
Abstract
Objectives
Mortality from respiratory and cardiovascular health conditions contributes largely to the total mortality that has been associated with exposure to PM2.5 in epidemiology studies. A mode of action (MoA) for these underlying morbidities has not been established, but it has been proposed that some effects of PM2.5 occur through activation of neural reflexes.
Materials and Methods
We critically reviewed the experimental studies of PM2.5 (including ambient PM2.5, diesel exhaust particles, concentrated ambient particles, diesel exhaust, and cigarette smoke) and neural reflex activation, and applied the principles of the International Programme on Chemical Safety (IPCS) MoA/human relevance framework to assess whether they support a biologically plausible and human-relevant MoA by which PM2.5 could contribute to cardiovascular and respiratory causes of death. We also considered whether the evidence from these studies supports a non-threshold MoA that operates at low, human-relevant PM2.5 exposure concentrations.
Results and Discussion
We found that the proposed MoA of neural reflex activation is biologically plausible for PM2.5-induced respiratory effects at high exposure levels used in experimental studies, but further studies are needed to fill important data gaps regarding the relevance of this MoA to humans at lower PM2.5 exposure levels. A role for the proposed MoA in PM2.5-induced cardiovascular effects is plausible for some effects but not others.
Conclusions
Further studies are needed to determine whether neural reflex activation is the MoA by which PM2.5 could cause either respiratory or cardiovascular morbidities in humans, particularly at the ambient concentrations associated with total mortality in epidemiology studies.
Introduction
Particulate matter (PM) consists of a mixture of solid particles and liquid droplets that comprises the particle phase of air pollution. PM is formed from a variety of natural and anthropogenic sources, including industrial processes, motor vehicles, fossil fuel combustion, burning of natural materials (e.g. wildfires), crustal and biological material, and chemical reactions of gaseous pollutants (e.g. sulfur oxides, nitrogen oxides, ammonia) (US EPA Citation2019). The numerous potential sources of PM result in a highly heterogenous chemical composition and particle size distribution for PM across locations and time periods.
PM is classified into different size fractions depending on the aerodynamic diameter of the particles it contains. The United States Environmental Protection Agency (US EPA) evaluates exposures and potential health effects for three main size fractions: PM10-2.5, known as coarse PM, consists of particles with a diameter of 2.5–10 μm; PM2.5, known as fine PM, consists of particles with a diameter less than 2.5 μm; and ultrafine particles (UFPs), also known as nanoparticles, have a diameter of less than 0.1 μm. The toxicity of PM depends on both its chemical composition and the size of particles. US EPA (2019) reviewed the epidemiology literature for each of the three main PM size fractions and concluded that there is a causal relationship between short-term (i.e.,up to one month) and long-term (i.e. one month or longer) exposure to PM2.5 and total (nonaccidental) mortality, based on positive associations of small magnitude reported across many of the reviewed studies (Prueitt et al. Citation2021). Mortality from cardiovascular and respiratory health conditions contributes largely to total mortality. US EPA (2019) stated that positive evidence for the effects of PM2.5 exposure on cardiovascular and respiratory parameters in experimental animal and controlled human exposure studies and related morbidities in epidemiology studies (particularly development and exacerbations of COPD and asthma, ischemic events, stroke, and heart failure) provides coherence and biological plausibility for the relationship between PM2.5 exposure and total mortality. US EPA (2019) also stated that there is evidence to support a concentration-response relationship for PM2.5 exposure and total mortality that is linear without a threshold.
A mode of action (MoA) is defined as a biologically plausible sequence of key events (i.e. observable steps) at the cellular, biochemical, or tissue level that result in a pathological outcome (Meek et al. Citation2014b). Several MoAs have been proposed for associations between PM2.5 exposure and the underlying cardiovascular and respiratory morbidities that contribute to cause-specific and total mortality, including: (1) respiratory tract injury resulting in inflammation and oxidative stress responses that lead to downstream respiratory or cardiovascular effects, and (2) activation of sensory nerves in the respiratory tract that trigger local reflex responses and modulate the autonomic nervous system (Watkinson et al. Citation2001; Schulz et al. Citation2005; Shannahan et al. Citation2012; Perez et al. Citation2015; US EPA Citation2019; Bhatnagar Citation2022). In addition, proposed MoAs for associations between PM2.5 and cardiovascular morbidities specifically include: (1) effects on ion channel function in myocardial cells, and (2) acceleration or exacerbation of atherosclerosis (Utell et al. Citation2002; Emmerechts and Hoylaerts, Citation2012). Our previous evaluation found that the MoA of acceleration or exacerbation of atherosclerosis is a biologically plausible MoA in experimental animal models, but the human relevance of the key events in this MoA is unclear (Prueitt et al. Citation2015).
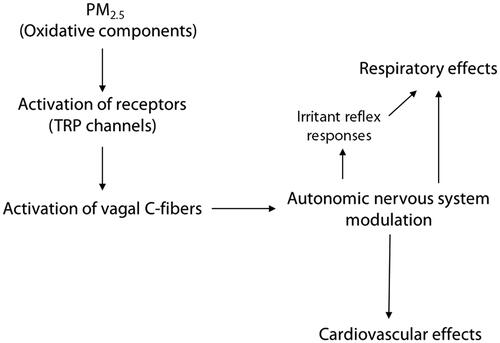
A proposed mode of action of neural reflex activation for both respiratory and cardiovascular effects following PM2.5 exposure is shown in . Inhalation of PM2.5 (particularly the oxidative components) can activate sensory receptors on the nerve terminals of fibers within the vagus nerve,such as vagal C-fibers, leading to modulation of the autonomic nervous system, includingthe triggering of reflex responses in the respiratory tract that are intended to decrease exposure to airway irritants (Undem and Sun Citation2020; US EPA Citation2019). These reflex responses include cough, bronchoconstriction, and shortness of breath (Perez et al. Citation2015), and their activationcan result inother consequences in the respiratory tract, such as airway inflammation and lung function decrements, which may be linked to respiratory morbidities such as asthma and exacerbation of COPD (US EPA Citation2019). Activation of irritant receptors on sensory nerves in the respiratory tract and modulation of the autonomic nervous system can also affect cardiovascular responses, with a shift toward increased sympathetic tone (Watkinson et al. Citation2001; Shannahan et al. Citation2012; Perez et al. Citation2015; US EPA Citation2019; Bhatnagar 2022). This type of shift can induce changes in heart rate (HR) and heart rate variability (HRV), increased blood pressure (BP), impaired vascular function, conduction abnormalities, and arrhythmia, which could lead to cardiovascular morbidities such as ischemic heart disease (IHD), stroke, or heart failure (Watkinson et al. Citation2001; Shannahan et al. Citation2012; Perez et al. Citation2015; US EPA Citation2019; Bhatnagar 2022).
Figure 1. Proposed mode of action of neural reflex activation for both respiratory and cardiovascular effects following PM2.5 exposure.
In this paper, we critically review the experimental studies of PM2.5 and neural reflex activation and assess whether they support a biologically plausible MoA by which PM2.5 could contribute to cardiovascular and respiratory causes of death. We also consider whether the evidence from these studies supports a non-threshold MoA that operates at low, human-relevant PM2.5 exposure concentrations.
Methods
We focused on identifying experimental studies that examined neural reflex activation in response to exposure to particles most relevant to ambient PM2.5. These included studies in experimental animals and humans with controlled exposures, such as chamber studies, with direct vagal nerve or respiratory exposures to ambient PM2.5, concentrated ambient particles (CAPs), diesel exhaust (DE), diesel exhaust particles (DEPs), or cigarette smoke that evaluated the effects of neural reflex manipulation on any type of respiratory or cardiovascular endpoint. Although DE and cigarette smoke are mixtures that contain not only fine particles but also gases, vapors, semi-volatile organic compounds, metals, and elemental carbon that could affect some of the responses to the mixtures (Fowles and Dybing Citation2003; Maricq Citation2007; Ris Citation2007; NTP Citation2016; Steiner et al. Citation2016), we included studies with exposure to DE and cigarette smoke because they contain high concentrations of PM2.5 and are major contributors to ambient PM2.5 (Maricq Citation2007; Ris Citation2007; Braun et al. Citation2019). Thus, these studies can still be informative for evaluating underlying mechanisms, particularly to corroborate the results of studies that used exposures to particles alone.
We performed literature searches for studies published through 26 October 2022, using PubMed and Scopus. We used the search terms: ((((‘Particulate matter’ OR ‘particulate’ OR ‘particles’ OR ‘particulates’ OR ‘Particulate Matter’[Mesh]) AND (‘2.5’ OR ‘diesel’ OR ultrafine OR ‘Ultrafine Particulate*’[Text Word] OR ‘PM2.5’[Text Word] OR ‘PM 2.5’[Text Word] OR ‘PM(2.5)’[Text Word])) AND ((‘controlled human’ OR ‘human challenge’ OR ‘Randomized Controlled Trial’ [Publication Type] OR ‘Randomized Controlled’[Text Word] OR animals OR rat OR rats OR mice OR mouse))) NOT ((‘cross-sectional’ OR ‘longitudinal’ OR ‘prospective’ OR ‘Cross-Sectional Studies’[Mesh] OR ‘Longitudinal Studies’[Mesh] OR ‘Prospective Studies’[Mesh]))) AND (‘sensory nerve*’[tw] OR ‘vagus nerve’ OR ‘vagal nerves’ OR ‘reflex response’ OR ‘neural reflex*’[tw] OR ‘C fiber’ OR ‘neural activation’ OR ‘reflexes’ OR ‘C-fiber’ OR ‘hypothalamic’ OR ‘neurogenic’ OR ‘nervous system’). We also identified additional studies from the reference lists of relevant studies and review articles identified in our searches, as well as from US EPA's most recent Integrated Science Assessment of Particulate Matter (US EPA Citation2019).
We reviewed the identified studies, incorporating study quality into the evaluation of the results, and applied the principles of the International Programme on Chemical Safety (IPCS) mode-of-action/human relevance framework (Boobis et al. Citation2008; Meek et al. Citation2014a, Citation2014b) to evaluate the plausibility and human relevance of the proposed MoA of neural activation-induced changes in respiratory and cardiovascular function following PM2.5 exposure. We identified the key events critical for the MoA and considered the consistency, essentiality, temporality, and exposure–response relationships of these key events, as well as the biological concordance with other available evidence. In addition, we assessed the relevance of the MoA to humans by examining doses used in experimental animal studies and considering key differences in these animal model studies with humans. We also identified data gaps and sources of uncertainty for the proposed MoA.
