
Abstract
Whole-exome sequencing (WES) is widely used in clinical settings; however, the exploration of its use in pharmacogenomic analysis remains limited. Our study compared the variant callings for 28 core absorption, distribution, metabolism and elimination genes by WES and array-based technology using clinical trials samples. The results revealed that WES had a positive predictive value of 0.71–0.92 and a sensitivity of single-nucleotide variants between 0.68 and 0.95, compared with array-based technology, for the variants in the commonly targeted regions of the WES and PhamacoScan™ assay. Besides the common variants detected by both assays, WES identified 200–300 exclusive variants per sample, totalling 55 annotated exclusive variants, including important modulators of metabolism such as rs2032582 (ABCB1) and rs72547527 (SULT1A1). This study highlights the potential clinical advantages of using WES to identify a wider range of genetic variations and enabling precision medicine.
Determining the right target, the right patient and the right dose is indispensable for successful precision medicine-driven drug development. One of the key elements of precision medicine is to understand the source of pharmacokinetic and pharmacodynamic (PK/PD) variability, which are important drivers for differences in drug response. Many intrinsic (e.g., age, sex, organ function and disease) and extrinsic (e.g., diet, concomitant medication use, smoking habits and physiochemical properties of the drug) factors have the potential to influence the absorption, distribution, metabolism and elimination (ADME) of a drug [Citation1,Citation2].
A patient’s genetic makeup is an important intrinsic factor that could contribute substantially to PK/PD variability. Drug-metabolizing enzymes such as CYP450 enzymes (e.g., CYP3A4, CYP2C8, CYP2D6), non-CYP450 enzymes (e.g., Uridine 5′-diphospho-glucuronosyltransferases [UGTs], aldehyde oxidase [AO], flavin monooxygenase [FMO]) and transporters (e.g., P-glycoprotein, Breast Cancer Resistance Protein [BCRP], organic anion transporting polypeptides [OATPs]) play important roles in the ADME process of a drug. Among these, polymorphisms in certain metabolizing enzymes (e.g., CYP2C9, CYP2C19, CYP2D6, CYP3A5, NAT2, UGT1A1) and transporters (e.g., BCRP, OATP1B1) have been extensively studied and reported in the literature [Citation3]. Polymorphisms in drug-metabolizing enzymes and transporters can significantly modulate the exposure of a drug [Citation4,Citation5], which can lead to dose adjustment in specific populations and has label implications [Citation6]. For example, approximately 8% of Caucasians and 3–8% of Black/African–Americans are considered poor metabolizers (i.e., individuals with decreased metabolism) of CYP2D6 substrates [Citation7–10]. Similarly, a reduced UGT1A1*28 enzyme activity is observed in approximately 20% of the Black/African–American population, 10% of the Caucasian and 2% of the Asian population [Citation7–10]. Among the approved drugs, the effects of the CYP2C19, CYP2D6 and CYP3A4 polymorphism on the exposure of proton pump inhibitors, antidepressants and statins, respectively, are all well-known examples [Citation7–10]. Polymorphisms in metabolic enzymes such as CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, UGT1A1, UGT2B17, NAT2, TPMT, DPYD and SLCO1B1 are reflected in many drug labels. In addition, CYP2C9, CYP2C19, CYP2D6, UGT1A1, NAT2 and TPMT are associated with recommendations for dose modification due to polymorphism [Citation6]. Hence, it is critical to incorporate the assessment of genetic polymorphism early in clinical development.
