
Abstract
Osteogenesis imperfecta (OI) is a rare heritable disease characterized by skeletal fragility, bone deformity, and growth retardation. In its classical and more common forms, it is caused by dominant mutations in collagen type I genes, COL1A1 and COL1A2. A wide range of clinical severity is described in OI, ranging from very mild to moderately severe, progressively deforming, and perinatal lethal. Mutations causing null allele and consequent synthesis of half of the amount of normal collagen are responsible for milder OI phenotypes, whereas point mutations altering amino acid sequence, thus affecting protein structure, lead to severe OI outcomes. Because no resolutive cures are so far available for OI patients and given the new advances in gene-targeting technology, genetic and cellular therapy represent an appealing option for OI treatment. In this review, we briefly summarized what has been done so far for classical OI in terms of novel regenerative approaches. The different strategies adopted to silence the mutant allele with the aim of converting severe qualitative defects to milder quantitative ones, and for transplanting normal multipotent cells to generate a mosaic condition, which in OI is associated to lack of clinical symptoms, are presented. The key issues that still need to be addressed for the effective clinical application of these strategies are critically discussed.
In context
Type I collagen is the most abundant protein in bone and its quantity and proper structure guarantee normal skeletal properties. It is constituted by two α1 and one α2 protein chains assembled in a triple helical motif. Mutations in one of the two genes encoding for the α chains are the main causes for osteogenesis imperfecta (OI), a heritable disorder characterized by skeletal fragility and deformity and growth delay. The defects can cause either a reduction in the normal amount of collagen, usually associated to a mild phenotype, or the synthesis of collagen with abnormal structure, generally causing a more severe outcome. No resolutive cures are available for OI. Novel approaches of gene and cellular therapy had been attempted. Gene therapy approaches are based on the suppression of the mutant gene that will convert a qualitative collagen type I defect into a quantitative one associated to a milder outcome. Cell therapy approaches are mainly based on transplantation of normal cells into OI bone to generate a mosaic of mutant and normal cells because OI mosaic subjects, that is, parents of OI affected children, do not present skeletal phenotype. In this review, we summarized and discussed the critical issues that still need to be addressed to make these approaches real treatment options.
Osteogenesis imperfecta (OI), usually referred as ‘brittle bone disease’, is a heritable disorder causing skeletal fragility and deformity, and associated to growth deficiency. The dominant OI forms are mainly caused by defects in the collagen type I genes COL1A1 and COL1A2, leading to an alteration in the amount or structure of type I collagen, the most abundant protein of bone extracellular matrix (ECM) (Citation1). These OI forms were originally classified into four types (I–IV) based on clinical and radiological data (Citation2). A null COL1A1 allele causes the mildest OI type I, characterized by half of the normal amount of structurally normal type I collagen and representing the major cause of hereditary juvenile osteoporosis (Citation3–Citation6). Point mutations in either COL1A1 or COL1A2 resulting in substitutions of a glycine residue or in an aberrant splicing are mainly responsible for the altered type I collagen structure found in the moderately severe OI type IV, progressive deforming OI type III and lethal OI type II (Citation7). More recently mutations in genes encoding for proteins involved in collagen type I post-translational modifications [CRTAP (Citation8), LEPRE1/P3H1 (Citation9), PPIB (Citation10)], folding [FKBP10 (Citation11), SERPINH1 (Citation12)], intracellular trafficking [SEC24D (Citation13)], and extracellular processing [BMP1 (Citation14)] had been described in recessive OI cases. Furthermore, genes involved in osteoblasts differentiation [WNT1 (Citation15), SP7 (Citation16), and CREB3L1 (Citation17)] had been linked to recessive OI. Recessive defects in SERPINF1 (Citation18), encoding for the pigment epithelium derived factor, and dominant mutations in interferon-induced-transmembrane protein 5 encoding gene (IFITM5) (Citation19) affect bone mineralization in rare cases of OI. The alteration of intracellular calcium homeostasis caused by loss-of-function of the trimeric monovalent cation channel TRIC-B encoded by TMEM38B (Citation20), and mutations in osteonectin gene (SPARC) (Citation21) are among the last causative defects identified as responsible for recessive OI. Whenever secreted, abnormal collagen is incorporated in the ECM where it affects cell–cell and cell–matrix interactions, leading to bone deformity and fragility (Citation7, Citation22). A fraction of mutant collagen is frequently retained inside the cells causing cellular stress that was hypothesized to modulate the OI phenotype (Citation23, Citation24). Physiotherapy, orthopedic surgery, and bisphosphonate administration are the most common interventions undertaken to ameliorate OI patient's quality of life because no resolutive cures are available for this disease (Citation1, Citation25). The antiresorptive drug denosumab, a receptor activator of nuclear factor kappa-B ligand (RANKL) antibody that impairs osteoclasts maturation, and two anabolic drugs stimulating osteoblast function, sclerostin antibody and the inhibitor of TGF-β signal transduction pathway, are now under investigation (Citation22, Citation26–Citation31) [for a more extensive review of these approaches, see (Citation32)].