Experimental studies relevant to the mode of action
Below, we evaluate the results of the studies we identified that assess the involvement of neural reflex activation in the respiratory and cardiovascular responses to PM2.5 exposure. These studies were conducted in several different experimental animal models (we did not identify any relevant studies conducted in humans) and used various sources of PM2.5 (e.g. DE, DEPs, CAPs, cigarette smoke). The studies assessed whether activation of neural reflexes or sensory nerve receptors associated with bronchopulmonary C-fibers mediates the effects of PM2.5 by either (1) blocking neural impulses and evaluating whether this attenuates or abolishes the PM2.5-induced responses or (2) activating neural impulses and evaluating whether this increases the PM2.5-induced responses.
C-fibers are vagal neurons that are found in many peripheral organ systems, including the smooth muscle cell layer of the respiratory tract, and they conduct the majority of afferent vagal input from the respiratory tract to the central nervous system (Rogerio et al. Citation2011; Undem and Sun Citation2020). C-fibers in the respiratory tract can be activated by a variety of stimuli, such as airborne irritants and mediators of inflammation, which can trigger reflex responses such as cough and shortness of breath (Canning et al. Citation2006; Adriaensen and Timmermans Citation2011; Undem and Sun Citation2020). These reflex responses are mediated by the release of neuropeptides (including substance P), which are released from the activated C-fibers (Barnes Citation1996; Canning et al. Citation2006). In experimental studies, neural impulses from bronchopulmonary C-fibers can be blocked by surgically severing the left and right vagus nerve (i.e. a bilateral cervical vagotomy) or by perivagal treatment with capsaicin. Capsaicin is a neuropeptide-depleting agent and strong respiratory irritant that, when applied directly to the area of the vagus nerve at high concentrations, inactivates C-fibers by stimulating massive secretion and depletion of neuropeptides, blocking the ability of C-fibers to conduct an action potential (Lin et al. Citation2010).
Bronchopulmonary C-fibers can also be stimulated by the activation of a specific family of neuronal receptors. Transient receptor potential (TRP) channels are a family of cation channels that act as environmental sensors for a variety of tissues and physiological processes (Silverman et al. Citation2020). The subfamilies of TRP vanilloid (TRPV) and TRP ankyrin (TRPA) channels are expressed on vagal neurons, such as C-fibers, and initiate the activation of these neurons when acted upon by endogenous and exogenous stimuli, including electrophiles and reactive oxygen species (ROS) (Robinsonet al. 2018; Xu et al. Citation2019; Undem and Sun Citation2020). It is hypothesized that the TRPV1, TRPV4, and TRPA1 channels may be involved in neural reflex responses to irritants in the respiratory tract that result in adverse respiratory and cardiovascular effects (Milici and Talavera Citation2021). The role of TRP channels can be evaluated in experimental studies by treatment with TRP channel agonists to activate these receptors, or by treatment with TRP channel antagonists to block their activation.
Capsaicin is an agonist and potent stimulant of TRPV1 channels, such that systemic administration of capsaicin (as opposed to perivagal application discussed above) activates TRPV1 channels on C-fibers (Silverman et al. Citation2020). Other activators of TRPV1 include bradykinin and acids (Canning et al. Citation2006). Capsazepine (CPZ) is a TRPV1 antagonist (Canning et al. Citation2006; Silverman et al. Citation2020), and various other pharmacological agents serve as agonists or antagonists to specific TRP channels or entire subfamilies of these channels. The studies reviewed below commonly employ systemic administration of these types of agents, in addition to vagotomy or perivagal capsaicin treatment, to evaluate the role of neural reflex activation in PM2.5-induced respiratory and cardiovascular responses.
Studies of respiratory effects with vagal nerve manipulation
We identified two studies conducted in rats that used vagal nerve manipulation via bilateral vagotomy to demonstrate that vagal nerves are responsible for reflex responses to cigarette smoke and DEPs on bradypnea (abnormally slow breathing rate), respiratory minute volume (RMV; the volume of air inhaled and/or exhaled in one minute), and lung inflammatory response (See ). These studies used small numbers of anesthetized animals that were not randomly selected nor weight-randomized into exposure groups and used only one concentration of particles. In addition, one of the studies used cigarette smoke, which is a mixture of particles, gases, and vapors (Lin et al. Citation2010), as the exposure and another used a non-physiological route of exposure to the respiratory tract (intratracheal instillation) (McQueen et al. Citation2007).
Table 1. Experimental animal studies of respiratory effects with vagal nerve manipulation.
Lin et al. (Citation2010) exposed anesthetized rats to 6 mL of 100% cigarette smoke via inhalation, and recorded ventilatory responses to 100% smoke before and after bilateral vagotomy. Respiratory frequency and expiratory duration were measured on a breath-by-breath basis. Cigarette smoke induced a bradypneic response, with animals showing an abnormally slow breathing rate and an increase in the apneic ratio (an index of the magnitude of the bradypneic response). Bilateral cervical vagotomy, serving to eliminate vagal reflexes, completely prevented the bradypneic response.
McQueen et al. (Citation2007) exposed anesthetized rats to 500 μg DEPs by intratracheal instillation once over a period of 15 s. A subset of animals wasbilaterally vagotomized prior to instillation, and another subset of animals was vagotomized 5 min before the end of the experiment (i.e. 6 h following DEP instillation). Animals treated with DEPs with intact vagus nerves had an increased RMV, reflecting an increase in the volume of air inhaled and/or exhaled from the lungs, and increased neutrophil counts in the lungs. The DEP-induced increases in RMV and neutrophil counts were not observed in animals bilaterally vagotomized prior to or 6 h after DEP treatment. Together, the results from the studies by Lin et al. (Citation2010) and McQueen et al. (Citation2007) suggest that elimination of vagal reflexes by severing of the vagus nerve eliminates the reflex respiratory responses to cigarette smoke and DEPs.
Studies of respiratory effects with pharmacological pretreatment to inhibit or activate C-fibers
Several studies examined the effects of PM2.5 on respiratory responses following pretreatment with one or more pharmacological agents to inhibit or activate C-fibers (See ). These studies used small numbers of animals, with most evaluating only one exposure concentration of PM2.5 and using non-physiological routes of exposure such as intratracheal instillation, intranasal instillation, or direct exposure of particles to the vagus nerve. Two studies used cigarette smoke as the exposure (Bergren Citation1985; Lin et al. Citation2010) and one study evaluated the effects of pharmacological pretreatment on respiratory responses to a mixture of PM2.5 and ozone, but not to PM2.5 alone (Lian et al. Citation2022). None of these studies randomized animals into exposure groups except for the studies by Wong et al. (Citation2003) and Lv et al. (Citation2016).
Table 2. Experimental Animal studies of respiratory effects with pharmacological pretreatment.
In the study described above, Lin et al. (Citation2010) exposed anesthetized rats to 6 mL of 100% cigarette smoke by inhalation and recorded the bradypneic response and activity of C-fibers before and after perivagal capsaicin treatment (to block conduction of the afferent vagus nerve fibers), pretreatment with the antioxidant N-acetylcysteine (NAC) to counteract ROS, or intravenous injection of the TRPA1 receptor antagonist HC-030031, the purinergic receptor P2X antagonist iso-Pyridoxalphosphate-6-azophenyl-2′,5′-disulfonate (iso-PPADS), or HC-030031 + iso-PPADS. P2X receptors are ionotropic sensory receptors (i.e. ligand-gated ion channels that open in response to the binding of a specific neurotransmitter) that transduce signals from ROS in the lungs (Lin et al. Citation2010). The mean peak activity of C-fibers in response to 100% cigarette smoke was significantly higher than the activity in response to 50% cigarette smoke, but the effects of the lower concentration of cigarette smoke on bradypneic response were not evaluated. Exposure to 100% cigarette smoke induced bradypnea and also stimulated C-fiber activity within the same time lag as the bradypneic response. Perivagal capsaicin treatment abolished the cigarette smoke-induced bradypneic response, as did pretreatment with the TRPA1 and P2X receptor antagonists HC-030031 and iso-PPADS. Both antagonists significantly attenuated the neural activity of C-fibers in response to cigarette smoke exposure, and this activity was further reduced by a combined treatment with HC-030031 and iso-PPADS, indicating that TRPA1 and P2X channels mediate cigarette smoke-induced C-fiber activity and bradypneic response. Bradypnea and C-fiber activation were also both prevented by NAC, suggesting ROS serves as a trigger for cigarette smoke-evoked stimulation of C-fibers and the bradypneic response.
In the other study described above, McQueen et al. (Citation2007) exposed anesthetized rats to 500 μg DEPs by a single intratracheal instillation, with or without pretreatment of animals with the muscarinic acetylcholine receptor (mAChR) antagonist atropine. Atropine acts as a competitive inhibitor against acetylcholine on structures innervated by postganglionic parasympathetic nerves, and the authors hypothesized that DEPs induce acute pulmonary inflammation through the action of mAChRs. Exposure to DEPs increased the neutrophil count in the lungs and the RMV, but pretreatment with atropine abolished these effects (McQueen et al.Citation2007). The authors also observed increased airway neutrophils in conscious rats exposed to DEPs, and this effect was reduced with pretreatment of the animals with atropine. This study indicates that pharmacological blockage of numerous receptor channels eliminates the C-fiber respiratory responses to DEPs of increased lung neutrophil count and RMV, and these responses act through mAChRs on parasympathetic nerves.
Wong et al. (Citation2003) examined respiratory tract inflammation and plasma extravasation (i.e. edema from neurogenic plasma leakage and a marker of neurogenic inflammation) in rats exposed by inhalation to DE containing either 35.3 ± 4.9 μg/m3 (low dose exposure; LDE) or 632.9 ± 47.6 μg/m3 (high dose exposure; HDE) of DEPs through a nose-only exposure chamber for 4 h per day, 5 days per week over 3 weeks. A third group of rats was exposed to diesel engine room air (with an unspecified concentration of DEPs), in which the rats were kept in their airflow cages beside the DEP exposure setup and were breathing room air. The authors hypothesized that DEP exposure induces neurogenic inflammation in the respiratory tract that is mediated by substance P released from C-fibers. Exposure to DEPs dose-dependently increased edema in the extrapulmonary (i.e. bronchi) and intrapulmonary (i.e. lung parenchyma) airways and exposure to HDE increased alveolar macrophage counts in the lungs and significantly reduced substance P protein expression in the lungs. Rats pretreated with an intraperitoneal dose of capsaicin (intended to stimulate C-fibers rather than ablate them) showed loss of substance P protein, confirming the efficiency of the capsaicin treatment, and also showed significantly higher airway edema in response to DEP exposure, indicating that C-fiber activation and substance P release increased DEP-induced neurogenic inflammation. Surprisingly, capsaicin pretreatment significantly decreased DEP-induced lung inflammation (i.e. number of particle-laden alveolar macrophages) in the HDE group. Thus, while capsaicin pretreatment exacerbated airway edema, indicating a role for C-fiber activation in DEP-induced neurogenic inflammation, capsaicin treatment also decreased lung inflammation histopathology, leading the authors to conclude that DEP-induced lung inflammation cannot be substantially attributed to substance P release from activated bronchopulmonary C-fibers.