Pharmacogenomic (PGx) assessment is a rapidly evolving approach in precision medicine which allows us to identify variants associated with exposure variability and to potentially facilitate tailored dosing to patients based on their genetic characteristics. Collecting pharmacogenomic information is now routinely implemented in clinical trials [Citation11,Citation12]. Germline single-nucleotide variants (SNVs) and copy number variations (CNVs), along with variable number tandem repeats and insertions/deletions, found in metabolic enzymes and transporters may have important functional roles in pharmacogenetics and phamacodynamics. Various methods such as SNV panel testing and next-generation sequencing (NGS) are being widely used to assess germline variations. The SNV panel testing is the most commonly used technology in pharmacogenomics practice, including the commercially available microarray platform, the PharmacoScan™ assay, which covers 4627 markers from 1191 genes associated with pharmacogenomic pathways. The PharmacoScan assay interrogates biallelic as well as multiallelic SNPs, insertions/deletions and CNVs in a single assay workflow. It focuses on genotyping highly predictive markers in genes like GSTM1 and various CYPs that are in highly homologous regions. Additionally, it offers CNV analysis, covering copy number states ranging from 0 to greater than 3, particularly for crucial ADME genes. The assay also provides star allele and translation tables for key actionable genes. Array-based assays effectively characterize the specific loci they are designed to target, often achieving higher accuracy within challenging genomic regions. However, array-based technology has a limited coverage of the genome. As such, it may miss important variants outside the targeted genes that could influence drug response. Recently, NGS technologies such as whole-exome sequencing (WES) have gained attention as promising tools for pharmacogenomic profiling [Citation13–16]. WES enables a thorough examination of genetic variants, both common and rare, associated with drug-treatment outcomes, as well as identifying genetic variants related to the pathophysiology of complex diseases such as cancers, neurological and psychological disorders and cardiovascular diseases [Citation17]. The germline genetic profile obtained through WES is of high importance for oncology translational research for two reasons: first, somatic mutations in tumor tissue can be called with high confidence if a matched blood sample is profiled in parallel; and second, it helps to understand any existing disease predisposition due to germline variants. This comprehensive understanding of genetic variation provides potential benefits for personalized drug treatment [Citation18]. Although WES is broadly applied clinically, especially in the area of clinical biomarkers, studies related to the application of this technique to pharmacogenomic analysis are still limited.
In this study, a systematic comparison of the variants associated with core ADME genes using two different techniques – WES and the array-based technology – was conducted.
Materials & methods
Study cohort
A total of 79 human blood samples from four phase I oncology clinical trials (NCT03724890, NCT03770689, NCT02516813 and NCT02278250) were included in this study [Citation19]. The study protocols were approved by independent ethics committees and relevant governmental authorities in each country, as per local requirement. The study was carried out in accordance with the Declaration of Helsinki and informed consent was obtained from each participant. DNA was extracted from peripheral blood using the DNAex-Flexigene Blood kit, as per the manufacturer’s instructions (Qiagen, MD, USA). DNA input for the following analysis was based on quantitation using Invitrogen™ Qubit™ products of dsDNA quantification.
Genotyping & whole-exome sequencing
A total of 150 ng input DNA per sample was analyzed by PharmacoScan array-based assay (Thermo Fisher Scientific, MA, USA). Arrays were hybridized, washed and imaged on an Applied Biosystems™ GeneTitan™ multichannel instrument controlled by GeneChip™ Command Console Software version 4.3. Data analysis was performed using software tools available with Applied Biosystems Axiom™ Analysis Suite version 3.1 to generate the SNV calls and a phenotypic report.
From the same set of samples, ImmunoID NeXT™ WES (Personalis Inc., Menlo Park, CA, USA) was performed using accuracy- and content-enhanced enrichment including library preparation and 2 × 150 base pairs sequencing on the NovaSeq instrument (Illumina, CA, USA), delivering a minimum of 20 Gb for the exome per sample. Short reads were aligned to reference genome GRCh37, using the Burrows–Wheeler Aligner tool, followed by variant calling using optimized reimplementation of Genome Analysis Toolkit (www.sentieon.com/). As the WES data were generated by aligning the .fastq files against the GRCh37 version of the human genome while the PharmacoScan array used the GRCh38 build, the WES variants were lifted over from GRCh37 to GRCh38 using the Picard LiftOver VCF tool. This tool takes the variants .vcf file, the UCSC chain file (a file that describes how positions in both builds correspond to each other) and reference genome (GRCh38) as the input. The chain file (hg19ToHg38.over.chain.gz) was downloaded from UCSC (http://hgdownload.soe.ucsc.edu/downloads.html). Moreover, to use the tool, the .vcf files had to be processed to modify the chromosome section. The .vcf files shared by Personalis WES were first modified to append ’chr’ to the chromosome number. This modified .vcf file was then used as input to the Picard LiftOver tool. Other input files required for this step were hg19ToHg38.over.chain file and Homo_sapiens_assembly38.fasta (reference sequence).