Gene therapy
Although the resolutive cure for dominant OI would be the correction of the DNA defect, for the more severe forms the silencing of the mutant allele may represent an interesting option. The specific suppression of the mutant allele, with the consequent haploinsufficiency, will convert a severe phenotype characterized by collagen structural defects to a mild OI, caused by a quantitative defect. Several attempts in this direction had been made using various antisense technologies such as antisense oligodeoxyribonucleotides (ODNs), ribozymes, short interfering RNA (siRNA), and short hairpin RNA (shRNA) (a–c) (Citation25). Although efficiency and specificity were not absolute, promising data were obtained for all the silencing tools tested. The majority of the published reports comes from studies using either in vitro or ex vivo approaches, and less data are available from in vivo studies in OI murine models. A selective 37–43% suppression of the mutant transcript associated to 44–47% decrease of the mutant protein was obtained using antisense ODNs against COL1A2 and only a 20% reduction of the normal mRNA was detected (Citation35). The antisense approach efficiency was confirmed in vivo in a mouse transgenic for a human COL1A1 mini gene in which the antisense sequence of the 3′ half of the COL1A1 mini gene was expressed under the same collagen promoter used for the transgene (Citation36). The hammerhead ribozymes were first successfully tested against synthetic collagenic targets in vitro, and no cleavage occurred using the normal transcript as target (Citation37). Ribozymes’ efficiency and allele-specific silencing against a mutant COL1A1 allele was then demonstrated ex vivo in a patient fibroblast line in which 50% suppression of the mutant allele was reported with no suppression of the wild-type allele (Citation38). Primary murine osteoblasts isolated from the transgenic mice expressing the human COL1A1 mini gene were also used to demonstrate the allele-specific silencing power of rybozymes, and no cleavage was observed in the endogenous allele (Citation39). shRNA molecules suppressed efficiently the mutant Col1a1 allele in primary fibroblasts from Brtl+/−, a model for classical OI carrying a G349C substitution in α1(I) (Citation40). In these cells, shRNAs subcloned in a lentiviral vector and stably integrated into the endogenous genome reduced the amount of mutant transcript by 57–65% versus 24–28% of wild-type transcripts. Mutant protein was reduced by 40% (Citation40). A mutation-independent approach using siRNA targeted heterozygous single nucleotide polymorphisms (SNPs) in COL1A2 (Citation41) or insertion/deletion polymorphisms (indels) in both the COL1A1 and COL1A2 genes (Citation42) in human bone-derived cells led to a 71% reduction in COL1A2 transcript when targeting SNPs and 65–78% reduction in COL1A1 and 26–49% COL1A2, respectively, when indels were targeted.
Fig. 1 Gene therapy strategies to target mutant collagen transcripts. (a) Antisense oligodeoxyribonucleotides (ODNs) are single-stranded DNA molecules that upon their entry into a cell are able to hybridize to their target RNA leading to its degradation through RNAase H or to directly inhibit protein translation. (b) Hammerhead ribozymes, synthetic RNA molecules containing a catalytic core and binding arms complementary to the mutant mRNA, specifically bind to the target mRNA leading to its degradation (Citation33). (c) Short interfering RNA (siRNA) can either be transfected into the target cells as synthetic double-stranded RNA or produced by plasmids coding for small hairpin RNA (shRNA) and stably incorporated into the endogenous genomic DNA. In the latter case, following transcription, the shRNAs are processed by Dicer to obtain siRNA. siRNAs are incorporated into the nuclease-containing multiprotein complex RISC (RNA-induced silencing complex) and one of the two siRNA strands, the guide strand, guides the RISC to its complementary target mRNA, which is then cleaved and degraded (Citation34).
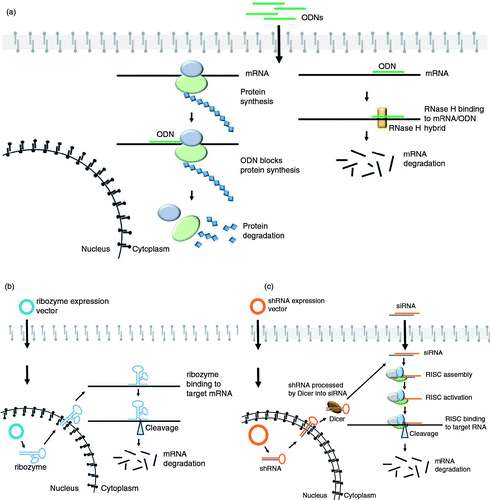
Given the repetitive nature of collagen sequence and the high number of mutations described so far (Citation7, Citation43), this latter strategy will definitely facilitate the design of gene silencing approaches for dominant OI reducing the number of siRNA to be designed and favoring the ones outside the region coding for the triple helix (Citation7).
