
Abstract
Atypical teratoid rhabdoid tumors (AT/RT) are rare and highly malignant CNS neoplasms primarily affecting children. Adult cases are extremely uncommon, with only approximately 92 reported. Spinal AT/RT in adults is particularly rare. Here, we present the case of a 50-year-old patient diagnosed with AT/RT of the spine. Initially, they were diagnosed and treated for a spinal ependymoma. However, after 10 years, a recurrence was detected through magnetic resonance imaging (MRI) and the tumor was reclassified as AT/RT. We discuss the significance of SMARCB1 gene mutations in diagnosing AT/RT and describe our unique treatment approach involving surgery, radiation and anti-PD1 therapy in this patient.
Plain language summary
Atypical teratoid rhabdoid tumors (AT/RT) are rare and serious cancers that affect the brain and spine, and mostly occur in children. AT/RT are rare in adults, with only about 92 cases reported. Our article tells the story of a 50-year-old patient, who was diagnosed with a spinal tumor, initially classified as an ependymoma. Ten years later, the tumor recurred, and was found on routine surveillance imaging. After pathological examination of the recurrent tumor, it was diagnosed as AT/RT. The initial tissue was re-examined, and the original tumor was reclassified as an AT/RT. We explain why a gene called SMARCB1 is important for diagnosing AT/RT. Additionally, we share details about the treatments utilized: including surgery, radiation, and medicines that stimulate the immune system to kill cancer cells. This case highlights the challenges and treatments for this rare cancer in adults.
Tweetable abstract
Rare spinal AT/RT case in a 50-year-old adult initially misdiagnosed as ependymoma. Recurrence led to reclassification. Highlights SMARCB1 mutations and treatment with surgery, radiation, and anti-PD1 therapy. #ATRT #SpinalTumor #RareCancer
Atypical teratoid/rhabdoid tumors (AT/RTs) are rare grade 4 CNS neoplasms as classified by the World Health Organization.
While predominantly identified in the pediatric population, there are instances where AT/RTs can manifest in adulthood, challenging the traditional perception of their occurrence.
In adults, AT/RT more frequently presents in the brain compared with the spine. AT/RT in the spine may exhibit a comparatively less aggressive nature, but this needs further validation.
The use of immunohistochemistry and/or next-generation sequencing, is crucial in the diagnosis of AT/RT. This diagnostic process relies on identifying SMARCB1 mutations, emphasizing the significance of molecular markers in confirming the tumor type.
Despite the absence of a standardized therapeutic approach, the course of treatment for AT/RT may involve surgical resection as the primary intervention, followed by a combination of radiation and chemotherapy.
Pembrolizumab, a checkpoint inhibitor, emerges as a consideration for potential treatment in cases of spinal AT/RT with a positive PD-L1 status. This novel therapeutic avenue introduces immunotherapy as a potential breakthrough in the management of these tumors.
For individuals diagnosed with AT/RT of the spine, establishing a routine of serial spine MRIs is imperative. This ongoing monitoring protocol is vital for the early detection of any signs of recurrence, facilitating timely intervention and management adjustments as needed.
AT/RTs are WHO grade 4 CNS neoplasms characterized genetically by biallelic inactivation of SMARCB1 (INI1/BAF47) or, more rarely, BRG1 [Citation1]. AT/RTs are composed of poorly differentiated cells and a variable number of rhabdoid cells and have the potential to differentiate along neuroepithelial, epithelial and mesenchymal lines [Citation1]. Tumors usually occur in the cerebellum and brainstem, with only a few reports of spine or spinal canal presentation [Citation2–4]. AT/RTs are predominantly found in the pediatric population: there are only about 92 reported cases of adult AT/RT in the literature [Citation5]. There have been fewer than 10 case reports on AT/RT in the spine for adults [Citation2,Citation5–7]. AT/RTs may also present in the ventricles [Citation8].