Orthogonal analysis
A comparison of variant calling by WES and array-based technologies in 28 core ADME pharmacogenes was performed (). Briefly, the coordinates of the 28 core genes were obtained from the GRCh38_latest_genomic.gtf file downloaded from the National Center for Biotechnology Information by mapping based on gene symbol. Regions probed in the PharmacoScan assay were taken from the PharmacoScan_96F.na36.r8.a3.annot.csv file, and the regions captured in the WES assay were taken from the WES assay browser extensible data (.bed) file. ADME gene regions that were interrogated by both assays were defined by the overlapping genomic regions of the WES enrichment kit and the probe regions of the SNP array.
A total of 79 blood samples from phase I oncology clinical trials were included for pharmacogenomic analysis using both ImmunoID NeXT™ WES and PharmacoScan™ assay. An orthogonal analysis was carried out to assess the concordance between WES and array-based assay in the commonly targeted regions within 28 core ADME genes. The exclusive variants that were unique to each technique were identified and followed by genotype-to-phenotype annotation.
ADME: Absorption, distribution, metabolism and elimination; SNP: Single-nucleotide polymorphism; SNV: Single-nucleotide variant; WES: Whole-exome sequencing.
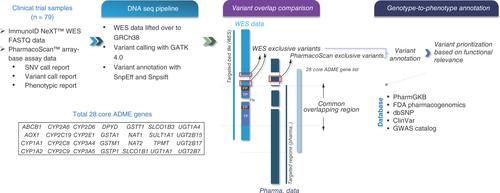
In the current project, we adopted the PharmacoScan assay as a benchmark assay, primarily due to its targeted focus on drug metabolism genes, comprehensive marker coverage, ability to identify highly predictive markers, inclusion of CNV analysis and provision of star allele and translation tables, all contributing to its potential for precision medication and clinical relevance. The positive predictive value (PPV) and sensitivity analyses of variants were done at sample level. Variants in the commonly targeted regions of WES and the PharmacoScan assay that were solely found in WES were defined as false positives, while variants not identified by WES but present in the array-based assay were defined as false negatives. Variants identified by both methods were defined as true positives. PPV was defined as the number of variants correctly identified by WES out of all variants detected by WES (true positive/[true positive + false positive]), while sensitivity referred to the number of variants correctly identified by WES out of all variants detected by the array-based assay (true positive/[true positive + false negative]).
Variant annotation of exclusive variants
The PharmacoScan unique variants of 28 core genes were identified and the variant annotations were generated through the data analysis using software tools available with the Applied Biosystems Axiom Analysis Suite version 3.1.
The variants only detected by WES of 28 core genes were identified, then were annotated using SnpEff, SIFT, GERP++ and MutationTaster, which annotated and predicted the effects of genetic variants on genes and proteins (such as amino acid changes) [Citation20–23]. Further, the variants were annotated using databases such as dbSNP (www.ncbi.nlm.nih.gov/snp/), ClinVar (www.ncbi.nlm.nih.gov/clinvar/) and GWAS (www.ebi.ac.uk/gwas/) Catalog using SnpSift. The inclusion of the dbSNP database helped in providing reference SNP cluster IDs to each of the exclusive WES variants. The annotations from PharmGKB and US FDA pharmacogenomics were also provided by taking a reference of the reference SNP cluster IDs or gene names, respectively (coming from the SnpEff tool). Given that PharmGKB provides annotation at multiple levels – namely, clinical annotation, functional assay annotation, phenotypic annotation and variant–drug association annotation [Citation24] – all such levels were considered to fetch all possible annotations for exclusive variants.