Hydroxyapatite/tricalcium phosphate powder-type I bovine fibrillar collagen matrices loaded with patients mesenchymal stem cells (MSCs) in which mutant COL1A1 or COL1A2 collagen genes were disrupted by adeno-associated virus vectors were placed under the skin of immunodeficient mice. Analysis of the implants revealed the presence of human osteoblasts able to form mineralized bone in vivo only in presence of human MSCs (Citation44, Citation45). No bone formation was detectable in cell-free implants, and neither human mitochondrial antigen nor human collagen was detectable in presence of implants loaded with murine MSCs. Combination of gene targeting in human MSCs isolated from OI patients and generation of induced pluripotent stem cells (iPSCs) has been used as proof of principle to target skeletal abnormalities. iPSCs derived from patients’ MSCs following in vitro correction were able to differentiate to osteoblast and to produce normal collagen and successfully formed bone in vivo (Citation46). The silencing of the mutant allele in induced patient's iPSCs able to differentiate to osteoblasts with subsequent return of the modified cells to the affected individual will represent an exciting avenue in OI treatment.
Cell-based therapy
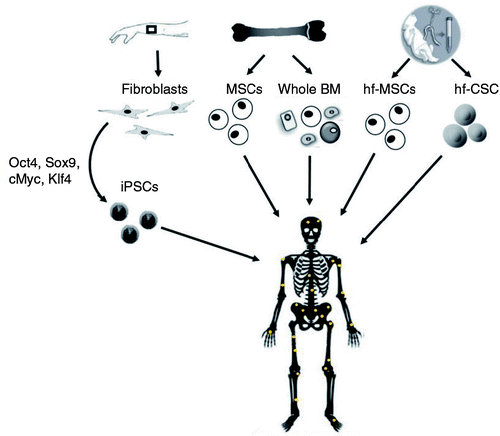
A novel and promising approach to treat OI is the transplantation into patients of normal stem cells with osteoblast differentiation potential. The scientific base of the treatment is the absence of clinical skeletal phenotype in mosaic carriers even in presence of a high number of mutant osteoblasts (Citation47). This observation suggested that even a small fraction of normal osteoprogenitor cells may lead to significant phenotypic benefits (). Feasibility trials for cell transplantation had been performed both in OI mice models and in patients. Because MSCs are able to differentiate into mesodermal lineages such as osteoblasts (Citation48, Citation49), whole BM or isolated MSCs had been tested as source of bone precursor cells.
Fig. 2 Potential sources of donor stem cells for OI cellular therapy based on their use in preclinical or clinical trials. iPSCs: induced pluripotent stem cells; MSCs: mesenchymal stem cells; BM: bone marrow; hf-MSCs: human fetal mesenchymal stem cells; hf-CSCs: human fetal chorionic stem cells.
Preclinical investigations
MSCs used in animal studies were generally modified to express the green fluorescent protein (GFP) for an easy detection, selected for positivity for putative MSCs markers and for the absence of markers associated with hematopoietic cells and macrophages (Citation50). In one case, MSCs were injected into normal mice and subsequently retrieved from bone, expanded in culture, and utilized for transplantation with the rationale that the use of cells selected for their liking to migrate in the skeletal tissues would allow a higher bone engraftment (Citation51).
Also cells from reporter mice expressing GFP either under the ubiquitously active cytomegalovirus promoter or the osteoblasts and osteocyte-specific collagen Col2.3 promoter were used to identify donor cells in the recipient mice (Citation52, Citation53). Human first-trimester fetal blood MSCs, selected by plastic adhesion and confirmed to express the specific MSCs markers (Citation54), as well as human fetal chorionic stem cells (hf-CSCs) (Citation55), easily isolated in high numbers from the placenta during ongoing pregnancy and containing a subfraction of cells expressing pluripotency embryonic stem cells markers, were also utilized.
In preclinical trials various OI murine models were used: a transgenic mice carrying a human COL1A1 mini gene (Citation56), the Brtl+/− mouse model of OI type IV (Citation57), and oim−/− mouse model of OI type III (Citation50–Citation55), and donor stem cells were transplanted either, in utero and/or in neonatal or in adult age. Also different cell delivery systems were used: from systemic injection throughout temporal or tail vein, to intraperitoneal or fetal liver injection, to direct infusion in the femoral cavity. Although the bone engraftment of donor cells into the host was generally low (0.3–28%), revealing the need to ameliorate either cell source and/or delivery system, it demonstrated the ability of the transplanted cells to home to bone, to differentiate into osteoblasts, to deposit normal matrix, and to improve bone structural properties (Citation50–Citation53, Citation56). In some cases also biomechanical properties were evaluated revealing a better bone matrix in the treated mice (Citation53, Citation57). Interestingly, in the Brtl+/− model, the BM transplantation decreased perinatal lethality further underlying its positive effect on modulating OI phenotype (Citation57). A pronounced reduction in fracture rate was detected using fetal blood MSCs in oim−/− mice (Citation54), and a higher stem cells engraftment in bone was obtained up regulating C-X-C chemokine receptor type 4 (CXCR-4) in the same cells (Citation58).