The clinical presentation of AT/RT depends on its location within the central nervous system (CNS). For instance, symptoms of a lesion in the posterior fossa or brainstem may include morning headaches, nausea, vomiting, loss of balance, coordination difficulties and abnormalities in eye movements. AT/RTs are unique in that they can metastasize outside of the CNS [Citation6]. The estimated 5-year survival rate is 32.2% [Citation9]. There is currently no standard of effective treatment for AT/RT. Maximal safe resection is generally the first-line option for AT/RT, followed by a combination of radiation and chemotherapy [Citation10].
The significance of this case lies in its rarity and the pivotal role it plays in refining diagnostic methodologies and treatment strategies for spinal neoplasms. This instance of AT/RT in an adult male, initially misdiagnosed as ependymoma due to limitations in molecular testing at the time, sheds light on the evolution of diagnostic accuracy. The recurrence of the neoplasm prompted deeper clinicopathologic analyses, resulting in a reclassification supported by the identification of a SMARCB1 mutation. This case highlights the role of SMARCB1 gene mutations in accurately diagnosing AT/RT, emphasizing the necessity of updated molecular approaches. Additionally, the utilization of anti-PD-1 therapy in the treatment regimen, after the detection of PD-L1 positivity by immunohistochemistry, represents a novel and promising therapeutic avenue in managing this rare spinal tumor. This case of AT/RT contributes significantly to the management of AT/RT, bridging diagnostic challenges and presenting a unique treatment approach that could impact future patient care paradigms. Importantly, this case report was structured using the CARE guidelines checklist [Citation11].
Patient information & case presentation
A 42-year-old man with no significant past medical history presented to clinic with new-onset bilateral lower extremity weakness and numbness from the T4 level down. He has a family history significant for prostate cancer, and arthritis. Regarding social history, the patient does not smoke and rarely drinks. He did not take any current medication.
Subsequent radiographic images demonstrated a lesion centered at the right T3–T4 neural foramen ( A & C). Histopathologically, the initial CT-guided biopsy was concerning for ependymoma. The lesion did not receive a grade, as the 2016 WHO CNS tumor guidelines suggested uncertain clinical utility for the histologic grading of ependymomas [Citation12]. Whole body PET CT scan and MRI of the brain and spine at that time showed no additional foci of disease. He underwent maximal safe resection followed by 6040 cGy of intensity-modulated radiation therapy with concurrent and adjuvant ‘off-label’ temozolomide chemotherapy. The patient continued with regular interval follow-up appointments and surveillance spinal imaging. He continued to endorse mild chronic thoracic back pain and was able to return to work full-time.
Figure 1. Magnetic resonance imaging of the spine.
(A) Post-contrast T1-weighted sagittal image of initial tumor. (B) T1 Dixon sagittal image of recurrent tumor (12 years later) with extramedullary extradural soft tissue mass between C7 and T1. (C) Post-contrast T1-weighted axial image of initial tumor. (D) STIR sagittal image of recurrent tumor.
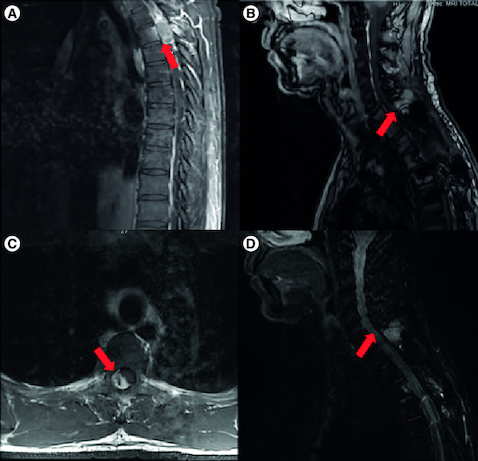
Unfortunately, 10 years after the initial diagnosis, routine full-spine imaging was concerning for neoplasm recurrence (B & D). At that time, he experienced baseline levels of chronic back pain, but denied any new symptoms and there were no new findings on neurological exam. A subsequent biopsy and maximal safe resection identified recurrent neoplasm. However, given the presence of a SMARCB1 mutation, the recurrent neoplasm as well as the original were reclassified as AT/RT. A CT of his chest, abdomen, and pelvis did not identify any disease outside of the CNS and MRI of the brain did not identify dissemination within the CNS. He went on to receive re-irradiation (2500 cGy) followed by 22 months of pembrolizumab with excellent tolerability until he developed presumed autoimmune hepatitis which resolved with discontinuation of therapy and prednisone. A summary of the clinical course and related therapies is provided in .