Lv et al. (Citation2016) recorded cough reflex sensitivity, airway hyperpermeability, and tracheal and parenchymal lung inflammation in guinea pigs exposed to either 80 mg/kg (low dose) or 320 mg/kg (high dose) PM2.5 through nasal instillation once per day for three weeks. The PM2.5 was collected from ambient air across five urban or suburban sites in Nanjing, China. PM2.5 exposure was associated with increased cough reflex sensitivity (indicated by reduced cough reflex threshold), mild to moderate and moderate-to-severe airway inflammation (low dose and high dose, respectively), increased airway hyperpermeability, and increased substance P and TRPV1 protein expression in the airways. Following exposure, a subset of guinea pigs was microinjected with capsaicin (intended to stimulate C-fibers) or the TRPV1 receptor antagonist CPZ. Stimulation of C-fibers by treatment with capsaicin induced a significantly higher cough reflex sensitivity than in animals treated with PM2.5 alone, and blockage of TRPV1 receptor activation by CPZ resulted in significantly lower cough reflex sensitivity (indicated by an increased cough reflex threshold) than in animals treated with PM2.5 alone. CPZ treatment to high-dose PM2.5 animals also relieved the PM2.5-induced airway inflammation and airway hyperpermeability (though there were no statistical analyses of these results) and was associated with reduced substance P protein expression, whereas capsaicin treatment was associated with increased substance P expression in PM2.5-treated animals. Collectively, the results indicate that TRPV1 activation promotes the cough reflex sensitivity, airway neurogenic inflammation, and airway hyperpermeability response to PM2.5 exposure.
Xu et al. (Citation2019) measured airway hyperresponsiveness to ACh challenge, airway resistance, lung inflammation, and lung tissue ROS in mice exposed to 7.8 mg/kg PM2.5 via intranasal instillation once per day for two days. The PM2.5 was collected from ambient air in an urban district in Shanghai, China. PM2.5 instillation significantly increased bronchial responsiveness, airway resistance, and lung inflammation in mice compared to vehicle controls. A subset of mice was pretreated with the TRPV1 antagonist AMG9810, the TRPA1 antagonist A967079, or both, and each of these treatments reduced the PM2.5-induced bronchial responsiveness and airway resistance to levels similar to vehicle controls. AMG9810 and A967079 also attenuated the PM2.5-induced increases in lung total cell count, macrophages, lymphocytes, neutrophils, and eosinophils, with A967079 and the combined pretreatment having the largest effect. In addition, AMG9810, A967079, or their combination attenuated the PM2.5-induced increase in lung inflammation score (based on histological changes) and the PM2.5-induced increase in lung tissue levels of malondialdehyde (MDA) and mitochondrial ROS, whereas pretreatment with A967079 alone reduced the PM2.5-induced increase in lung tissue hydrogen peroxide levels. Together, the results show that inhibition of TRPA1 channels or combined inhibition of TRPA1 and TRPV1 channels resulted in a greater inhibitory effect on lung inflammation and injury, oxidative stress, and airway hyperresponsiveness than inhibition of TRPV1 channels, suggesting that TRPA1 channels play a more prominent role in reflex respiratory responses compared to TRPV1 (Xu et al. Citation2019).
Bergren (1985) examined the effects of cigarette smoke on airway hyperresponsiveness by exposing ovalbumin (OVA)-sensitized and non-sensitized (NS) guinea pigs to cigarette smoke for 30 min daily in a whole-body inhalation chamber at an average concentration of 5.3 ± 0.1 mg/L for 120 to 156 days. Following the end of exposure to cigarette smoke, the guinea pigs were deeply anesthetized and challenged with capsaicin and bradykinin, both by intravenous injection and aerosolization of capsaicin with subsequent inspiration through a ventilator. Systemic capsaicin and bradykinin act as selective agents for the activation of C-fibers. Chronic cigarette smoke exposure enhanced the intravenous capsaicin- and bradykinin-induced increase in C-fiber activity and airway hyperresponsiveness (as indicated by increased tracheal pulmonary pressure) in OVA-sensitized guinea pigs, but not in NS guinea pigs. Aerosolized capsaicin-induced increases in tracheal pulmonary pressure were also enhanced in cigarette smoke-exposed, OVA-sensitized animals. Overall, this study indicates that C-fibers were hyperactivated by OVA sensitization and cigarette smoke, and suggests that the airway hyperresponsiveness induced by this exposure may be modulated by the resulting hyperactivation of C-fibers as an underlying key event.
Robinson et al. (Citation2018) recorded impulses from C-fibers in anesthetized guinea pigs pretreated with the TRPA1 antagonist Janssen 130, the TRPV1 antagonist Xention D0501, or the TRPV4 antagonist GSK2193874, and challenged with an aerosolized 1 μg/mL dose of DEP organic extract (DEP-OE). A subset of animals was also exposed intratracheally to whole DEPs without TRPA1 or TRPV1 blockage. DEP exposure (10 μg/mL) activated C-fibers, and a range of DEP exposures (0.1–100 μg/mL) induced a concentration-dependent depolarization (i.e. activation) of isolated guinea pig vagus nerves. Intratracheal DEP-OE also depolarized the vagus nerve in a concentration-dependent manner similar to DEPs, indicating the organic components of DEPs are responsible for activation of sensory nerves. Administration of the TRPA1 antagonist Janssen 130 or the antioxidant NAC inhibited aerosolized DEP-OE-induced impulses in the isolated guinea pig vagus nerve, whereas the TRPV1 and TRPV4 antagonists had no effect. Janssen 130 and NAC also inhibited DEP-OE-induced impulses in isolated human vagus nerve tissue. Separate experiments with TRPA1 null mice showed inhibition of DEP-OE-induced impulses, but no significant differences in responses in TRPV1 and TRPV4 null mice compared to wild-type mice. Janssen 130 also inhibited the increased tracheal pressure induced by exposure of isolated guinea pig vagus nerves to aerosolized DEP-OE (though the TRPV1 and TRPV4 antagonists were not evaluated for effects on tracheal pressure). This study indicates that TRPA1 is a primary channel targeted by DEP-OE and is involved in reflex respiratory responses, likely through an oxidative stress mechanism.
Lian et al. (Citation2022) exposed OVA-sensitized and non-sensitized mice to 20 μg PM2.5(collected from ambient air in Wuhan, China) via intratracheal instillation on days 12, 15, and 18 of the experiment and examined allergic asthma endpoints on day 26. The study also included groups exposed to 0.5 ppm ozone via inhalation or a combination of 20 μg PM2.5 and 0.5 ppm ozone from days 12–18. Combined exposure to PM2.5 and ozone (but not to PM2.5 or ozone alone) following OVA challenge resulted in significant increases in lung allergic inflammation, histopathological lesions, oxidative stress, airway hyperresponsiveness, and TRPV1 and substance P expression. The TRPV1 antagonist CPZ was administered to a subset of animals in the PM2.5 + ozone combined exposure group on days 12–18 to investigate the role of the TRPV1 channels in allergic asthma. CPZ reduced the synergistic effects of PM2.5 and ozone on allergic inflammatory responses, histopathological changes, oxidative stress, and airway hyperresponsiveness. Collectively, the results suggest that neurons innervating the lung and expressing TRPV1, such as C-fibers, are responsible for aggravated allergic asthma following combined PM2.5 and O3 exposure.
A study by Weng et al. (Citation2013) provides further evidence that neural receptors and ROS are involved in C-fiber activation. The authors exposed anesthetized rats to 6 mL of 50% or 100% cigarette smoke (with the 50% cigarette smoke generated by mixing the 100% cigarette smoke with an equal volume of air) via inhalation from a syringe and measured activity of lung vagal C-fiber afferents before and after pretreatment with the TRPV1 receptor antagonist CPZ, the hydroxyl radical scavenger dimethylthiourea (DMTU), the P2X receptor antagonist iso-PPADS, or CPZ + iso-PPADS. Cigarette smoke exposure induced significant increases in C-fiber impulses in a concentration-dependent manner. Pretreatment with DMTU attenuated the C-fiber response to 100% cigarette smoke, indicating that the hydroxyl radical, which is a major component of ROS, mediates the cigarette smoke-induced C-fiber response. Pretreatment with CPZ or iso-PPADS also significantly reduced C-fiber impulses induced by 100% cigarette smoke, and this suppression of C-fiber activity was further reduced by pretreatment with the combination of CPZ and iso-PPADS. Collectively, these results indicate that the TRPV1 and P2X receptors are critical for activating C-fibers in response to exposure to cigarette smoke, and that ROS (including the hydroxyl radical) mediates this activation.
Together, the studies that used pharmacological treatments to evaluate whether PM2.5-induced respiratory responses are modulated by neural activation indicate that parasympathetic vagal nerves such as C-fibers mediate reflex respiratory responses to PM2.5, and that ROS may be a trigger for their activation. The studies also indicate that activation of TRPV1 and TRPA1 channels on these nerves facilitates the PM2.5-induced effects on a variety of respiratory endpoints, including cough reflex sensitivity, airway hyperresponsiveness, lung inflammation, and oxidative stress in the lung.
Studies of cardiovascular effects with vagal nerve manipulation
We identified four studies that examined the effects of PM2.5 on cardiovascular endpoints following vagal nerve manipulation via bilateral vagotomy (See ). These studies used small numbers of animals that were not randomized into exposure groups and each evaluated only one exposure concentration of particles. One of these studies used cigarette smoke (Matran et al. Citation1990) and two used DE (Hazari et al. Citation2011; Carll et al. Citation2013) as surrogates for PM2.5. In addition, one of the studies used intratracheal instillation as the exposure route (McQueen et al. Citation2007).
Table 3. Experimental animal studies of cardiovascular effects with vagal nerve manipulation.
McQueen et al. (Citation2007) exposed anesthetized rats to 500 μg DEPs via a single intratracheal instillation and subsets of animals were bilaterally vagotomized prior to instillation or 6 h following DEP instillation. The authors examined the effects of DEPs on blood pressure (BP), heart rate (HR), and heart rate variability (HRV) during the 6-h period after instillation. Neither instillation of DEPs nor bilateral vagotomy conducted prior to instillation altered mean BP, HR, or HRV. The authors credited several possible reasons for the lack of cardiovascular changes, including the DEP stimulus being inadequate to produce a response, an exposure period that was too short, or that the anesthesia itself may have affected the results.