Clinical investigations
Clinical trials using whole BM or fractionated MSCs were also attempted in children with severe OI (Citation59–Citation63) (). In two pilot studies, the transplant of allogeneic BM obtained from Human Leukocyte Antigen (HLA)-compatible donors resulted in donor cells engraftment in three of the six treated patients favoring growth velocity and total body bone mineral content (Citation62, Citation63). Donor BM-derived MSCs obtained from the same BM donors of the first transplantation were infused into the six affected children previously treated by standard bone marrow transplantation (BMT). Donor MSCs engrafted in bone in five patients and caused an acceleration of growth rate and an increase in total body bone mineral content suggesting that post-transplant treatment with MSCs may enhance the effects of BMT (Citation60). In utero transplantation with HLA-mismatched fetal MSCs revealed successful engraftment in a fetus diagnosed with OI and led to a normal growth rate and psychomotor development until 2 years of age (Citation61). This patient and another OI proband who received as well prenatal human fetal MSCs (hfMSCs) transplantation were also treated postnatally with same-donor MSCs and a reduction in the fracture incidence and a normalization of the growth rate were reported (Citation59). A deep evaluation of bone engraftment, long-term effects and the identification of the best cell source await further studies.
Table 1 Cellular therapy in OI patients
A critical issue that will need further investigation is to define the most effective time for cell transplantation. Being OI, an inborn disease the in utero treatment seems the most appropriate. On the other hand, what really counts is the number of donor cells able to engraft in the host and, based on the available data, it does not seem to be correlated with an early time of transplantation. Importantly, a prenatal infusion will allow the use of HLA-mismatched donor cells which are easier to obtain, and the use of the same-donor cells postnatally will be a further benefit.
Of relevance also will be to clarify what is really responsible for the detected improvement on bone properties after transplantation both in mice and in humans. Using in utero BM transplantation in Brtl+/− mice, we recently demonstrated that a low cell engraftment (~2%) was able to produce over 20% of normal collagen that may explain the amelioration in bone phenotype (Citation57). On the other hand, a low percentage of donor-derived osteoblasts in the host suggests that soluble factors secreted by donor MSCs may positively act on the endogenous cellular environment (Citation64).
Bone delivery: a challenge for OI gene/cellular therapy
The challenge for OI gene and stem cells therapy will be to specifically target the bone tissue. To date a high therapeutic dose of systemically administered modifying molecule or a high number of transplanted cells would be needed to stimulate sufficient bone formation and may carry a high risk for adverse effects in non-skeletal tissues (Citation65).
For systemic delivery of therapeutic nucleic acids, viral vectors have been used because of their high transfection efficacy, but the severe safety risks due to their oncogenic potential and their inflammatory and immunogenic effects prevent them from repeated administration (Citation66, Citation67). siRNA delivery systems made in the last years a rapid progress, but bone remains a difficult target tissue to be reached (Citation68), although research is particularly active in this field. Atelocollagen-mediated systemic administration of siRNAs designed for inhibition of bone metastasis resulted in an efficient inhibition of metastatic tumor growth revealing its capacity to target bone (Citation69). The complexation of siRNAs with polyethylenimine efficiently stabilized siRNAs and, upon systemic administration, led to the delivery of the siRNAs into different organs, but no data on specific bone targeting are available jet (Citation70, Citation71).
Recently, a bone targeting system involving dioleoyl trimethylammonium propane-based cationic liposomes attached to six repetitive sequences of the tripeptide Asp-Ser-Ser has been successful in delivering siRNAs specifically to bone-formation surfaces (Citation72). Even more recently Zhang et al. developed aptamer-functionalized lipid nanoparticles for in vivo siRNA delivery specifically to osteoblasts cells, a system that seems to overcome the potentially harmful off target effect of the cationic liposomes (Citation73).
To date, the homing percentage of donor stem cells into recipient bone is not considered enough for effective therapy. This issue has become the major drawback for OI cellular therapy (Citation65). Only if genetically modified (Citation74), in vitro pretreated with specific drugs (Citation75), or in response to injuries (Citation76), MSCs showed a better engraftment into bone. Because over expression of α4 integrin on MSCs was reported to increase their homing to bone (Citation77), a novel method has been recently developed taking advantage of the high bone affinity of the bisphosphonate alendronate (Ale). Alendronate has been linked to a specific peptidomimetic ligand (LLP2A) of the integrin α4β1 present on the MSC surface (Citation78, Citation79). A single intravenous injection of LLP2A-Ale-MSCs was able to increase MSCs homing to bone, and bone mass in vivo (Citation78).