Table 1. Comprehensive overview of the patient's clinical course, detailing the initial diagnosis, treatment modalities employed at various stages and the response to therapies.
Diagnostic assessment
Given the patient's history of spinal ependymoma, the MRI findings of a new enhancing lesion 10 years after initial diagnosis, measuring 27 × 29 × 34 mm between C7 and T1 raised concern for recurrence (). The tissue from the biopsy had histological characteristics of ependymoma. However, the patient underwent subsequent maximal safe resection and further pathological analyses demonstrated loss of nuclear staining for INI1/BAF47 (). SMARCB1 mutation was confirmed with FoundationOne testing. FoundationOne analysis also identified BCORL1 and PRKAR1A mutations, the significance of which is unknown in the case of AT/RT. In house PD-L1 testing by immunohistochemistry (22C3 antibody clone) was positive.
Figure 2. The patient's atypical teratoid rhabdoid tumor (AT/RT) on histology.
H&E sections demonstrate a malignant neoplasm characterized by cells with vesicular chromatin and visible nucleoli in both the initial resection and the re-resection (A, C). Immunohistochemistry for BAF47 (INI1) shows loss of expression in tumor cells in both the initial resection (B) and the re-resection (D). Immunohistochemistry performed on the re-resection was positive for vimentin (E), smooth muscle actin (F), and EMA (G). PDL-1 (22C3) performed on the re-resection was positive in approximately 30% of neoplastic cells (H).
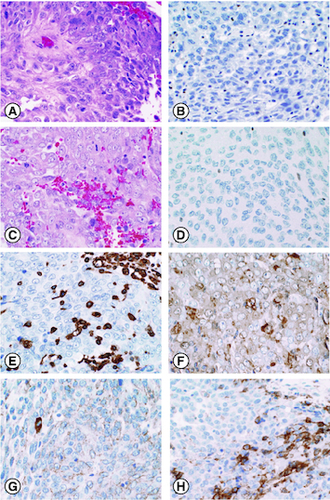
The decision was made to re-examine the initial neoplasm specimen. AT/RT had not been in the differential diagnosis, as it was considered a pediatric neoplasm, very rare in adults and even more so in the spine. INI1/BAF47 immunohistochemistry was performed on the original resected neoplasm, and this also showed loss of nuclear INI1 staining, indicating that the recurrent neoplasm and the original were likely the same.
Differential diagnosis
The differential diagnosis for the patient's clinical presentation and radiographical findings were heavily influenced by the pathologic examinations and immunohistochemical findings of the surgical resection of the neoplasm. For the neoplasm of the initial resection, the microscopic examination depicted poorly differentiated epithelioid neoplasm with numerous mitotic figures and foci of abnormal bone within the neoplasm was also seen. Given the poorly differentiated features, immunohistochemistry stains were performed to assess for metastatic carcinoma, hematopoietic neoplasm, melanoma, or a neuroectodermal neoplasm.
Metastatic carcinoma was unlikely since immunohistochemistry for cytokeratin was negative. Negative immunohistochemistry for CD45 and non-lymphoid impression on flow cytometry indicated the neoplasm was not a hematopoietic origin. Negative immunohistochemistry for MART-1 and HMB-45 made melanoma less likely. Immunohistochemistry was positive, however, for S-100, EMA, and GFAP. Additionally, scattered individual cells were positive for smooth muscle actin (SMA) and neurofilament. The positive S-100, EMA, and GFAP immunostains supported a neuroectodermal origin, which broadened the differential diagnosis to include malignant meningioma, anaplastic ependymoma, and a malignment peripheral nerve sheath tumor. Subsequently, the neoplasm was sent out for an additional expert opinion and electron microscopy was completed to form a more definitive diagnosis. The ultrastructural features of the tissue found using electron microscopy were indicative of glial differentiation, supporting a diagnosis of anaplastic ependymoma.