Results
Comparison of variants in whole-exome sequencing & array-based technology
Taking the variants identified by the PharmacoScan assay as standard, PPV and sensitivity of the WES-based SNV detection over the commonly targeted regions for 28 core ADME genes was calculated. Compared with PharmacoScan, the PPV of ImmunoID NeXT WES ranged from 0.71 to 0.92 and the sensitivity of SNVs ranged from 0.68 to 0.95 in the 28 core ADME gene regions. A total of 160 common variants were detected in both assays.
In the uncommonly targeted regions, approximately 200–300 SNVs per sample were exclusively identified by WES within 28 ADME gene regions and not by PharmacoScan (Supplementary Table 1), giving a total of 1136 unique variants across all samples. On the other hand, 40–80 variants per sample were exclusively identified by the PharmacoScan assay (Supplementary Table 2).
Array-based technology exclusive variant prioritization
In total, 31 exclusive PharmacoScan variants that were not identified by WES analysis were thoroughly annotated. Out of these, 12 variants of the CYP2C9, CYP2D6, UGT1A1 or UGT2B7 genes were identified and prioritized based on gene activity filters (either increased or decreased) and phenotype call (i.e., poor metabolizer, rapid metabolizer or intermediate metabolizer; ).
Table 1. Comprehensive summary of exclusive variants identified by PharmacoScan™.
Whole-exome sequencing exclusive variant prioritization
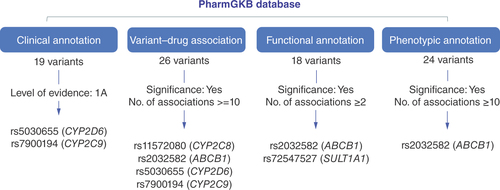
Among 1136 exclusive WES variants falling within the core ADME genes which were not detected by PharmacoScan, a total of 55 variants could be annotated using the PharmGKB database ( & Supplementary Table 3). A total of 26 variants were annotated at the variant–drug level, 18 at the variant–functional assay level, 19 had clinical annotations and 24 had variant–phenotype annotations. Variants with the highest level of evidence (1A), indicating a high degree of confidence in the relationship between the gene variant and the drug response, were prioritized at the clinical level. This prioritization led to the identification of rs5030655 (CYP2D6) and rs7900194 (CYP2C9) as clinically significant. Meanwhile, for variants with significant variant–drug associations, the maximum number of drug/phenotypic associations were considered for prioritization at the variant–drug association, functional annotation and phenotypic level. This led to the identification of rs2032582 (ABCB1), rs5030655 (CYP2D6), rs11572080 (CYP2C8), rs7900194 (CYP2C9) and rs72547527 (SULT1A1) as important modifiers for metabolism, dosage, PK or toxicity based on factors such as phenotypic category, evidence level, significance of association and maximum number of drug associations.
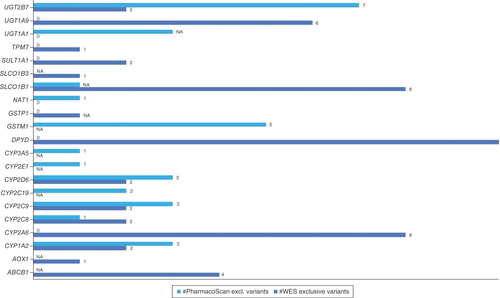
Overall, the number of variants exclusively identified by WES exceeded the number of exclusive PharmacoScan variants. WES picked up exclusively eight variants from CYP2A6, which are associated with metabolism of CYP2A6 substrates, while PharmacoScan did not identify any exclusive variant for this enzyme. CYP2A6 is known for its role in the metabolism of nicotine, some hormones and several therapeutic drugs. It is also involved in the activation of certain procarcinogens into carcinogenic compounds [Citation25]. Because of the variability in the genetic expression of CYP2A6 among individuals, there is significant interest in understanding how this enzyme affects the metabolism of drugs and other substances, as this can impact personalized medicine and the optimization of drug dosing. A similar observation was made for DPYD, where WES identified 12 variants related to metabolism/PK and toxicity while PharmacoScan picked up none. For TPMT, one variant (related to metabolism/PK) was identified by WES while none was captured from PharmacoScan. Also, for SLCO1B1, seven variants having associations with metabolism/PK functioning and one variant having an influence on toxicity were detected by WES only ().
