N-terminal fragment of cardiac myosin binding protein-C triggers pro-inflammatory responses in vitro. J Mol Cell Cardiol. 2016 Oct;99:47–56.
- Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res. 2016 Jun 24;119(1):91–112.
- Frangogiannis NG, Rosenzweig A. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012 Jan 6;110(1):159–173.
- Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ Res. 2011 Apr 29;108(9):1122–1132.
- Bujak M, Dobaczewski M, Chatila K, et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008 Jul;173(1):57–67. doi: 10.2353/ajpath.2008.070974
- Müller J, Gorressen S, Grandoch M, et al. Interleukin-6-dependent phenotypic modulation of cardiac fibroblasts after acute myocardial infarction. Basic Res Cardiol. 2014;109(6):440. doi: 10.1007/s00395-014-0440-y
- Lindsey ML, Bolli R, Canty JM Jr, et al. Guidelines for experimental models of myocardial ischemia and infarction. Am J Physiol Heart Circ Physiol. 2018 Apr 1;314(4):H812–H838.
- Armstrong PW, Collen D, Antman E. Fibrinolysis for acute myocardial infarction: the future is here and now. Circulation. 2003 May 27;107(20):2533–2537.
- Virani SS, Alonso A, Benjamin EJ, et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation. 2020 Mar 3;141(9):e139–e596.
- Heusch G, Gersh BJ. The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. Eur Heart J. 2017 Mar 14;38(11):774–784.
- Eltzschig HK, Bratton DL, Colgan SP. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discov. 2014 Nov;13(11):852–869.
- Lin L, Knowlton AA. Innate immunity and cardiomyocytes in ischemic heart disease. Life Sci. 2014 Mar 28;100(1):1–8.
- Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014 May;11(5):255–265.
- Mitchell JA, Ryffel B, Quesniaux VF, et al. Role of pattern-recognition receptors in cardiovascular health and disease. Biochem Soc Trans. 2007 Dec;35(Pt 6):1449–1452. doi: 10.1042/BST0351449
- Suetomi T, Willeford A, Brand CS, et al. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca(2+)/Calmodulin-Dependent Protein Kinase II δ Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation. 2018 Nov 27;138(22):2530–2544.
- Christia P, Frangogiannis NG. Targeting inflammatory pathways in myocardial infarction. Eur J Clin Invest. 2013 Sep;43(9):986–995.
- Turillazzi E, Di Paolo M, Neri M, et al. A theoretical timeline for myocardial infarction: immunohistochemical evaluation and western blot quantification for Interleukin-15 and Monocyte chemotactic protein-1 as very early markers. J Transl Med. 2014 Jul 2;12(1):188.
- Ortega-Gómez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med. 2013 May;5(5):661–674.
- Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, et al. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res Cardiol. 2018 Jun 4;113(4):26.
- Frantz S, Bauersachs J, Ertl G. Post-infarct remodelling: contribution of wound healing and inflammation. Cardiovasc Res. 2009 Feb 15;81(3):474–481.
- Riley JS, Quarato G, Cloix C, et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. Embo j. 2018 Sep 3;37(17). doi: 10.15252/embj.201899238
- Lam AR, Bert NL, Ho SS, et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res. 2014 Apr 15;74(8):2193–2203.
- Matsumoto K, Obana M, Kobayashi A, et al. Blockade of NKG2D/NKG2D ligand interaction attenuated cardiac remodelling after myocardial infarction. Cardiovasc Res. 2019 Mar 15;115(4):765–775.
- Toldo S, Mauro AG, Cutter Z, et al. The NLRP3 Inflammasome Inhibitor, OLT1177 (Dapansutrile), Reduces Infarct Size and Preserves Contractile Function After Ischemia Reperfusion Injury in the Mouse. J Cardiovasc Pharmacol. 2019 Apr;73(4):215–222. doi: 10.1097/FJC.0000000000000658
- Marchetti C, Toldo S, Chojnacki J, et al. Pharmacologic Inhibition of the NLRP3 Inflammasome Preserves Cardiac Function After Ischemic and Nonischemic Injury in the Mouse. J Cardiovasc Pharmacol. 2015 Jul;66(1):1–8. doi: 10.1097/FJC.0000000000000247
- Liu Y, Lian K, Zhang L, et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res Cardiol. 2014;109(5):415. doi: 10.1007/s00395-014-0415-z.