10 years later, MRI images of the cervical and thoracic spine demonstrated an enlarging soft tissue mass at C7 and T1. Clinically, this was concerning for recurrent tumor, presumably ependymoma, or treatment effect given the patients prior history of radiation. The recurrent lesion was resected, and the tissue was sent to pathology for identification. The neoplasm was poorly differentiated and characterized by cells with vesicular chromatin, prominent nucleoli and a high mitotic rate. Immunohistochemistry for GFAP and S-100 was negative, ruling out an ependymoma. Although classic rhabdoid cells were absent, the tissue was positive for EMA (membranous), SMA, and vimentin, and there was a loss of nuclear staining for BAF47 (INI1). These findings supported a diagnosis of atypical teratoid/rhabdoid tumor (AT/RT).
AT/RT was not initially considered in the differential because it is considered a pediatric neoplasm, and it is very rare in adults, especially in the spine. Since the recurrent neoplasm was histologically similar to the initial biopsy specimen, nuclear INI1 staining was completed on the original resected neoplasm and it also showed loss of nuclear INI1 staining, which indicates the original and recurrent neoplasms were both consistent with AT/RT. A comprehensive summary of the differential diagnosis is presented in .
Table 2. Concise overview of the patient's differential diagnosis, covering considerations for both initial and recurrent tumors, immunohistochemistry findings and final diagnosis.
Therapeutic interventions
After the initial diagnosis of an ependymoma, the patient was treated with T1–T4 radiation therapy (total dose: 6040 cGy) with off-label concurrent chemotherapy, temozolomide 75 mg/m2 daily, for 42 days. The patient demonstrated marked improvements with the treatment course and serial spine MRI monitoring was initiated.
At the time of recurrence, the treatment course consisted of C7-T1 gross-total resection with C5-T1 fusion and re-irradiation with a total of 2500 cGy delivered in five daily fractions. Traditional high dose chemotherapies were considered. Given poor tolerability of multi-drug chemotherapy regimens in adults and presence of PD-L1 expression, the decision was made to treat with pembrolizumab, 200 mg IV every 3 weeks. The patient tolerated the systemic therapy well for 22 continuous months with radiographic stability. Treatment was discontinued due to abnormal liver function tests concerning for pembrolizumab-induced hepatitis.
Follow-up & outcomes
The patient is clinically and radiographically stable 14 years after his initial diagnosis and 5 years after recurrence. He is monitored with regular full-spine MRIs. Liver function tests remain normal with the discontinuation of pembrolizumab.
He completed 5 months of physical therapy for neck pain and decreased strength with modest improvement. He is being treated with zoledronic acid infusions every 6 months, along with oral vitamin D3 and calcium supplementation to optimize his vertebral bone health. Ultimately, the patient was able to resume jogging and work in food services.
Discussion
This case of AT/RT highlights the rare occurrence in an adult patient in the spine. The observed less aggressive nature of adult spinal AT/RT in our case further emphasizes the atypical nature of this lesion. Our case sheds light on check point inhibition as a potential therapeutic avenue for adult spinal AT/RT with confirmed PD-L1 expression. It also demonstrates the importance of using radiographic imaging to monitor for recurrence.