Overview of the exclusive performance of WES can also be observed from the image for CYP2A6, DPYD, TPMT.
NA: Genes for which no annotation data exist. ’0’: No variants detected by the technique; WES: Whole-exome sequencing.
Discussion
WES has already become an integral part of patients’ characterization in clinical trials, especially in oncology [Citation26–28]. Given that blood collection for use in WES-based analyses of tumor genotypes has become clinical standard practice, further utility of the germline variation information from these blood samples would be attractive both from a sample collection/utility perspective and for analysis cost reduction. However, the use of WES as a standard method for pharmacogenomic testing has not been established. In this study we used 79 blood pharmacogenomic samples collected from four clinical trials to compare the ImmunoID NeXT WES readouts versus standard pharmacogenomic testing by a microarray-based technology, PharmacoScan. It was found that WES detected relevant SNVs with high PPV and sensitivity when compared with the standard microarray-based SNV detection assay for pharmacogenomic profiling, indicating that WES has a high accuracy for detecting SNVs in the targeted ADME genes. This is consistent with a previous study comparing between WES and the iPLEX® ADME PGx 36-gene panel (Agena Bioscience, CA, USA) conducted on a smaller sample size of 12 samples [Citation29]. Chua et al. also revealed that the majority of variant calls derived from WES were in agreement with those obtained from 192 polymorphisms covered by the iPLEX ADME PGx panel, resulting in a concordance rate of approximately 90%. PharmacoScan is a microarray-based platform designed specifically for pharmacogenomic studies. It focuses on specific genes and variants known to be associated with drug metabolism and response. In contrast, WES aims to capture the entire exome, which includes all protein-coding regions of the genome. Differences in the variant calls (sensitivity) are not unexpected and could be attributed to the different assay technologies with varying assay principles and sensitivities. Exome sequencing has several limitations, including missing noncoding variants and difficulties in sequencing specific genomic regions, such as regions with high homology, repetitive regions and GC-rich regions, for which the PharmacoScan assay is specifically designed. For example, CYP2D6 presents a unique challenge for sequencing technologies, largely due to its genetic complexity and the presence of highly similar pseudogenes, such as CYP2D7 and CYP2D8, in its vicinity [Citation30]. These factors can lead to inappropriate capture and mismapping of reads, which are the potential reasons for the absence of these variants in the WES data. The homology between CYP2D6 and its pseudogenes complicates accurate variant calling, as reads may be incorrectly assigned to the pseudogenes instead of the target gene.
Because WES analyses, in contrast to oligonucleotide-based array technology, cover all exonic regions of the human genome, it is expected that additional variants of the ADME genes could be identified. Using PharmGKB [Citation31], one of the most comprehensive and widely known databases for pharmacogenomics, as an SNV annotation source, we showed that those additionally identified variants could be important for assessment of the metabolic activities of the 28 core ADME proteins for individual patients. Furthermore, WES identified a significantly higher number of unique variants in the 28 core ADME gene regions compared with array-based technology. This suggests that WES has the potential to reveal additional pharmacogenetic biomarkers and may facilitate the discovery of novel loci that may be relevant to PK variability. We plan to conduct separate confirmatory tests to validate the unique variants detected by WES in future work.
Although there is a need for further development in the functional annotation of ADME gene variants, WES-based pharmacogenomic tests show promise for continued investigation. The strong end-user support and continuous improvement are crucial for the success of WES-based technology in pharmacogenomics. To advance this field, the potential areas of focus may include: first, thorough analysis of the variants generated by WES, including CNV and HLA mapping, and their effects on ADME; and second, establishing an NGS-based pharmacogenomic analysis solution that covers sequencing, annotation and interpretation of pharmacogenomic variants. We envision such a ’fit for purpose’ technology application to detect germline polymorphisms and perform pharmacogenomic profiling in future clinical trials with an aim to significantly enhance the efficiency and accuracy of pharmacogenomic analysis.