Hazari et al. (Citation2011) exposed spontaneously hypertensive rats to DE containing 150 μg/m3 PM2.5 for four hours via inhalation in an exposure chamber and assessed arrhythmogenesis (by challenging animals with a continuous intravenous infusion of the arrhythmogenic drug aconitine) and changes in cardiac function (HR, HRV, QRS duration, and ST segment duration). Immediately after the DE exposure, a subset of rats underwent bilateral vagotomy. The authors reported that exposure to DE produced a slight increase in HR, increases in HRV measures and QRS duration, a decrease in ST segment duration, and a heightened sensitivity to arrhythmia. The DE-induced increase in HR was exacerbated by bilateral vagotomy, whereas the DE-induced increases in HRV measures were abolished by bilateral vagotomy, except for the increase in the low frequency/high frequency (LF/HF) ratio, which is an estimate of the balance between sympathetic and vagal (parasympathetic) activity. Bilateral vagotomy also prevented the DE-induced increase in QRS duration, but not the DE-induced decrease in ST segment duration, and only partially prevented the DE-induced heightened sensitivity to arrhythmia (Hazari et al. Citation2011).
Carll et al. (Citation2013) exposed spontaneously hypertensive heart failure rats to 500 μg/m3 DE for four hours by inhalation in an exposure chamber and evaluated changes in cardiac contractility, relaxation, and HR. At 24 h after exposure, the rats were anesthetized and infused with dobutamine twice (for a cardiac stress test), and a subset of rats underwent bilateral vagotomy in between infusions. Exposure to DE decreased contractility and slowed left ventricle (LV) relaxation but did not affect HR before infusion. Vagotomy increased HR and contractility compared to pre-dobutamine infusion in both the unexposed control and DE-exposed groups, which indicates parasympathetic signaling by the vagus nerve was successfully impaired, but vagotomy had no effect on LV relaxation or HR deceleration in DE-exposed animals. This study indicates that the effects of DE exposure on HR and contractility act through sympathetic nervous system modulation.
Matran et al. (Citation1990) exposed anesthetized pigs to the smoke from one cigarette (with or without a filter that removed the PM from the smoke) by inhalation for 2 min and assessed vascular responses in three different types of surgically-exposed arteries in vivo: the nasal cavity sphenopalatine artery (SPA), superior laryngeal artery (SLA), and bronchial artery (BA). Exposure to cigarette smoke (either filtered or unfiltered) decreased the vascular resistance of the SPA, SLA, and BA, with the largest decrease in the BA. Decreased vascular resistance indicates an increase in vasodilatation, which is widening of the blood vessels that results in increased bloodflow. Because the results were similar with both filtered and unfiltered cigarette smoke, the effects on vascular resistance were likely attributable to the vapor phase of the smoke rather than the particulate phase. This is consistent with other studies indicating that nitrogen oxide and carbon monoxide in the vapor phase of cigarette smoke are responsible for pulmonary vasodilatation (Zhu and Parmley Citation1995). Matran et al. (Citation1990) also evaluated vascular resistance in the BA in a subset of animals that underwent bilateral vagotomy to sever the vagus nerve prior to exposure. In these animals, the decrease in vascular resistance was unchanged by vagotomy, indicating that an afferent vagal pathway is not likely to be involved in the effects of cigarette smoke on vasodilatation in the BA. The vasodilatation effects were induced by the vapor phase of the smoke rather than the particulates, however, so this study indicates that particulates may not be as potent as the vapor phase in activating neural reflexes.
Together, these studies demonstrate that vagal signaling may not mediate PM2.5-induced changes in HR, LV relaxation, or vascular resistance of bronchial arteries, but may affect PM2.5-induced changes in HRV, QRS duration, and sensitivity to arrhythmias.
Studies of cardiovascular effects with pharmacological pretreatment to inhibit or activate C-fibers
Several studies evaluated the effects of PM2.5 on cardiovascular responses following pretreatment with one or more pharmacological agents to inhibit or activate C-fibers (See ). All of these studies used small numbers of animals that were not randomized into exposure groups and each evaluated only one exposure concentration of PM2.5. Two studies used the non-physiological exposure route of intratracheal instillation (McQueen et al. Citation2007; Robertson et al. Citation2014). Two studies used cigarette smoke (Matran et al. Citation1990; Lin et al. Citation2010) and two used DE (Hazari et al. Citation2011; Carll et al. Citation2013) as the PM2.5 exposure.
Table 4. Experimental animal studies of cardiovascular effects with pharmacological pretreatment.
As discussed above, McQueen et al. (Citation2007) exposed anesthetized rats to 500 μg DEPs via a single intratracheal instillation. A subset of animals was pretreated with atropine 10–20 min before instillation to block parasympathetic signaling, and physiological changes in BP, HR, and HRV were monitored throughout the six-hour period following instillation. As discussed above, instillation of DEPs did not induce alterations in BP, HR, or HRV, and the authors note that BP was also not affected by atropine pretreatment. Due to the lack of cardiovascular effects from DEP exposure, this study does not provide evidence as to whether cardiovascular effects of PM2.5 operate through activation of neural reflexes.
Carll et al. (Citation2013) exposed spontaneously hypertensive heart failure rats to 500 μg/m3 DE for four hours by inhalation and assessed effects on HR, BP, pre-ejection period (PEP, a contractility index), and the HRV indices of standard deviation of the RR interval (SDNN), LF/HF ratio, and square root of the mean of squared differences of adjacent RR intervals (RMSSD), both during exposure and in treadmill stress tests conducted 4 and 21 h after exposure. Subgroups of animals were treated with atropine or atenolol prior to DE exposure to investigate the effects of parasympathetic and sympathetic signaling, respectively, on cardiac response to DE. DE exposure increased HR, mean arterial blood pressure, and cardiac contractility (as indicated by reduced PEP) during the exposure period, and atenolol pretreatment (but not atropine pretreatment) prevented these DE-induced effects. In the treadmill stress test 4 h after exposure, DE significantly increased SDNN and RMSSD but had no effect on HR or LF/HF ratio. Atropine pretreatment did not significantly affect any of these parameters, while atenolol pretreatment significantly decreased HR but had no effect on the HRV parameters. In the treadmill stress test 21 h after exposure, DE exposure increased HR and LF/HF and decreased RMSSD (but had no effect on SDNN), and there were no significant effects on these parameters with either atropine or atenolol pretreatment. There were no significant effects of DE exposure on arrhythmia frequency, HR increase, HR decrease, HR recovery time, or ECG morphology during the treadmill stress tests. There were also no effects of DE on measures of oxidative stress or inflammation in blood, lung lavage fluid, lung tissue, or heart tissue collected 24 h after DE exposure cessation. Overall, this study shows that DE-induced effects on HR, BP, and cardiac contractility act through sympathetic, rather than parasympathetic, signaling in spontaneous hypertensive heart failure rats.
Matran et al. (Citation1990) exposed pigs to the smoke from one cigarette (with or without a filter to remove the PM) by inhalation for 2 min and assessed vascular responses in the SPA, SLA, and BA, with or without pretreatment with atropine to block vagal activation of the heart via parasympathetic signaling, the anesthetic lignocaine to block stimulation of sensory irritant receptors, or systemic capsaicin, all administered via subcutaneous injection. Pretreatment with systemic capsaicin had no effect on the cigarette smoke-induced decreases in vascular resistance in the SPA, SLA, and BA, but did significantly attenuate the local integrated blood flow increase (a marker of dilatation) in the SPA (with no changes observed in the SLA and BA). Atropine and lignocaine also had no effect on cigarette smoke-induced decreases in vascular resistance in the SPA, SLA, or BA. The results from this study suggest that C-fibers or other afferent vagal pathways are not likely involved in the cigarette smoke-induced vasodilatation in the SLA (larynx) and BA (bronchi), but the authors stated that C-fibers may be involved in the response in the SPA (nasal mucosa), suggesting that the systemic capsaicin treatment used in this study was intended to block C-fibers rather than stimulate them. As discussed above, however, the fact that results for vascular resistance were similar whether filtered or unfiltered cigarette smoke was used suggests that the effects were attributable to the vapor phase of the smoke rather than the particulate phase, indicating that particulates may not be as potent as the vapor phase in activating neural reflexes.
Lin et al. (Citation2010) evaluated changes in arterial BP and HR in anesthetized rats exposed to 6 mL of 100% cigarette smoke by inhalation, before and after perivagal capsaicin treatment to block conduction of the afferent vagus nerve fibers, pretreatment with the antioxidant NAC to counteract ROS, or intravenous injection of the TRPA1 receptor antagonist HC030031, the P2X receptor antagonist iso-PPADS, or HC-030031 + iso-PPADS. Cigarette smoke exposure resulted in transient decreases in mean arterial BP and HR, and these effects were attenuated with perivagal capsaicin treatment, as well as treatment with NAC, HC-030031, iso-PPADS, or HC-030031 + iso-PPADS. These results suggest that cigarette smoke-induced decreases in BP and HR are mediated by activation of TRPA1 and P2X receptors on C-fibers and that ROS may serve as the initiating event.
As discussed above, Hazari et al. (Citation2011) exposed spontaneously hypertensive rats to DE containing 150 μg/m3 PM2.5 for four hours via inhalation and assessed HR, HRV, QRS duration, ST segment duration, and sensitivity to arrhythmias from aconitine challenge. Prior to exposure to DE, a subset of rats were pretreated with the TRPA1 antagonist HC030031, the general TRP antagonist ruthenium red, or the TRPV1 antagonist SB366791. Another subset of rats was treated with atropine immediately after the DE exposure to examine effects of parasympathetic signaling. Pretreatment with the TRPA1 antagonist, but not the other agents, prevented the DE-induced increase in HR. Pretreatment with the TRPA1, general TRP, and TRPV1 antagonists, as well as treatment with atropine, prevented the DE-induced increase in QRS duration, and atropine prevented the DE-induced increases in HRV measures. Exposure to DE also produced a decrease in the ST segment duration, and this effect was prevented by the TRPA1, general TRP, and TRPV1 antagonists but not atropine. DE exposure also induced an increase in sensitivity to arrhythmia, and this effect was abolished by pretreatment with the TRPA1 and general TRP antagonists, was partially reduced by the TRPV1 antagonist, and was not affected by atropine. Taken together, the results from this study indicate that TRPA1 and TRPV1 receptors mediate PM2.5-induced changes in cardiac function and proarrhythmic cardiac responses, with effects on HRV and QRS duration involving parasympathetic signaling, and that TRPA1 specifically mediates PM2.5-induced effects on HR.