AT/RT are malignant neoplasms that can occur anywhere in the brain and spinal cord, but they primarily occur in the cerebellum or brain stem of pediatric patients [Citation6]. However, the most common location for adult AT/RT patients is within the sellar region [Citation10]. The biology of AT/RT tumors can vary based on their location. This particular case emphasizes the potential differences between spinal AT/RT and cerebral AT/RT [Citation13,Citation14]. The first spinal case of AT/RT was reported in 1994 [Citation15]. A two-year old girl was initially diagnosed with an intradural extramedullary malignant rhabdoid tumor. The neoplasm met diagnostic criteria through histologic, immunologic, and ultrastructural testing. Since the initial publication of spinal AT/RT, there have been several other documented cases of spinal AT/RT in children aged 7–17 years [Citation16]. The majority of the reported cases were focal lesions that were intradural, and most of the patients died within 10 months of the diagnosis. AT/RT neoplasms are historically considered aggressive tumors with a survival time of less than 1 year.
A systematic review of 39 articles comprised of a total of 50 adult patients with AT/RT found that 94% of the neoplasms were intracranial and 6% of neoplasms were in the spine. Regarding prognosis, 62% of patients passed away due to their disease [Citation17]. The average time of death after initial diagnosis is about 20 months, and the range was 0–168 months. Out of the 50 adult patients, 24 patients reported recurrence of AT/RT with an average time of 12.4 months to a reported recurrence. This demonstrates that some individuals may have aggressive AT/RTs. Our patient in this case having the latter.
There is currently no standard of effective treatment for AT/RT. Studies demonstrated the use of surgery, radiotherapy, chemotherapy, and high-dose chemotherapy with stem cell transplant to treat AT/RT [Citation10,Citation17–19]. Overall survival for patients with gross surgical resection is varied among studies [Citation20–22]. However, early initiation of chemotherapy and radiotherapy improved the prognosis of patients with AT/RT [Citation18]. There is an increased emphasis on studies to assess the effectiveness of molecularly targeted therapies for AT/RTs. AT/RTs are thought to result from chromatin remodeling due to the inactivation of SMARCB1 or SMARCA4, histone modification due to reduced regulation of H3K27, and DNA methylation due to dysregulation of Notch/sonic hedgehog (ATRT-SHH), tyrosinase enzyme (ATRT-TYR), and the MYC oncogene (ATRT-MYC) [Citation21,Citation23]. Inhibitors used in targeted therapy clinical trials to treat AT/RT include mTOR (drug: Sirolimus), EZH2 (drug: Tazemetostat), and CDK4/6 (drug: Ribociclib) [Citation24–26].
Currently, there is a Phase II trial sponsored by the National Cancer Institute assessing the use of atezolizumab, a drug that inhibits programmed death-ligand 1, to treat relapsed or refractory SMARCB1 or SMARCA4 deficient tumors [Citation27]. Programmed death-ligand 1 (PD-L1) inhibitors have demonstrated effectiveness in treating cancers like squamous non-small-cell lung cancer, urothelial carcinomas, and microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors [Citation28]. Pembrolizumab is another type of PD-L1 checkpoint inhibitor, and it was effective in halting the progression of recurrent spinal AT/RT in our patient. Checkpoint inhibitors are a potential treatment option for spinal AT/RT with confirmed PD-L1 expression.
In this specific case, the use of imaging played a crucial role in the timely identification of recurrence within the spinal tumor. The remarkable role played by imaging in discerning the recurrence since the biological activity of AT/RT tumors may be unpredictable. Imaging valuable and reliable tool for ongoing monitoring in patients diagnosed with AT/RT in the spine. The application of full spine MRIs, in particular, proves advantageous in providing a comprehensive assessment of the spinal region, allowing for the early detection and management of any potential recurrence. However, there are minimal evidence-based guidelines for monitoring the brain or other sites of distant recurrence, so a brain MRI will only be performed in the event of new symptoms or disease progression.