Assessment of pharmacogenetic variability is a continuous effort throughout drug development. The enzymes responsible for the metabolism of an investigational drug are typically screened early during candidate selection. It would be critical to integrate pharmacogenetic analysis in early clinical trials for characterization of PK/PD variability. In addition, in oncology clinical trials, having pharmacogenetic data along with germline variant (PD and/or disease) information could be highly informative to optimize the dose required for a favorable benefit/risk profile in proof-of-concept studies. Although the numbers of subjects enrolled in phase I trials are generally limited, data from these studies could provide preliminary evidence of the effect of pharmacogenetics on PK/PD variability. Furthermore, this would contribute to pooled analyses with later clinical trials (e.g., phase II/III trials) by incorporating these data in population PK models to understand the effect of genetic variability on exposure and efficacy/safety. WES has been utilized in oncology clinical trials to generate comprehensive molecular profiling and a multidimensional view of the tumor [Citation32]. Meanwhile, growing evidence has demonstrated that gene polymorphisms have the potential to influence drug activity and that pharmacogenomics influences the efficacy of drugs in additional ways, both in specific areas and probably beyond. Thus, conducting genomic profiling of blood samples in these trials is in high demand. Therefore, the integrated analytical approach using WES will be of high scientific value in clinical trials.
Conclusion
Collectively, WES has promising and feasible clinical utility in identifying pharmacogenetic variants and expanding the understanding of the role of genetic variability. With more research conducted in this area, the efficiency and accuracy of pharmacogenomic analysis using WES can be further enhanced. This will ultimately contribute to the development of more effective and personalized treatment strategies for patients, paving the way for precision medicine-driven drug development.
Precision medicine in drug development requires identifying the right target, patient and dose, focusing on pharmacokinetic and pharmacodynamic variability.
Whole-exome sequencing (WES), a technique that sequences all protein-coding genes in the genome, has gained attention as a promising tool for pharmacogenomic profiling, providing a comprehensive view of an individual’s genetic variations, including those affecting drug metabolism and response.
This study analyzed 79 blood samples from four phase I oncology clinical trials, employing both PharmacoScan™ array and ImmunoID NeXT™ WES, to conduct a comparative analysis.
WES showed high positive predictive value and sensitivity in identifying single-nucleotide variants in absorption, distribution, metabolism and elimination genes compared with standard array-based technology.
WES identified a significantly higher number of unique variants in core absorption, distribution, metabolism and elimination genes, suggesting its potential in revealing additional pharmacogenetic biomarkers.
Differences in variant calls between WES and array technology can be attributed to their varying principles and sensitivities.
The study suggests that WES-based pharmacogenomics tests show promise for enhancing efficiency and accuracy in identifying genetic variability impacting drug response.
Overall, WES demonstrates promising clinical utility in pharmacogenetic variant identification and advancing personalized treatment strategies in precision medicine.
Author contributions
D Wang conceptualized, designed and administered the project, performed data analysis and interpretation and wrote the manuscript. J Bolleddula designed the project, was involved in data analysis and interpretation and made substantial contribution to composing the manuscript. A Coenen-Stass contributed to data analysis and interpretation. T Grombacher contributed to data analysis and interpretation and also wrote the manuscript. J Dong designed the project, was involved in data interpretation and revised the manuscript critically for important intellectual content. J Scheuenpflug designed the project and provided scientific expertise. G Locatelli conceived and designed the study, supervised the project, wrote the manuscript and acquired the funding. Z Feng supervised the project, contributed to the conceptualization and design of the study, was involved in data interpretation and also contributed to writing the manuscript. All authors contributed to the review process and approved the final version.
Financial disclosure
This research was supported by the Merck (CrossRef Funder ID: 10.13039/100009945). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Competing interests disclosure
D Wang, J Bolleddula, J Dong and Z Feng are employees of EMD Serono Research & Development Institute, Inc., Billerica, MA, USA, an affiliate of Merck KGaA and own stock in Merck. A Coenen-Stass, T Grombacher, J Scheuenpflug and G Locatelli are employees of Merck Healthcare KGaA, Darmstadt, Germany.