- Myung J, Beom JH, Kim JH, et al. Recombinant Klotho Protein Ameliorates Myocardial Ischemia/Reperfusion Injury by Attenuating Sterile Inflammation. Biomedicines. 2022 Apr 13;10(4). 894 10.3390/biomedicines10040894
- Veltman D, Wu M, Pokreisz P, et al. Clec4e-Receptor Signaling in Myocardial Repair After Ischemia-Reperfusion Injury. JACC Basic Transl Sci. 2021 Aug;6(8):631–646. doi: 10.1016/j.jacbts.2021.07.001
- Levy D, Garrison RJ, Savage DD, et al. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990 May 31;322(22):1561–1566.
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006 Aug;7(8):589–600.
- Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. 2015 Mar 27;116(7):1254–1268.
- Chen WY, Hong J, Gannon J, et al. Myocardial pressure overload induces systemic inflammation through endothelial cell IL-33. Proc Natl Acad Sci U S A. 2015 Jun 9;112(23):7249–7254.
- O’Brien M, Baicu CF, Van Laer AO, et al. Pressure overload generates a cardiac-specific profile of inflammatory mediators. Am J Physiol Heart Circ Physiol. 2020 Aug 1;319(2):H331–H340.
- Bossuyt J, Helmstadter K, Wu X, et al. Ca2+/calmodulin-dependent protein kinase IIdelta and protein kinase D overexpression reinforce the histone deacetylase 5 redistribution in heart failure. Circ Res. 2008 Mar 28;102(6):695–702.
- Sossalla S, Fluschnik N, Schotola H, et al. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res. 2010 Oct 29;107(9):1150–1161.
- Willeford A, Suetomi T, Nickle A, et al. CaMKIIdelta-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight. 2018 Jun 21;3(12). doi: 10.1172/jci.insight.97054
- Higashikuni Y, Liu W, Numata G, et al. NLRP3 Inflammasome Activation Through Heart-Brain Interaction Initiates Cardiac Inflammation and Hypertrophy During Pressure Overload. Circulation. 2023 Jan 24;147(4):338–355.
- Suetomi T, Willeford A, Brand CS, et al. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca(2+)/Calmodulin-Dependent Protein Kinase II delta Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation. 2018 Nov 27;138(22):2530–2544. doi: 10.1161/CIRCULATIONAHA.118.034621
- Yoshida K, Abe K, Ishikawa M, et al. Inhibition of TLR9-NF-κB-mediated sterile inflammation improves pressure overload-induced right ventricular dysfunction in rats. Cardiovasc Res. 2019 Mar 1;115(3):658–668.
- Omiya S, Omori Y, Taneike M, et al. Cytokine mRNA Degradation in Cardiomyocytes Restrains Sterile Inflammation in Pressure-Overloaded Hearts. Circulation. 2020 Feb 25;141(8):667–677.
- Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia-and insulin-resistance-induced heart disease. Diabetologia. 2018 Jan;61(1):21–28.
- Hofmann U, Frantz S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ Res. 2015 Jan 16;116(2):354–367.
- McMaster WG, Kirabo A, Madhur MS, et al. Inflammation, immunity, and hypertensive end-organ damage. Circ Res. 2015 Mar 13;116(6):1022–1033.
- SantaCruz-Calvo S, Bharath L, Pugh G, et al. Adaptive immune cells shape obesity-associated type 2 diabetes mellitus and less prominent comorbidities. Nat Rev Endocrinol. 2022 Jan;18(1):23–42. doi: 10.1038/s41574-021-00575-1
- Mori J, Alrob OA, Wagg CS, et al. ANG II causes insulin resistance and induces cardiac metabolic switch and inefficiency: a critical role of PDK4. Am J Physiol Heart Circ Physiol. 2013 Apr 15;304(8):H1103–H1113.
- Tschope C, Walther T, Escher F, et al. Transgenic activation of the kallikrein-kinin system inhibits intramyocardial inflammation, endothelial dysfunction and oxidative stress in experimental diabetic cardiomyopathy. FASEB J. 2005 Dec;19(14):2057–2059. doi: 10.1096/fj.05-4095fje
- Luo B, Li B, Wang W, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One. 2014;9(8):e104771. doi: 10.1371/journal.pone.0104771
- Luo B, Li B, Wang W, et al. Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model. Cardiovasc Drugs Ther. 2014 Feb;28(1):33–43. doi: 10.1007/s10557-013-6498-1
- Ye Y, Bajaj M, Yang HC, et al. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc Drugs Ther. 2017 Apr;31(2):119–132. doi: 10.1007/s10557-017-6725-2
- Shen L, Li L, Li M, et al. Silencing of NOD2 protects against diabetic cardiomyopathy in a murine diabetes model. Int J Mol Med. 2018 Dec;42(6):3017–3026. doi: 10.3892/ijmm.2018.3880
- Prieto P, Vallejo-Cremades MT, Benito G, et al. NOD1 receptor is up-regulated in diabetic human and murine myocardium. Clin Sci (Lond). 2014 Dec;127(12):665–677. doi: 10.1042/CS20140180
- Li X, Du N, Zhang Q, et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. 2014 Oct 23;5(10):e1479–e1479.