Other delivery systems based on nanospheres or nanofibers are currently under investigation. Surface-modified nanospheres have been utilized for targeting drugs and diagnostic agents to the bone while extending their circulation time in the blood stream (Citation80). In particular, polymeric nanoparticles consisting of poly-d,l-lactic-co-glycolic acid, polyethylene glycol, and the target ligand alendronate showed retention and accumulation in bone in vivo (Citation81). Nanofiber scaffolds have also shown promising results in delivering bioactive agents and stem cells for bone regeneration and in promoting stem cell differentiation (Citation82).
Future perspective and conclusions
Correction of the mutant allele in patient cells followed by their transplantation back to the affected individual would represent the most attractive avenue for the treatment of classical dominant OI. The use of iPSCs obtained in vitro from patients skin fibroblasts instead of MSCs, which are difficult to isolate and to maintain multipotent in vitro (Citation83), will represent a challenge in OI treatment. iPSCs will allow higher amount of transplantable cells and will eliminate the need for the availability of HLA-matched donors allowing the treatment of all the patients. New technologies for genetic correction such as CRISPR/Cas system, TALEN, and zinc-finger nucleases may represent a step forward in DNA correction in adult cells, although their use for the highly repetitive GC-rich collagen genes will need optimization (Citation84).
The increase of stem cells bone-specific homing together with a better knowledge of the best stem cells source and the understanding of transplantation long-term effects remain the main challenges for the development of combined gene-cell therapy for OI.
The increasing number of genes whose mutations cause OI raises the question about the existence of a common therapy for all the OI types. The correction of the gene defect and cell transplantation will potentially benefit all OI forms, but less invasive and laborious approaches may be identified based on the affected metabolic pathway.
Conflict of interest
The authors have not received any funding or benefits from industry or elsewhere to conduct this study.
Funding
The work was supported by Fondazione Cariplo [grant n. 2013-0612], Telethon [grant n. GGP13098] and the European Community, FP7, “Sybil” project [grant n. 602300].
References
- Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. 2011; 7: 540-57.
- Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979; 16: 101-16.
- Krassas GE. Idiopathic juvenile osteoporosis. Ann N Y Acad Sci. 2000; 900: 409-12.
- Slayton RL, Deschenes SP, Willing MC. Nonsense mutations in the COL1A1 gene preferentially reduce nuclear levels of mRNA but not hnRNA in osteogenesis imperfecta type I cell strains. Matrix Biol. 2000; 19: 1-9.
- Fahiminiya S, Majewski J, Al-Jallad H, Moffatt P, Mort J, Glorieux FH. Osteoporosis caused by mutations in PLS3: clinical and bone tissue characteristics. J Bone Miner Res. 2014; 29: 1805-14.
- Palomo T, Al-Jallad H, Moffatt P, Glorieux FH, Lentle B, Roschger P. Skeletal characteristics associated with homozygous and heterozygous WNT1 mutations. Bone. 2014; 67: 63-70.
- Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007; 28: 209-21.
- Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006; 127: 291-304.
- Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet. 2007; 39: 359-65.
- van Dijk FS, Nesbitt IM, Zwikstra EH, Nikkels PG, Piersma SR, Fratantoni SA. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet. 2009; 85: 521-7.
- Alanay Y, Avaygan H, Camacho N, Utine GE, Boduroglu K, Aktas D. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2010; 86: 551-9.
- Christiansen HE, Schwarze U, Pyott SM, AlSwaid A, Al Balwi M, Alrasheed S. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet. 2010; 86: 389-98.
- Garbes L, Kim K, Riess A, Hoyer-Kuhn H, Beleggia F, Bevot A. Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am J Hum Genet. 2015; 96: 432-9.
- Martinez-Glez V, Valencia M, Caparros-Martin JA, Aglan M, Temtamy S, Tenorio J. Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum Mutat. 2012; 33: 343-50.
- Pyott SM, Tran TT, Leistritz DF, Pepin MG, Mendelsohn NJ, Temme RT. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet. 2013; 92: 590-7.
- Lapunzina P, Aglan M, Temtamy S, Caparros-Martin JA, Valencia M, Leton R. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am J Hum Genet. 2010; 87: 110-14.
- Symoens S, Malfait F, D'Hondt S, Callewaert B, Dheedene A, Steyaert W. Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J Rare Dis. 2013; 8: 154
- Becker J, Semler O, Gilissen C, Li Y, Bolz HJ, Giunta C. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2011; 88: 362-71.
- Semler O, Garbes L, Keupp K, Swan D, Zimmermann K, Becker J. A mutation in the 5’-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet. 2012; 91: 349-57.