The authors have no other competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript apart from those disclosed. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Writing disclosure
No writing assistance was utilized in the production of this manuscript.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
Supplementary Table 4
Download PDF (595.1 KB)Supplementary Table 1
Download PDF (5.1 MB)Supplementary Table 2
Download PDF (26.4 MB)Acknowledgments
The authors would like to thank Vittorio D’Urso, NilsGrabole and Rene Eickhoff of Merck Healthcare KGaA, for providing support during the study. The authors are grateful to Florian Lorenz of Merck Healthcare KGaA, and Nisha Sheikh of Merck Serono Ltd., Feltham, UK, an affiliate of Merck KGaA, for their editorial assistance. A preliminary analysis of the dataset used in the current manuscript was presented as a poster (# M0930-10-59) at the American Association of Pharmaceutical Scientists 2022.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2217/pgs-2023-0243
Data availability statement
Any requests for data by qualified scientific and medical researchers for legitimate research purposes will be subject to Merck’s (CrossRef Funder ID: 10.13039/100009945) Data Sharing Policy. All requests should be submitted in writing to Merck’s data sharing portal (https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html). When Merck has a co-research, co-development, or co-marketing or co-promotion agreement, or when the product has been out-licensed, the responsibility for disclosure might be dependent on the agreement between parties. Under these circumstances, Merck will endeavor to gain agreement to share data in response to requests.
Reference
- Huang SM , TempleR. Is this the drug or dose for you? Impact and consideration of ethnic factors in global drug development, regulatory review, and clinical practice. Clin. Pharmacol. Ther.84(3), 287–294 (2008).
- Reyner E , LumB , JingJ , KagedalM , WareJA , DickmannLJ. Intrinsic and extrinsic pharmacokinetic variability of small molecule targeted cancer therapy. Clin. Transl. Sci.13(2), 410–418 (2020).
- Ma Q , LuAY. Pharmacogenetics, pharmacogenomics, and individualized medicine. Pharmacol. Rev.63(2), 437–459 (2011).
- Sim SC , KacevskaM , Ingelman-SundbergM. Pharmacogenomics of drug-metabolizing enzymes: a recent update on clinical implications and endogenous effects. Pharmacogenomics J.13(1), 1–11 (2013).
- Li J , BluthMH. Pharmacogenomics of drug metabolizing enzymes and transporters: implications for cancer therapy. Pharmgenomics Pers. Med.4, 11–33 (2011).
- US Food and Drug Administration . Table of pharmacogenomic biomarkers in drug labeling. www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling
- Annotation of FDA Label for siponimod and CYP2C9. www.accessdata.fda.gov/drugsatfda_docs/label/2023/209884s015lbl.pdf
- El Rouby N , LimaJJ , JohnsonJA. Proton pump inhibitors: from CYP2C19 pharmacogenetics to precision medicine. Expert Opin. Drug Metab. Toxicol.14(4), 447–460 (2018).
- Stingl J , VivianiR. Polymorphism in CYP2D6 and CYP2C19, members of the cytochrome P450 mixed-function oxidase system, in the metabolism of psychotropic drugs. J. Intern. Med.277(2), 167–177 (2015).
- Tornio A , BackmanJT. Cytochrome P450 in pharmacogenetics: an update. Adv. Pharmacol.83, 3–32 (2018).
- Bienfait K , ChhibberA , MarshallJCet al. Current challenges and opportunities for pharmacogenomics: perspective of the Industry Pharmacogenomics Working Group (I-PWG). Hum. Genet.141(6), 1165–1173 (2022).
- Medwid S , KimRB. Implementation of pharmacogenomics: where are we now?Br. J. Clin. Pharmacol. doi: 10.1111/bcp.15591 (2022) .
- Arbitrio M , SciontiF , DiMartino MTet al. Pharmacogenomics biomarker discovery and validation for translation in clinical practice. Clin. Transl. Sci.14(1), 113–119 (2021).