- Xie Y, Huang Y, Ling X, et al. Chemerin/CMKLR1 Axis Promotes Inflammation and Pyroptosis by Activating NLRP3 Inflammasome in Diabetic Cardiomyopathy Rat. Front Physiol. 2020;11:381.
- Tang D, Kang R, Cheh CW, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010 Sep 23;29(38):5299–5310.
- Liu L, Yang M, Kang R, et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia. 2011 Jan;25(1):23–31. doi: 10.1038/leu.2010.225
- Bianchi ME. HMGB1 loves company. J Leukoc Biol. 2009 Sep;86(3):573–576.
- Shi CS, Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem. 2008 Nov 28;283(48):33175–33182.
- Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010 May 25;3(123):ra42.
- Zalckvar E, Berissi H, Mizrachy L, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009 Mar;10(3):285–292. doi: 10.1038/embor.2008.246
- Kang R, Zeh HJ, Lotze MT, et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011 Apr;18(4):571–580. doi: 10.1038/cdd.2010.191
- Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010 Jan;11(1):55–62. doi: 10.1038/ni.1823
- Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008 Nov 13;456(7219):2648.
- Shi CS, Shenderov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012 Jan 29;13(3):255–263.
- Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011 Mar;12(3):222–230. doi: 10.1038/ni.1980
- Jounai N, Kobiyama K, Shiina M, et al. NLRP4 negatively regulates autophagic processes through an association with beclin1. J Immunol. 2011 Feb 1;186(3):1646–1655.
- Dupont N, Jiang S, Pilli M, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011 Nov 8;30(23):4701–4711.
- Pellegrini L, Foglio E, Pontemezzo E, et al. HMGB1 and repair: focus on the heart. Pharmacol Ther. 2019 Apr;196:160–182.
- Volz HC, Seidel C, Laohachewin D, et al. HMGB1: the missing link between diabetes mellitus and heart failure. Basic Res Cardiol. 2010 Nov;105(6):805–820. doi: 10.1007/s00395-010-0114-3
- Banoth B, Cassel SL. Mitochondria in innate immune signaling. Transl Res. 2018 Dec;202:52–68.
- Kitahara T, Takeishi Y, Harada M, et al. High-mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovasc Res. 2008 Oct 1;80(1):40–46.
- Bangert A, Andrassy M, Müller AM, et al. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc Natl Acad Sci U S A. 2016 Jan 12;113(2):E155–64.
- Zhao W, Li Y, Jia L, et al. Atg5 deficiency-mediated mitophagy aggravates cardiac inflammation and injury in response to angiotensin II. Free Radic Biol Med. 2014 Apr;69:108–115.
- Horckmans M, Ring L, Duchene J, et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. 2017 Jan 14;38(3):187–197.
- Riffelmacher T, Clarke A, Richter FC, et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity. 2017 Sep 19;47(3):466–480 e5.
- Reglero-Real N, Perez-Gutierrez L, Yoshimura A, et al. Autophagy modulates endothelial junctions to restrain neutrophil diapedesis during inflammation. Immunity. 2021 Sep 14;54(9):1989–2004 e9.
- Steffens S, Nahrendorf M, Madonna R. Immune cells in cardiac homeostasis and disease: emerging insights from novel technologies. Eur Heart J. 2022 Apr 19;43(16):1533–1541.
- Javaheri A, Bajpai G, Picataggi A, et al. TFEB activation in macrophages attenuates postmyocardial infarction ventricular dysfunction independently of ATG5-mediated autophagy. JCI Insight. 2019 Nov 1;4(21). doi: 10.1172/jci.insight.127312
- Gong N, Yang X, Li X, et al. MicroRNA-590-3p relieves hypoxia/reoxygenation induced cardiomyocytes apoptosis and autophagy by targeting HIF-1α. Exp Ther Med. 2021 Oct;22(4):1077. doi: 10.3892/etm.2021.10511
- Li Z, Song Y, Liu L, et al. miR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. 2017 Jul;24(7):1205–1213. doi: 10.1038/cdd.2015.95
- Ning S, Li Z, Ji Z, et al. MicroRNA‑494 suppresses hypoxia/reoxygenation‑induced cardiomyocyte apoptosis and autophagy via the PI3K/AKT/mTOR signaling pathway by targeting SIRT1. Mol Med Rep. 2020 Dec;22(6):5231–5242. doi: 10.3892/mmr.2020.11636
- Qiu R, Li W, Liu Y. MicroRNA-204 protects H9C2 cells against hypoxia/reoxygenation-induced injury through regulating SIRT1-mediated autophagy. Biomed Pharmacother. 2018 Apr;100:15–19.