Robertson et al. (Citation2014) examined effects on BP, ventricular arrhythmia, heart tissue edema, and reperfusion injury in the heart in anesthetized rats exposed to DEPs (0.5 mg) once via intratracheal installation for 6 h, with or without co-treatment with the TRPV1 antagonist AMG 9810. The authors reported that DEPs induced increases in BP, duration of arrhythmia after induction of myocardial ischemia (i.e. restriction of blood flow to the heart), heart tissue edema, and infarct size after reperfusion injury in both in vivo hearts and in hearts isolated from DEP-exposed rats that underwent ischemia induction and reperfusion ex vivo. DEP exposure did not induce inflammatory cell recruitment to the heart, but did increase oxidative stress and apoptosis of cardiomyocytes and reduced cardiomyocyte viability. Co-treatment with AMG 9810 attenuated the DEP-induced increases in BP and arrhythmias and prevented the reperfusion-induced myocardial injury (i.e. increased infarct size, increased apoptosis, and reduced cardiomyocyte viability) observed with DEP exposure for in vivo hearts. The authors did not report the statistical significance of the effects of AMG 9810 on arrhythmia and cardiomyocyte apoptosis, however. These results indicate that TRPV1 activation underlies the DEP-induced increases in BP, arrhythmia, and myocardial injury.
Ghelfi et al. (Citation2008) investigated effects on cardiac function in rats exposed by inhalation to CAPs generated using the Harvard Ambient Particle Concentrator in Boston, Massachusetts (mean PM2.5 mass concentration: 218 ± 23 mg/m3), once for 5 h (24 rats per group) or repeatedly for 5 h per day over 4 months (8 rats per group). For the single exposure study, a subset of rats was pretreated with the TRPV1 antagonist CPZ via intraperitoneal (IP) injection or at a concentration of 500 μM for 20 min via inhalation as an aerosol. Exposure to CAPs induced oxidative stress in the heart (as measured by in situ chemiluminescence in heart tissue), increased oxidized lipids in heart tissue, and induced heart edema, and pretreatment with CPZ by IP injection or aerosol inhalation prevented all of these effects. For the repeated exposure study, a subset of rats was pretreated with CPZ via IP injection and HR was the only endpoint measured. Exposure to CAPs decreased HR, and CPZ pretreatment abolished this effect. The authors did not report whether any of the effects of CPZ pretreatment were significantly different from the effects of exposure to CAPs alone. The findings from this study suggest that CAPs activate TRPV1, which mediates alterations in cardiovascular endpoints such as oxidative stress and HR.
Together, the studies that used pharmacological treatments to evaluate whether PM2.5-induced cardiovascular responses are modulated by neural activation indicate that most responses are mediated through sympathetic signaling, but that parasympathetic signaling is involved in changes in HRV and QRS duration. The activation of TRPV1 and TRPA1 channels facilitates the PM2.5-induced effects on several cardiovascular endpoints, including HR, BP, sensitivity to arrhythmias, QRS duration, ST segment duration, cardiac injury, and cardiac oxidative stress, but these channels do not appear to facilitate effects on HRV. The studies also indicate that P2X receptors and ROS are also mediators in the neural responses to PM2.5.
Evaluation of the proposed mode of action
In this section, we evaluate the plausibility of the activation of neural reflexes as the MoA for the relationship between PM2.5 and mortality induced by altered respiratory and cardiovascular function. We conduct our evaluation in accordance with the principles of the IPCS mode-of-action/human relevance framework (Boobis et al. Citation2008; Meek et al.Citation2014a, Citation2014b). In line with this framework, we evaluate the consistency, essentiality, temporality, and exposure–response relationships of the key events underlying the toxic responses. In addition, we evaluate the biological concordance of the proposed MoA with other available evidence to inform the biological plausibility of the MoA. Then, we evaluate the relevance of the MoA to humans, considering exposure levels at which the key events occur and any differences in the key events between animals and humans. Finally, we identify study uncertainties and data gaps in the literature.
Consistency of effects
Respiratory studies
The respiratory studies using bilateral vagotomy and perivagal capsaicin treatment to block vagal nerve activation showed that cigarette smoke-induced bradypneic responses and DEP-induced RMV and inflammation are mediated through vagal signaling in different animal strains at variable exposures (McQueen et al. Citation2007; Lin et al. Citation2010). In addition, McQueen et al. (Citation2007) showed that the DEP-induced effects on RMV, as well as lung inflammation, are modulated by activation of parasympathetic signaling.
Studies using aerosolized or systemic capsaicin treatment to activate TRPV1 receptors consistently showed evidence of C-fiber stimulation in mediating respiratory responses to different types of PM2.5 (cigarette smoke, DEPs, ambient PM2.5, and PM2.5 + ozone) and across different species (Bergren 1985; Wong et al. Citation2003; Lvet al. Citation2016). However, because Wong et al. (Citation2003) reported that systemic capsaicin treatment exacerbated DEP-induced neurogenic inflammation when measured by plasma extravasation in the respiratory tract, but did not affect lung inflammation when measured as the number of particle-laden macrophages in the lung, the authors concluded that C-fibers did not dominate the inflammatory responses to DEPs.
In the studies that utilized TRP antagonists to block TRP signaling, it was shown that TRPA1 and TRPV1 receptors are involved in the PM2.5-induced respiratory responses across different species and types of PM2.5, including cigarette smoke, DEP-OE, and ambient PM2.5(Lin et al. Citation2010; Lv et al. Citation2016; Robinson et al. Citation2018; Xu et al. Citation2019; Lian et al. Citation2022). In addition, the studies by Robinson et al. (Citation2018) and Xu et al. (Citation2019) both showed that TRPA1 plays a larger role in the respiratory and inflammatory reflex responses to particulates. In contrast, the Weng et al. (Citation2013) study showed that cigarette smoke-induced C-fiber impulses are mediated by TRPV1 channels in rats (using the TRPV1 antagonist CPZ), whereas Robinson et al. (Citation2018) demonstrated that the TRPV1 antagonist Xention D0501 had no impact on DEP-OE-evoked impulses in the isolated guinea pig vagus nerve. This may be attributable to different exposures (cigarette smoke vs. DEP-OE), different TRPV1 antagonists, or both.
Two studies also utilized the P2X antagonist iso-PPADS. The findings from these studies provide evidence that P2X receptors are involved in the stimulation of C-fibers and the induction of the bradypneic response by cigarette smoke (Linet al. Citation2010; Weng et al. Citation2013). Because P2X receptors play an important role in the sensory transduction of pulmonary ROS, these findings are consistent with other studies indicating that the oxidative components are the primary mediators involved in neural reflex responses to inhaled particulates from cigarette smoke and DEPs (Lin et al. Citation2010; Weng et al. Citation2013; Robinson et al. Citation2018). Collectively, these animal exposure models using antioxidants and free radical scavengers provide consistent evidence that ROS are important mediators in the C-fiber response to inhaled cigarette smoke and DEPs.
Cardiovascular studies
The cardiovascular studies using bilateral vagotomy and perivagal capsaicin treatment to block vagal nerve signaling reported inconsistent results with respect to effects on PM2.5-induced cardiovascular endpoints. The bilateral vagotomy studies demonstrate that vagal signaling may affect DE-induced changes in HRV, QRS duration, and sensitivity to arrhythmias in spontaneously hypertensive rats (Hazari et al. Citation2011), but may not mediate DE-induced increases in HR in spontaneously hypertensive rats (Hazari et al. Citation2011), DE-induced slowing of LV relaxation in spontaneously hypertensive heart failure rats (Carll et al. Citation2013), or cigarette smoke-induced changes in vascular resistance of bronchial arteries in pigs (Matran et al. Citation1990). In contrast, perivagal capsaicin treatment attenuated cigarette smoke-induced transient increases in HR and arterial blood pressure in rats (Lin et al. Citation2010), though we note these studies used different animal models and different types of PM2.5 exposure.
Studies that used atropine or atenolol to investigate whether PM2.5-induced cardiovascular effects are modulated by parasympathetic or sympathetic signaling also showed inconsistent effects. Atenolol, but not atropine, prevented the DE-induced effects on HR, BP, and cardiac contractility in spontaneous hypertensive heart failure rats, indicating that these effects are mediated by sympathetic, but not parasympathetic signaling (Carll et al. Citation2013). By contrast, parasympathetic signaling was not implicated in the effects of cigarette smoke on vascular resistance in pigs (Matran et al. Citation1990) or the effects of DE exposure on HR, ST segment duration, or sensitivity to arrhythmia in spontaneously hypertensive rats (Hazari et al. Citation2011), as atropine treatment had no effect on these endpoints. Atropine prevented the DE-induced increase in HRV and QRS duration in spontaneously hypertensive rats, however, indicating that these effects are induced through parasympathetic signaling (Hazari et al. Citation2011).
One study used systemic capsaicin treatment to activate TRPV1 receptors and showed that this activation had no effect on the cigarette smoke-induced decrease in vascular resistance in pigs, consistent with the lack of vagal signaling for these effects with bilateral vagotomy (Matran et al. Citation1990). The studies that used TRP antagonists generally showed that TRPA1 receptors mediate cigarette smoke-induced effects on HR (Lin et al. Citation2010), TRPV1 receptors mediate effects of DEPs or CAPs on cardiac injury and oxidative stress (Ghelfi et al. Citation2008; Robertson et al. Citation2014), and both types of receptors mediate effects of DE, DEPs, or cigarette smoke on BP, sensitivity to arrhythmias, QRS duration, and ST segment duration (Lin et al. Citation2010; Hazari et al. Citation2011; Robertson et al. Citation2014). Neither TRPA1 nor TRPV1 receptors were shown to mediate effects on HRV (Hazari et al. Citation2011).
One study evaluated the effects of P2X receptors and NAC on the transient increases in HR and BP induced by cigarette smoke in rats (Lin et al. Citation2010). These inhibitors attenuated the effects on HR and BP, indicating that P2X receptors and ROS are mediators in the neural responses and ultimate CV effects from cigarette smoke.
Essentiality
Essentiality considers the evidence supporting the necessary key events for a proposed MoA. Many of the reviewed studies directly evaluated essentiality by blocking ROS production, C-fiber activation, TRP signaling, or vagal reflexes through vagal manipulation or pharmacological treatments. These studies showed that blockage of these key events attenuated or eliminated downstream respiratory effects and, in some cases, cardiovascular effects. Thus, the studies provide support that these key events are essential in the proposed MoA for respiratory and, possibly, cardiovascular effects.
Temporality
With regard to the temporal relationships of the key events in the proposed MoA, the experimental animal studies indicate that C-fiber impulses and respiratory bradypneic responses follow a similar time-course, indicative of a common mechanistic origin. Activation of neural reflex responses involves several steps that must occur sequentially to generate a physiological response.