- Tafazoli A , VanDer Lee M , SwenJJet al. Development of an extensive workflow for comprehensive clinical pharmacogenomic profiling: lessons from a pilot study on 100 whole exome sequencing data. Pharmacogenomics J.22(5–6), 276–283 (2022).
- Aboul-Soud MaM , AlzahraniAJ , MahmoudA. Decoding variants in drug-metabolizing enzymes and transporters in solid tumor patients by whole-exome sequencing. Saudi J. Biol. Sci.28(1), 628–634 (2021).
- Wang L , SchererSE , BielinskiSJet al. Implementation of preemptive DNA sequence-based pharmacogenomics testing across a large academic medical center: the Mayo-Baylor RIGHT 10K Study. Genet. Med.24(5), 1062–1072 (2022).
- Lanillos J , CarcajonaM , MaiettaP , AlvarezS , Rodriguez-AntonaC. Clinical pharmacogenetic analysis in 5001 individuals with diagnostic exome sequencing data. NPJ Genom. Med.7(1), 12 (2022).
- Suwinski P , OngC , LingMHT , PohYM , KhanAM , OngHS. Advancing personalized medicine through the application of whole exome sequencing and big data analytics. Front. Genet.10, 49 (2019).
- Samuels M , FalkeniusJ , Bar-AdVet al. A phase 1 study of the DNA-PK inhibitor peposertib in combination with radiation therapy with or without cisplatin in patients with advanced head and neck tumors. Int. J. Radiat. Oncol. Biol. Phys.118(3), 743–756 (2023).
- Cingolani P , Platts, A , Wangleet al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 6(2), 80–92 (2012).
- Cingolani P . SnpSift. [Software] (2012). http://snpeff.sourceforge.net/SnpSift.html
- Davydov E V , GoodeD L , SirotaMet al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLOS Computational Biology6(12), e1001025 (Year).
- Schwarz J M , RödelspergerC , SchuelkeM , SeelowD. MutationTaster evaluates disease-causing potential of sequence alterations. Nature Methods7(8), 575–576 (2010).
- Gong L , Whirl-CarrilloM , KleinTE. PharmGKB, an integrated resource of pharmacogenomic knowledge. Curr. Protoc.1(8), e226 (2021).
- McDonagh EM , WassenaarC , DavidSPet al. PharmGKB summary: very important pharmacogene information for cytochrome P-450, family 2, subfamily A, polypeptide 6. Pharmacogenet. Genomics22(9), 695–708 (2012).
- Lopacinska-Joergensen J , OliveiraD , PoulsenTS , HoegdallCK , HoegdallEV. Somatic variants in DNA damage response genes in ovarian cancer patients using whole-exome sequencing. Anticancer Res.43(5), 1891–1900 (2023).
- Wen L , BrittonCJ , GarjeR , DarbroBW , PackiamVT. The emerging role of somatic tumor sequencing in the treatment of urothelial cancer. Asian J. Urol. 8(4), 391–399 (2021).
- Gray SW , ParkER , NajitaJet al. Oncologists’ and cancer patients’ views on whole-exome sequencing and incidental findings: results from the CanSeq study. Genet. Med.18(10), 1011–1019 (2016).
- Chua EW , CreeSL , TonKNet al. Cross-comparison of exome analysis, next-generation sequencing of amplicons, and the iPLEX(®) ADME PGx panel for pharmacogenomic profiling. Front. Pharmacol.7, 1 (2016).
- Taylor C , CrosbyI , YipV , MaguireP , PirmohamedM , TurnerRM. A Review of the Important Role of CYP2D6 in Pharmacogenomics. Genes (Basel)11(11), 1295 (2020).
- Barbarino JM , Whirl-CarrilloM , AltmanRB , KleinTE. PharmGKB: a worldwide resource for pharmacogenomic information. Wiley Interdiscip. Rev. Syst. Biol. Med.10(4), e1417 (2018).
- Donoghue MTA , SchramAM , HymanDM , TaylorBS. Discovery through clinical sequencing in oncology. Nat. Cancer1(8), 774–783 (2020).