- Volodarsky M, Markus B, Cohen I, Staretz-Chacham O, Flusser H, Landau D. A deletion mutation in TMEM38B associated with autosomal recessive osteogenesis imperfecta. Hum Mutat. 2013; 34: 582-6 [PubMed Abstract]
- Mendoza-Londono R, Fahiminiya S, Majewski J, Tetreault M, Nadaf J, Kannu P. Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am J Hum Genet. 2015; 96: 979-85.
- Grafe I, Yang T, Alexander S, Homan EP, Lietman C, Jiang MM. Excessive transforming growth factor-beta signaling is a common mechanism in osteogenesis imperfecta. Nat Med. 2014; 20: 670-5.
- Forlino A, Tani C, Rossi A, Lupi A, Campari E, Gualeni B. Differential expression of both extracellular and intracellular proteins is involved in the lethal or nonlethal phenotypic variation of BrtlIV, a murine model for osteogenesis imperfecta. Proteomics. 2007; 7: 1877-91.
- Bianchi L, Gagliardi A, Gioia R, Besio R, Tani C, Landi C. Differential response to intracellular stress in the skin from osteogenesis imperfecta Brtl mice with lethal and non lethal phenotype: a proteomic approach. J Proteomics. 2012; 75: 4717-33.
- Shapiro JR, Rowe DW. Genetic approach to treatment of osteogenesis imperfecta in Osteogenesis imperfecta. 1st ed2013London, UKElsevier Science and Technology
- Bargman R, Huang A, Boskey AL, Raggio C, Pleshko N. RANKL inhibition improves bone properties in a mouse model of osteogenesis imperfecta. Connect Tissue Res. 2010; 51: 123-31.
- Hoyer-Kuhn H, Semler O, Schoenau E. Effect of Denosumab on the Growing Skeleton in Osteogenesis Imperfecta. J Clin Endocrinol Metab 2014; 99: 3954-5 [PubMed Abstract]10.1210/jc.2014-3072
- Roschger A, Roschger P, Keplingter P, Klaushofer K, Abdullah S, Kneissel M. Effect of sclerostin antibody treatment in a mouse model of severe osteogenesis imperfecta. Bone. 2014; 66: 182-8.
- Semler O, Netzer C, Hoyer-Kuhn H, Becker J, Eysel P, Schoenau E. First use of the RANKL antibody denosumab in osteogenesis imperfecta type VI. J Musculoskelet Neuronal Interact. 2012; 12: 183-8 [PubMed Abstract]
- Sinder BP, Eddy MM, Ominsky MS, Caird MS, Marini JC, Kozloff KM. Sclerostin antibody improves skeletal parameters in a Brtl/+ mouse model of osteogenesis imperfecta. J Bone Miner Res. 2012; 28: 73-80.
- Sinder BP, White LE, Salemi JD, Ominsky MS, Caird MS, Marini JC. Adult Brtl/+ mouse model of osteogenesis imperfecta demonstrates anabolic response to sclerostin antibody treatment with increased bone mass and strength. Osteoporos Int. 2014; 25: 2097-107.
- Besio R, Forlino A. Treatment options for osteogenesis imperfecta. Expert Opin Orphan D. 2015; 3: 165-81.
- Grassi G, Marini JC. Ribozymes: structure, function, and potential therapy for dominant genetic disorders. Ann Med. 1996; 28: 499-510.
- Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000; 404: 293-6.
- Wang Q, Marini JC. Antisense oligodeoxynucleotides selectively suppress expression of the mutant alpha 2(I) collagen allele in type IV osteogenesis imperfecta fibroblasts. A molecular approach to therapeutics of dominant negative disorders. J Clin Invest. 1996; 97: 448-54.
- Khillan JS, Li SW, Prockop DJ. Partial rescue of a lethal phenotype of fragile bones in transgenic mice with a chimeric antisense gene directed against a mutated collagen gene. Proc Natl Acad Sci U S A. 1994; 91: 6298-302.
- Grassi G, Forlino A, Marini JC. Cleavage of collagen RNA transcripts by hammerhead ribozymes in vitro is mutation-specific and shows competitive binding effects. Nucleic Acids Res. 1997; 25: 3451-8.
- Dawson PA, Marini JC. Hammerhead ribozymes selectively suppress mutant type I collagen mRNA in osteogenesis imperfecta fibroblasts. Nucleic Acids Res. 2000; 28: 4013-20.
- Toudjarska I, Kilpatrick MW, Niu J, Wenstrup RJ, Tsipouras P. Delivery of a hammerhead ribozyme specifically downregulates mutant type I collagen mRNA in a murine model of osteogenesis imperfecta. Antisense Nucleic Acid Drug Dev. 2001; 11: 341-6.
- Rousseau J, Gioia R, Layrolle P, Lieubeau B, Heymann D, Rossi A. Allele-specific Col1a1 silencing reduces mutant collagen in fibroblasts from Brtl mouse, a model for classical osteogenesis imperfecta. Eur J Hum Genet. 2013; 22: 667-74.