For PM2.5 to directly stimulate a C-fiber, PM2.5 must first be inhaled, and inflammatory mediators (e.g. ROS) must interact with their cognate receptors in the neural membrane to form a mediator-receptor interaction. Multiple animal studies indicate oxidative components of PM2.5 to be the primary mediators responsible for physiological responses, as pretreatment with ROS scavengers (i.e. DMTU, NAC) significantly reduces CSLVA impulses and changes in respiratory and cardiac function (Wong et al. Citation2003; Weng et al. Citation2013; Ghelfi et al. Citation2008; Lin et al. Citation2010). Cardiovascular studies also indicate that cardiac oxidative stress and electrophysiological changes are mediated by pulmonary reflexes (McQueen et al. Citation2007; Lin et al. Citation2010). This highlights that oxidative stress is a key event in the MoA and cardiovascular responses are dependent on respiratory responses.
Studies in guinea pigs, rats, and mice support a role for the key events of TRPA1 and TRPV1 agonism by ROS in the activation of C-fibers in the proposed MoA, as animal studies indicate that pretreatment with pharmacological TRPA1 and TRPV1 antagonists reduces impulses recorded from particulate-stimulated C-fibers (Lin et al. Citation2010; Weng et al. Citation2013; Robinson et al. Citation2018), eliminates bradypneic responses, and reduces changes in RMV, airway resistance, airway responsiveness, HRV, and changes in systolic blood pressure (Ghelfi et al. Citation2008; Lin et al. Citation2010; Hazari et al. Citation2011; Robertson et al. Citation2014; Lv et al. Citation2016; Robinson et al. Citation2018; Xu et al. Citation2019; Lian et al. Citation2022). These results highlight that C-fiber activation, and corresponding respiratory and cardiovascular reflex responses, depends upon TRPA1 and TRPV1 channel activation by the oxidative components of PM2.5.
Overall, the results of the reviewed studies provide support that ROS, TRP channel activation, and activation of C-fibers are key events that are consistent with the postulated sequence of events in the proposed MoA, with clear involvement in changes in lung and cardiovascular function.
Exposure-response relationships
The majority of studies did not examine the role of neural signaling on the effects of more than one exposure concentration of PM2.5 on respiratory or cardiac function. Only two studies with respiratory endpoints evaluated more than one PM2.5 concentration. Wong et al. (Citation2003) exposed rats to two concentrations of DEPs, 35.3 ± 4.9 μg/m3 or 632.9 ± 47.6 μg/m3, as well as to an unreported but presumably lower concentration of DEPs in diesel engine room air, and demonstrated a dose-dependent increase in plasma extravasation in DEP-exposed rats. Pretreatment with systemic capsaicin to activate C-fiber signaling exacerbated the plasma extravasation at all DEP concentrations, indicating that neural signaling was involved in this response to DEP across a range of concentrations. Similarly, Lv et al. (Citation2016) exposed guinea pigs to 80 or 320 mg/kg PM2.5 by intranasal instillation and the observed increases in cough reflex sensitivity with both exposures were exacerbated by activation of C-fibers and attenuated when C-fiber signaling through TRPV1 receptors was blocked. All other studies examined neural signaling using only one PM2.5 exposure concentration, and the dose regimens differed significantly across studies (type of PM2.5, concentration and duration of exposure, route of exposure, species of experimental animal) so we could not evaluate exposure-response relationships across studies.The limited exposure-response data within and across studies also does not allow for the identification of a threshold for the proposed MoA.
A proposed MoA is not supported if the early key events only occur at exposures higher than those that induce the apical effect (Meek et al. Citation2014a). The PM2.5-induced respiratory and cardiovascular morbidities that underlie reported associations with mortality in epidemiology studies have been reported to occur at much lower exposures than those used in the experimental animal studies of key events reviewed here (US EPA Citation2019), so it is unclear whether the key events occur at the same or lower exposures than these morbidities.
Biological concordance
Biological concordance is the consistency of the proposed MoA with the current biological knowledge of the toxicological outcome (Meek et al. Citation2014a). In this section, we consider whether the proposed MoA of PM2.5-induced neural activation is consistent with what is known about respiratory and cardiovascular morbidities associated with PM2.5 exposure.
Many of the respiratory and cardiovascular morbidities that have been associated with PM2.5 exposure in epidemiology studies are related to the activation of inflammatory pathways, either in target tissues or systemically (Hancoxet al. Citation2007). Additionally, it has been proposed that inflammation mediates the relationship between lung function decrements and the risk of cardiovascular outcomes (Sin and Man Citation2003). This theory is substantiated by several studies that have documented high levels of circulating inflammatory markers in the serum of patients with COPD and increases in systemic inflammation that have been associated with increased risk of cardiovascular disease (Libby et al. Citation2002; Hancox et al. Citation2007). Furthermore, increases in C-reactive protein (CRP) levels, a well-known inflammatory marker, have been observed in patients with asthma and cardiovascular disease (Patel et al. Citation2007; Halvani et al. Citation2012).
TRPA1 and TRPV1 channels play an important role in inflammatory responses and airway hyperresponsivenessin humans (Xu et al. Citation2019). The studies reviewed here showed that stimulation of bronchopulmonary C-fibers by PM2.5 activates TRP ion channels that regulate sympathetic responses in the lung and heart. TRPA1-induced inflammation has been suggested to be a contributing factor in the development of COPD (Andrè et al. Citation2008). While expression of TRPV1 is lower in the airway compared to TRPA1, inhibition of TRPV1 also reduces lung tissue inflammation (Xuet al. Citation2019). Furthermore, human data indicate that TRPV1 mRNA expression is increased in patients with emphysema and single nucleotide polymorphisms (SNPs) in TRPV1 are associated with childhood asthma (Cantero-Recasens et al. Citation2010; Baxter et al. Citation2012).
In addition to inflammation, TRP channel activation related to ROS is likely a mediator for observed respiratory and cardiovascular responses to PM2.5. Exposure to PM2.5 increases ROS formation, which activates TRP channels on bronchopulmonary C-fibers, subsequently altering parasympathetic signaling (Xu et al. Citation2019). More specifically, capsaicin, acidic pH, and oxidative stress activate TRPV1 while superoxide, hydrogen peroxide, and the products of lipid peroxidation activate TRPA1 (Gültekin et al. Citation2018; Trevisan et al. Citation2016). Several studies support the biological plausibility of ROS as a mediator in the proposed MoA. As discussed above, pretreatment with the antioxidant NAC prevented bradypnea and C-fiber activation, which suggests that ROS serves as a trigger for PM2.5-induced stimulation of C-fibers (Lin et al. Citation2010). In addition, pretreatment with the hydroxide radical scavenger DMTU reduced vagal responses to cigarette smoke (Weng et al. Citation2013). Robinson et al. (Citation2018) also demonstrated that the organic component of DEPs (DEP-OE) depolarized the vagus nerve in a similar manner to whole DEPs, whereas the particulate carbon core (par-DEPs) remaining after the extraction of organic components did not induce a response. Ethanol and n-butyl chloride/hexane extracts activated TRPA1 in isolated HEK-293 cells, with the ethanol extract being substantially more potent than whole DEPs (Deering-Rice et al. Citation2011). Finally, increases in markers of oxidative stress have been observed in patients with COPD and cardiovascular disease (Ho et al. Citation2013; McGuinness and Sapey Citation2017). Taken together, there is substantial evidence that PM2.5 exposure increases ROS and activates TRP channels, which increases inflammation that is associated with intermediate morbidities (e.g. asthma, COPD, CVD) that underlie mortality.
Evaluation of human relevance of proposed mode of action
In this section, we evaluate the relevance of the proposed MoA to humans through consideration of the exposure levels used in the experimental animal studies, differences between humans and experimental animals, and whether these factors support or exclude human relevance of the MoA.
All of the studies discussed in this review were conducted using experimental animal models. This is largely because many of the key events of the proposed MoA can only be investigated using experimental animals for ethical reasons. While the effects of these methods cannot be validated with human studies, other evidence exists that supports the role of TRP channels in regulating respiratory and cardiovascular endpoints, as described above.
Some of the experimental animal studies reviewed here used the human-relevant exposure route of inhalation, but many others used non-physiological routes, including intratracheal or intranasal instillation or direct exposure to the vagus nerve. The evidence supporting the proposed MoA for PM2.5-induced respiratory effects was consistent across the studies using inhalation as the exposure route, as well as the studies using non-physiological routes, suggesting that the proposed MoA is plausible regardless of the exposure route.
The relevance of the exposure levels of PM2.5 used in the experimental animal studies to human exposures to PM2.5 is unclear. The current primary annual National Ambient Air Quality Standard (NAAQS) for PM2.5 is an annual mean concentration of 12 μg/m3, averaged over three years (US EPA Citation2020). Recent studies have shown that global annual average PM2.5 concentrations were 35 μg/m3 in urban areas in 2019 (Southerland et al. Citation2022). The lowest inhalation exposure to PM2.5 in the studies reviewed here was 35.3 μg/m3 PM2.5, with all other inhalation studies using PM2.5 exposure concentrations ranging from 150 to 5,300,000 μg/m3. In the studies using cigarette smoke, exposure was typically to the smoke from one cigarette (6 mL) inhaled within a few breaths. It has been shown that reference cigarettes, which are used as a standard in laboratory research, produce smoke that contains, on average, 918 μg/m3 of PM2.5 (Braun et al. Citation2019). These exposure concentrations are far higher than the PM2.5 NAAQS (12 μg/m3), and all but one (35.3 μg/m3) are far higher than typical exposures to humans in ambient air, even in urban environments. These exposures also occurred over shorter time periods than the chronic human exposures to ambient PM2.5, effectively resulting in enormous peak exposures that aren’t comparable to typical ambient exposures.
The studies that used non-physiological routes of exposure did not attempt to correlate the exposure levels to human inhalation exposures, and it would be difficult to do so. Extrapolation of exposures from these routes has limitations, including the fact that these routes result in 100% particle deposition. Such deposition cannot be assumed for human exposures to ambient PM2.5, which include a broad range of particle sizes that can result in differential deposition and internal dose values. Particle deposition in humans varies by particle size and anatomical location along the airway. Mathematical simulation modeling has shown that total inhalation deposition of fine particles in humans ranges from 8.5%–68.4% (Sturm Citation2020).
Overall, the human relevance of the studies reviewed here is unclear, as the majority of studies used high exposures that are not relevant to typical ambient exposures.The lack of studies at lower PM2.5 exposures does not negate the involvement of the key events in the proposed MoA at more typical human-relevant exposures, but further studies using lower exposure levels and human-relevant exposure routes are needed before this can be established.