Conclusion
All in all, our case report sheds light on the uncommon occurrence of atypical teratoid/rhabdoid tumors (AT/RT) in adults, particularly within the spinal region. Despite the rarity of adult spinal AT/RT, our findings highlight the importance of considering this diagnosis in the adult population, extending beyond the typical pediatric demographic. Furthermore, the exploration of Pembrolizumab, a checkpoint inhibitor, as a potential therapeutic avenue for adult spinal AT/RT with confirmed PD-L1 expression introduces a novel perspective on treatment strategies. The observed less aggressive nature of adult spinal AT/RT, as evidenced by our single case, challenges existing perceptions and prompts a reevaluation of clinical course and prognosis. Importantly, our report underscores the necessity for vigilant monitoring through serial spine MRIs in patients diagnosed with AT/RT of the spine to promptly identify and manage potential recurrences. This case contributes valuable insights that may inform future research, clinical approaches and therapeutic considerations for adult spinal AT/RT.
Author contributions
AJ Charles: conception and design, acquisition of data, interpretation of data, manuscript writing; VL Smith: acquisition, analysis, and interpretation of data; CR Goodwin: manuscript writing and editing; MO Johnson: conception and design, interpretation of data, manuscript writing and editing.
Financial disclosure
The authors have no financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Writing disclosure
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained verbal and written informed consent from the patient/patients for the inclusion of their medical and treatment history within this case report.
Competing interests disclosure
CR Goodwin received grants from the Robert Wood Johnson Harold Amos Medical Faculty Development Program, the Federal Food and Drug Administration, and the NIH 1R01DE031053-01A1, acted as a consultant for Stryker and Medtronic and is Deputy Editor for Spine. Patent application/invention disclosures outside of the current work. The authors have no other competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reference
- WHO Classification of tumors Editorial Board. Central Nervous System tumors. International Agency for Research on Cancer, Lyon, France. https://publications.iarc.fr/Book-And-Report-Series/Who-Classification-Of-Tumours/Central-Nervous-System-Tumours-2021
- Li L, Patel M, Nguyen HS, Doan N, Sharma A, Maiman D. Primary atypical teratoid/rhabdoid tumor of the spine in an adult patient. Surgl. Neurol. Int. 7, 27 (2016).
- Chao MF, Su Y-F, Jaw T-S, Chiou S-S, Lin C-H. Atypical teratoid/rhabdoid tumor of lumbar spine in a toddler child. Spinal Cord Ser. Cases. 3, 16026 (2017).
- Amit A, Vats A, Shoakazemi A, Herron B, McCarthy A, McConnell RS. Dumbell atypical teratoid/rhabdoid tumour (AT/RT) of the cervical spine. Br. J. Neurosurg. 34(3), 339–341 (2020).
- Liebigt S, Florschütz A, Arndt N, Stock K, Renner C. Atypical teratoid/rhabdoid tumor of the pineal region in a young adult male patient: case report and review of the literature. J. Neurol. Surg. A Cent Eur. Neurosurg. 78(1), 92–98 (2017).
- Broggi G, Gianno F, Shemy DT et al. Atypical teratoid/rhabdoid tumor in adults: a systematic review of the literature with meta-analysis and additional reports of 4 cases. J. Neurooncol. 157(1), 1–14 (2022).
- Zarovnaya EL, Pallatroni HF, Hug EB et al. Atypical teratoid/rhabdoid tumor of the spine in an adult: case report and review of the literature. J. Neurooncol. 84(1), 49–55 (2007).
- Karim A, Shaikhyzada K, Suleimenova A, Ibraimov B, Nurgaliev D, Poddighe D. Case report: atypical teratoid/rhabdoid tumor of the lateral ventricle in a male adolescent (case-based review and diagnostic challenges in developing countries). Front. Oncol. 12, 985862 (2022).
- Truitt G, Gittleman H, Leece R et al. Partnership for defining the impact of 12 selected rare CNS tumors: a report from the CBTRUS and the NCI-CONNECT. J. Neurooncol. 144(1), 53–63 (2019).
- Peng AJ, Fan SC, Chen YX et al. Atypical teratoid/rhabdoid tumor in adult: case series and an integrated survival analysis. Br. J. Neurosurg. doi:10.1080/02688697.2021.1885620 (2021) ( Epub ahead of print).