- Lindahl K, Rubin CJ, Kindmark A, Ljunggren O. Allele dependent silencing of COL1A2 using small interfering RNAs. Int J Med Sci. 2008; 5: 361-5.
- Lindahl K, Kindmark A, Laxman N, Astrom E, Rubin CJ, Ljunggren O. Allele dependent silencing of collagen type I using small interfering RNAs targeting 3'UTR Indels – a novel therapeutic approach in osteogenesis imperfecta. Int J Med Sci. 2013; 10: 1333-43.
- Dalgleish R. The Human Collagen Mutation Database 1998. Nucleic Acids Res. 1998; 26: 253-5.
- Chamberlain JR, Deyle DR, Schwarze U, Wang P, Hirata RK, Li Y. Gene targeting of mutant COL1A2 alleles in mesenchymal stem cells from individuals with osteogenesis imperfecta. Mol Ther. 2008; 16: 187-93.
- Chamberlain JR, Schwarze U, Wang PR, Hirata RK, Hankenson KD, Pace JM. Gene targeting in stem cells from individuals with osteogenesis imperfecta. Science. 2004; 303: 1198-201.
- Deyle DR, Khan IF, Ren G, Wang PR, Kho J, Schwarze U. Normal collagen and bone production by gene-targeted human osteogenesis imperfecta iPSCs. Mol Ther. 2012; 20: 204-13.
- Cabral WA, Marini JC. High proportion of mutant osteoblasts is compatible with normal skeletal function in mosaic carriers of osteogenesis imperfecta. Am J Hum Genet. 2004; 74: 752-60.
- Grove JE, Bruscia E, Krause DS. Plasticity of bone marrow-derived stem cells. Stem Cells. 2004; 22: 487-500.
- Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997; 4: 71-4.
- Li F, Wang X, Niyibizi C. Distribution of single-cell expanded marrow derived progenitors in a developing mouse model of osteogenesis imperfecta following systemic transplantation. Stem Cells. 2007; 25: 3183-93.
- Wang X, Li F, Niyibizi C. Progenitors systemically transplanted into neonatal mice localize to areas of active bone formation in vivo: implications of cell therapy for skeletal diseases. Stem Cells. 2006; 24: 1869-78.
- Pauley P, Matthews BG, Wang L, Dyment NA, Matic I, Rowe DW. Local transplantation is an effective method for cell delivery in the osteogenesis imperfecta murine model. Int Orthop. 2014; 38: 1955-62.
- Li F, Wang X, Niyibizi C. Bone marrow stromal cells contribute to bone formation following infusion into femoral cavities of a mouse model of osteogenesis imperfecta. Bone. 2010; 47: 546-55.
- Guillot PV, Abass O, Bassett JH, Shefelbine SJ, Bou-Gharios G, Chan J. Intrauterine transplantation of human fetal mesenchymal stem cells from first-trimester blood repairs bone and reduces fractures in osteogenesis imperfecta mice. Blood. 2008; 111: 1717-25.
- Jones GN, Moschidou D, Abdulrazzak H, Kalirai BS, Vanleene M, Osatis S. Potential of human fetal chorionic stem cells for the treatment of osteogenesis imperfecta. Stem Cells Dev. 2014; 23: 262-76.
- Pereira RF, O'Hara MD, Laptev AV, Halford KW, Pollard MD, Class R. Marrow stromal cells as a source of progenitor cells for nonhematopoietic tissues in transgenic mice with a phenotype of osteogenesis imperfecta. Proc Natl Acad Sci U S A. 1998; 95: 1142-7.
- Panaroni C, Gioia R, Lupi A, Besio R, Goldstein SA, Kreider J. In utero transplantation of adult bone marrow decreases perinatal lethality and rescues the bone phenotype in the knockin murine model for classical, dominant osteogenesis imperfecta. Blood. 2009; 114: 459-68.
- Jones GN, Moschidou D, Lay K, Abdulrazzak H, Vanleene M, Shefelbine SJ. Upregulating CXCR4 in human fetal mesenchymal stem cells enhances engraftment and bone mechanics in a mouse model of osteogenesis imperfecta. Stem Cells Transl Med. 2012; 1: 70-8.
- Gotherstrom C, Westgren M, Shaw SW, Astrom E, Biswas A, Byers PH. Pre- and postnatal transplantation of fetal mesenchymal stem cells in osteogenesis imperfecta: a two-center experience. Stem Cells Transl Med. 2014; 3: 255-64.
- Horwitz EM, Gordon PL, Koo WK, Marx JC, Neel MD, McNall RY. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: implications for cell therapy of bone. Proc Natl Acad Sci U S A. 2002; 99: 8932-7.
- Le Blanc K, Gotherstrom C, Ringden O, Hassan M, McMahon R, Horwitz E. Fetal mesenchymal stem-cell engraftment in bone after in utero transplantation in a patient with severe osteogenesis imperfecta. Transplantation. 2005; 79: 1607-14.