Data gaps
There remain several critical data gaps in understanding the proposed MoA by which PM2.5 may induce respiratory and cardiovascular morbidities contributing to associated mortalities. First, the reviewed studies generally did not evaluate exposure-response trends for evaluation of potential thresholds. Many of the studies only assessed a single high concentration of PM2.5, CAPs, cigarette smoke, or DEPs. In addition, the exposure paradigm varied significantly across studies in terms of the type of PM2.5, exposure duration and concentration, and experimental animal species and strain, precluding an evaluation of exposure-response relationships across studies. In studies where exposure-response was considered, the concentrations used may not resemble typical real-world environmental exposures. For example, Wong et al. (Citation2003) evaluated two concentrations of DEPs, 35.3 μg/m3 and 632.9 μg/m3. While the lower exposure level in this study is reflective of the reported global average urban PM2.5 concentration of 35 μg/m3 noted above, the higher concentration of 632.9 μg/m3 is far higher than typical ambient concentrations of PM2.5 in the absence of exceptional events that can have extremely high peak exposures, such as wildfires. This is true even in the most densely urbanized environments in the world, such as the Chinese city of Guangzhou, where maximum PM2.5 concentrations generally do not exceed 167 μg/m3(Jahn et al. Citation2013). Mechanistic studies of neural reflex and TRP channel activation at more typical environmentally-relevant exposures are needed to address this. However, the high exposure studies could potentially be used to inform the MoA for respiratory or cardiovascular effects in humans resulting from extremely high peakexposures to PM2.5 from events such as wildfires.
The lack of exposure-response data is complicated by a lack of extended exposure data. Many of the studies considered here only assessed a single exposure. Typical environmental exposures occur over a lifetime, at much lower doses than those considered in most experimental animal models. Exposure to PM2.5 occurs on a daily basis from both indoor and outdoor sources. It is unclear whether a single, high exposure to PM2.5 can predict chronic low-exposure effects via the proposed MoA, such that short-term peaks in ambient PM2.5 exposure could cause respiratory and cardiovascular effects in humans via the proposed MoA. Mechanistic studies of neural reflex stimulation of the respiratory and cardiovascular systems with a focus on environmentally-relevant, extended exposure durations are necessary to determine if the key events in the proposed MoA occur in situations relevant to typical human exposures.
Finally, the evidence regarding whether the proposed MoA is involved in PM2.5-induced cardiovascular morbidities is inconsistent as to the role of vagal signals in the regulation of heart function and requires further investigation. The studies provide evidence to suggest that vagal signaling through TRP receptor activation may regulate certain PM2.5-induced cardiovascular effects but not others. Because the data are inconsistent, there is a need for more studies with cardiovascular endpoints to resolve the inconsistency, and this represents a gap in the literature. In addition, future models investigating the connection between respiratory reflex responses and changes in cardiovascular function are needed to address this further.
Conclusions
We found that studies in experimental animals provide relatively consistent evidence that PM2.5-induced respiratory responses are mediated by a neural pathway, and demonstrated essentiality of multiple early key events. ROS appear to be the primary signal for generating the neural response, as antioxidants and free radical scavengers abolish PM2.5-induced lung reflex responses. Studies also showed consistent evidence that activation of TRPA1 and TRPV1 ion channels and associated pro-inflammatory responses are essential key events in the proposed MoA for respiratory effects. The results are also consistent with the postulated temporality of the key events in the MoA. The evidence across the experimental studies is concordant and indicates that neural reflex activation through stimulation of TRP ion channels is a biologically plausible MoA by which PM2.5 could cause respiratory morbidities. Exposure-response relationships and thresholds for the key events cannot be determined, as the majority of studies used only one exposure level that was much higher than environmentally-relevant exposures that have been shown to be associated with total mortality in epidemiology studies. Because of this, the human relevance of the key events in the proposed MoA is unclear. Further studies evaluating exposure-response relationships and chronic exposures, with an emphasis on relating the findings to typical exposures in humans, are needed to address data gaps.
The studies that evaluated cardiovascular endpoints provide inconsistent evidence for a role of the proposed MoA. Evidence from these studies suggests that vagal signaling through TRP receptor activation may regulate certain PM2.5-induced changes in cardiovascular function, but other cardiovascular effects may not be mediated through this MoA, suggesting that a different MoA may be more plausible.
We conclude that, based on the currently available experimental evidence, neural reflex activation is a biologically plausible MoA by which PM2.5 can cause respiratory and certain cardiovascular morbidities in experimental animal models and may be relevant to humans when exposed to extremely high peak levels of PM2.5, but further studies are needed to determine whether this occurs in humans exposed to typical ambient PM2.5 concentrations.
Disclosure statement
All of the authors are employed by Gradient, an independent environmental risk science consulting firm. The work reported in this paper was conducted during the normal course of employment, with financial support from the American Petroleum Institute (API) and was reviewed by members of API while in preparation. This paper is the professional work product of the authors, and the opinions and conclusions within are not necessarily those of their employers or the financial sponsor of the work.
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- Adriaensen D, Timmermans JP. 2011. Breath-taking complexity of vagal C-fibre nociceptors: implications for inflammatory pulmonary disease, dyspnoea and cough. J Physiol. 589(Pt. 1):3–4. doi:10.1113/jphysiol.2010.201434.
- Andrè E, Campi B, Materazzi S, Trevisani M, Amadesi S, Massi D, Creminon C, Vaksman N, Nassini R, Civelli M, et al. 2008. Cigarette smoke-induced neurogenic inflammation is mediated by alpha,beta-unsaturated aldehydes and the TRPA1 receptor in rodents. J Clin Invest. 118(7):2574–2582. doi:10.1172/JCI34886.
- Barnes PJ. 1996. Neuroeffector mechanisms: the interface between inflammation and neuronal responses. J. Allergy Clin. Immunol. 98(5):S73–S83. doi:10.1016/S0091-6749(96)70020-9.
- Baxter MD, Birrell MA, Belvisi MG. 2012. The role of transient receptor potential vanilloid 1 (TRPV1) in tobacco smoke induced airway inflammation. Am. J. Respir. Crit. Care Med. 185:A6410. Presented at the American Thoracic Society International Conference, San Francisco, CA, May 18–23. doi:10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A6410.
- Bergren DR. 1985. Enhanced lung C-fiber responsiveness in sensitized adult guinea pigs exposed to chronic tobacco smoke. J Appl Physiol (1985). 91(4):1645–1654. doi:10.1152/jappl.2001.91.4.1645.
- Bhatnagar A. 2022. Cardiovascular effects of particulate air pollution. Annu Rev Med. 73(1):393–406. doi:10.1146/annurev-med-042220-011549.
- Boobis AR, Doe JE, Heinrich-Hirsch B, Meek ME, Munn S, Ruchirawat M, Schlatter J, Seed J, Vickers C. 2008. IPCS framework for analyzing the relevance of a noncancer mode of action for humans. Crit Rev Toxicol. 38(2):87–96. doi:10.1080/10408440701749421.
- Braun M, Koger F, Klingelhöfer D, Müller R, Groneberg D. 2019. Particulate matter emissions of four different cigarette types of one popular brand: influence of tobacco strength and additives. IJERPH. 16(2):263. doi:10.3390/ijerph16020263.
- Canning BJ, Mori N, Mazzone SB. 2006. Vagal afferent nerves regulating the cough reflex. Respir Physiol Neurobiol. 152(3):223–242. doi:10.1016/j.resp.2006.03.001.
- Cantero-Recasens G, Gonzalez JR, Fandos C, Duran-Tauleria E, Smit LA, Kauffmann F, Antó JM, Valverde MA. 2010. Loss of function of transient receptor potential vanilloid 1 (TRPV1) genetic variant is associated with lower risk of active childhood asthma. J Biol Chem. 285(36):27532–27535. doi:10.1074/jbc.C110.159491.
- Carll AP, Hazari MS, Perez CM, Krantz QT, King CJ, Haykal-Coates N, Cascio WE, Costa DL, Farraj AK. 2013. An autonomic link between inhaled diesel exhaust and impaired cardiac performance: insight from treadmill and dobutamine challenges in heart failure-prone rats. Toxicol Sci. 135(2):425–436. doi:10.1093/toxsci/kft155.
- Deering-Rice CE, Romero EG, Shapiro D, Hughen RW, Light AR, Yost GS, Veranth JM, Reilly CA. 2011. Electrophilic components of diesel exhaust particles (DEP) activate transient receptor potential ankyrin-1 (TRPA1): a probable mechanism of acute pulmonary toxicity for DEP. Chem Res Toxicol. 24(6):950–959. doi:10.1021/tx200123z.
- Emmerechts J, Hoylaerts MF. 2012. The effect of air pollution on haemostasis. Hamostaseologie. 32(1):5–13. doi:10.5482/ha-1179.
- Fowles J, Dybing E. 2003. Application of toxicological risk assessment principles to the chemical constituents of cigarette smoke. Tob. Control. 12(4):424–430. doi:10.1136/tc.12.4.424.
- Ghelfi E, Rhoden CR, Wellenius GA, Lawrence J, Gonzalez-Flecha B. 2008. Cardiac oxidative stress and electrophysiological changes in rats exposed to concentrated ambient particles are mediated by TRP-dependent pulmonary reflexes. Toxicol Sci. 102(2):328–336. doi:10.1093/toxsci/kfn005.
- Gültekin F, Nazıroğlu M, Savaş HB, Çiğ B. 2018. Calorie restriction protects against apoptosis, mitochondrial oxidative stress and increased calcium signaling through inhibition of TRPV1 channel in the hippocampus and dorsal root ganglion of rats. Metab Brain Dis. 33(5):1761–1774. doi:10.1007/s11011-018-0289-0.
- Halvani A, Tahghighi F, Nadooshan HH. 2012. Evaluation of correlation between airway and serum inflammatory markers in asthmatic patients. Lung India. 29(2):143–146. doi:10.4103/0970-2113.95317.
- Hancox RJ, Poulton R, Greene JM, Filsell S, McLachlan CR, Rasmussen F, Taylor DR, Williams MJ, Williamson A, Sears MR. 2007. Systemic inflammation and lung function in young adults. Thorax. 62(12):1064–1068. doi:10.1136/thx.2006.076877.
- Hazari MS, Haykal-Coates N, Winsett DW, Krantz QT, King C, Costa DL, Farraj AK. 2011. TRPA1 and sympathetic activation contribute to increased risk of triggered cardiac arrhythmias in hypertensive rats exposed to diesel exhaust. Environ Health Perspect. 119(7):951–957. doi:10.1289/ehp.1003200.
- Ho E, Karimi Galougahi K, Liu CC, Bhindi R, Figtree GA. 2013. Biological markers of oxidative stress: applications to cardiovascular research and practice. Redox Biol. 1(1):483–491. doi:10.1016/j.redox.2013.07.006.