- Riley DS, Barber MS, Kienle GS et al. CARE guidelines for case reports: explanation and elaboration document. J. Clin. Epidemiol. 89, 218–235 (2017).
- Louis DN, Perry A, Reifenberger G et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131(6), 803–820 (2016).
- Rauschenbach L. Spinal cord tumor microenvironment. Adv. Exp. Med. Biol. 1226, 97–109 (2020).
- Karsy M, Guan J, Sivakumar W, Neil JA, Schmidt MH, Mahan MA. The genetic basis of intradural spinal tumors and its impact on clinical treatment. Neurosurg Focus. 39(2), E3 (2015).
- Rosemberg S, Menezes Y, Sousa MR, Plese P, Ciquini O. Primary malignant rhabdoid tumor of the spinal dura. Clin. Neuropathol. 13(4), 221–224 (1994).
- Moeller KK, Coventry S, Jernigan S, Moriarty TM. Atypical teratoid/rhabdoid tumor of the spine. Am. J. Neuroradiol. 28(3), 593–595 (2007).
- Chan V, Marro A, Findlay JM, Schmitt LM, Das S. A systematic review of atypical teratoid rhabdoid tumor in adults. Front. Oncol. 8, 567 (2018).
- Ginn KF, Gajjar A. Atypical teratoid rhabdoid tumor: current therapy and future directions. Front Oncol. 2, 114 (2012).
- Major K, Daggubati LC, Mau C, Zacharia B, Glantz M, Pu C. Sellar atypical teratoid/rhabdoid tumors (AT/RT): a systematic review and case illustration. Cureus. 14(7), e26838 (2022).
- Park M, Han JW, Hahn SM et al. Atypical teratoid/rhabdoid tumor of the central nervous system in children under the age of 3 years. Cancer Res. Treat. 53(2), 378–388 (2021).
- Zhang C, Li H. Molecular targeted therapies for pediatric atypical teratoid/rhabdoid tumors. Pediatr Investig. 6, 111–122 (2022).
- Ren Y-M, Wu X, You C, Zhang Y-K, Li Q, Ju Y. Multimodal treatments combined with gamma knife surgery for primary atypical teratoid/rhabdoid tumor of the central nervous system: a single-institute experience of 18 patients. Childs Ner. Syst. 34(4), 627–638 (2017).
- Torchia J, Golbourn B, Feng S et al. Integrated (epi)-genomic analyses identify subgroup-specific therapeutic targets in CNS rhabdoid tumors. Cancer Cell. 30(6), 891–908 (2016).
- Cash T, Jonus HC, Tsvetkova M et al. A Phase I study of simvastatin in combination with topotecan and cyclophosphamide in pediatric patients with relapsed and/or refractory solid and CNS tumors. Pediatr. Blood Cancer 70(8), e30405 (2023).
- Epizyme, Inc. A Phase I study of the EZH2 inhibitor tazemetostat in pediatric subjects with relapsed or refractory INI1-negative tumors or synovial sarcoma. clinicaltrials.gov, Bethesda, MD, USA (2021). https://classic.clinicaltrials.gov/ct2/show/NCT02601937
- DeWire MD, Fuller C, Campagne O et al. A Phase I and surgical study of ribociclib and everolimus in children with recurrent or refractory malignant brain tumors: a pediatric brain tumor consortium study. Clin Cancer Res. 27(9), 2442–2451 (2021).
- National Cancer Institute. A Phase I/II study of tiragolumab (NSC# 827799) and Atezolizumab (NSC# 783608) in Patients With Relapsed or Refractory SMARCB1 or SMARCA4 Deficient Tumors. clinicaltrials.gov, Bethesda, MD, USA (2023). https://clinicaltrials.gov/ct2/show/NCT05286801
- Ai L, Chen J, Yan H et al. Research status and outlook of PD-1/PD-L1 inhibitors for cancer therapy. Drug Des Devel Ther. 14, 3625–3649 (2020).