- Horwitz EM, Prockop DJ, Gordon PL, Koo WW, Fitzpatrick LA, Neel MD. Clinical responses to bone marrow transplantation in children with severe osteogenesis imperfecta. Blood. 2001; 97: 1227-31.
- Horwitz EM, Prockop DJ, Fitzpatrick LA, Koo WWK, Gordon PL, Neel M. Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat Med. 1999; 5: 309-13.
- Prockop DJ, Kota DJ, Bazhanov N, Reger RL. Evolving paradigms for repair of tissues by adult stem/progenitor cells (MSCs). J Cell Mol Med. 2010; 14: 2190-9.
- Gao J, Dennis JE, Muzic RF, Lundberg M, Caplan AI. The dynamic in vivo distribution of bone marrow-derived mesenchymal stem cells after infusion. Cells Tissues Organs. 2001; 169: 12-20.
- Lehrman S. Virus treatment questioned after gene therapy death. Nature. 1999; 401: 517-18.
- Sun JY, Anand-Jawa V, Chatterjee S, Wong KK. Immune responses to adeno-associated virus and its recombinant vectors. Gene Ther. 2003; 10: 964-76.
- Oliveira S, Storm G, Schiffelers RM. Targeted delivery of siRNA. J Biomed Biotechnol. 2006; 2006: 63675
- Takeshita F, Minakuchi Y, Nagahara S, Honma K, Sasaki H, Hirai K. Efficient delivery of small interfering RNA to bone-metastatic tumors by using atelocollagen in vivo. Proc Natl Acad Sci U S A. 2005; 102: 12177-82.
- Aigner A. Delivery systems for the direct application of siRNAs to induce RNA interference (RNAi) in vivo. J Biomed Biotechnol. 2006; 2006: 71659
- Urban-Klein B, Werth S, Abuharbeid S, Czubayko F, Aigner A. RNAi-mediated gene-targeting through systemic application of polyethylenimine (PEI)-complexed siRNA in vivo. Gene Ther. 2005; 12: 461-6.
- Zhang G, Guo B, Wu H, Tang T, Zhang BT, Zheng L. A delivery system targeting bone formation surfaces to facilitate RNAi-based anabolic therapy. Nat Med. 2012; 18: 307-14.
- Liang C, Guo B, Wu H, Shao N, Li D, Liu J. Aptamer-functionalized lipid nanoparticles targeting osteoblasts as a novel RNA interference-based bone anabolic strategy. Nat Med. 2015; 21: 288-94.
- Marquez-Curtis LA, Janowska-Wieczorek A. Enhancing the migration ability of mesenchymal stromal cells by targeting the SDF-1/CXCR4 axis. Biomed Res Int. 2013; 2013: 561098
- Mukherjee S, Raje N, Schoonmaker JA, Liu JC, Hideshima T, Wein MN. Pharmacologic targeting of a stem/progenitor population in vivo is associated with enhanced bone regeneration in mice. J Clin Invest. 2008; 118: 491-504 [PubMed Abstract] [PubMed CentralFull Text]
- Granero-Molto F, Weis JA, Miga MI, Landis B, Myers TJ, O'Rear L. Regenerative effects of transplanted mesenchymal stem cells in fracture healing. Stem Cells. 2009; 27: 1887-98.
- Kumar S, Ponnazhagan S. Bone homing of mesenchymal stem cells by ectopic alpha 4 integrin expression. FASEB J. 2007; 21: 3917-27.
- Guan M, Yao W, Liu R, Lam KS, Nolta J, Jia J. Directing mesenchymal stem cells to bone to augment bone formation and increase bone mass. Nat Med. 2012; 18: 456-62.
- Yao W, Lane NE. Targeted delivery of mesenchymal stem cells to the bone. Bone. 2015; 70: 62-5.
- Park YJ, Nah SH, Lee JY, Jeong JM, Chung JK, Lee MC. Surface-modified poly(lactide-co-glycolide) nanospheres for targeted bone imaging with enhanced labeling and delivery of radioisotope. J Biomed Mater Res A. 2003; 67: 751-60.
- Swami A, Reagan MR, Basto P, Mishima Y, Kamaly N, Glavey S. Engineered nanomedicine for myeloma and bone microenvironment targeting. Proc Natl Acad Sci U S A. 2014; 111: 10287-92.
- Zhang Z, Hu J, Ma PX. Nanofiber-based delivery of bioactive agents and stem cells to bone sites. Adv Drug Deliv Rev. 2012; 64: 1129-41.
- Morikawa S, Mabuchi Y, Kubota Y, Nagai Y, Niibe K, Hiratsu E. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med. 2009; 206: 2483-96.
- van der Oost J. Molecular biology. New tool for genome surgery. Science. 2013; 339: 768-70 [PubMed Abstract]