- Jahn HJ, Kraemer A, Chen XC, Chan CY, Engling G, Ward TJ. 2013. Ambient and personal PM2.5 exposure assessment in the Chinese megacity of Guangzhou. Atmos. Environ.74:402–411. 74:402–411. doi:10.1016/j.atmosenv.2013.04.011.
- Lian Z, Qi H, Liu X, Zhang Y, Xu R, Yang X, Zeng Y, Li J. 2022. Ambient ozone, and urban PM2.5 co-exposure, aggravate allergic asthma via transient receptor potential vanilloid 1-mediated neurogenic inflammation. Ecotoxicol Environ Saf. 243:114000. doi:10.1016/j.ecoenv.2022.114000.
- Libby P, Ridker PM, Maseri A. 2002. Inflammation and atherosclerosis. Circulation. 105(9):1135–1143. doi:10.1161/hc0902.104353.
- Lin YS, Hsu CC, Bien MY, Hsu HC, Weng HT, Kou YR. 2010. Activations of TRPA1 and P2X receptors are important in ROS-mediated stimulation of capsaicin-sensitive lung vagal afferents by cigarette smoke in rats. J Appl Physiol (1985). 108(5):1293–1303. doi:10.1152/japplphysiol.01048.2009.
- Lv H, Yue J, Chen Z, Chai S, Cao X, Zhan J, Ji Z, Zhang H, Dong R, Lai K. 2016. Effect of transient receptor potential vanilloid-1 on cough hypersensitivity induced by particulate matter 2.5. Life Sci. 151:157–166. doi:10.1016/j.lfs.2016.02.064.
- Maricq MM. 2007. Chemical characterization of particulate emissions from diesel engines: a review. Aerosol Sci. 38:1079–1118.
- Matran R, Alving K, Lundberg JM. 1990. Cigarette smoke, nicotine and capsaicin aerosol-induced vasodilatation in pig respiratory mucosa. Br J Pharmacol. 100(3):535–541. doi:10.1111/j.1476-5381.1990.tb15842.x.
- McGuinness AJ, Sapey E. 2017. Oxidative stress in COPD: sources, markers, and potential mechanisms. J Clin Med. 6(2):21. doi:10.3390/jcm6020021.
- McQueen DS, Donaldson K, Bond SM, McNeilly JD, Newman S, Barton NJ, Duffin R. 2007. Bilateral vagotomy or atropine pre-treatment reduces experimental diesel-soot induced lung inflammation. Toxicol Appl Pharmacol. 219(1):62–71. doi:10.1016/j.taap.2006.11.034.
- Meek ME, Boobis A, Cote I, Dellarco V, Fotakis G, Munn S, Seed J, Vickers C. 2014b. New developments in the evolution and application of the WHO/IPCS framework on mode of action/species concordance analysis. J Appl Toxicol. 34(1):1–18. doi:10.1002/jat.2949.
- Meek ME, Palermo CM, Bachman AN, North CM, Lewis RJ. 2014a. Mode of action human relevance (species concordance) framework: evolution of the Bradford Hill considerations and comparative analysis of weight of evidence. J Appl Toxicol. 34(6):595–606. doi:10.1002/jat.2984.
- Milici A, Talavera K. 2021. TRP channels as cellular targets of particulate matter. Int J Mol Sci. 22(5):2783. doi:10.3390/ijms22052783.
- [NTP] National Toxicology Program . 2016. Tobacco-related exposures. In Report on carcinogens. 14th ed. US Dept. of Health and Human Services, Public Health Service, National Toxicology Program. p. 6. https://ntp.niehs.nih.gov/ntp/roc/content/profiles/tobaccorelatedexposures.pdf
- Patel DN, King CA, Bailey SR, Holt JW, Venkatachalam K, Agrawal A, Valente AJ, Chandrasekar B. 2007. Interleukin-17 stimulates C-reactive protein expression in hepatocytes and smooth muscle cells via p38 MAPK and ERK1/2-dependent NF-kappaB and C/EBPbeta activation. J Biol Chem. 282(37):27229–27238. doi:10.1074/jbc.M703250200.
- Perez CM, Hazari MS, Farraj AK. 2015. Role of autonomic reflex arcs in cardiovascular responses to air pollution exposure. Cardiovasc Toxicol. 15(1):69–78. doi:10.1007/s12012-014-9272-0.
- Prueitt RL, Cohen JM, Goodman JE. 2015. Evaluation of atherosclerosis as a potential mode of action for cardiovascular effects of particulate matter. Regul Toxicol Pharmacol. 73(Suppl. 2):S1–S15. doi:10.1016/j.yrtph.2015.09.034.
- Prueitt RL, Li W, Edwards L, Zhou J, Goodman JE. 2021. Systematic review of the association between long-term exposure to fine particulate matter and mortality. Int J Environ Health Res. 32(8):1647–1685. doi:10.1080/09603123.2021.1901864.
- Ris C. 2007. U.S. EPA health assessment for diesel engine exhaust: a review. Inhal Toxicol. 19 Suppl 1(sup1):229–239. doi:10.1080/08958370701497960.
- Robertson S, Thomson AL, Carter R, Stott HR, Shaw CA, Hadoke PW, Newby DE, Miller MR, Gray GA. 2014. Pulmonary diesel particulate increases susceptibility to myocardial ischemia/reperfusion injury via activation of sensory TRPV1 and beta-1 adrenoreceptors. Part Fibre Toxicol. 11:12. doi:10.1186/1743-8977-11-12.
- Robinson RK, Birrell MA, Adcock JJ, Wortley MA, Dubuis ED, Chen S, McGilvery CM, Hu S, Shaffer MSP, Bonvini SJ, et al. 2018. Mechanistic link between diesel exhaust particles and respiratory reflexes. J Allergy Clin Immunol. 141(3):1074–1084.e9. doi:10.1016/j.jaci.2017.04.038.
- Rogerio AP, Andrade EL, Calixto JB. 2011. C-fibers, but not the transient potential receptor vanilloid 1 (TRPV1), play a role in experimental allergic airway inflammation. Eur J Pharmacol. 662(1-3):55–62. doi:10.1016/j.ejphar.2011.04.027.
- Schulz H, Harder V, Ibald-Mulli A, Khandoga A, Koenig W, Krombach F, Radykewicz R, Stampfl A, Thorand B, Peters A. 2005. Cardiovascular effects of fine and ultrafine particles. J Aerosol Med. 18(1):1–22. doi:10.1089/jam.2005.18.1.
- Shannahan JH, Kodavanti UP, Brown JM. 2012. Manufactured and airborne nanoparticle cardiopulmonary interactions: A review of mechanisms and the possible contribution of mast cells. Inhal Toxicol. 24(5):320–339. doi:10.3109/08958378.2012.668229.
- Silverman HA, Chen A, Kravatz NL, Chavan SS, Chang EH. 2020. Involvement of neural transient receptor potential channels in peripheral inflammation. Front Immunol. 11:590261. doi:10.3389/fimmu.2020.590261.
- Sin DD, Man SF. 2003. Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation. 107(11):1514–1519. doi:10.1161/01.cir.0000056767.69054.b3.
- Southerland VA, Brauer M, Mohegh A, Hammer MS, van Donkelaar A, Martin RV, Apte JS, Anenberg SC. 2022. Global urban temporal trends in fine particulate matter (PM2.5) and attributable health burdens: estimates from global datasets. Lancet Planet Health. 6(2):e139–e146. doi:10.1016/S2542-5196(21)00350-8.
- Steiner S, Bisig C, Petri-Fink A, Rothen-Rutishauser B. 2016. Diesel exhaust: current knowledge of adverse effects and underlying cellular mechanisms. Arch Toxicol. 90(7):1541–1553. doi:10.1007/s00204-016-1736-5.
- Sturm R. 2020. Modelling the deposition of fine particulate matter (PM2.5) in the human respiratory tract. AME Med J. 5:14–14. doi:10.21037/amj.2020.03.04.
- Trevisan G, Benemei S, Materazzi S, De Logu F, De Siena G, Fusi C, Fortes Rossato M, Coppi E, Marone IM, Ferreira J, et al. 2016. TRPA1 mediates trigeminal neuropathic pain in mice downstream of monocytes/macrophages and oxidative stress. Brain. 139(Pt 5):1361–1377. doi:10.1093/brain/aww038.
- Undem BJ, Sun H. 2020. Molecular/ionic basis of vagal bronchopulmonary C-fiber activation by inflammatory mediators. Physiology (Bethesda). 35(1):57–68. doi:10.1152/physiol.00014.2019.
- US EPA. 2019. Integrated Science Assessment for Particulate Matter (Final). EPA/600/R-19/188. 1967p., December; [accessed 2020 March 17]. https://cfpub.epa.gov/ncea/isa/recordisplay.cfm?deid=347534.
- US EPA. 2020. Review of the national ambient air quality standards for particulate matter (Final action). Fed. Reg. 85(244):82684–82748. 40 CFR 50., December 18.
- Utell MJ, Frampton MW, Zareba W, Devlin RB, Cascio WE. 2002. Cardiovascular effects associated with air pollution: potential mechanisms and methods of testing. Inhal Toxicol. 14(12):1231–1247. doi:10.1080/08958370290084881.
- Watkinson WP, Campen MJ, Nolan JP, Costa DL. 2001. Cardiovascular and systemic responses to inhaled pollutants in rodents: effects of ozone and particulate matter. Environ Health Perspect. 109(Suppl. 4):539–546. doi:10.1289/ehp.01109s4539.
- Weng WH, Hsu CC, Chiang LL, Lin YJ, Lin YS, Su CL. 2013. Role of TRPV1 and P2X receptors in the activation of lung vagal C-fiber afferents by inhaled cigarette smoke in rats. Mol Med Rep. 7(4):1300–1304. doi:10.3892/mmr.2013.1300.
- Wong SS, Sun NN, Keith I, Kweon CB, Foster DE, Schauer JJ, Witten ML. 2003. Tachykinin substance P signaling involved in diesel exhaust-induced bronchopulmonary neurogenic inflammation in rats. Arch Toxicol. 77(11):638–650. doi:10.1007/s00204-003-0485-4.
- Xu M, Zhang Y, Wang M, Zhang H, Chen Y, Adcock IM, Chung KF, Mo J, Zhang Y, Li F. 2019. TRPV1 and TRPA1 in lung inflammation and airway hyperresponsiveness induced by fine particulate matter (PM2.5). Oxid Med Cell Longev. 2019:7450151–7450115. doi:10.1155/2019/7450151.
- Zhu BQ, Parmley WW. 1995. Hemodynamic and vascular effects of active and passive smoking. Am Heart J. 130(6):1270–1275. doi:10.1016/0002-8703(95)90154-x.