- Wang F, Min X, Hu SY, et al. Hypoxia/reoxygenation-induced upregulation of miRNA-542-5p aggravated cardiomyocyte injury by repressing autophagy. Hum Cell. 2021 Mar;34(2):349–359. doi: 10.1007/s13577-020-00466-z
- Wu K, Chen Y, Wang D, et al. MicroRNA-520d-3p alleviates hypoxia/reoxygenation-induced damage in human cardiomyocytes by targeting ATG-12. J Thromb Thrombolysis. 2021 Aug;52(2):429–439. doi: 10.1007/s11239-020-02352-9
- Zhang Y, Zhan B, Hu Y, et al. Sevoflurane inhibits the apoptosis of hypoxia/reoxygenation-induced cardiomyocytes via regulating miR-27a-3p-mediated autophagy. J Pharm Pharmacol. 2021 Oct 7;73(11):1470–1479.
- Zhou K, Xu Y, Wang Q, et al. Overexpression of miR-431 attenuates hypoxia/reoxygenation-induced myocardial damage via autophagy-related 3. Acta Biochim Biophys Sin (Shanghai). 2021 Feb 4;53(2):140–148.
- Hu X, Zhang K, Chen Z, et al. The HMGB1‑IL‑17A axis contributes to hypoxia/reoxygenation injury via regulation of cardiomyocyte apoptosis and autophagy. Mol Med Rep. 2018 Jan;17(1):336–341. doi: 10.3892/mmr.2017.7839
- Lu S, Yu L, Liu H. Trimetazidine alleviates hypoxia/reoxygenation-induced apoptosis in neonatal mice cardiomyocytes via up-regulating HMGB1 expression to promote autophagy. J Recept Signal Transduct Res. 2021 Apr;41(2):170–179.
- Luo C, Zhang Y, Guo H, et al. Ferulic Acid Attenuates Hypoxia/Reoxygenation Injury by Suppressing Mitophagy Through the PINK1/Parkin Signaling Pathway in H9c2 Cells. Front Pharmacol. 2020;11:103.
- Sun L, Zhao M, Yang Y, et al. Acetylcholine Attenuates Hypoxia/Reoxygenation Injury by Inducing Mitophagy Through PINK1/Parkin Signal Pathway in H9c2 Cells. J Cell Physiol. 2016 May;231(5):1171–1181. doi: 10.1002/jcp.25215
- Chen X, Li X, Zhang W, et al. Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-κB pathway. Metabolism. 2018 Jun;83:256–270.
- Takikawa T, Ohashi K, Ogawa H, et al. Adipolin/C1q/Tnf-related protein 12 prevents adverse cardiac remodeling after myocardial infarction. PLoS One. 2020;15(12):e0243483. doi: 10.1371/journal.pone.0243483
- Jin AP, Zhang QR, Yang CL, et al. Up-regulation of CTRP12 ameliorates hypoxia/re-oxygenation-induced cardiomyocyte injury by inhibiting apoptosis, oxidative stress, and inflammation via the enhancement of Nrf2 signaling. Hum Exp Toxicol. 2021 Dec;40(12):2087–2098. doi: 10.1177/09603271211021880
- Oka T, Hikoso S, Yamaguchi O, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012 May 10;485(7397):251–255.
- Stunz LL, Lenert P, Peckham D, et al. Inhibitory oligonucleotides specifically block effects of stimulatory CpG oligonucleotides in B cells. Eur J Immunol. 2002 May;32(5):1212–1222. doi: 10.1002/1521-4141(200205)32:5<1212::AID-IMMU1212>3.0.CO;2-D
- Poole AC, Thomas RE, Yu S, et al. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One. 2010 Apr 7;5(4):e10054.
- Xu A, Sweeney G. Emerging role of autophagy in mediating widespread actions of ADIPOQ/adiponectin. Autophagy. 2015 Apr 3;11(4):723–724.
- Caselli C, D’Amico A, Cabiati M, et al. Back to the heart: the protective role of adiponectin. Pharmacol Res. 2014 Apr;82:9–20.
- Luo Y, Liu M. Adiponectin: a versatile player of innate immunity. J Mol Cell Biol. 2016 Apr;8(2):120–128.
- Yamaguchi N, Argueta JG, Masuhiro Y, et al. Adiponectin inhibits Toll-like receptor family-induced signaling. FEBS Lett. 2005 Dec 19;579(30):6821–6826.
- Park PH, Huang H, McMullen MR, et al. Activation of cyclic-AMP response element binding protein contributes to adiponectin-stimulated interleukin-10 expression in RAW 264.7 macrophages. J Leukoc Biol. 2008 May;83(5):1258–1266. doi: 10.1189/jlb.0907631
- Ohashi K, Parker JL, Ouchi N, et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J Biol Chem. 2010 Feb 26;285(9):6153–6160.
- Hui X, Gu P, Zhang J, et al. Adiponectin Enhances Cold-Induced Browning of Subcutaneous Adipose Tissue via Promoting M2 Macrophage Proliferation. Cell Metab. 2015 Aug 4;22(2):279–290.
- Liu Y, Vu V, Sweeney G. Examining the Potential of Developing and Implementing Use of Adiponectin-Targeted Therapeutics for Metabolic and Cardiovascular Diseases. Front Endocrinol (Lausanne). 2019;10:842.
- Zhang L, Yuan M, Zhang L, et al. Adiponectin alleviates NLRP3-inflammasome-mediated pyroptosis of aortic endothelial cells by inhibiting FoxO4 in arteriosclerosis. Biochem Biophys Res Commun. 2019 Jun 18;514(1):266–272.
- Ren J, Xu X, Wang Q, et al. Permissive role of AMPK and autophagy in adiponectin deficiency-accentuated myocardial injury and inflammation in endotoxemia. J Mol Cell Cardiol. 2016 Apr;93:18–31.
- Everett BM, MacFadyen JG, Thuren T, et al. Inhibition of Interleukin-1beta and Reduction in Atherothrombotic Cardiovascular Events in the CANTOS Trial. J Am Coll Cardiol. 2020 Oct 6;76(14):1660–1670.
- Klein AL, Imazio M, Cremer P, et al. Phase 3 Trial of Interleukin-1 Trap Rilonacept in Recurrent Pericarditis. N Engl J Med. 2021 Jan 7;384(1):31–41.
- Abbate A, Wohlford GF, Del Buono MG, et al. Interleukin-1 blockade with anakinra and heart failure following ST-segment elevation myocardial infarction: results from a pooled analysis of the VCUART clinical trials. Eur Heart J Cardiovasc Pharmacother. 2022 Aug 11;8(5):503–510.
- Abbate A, Trankle CR, Buckley LF, et al. Interleukin-1 Blockade Inhibits the Acute Inflammatory Response in Patients With ST-Segment-Elevation Myocardial Infarction. J Am Heart Assoc. 2020 Mar 3;9(5):e014941.
- Farag A, Mandour AS, Kaneda M, et al. Effect of trehalose on heart functions in rats model after myocardial infarction: assessment of novel intraventricular pressure and heart rate variability. Front Cardiovasc Med. 2023;10:1182628.
- Jamialahmadi T, Emami F, Bagheri RK, et al. The effect of trehalose administration on vascular inflammation in patients with coronary artery disease. Biomed Pharmacother. 2022 Mar;147:112632.
- Oesterle A, Laufs U, Liao JK. Pleiotropic Effects of Statins on the Cardiovascular System. Circ Res. 2017 Jan 6;120(1):229–243.
- Andres AM, Hernandez G, Lee P, et al. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid Redox Signal. 2014 Nov 10;21(14):1960–1973.
- Li X, Liu J, Lu Q, et al. AMPK: a therapeutic target of heart failure-not only metabolism regulation. Biosci Rep. 2019 Jan 31;39(1):BSR20181767.
- Wang B, Nie J, Wu L, et al. AMPKalpha2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ Res. 2018 Mar 2;122(5):712–729.
- Koyani CN, Plastira I, Sourij H, et al. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Pharmacol Res. 2020 Aug;158:104870.
- Cusi K, Alkhouri N, Harrison SA, et al. Efficacy and safety of PXL770, a direct AMP kinase activator, for the treatment of non-alcoholic fatty liver disease (STAMP-NAFLD): a randomised, double-blind, placebo-controlled, phase 2a study. Lancet Gastroenterol Hepatol. 2021 Nov;6(11):889–902. doi: 10.1016/S2468-1253(21)00300-9
- Stephan Y, Souille M, Larcheveque M, et al. Both short-and long-term treatment with the direct AMP kinase activator PXL770 improves cardiac function in ZSF-1 rats. Eur Heart J. 2021;42(1):ehab724.1050. doi: 10.1093/eurheartj/ehab724.1050