Abstract
Chronic obstructive pulmonary disease (COPD) is a major public health problem worldwide. COPD is strongly related to cigarette smoke exposure, but not all smokers develop the disease. It is thought that COPD progresses slowly over time stimulated by environmental exposures, including free radicals from cigarette smoke, which ultimately establish chronic inflammation and result in a progressive destruction of lung tissues. COPD is known to occur in family clusters, which has prompted interest in determining genetic risk factors for the disease. Several genetic studies have identified an association between extracellular superoxide dismutase (ECSOD) polymorphisms and risk for developing COPD. ECSOD is an antioxidant protein that scavenges superoxide free radicals from cigarette smoke and protects the lungs from free radical damage and chronic inflammation.
INTRODUCTION
Chronic Obstructive Pulmonary Disease (COPD) is the fourth leading cause of death in the world and is a major public health problem due to its associated disability and consumption of health care resources. It is primarily related to tobacco smoke exposure, although other inhalational exposures appear to increase the risk of disease. However, not all smokers will develop COPD. A recent study found that only 25% of smokers developed COPD in a Danish population (Citation[1]). This suggests that individuals have varying susceptibility to develop the disease.
COPD prevalence rises with increasing age and is known to occur in family clusters. Increased risk of COPD in smokers' relatives and recognition of a specific genetic mutation, alpha 1 antitrypsin deficiency, that leads to COPD, have excited interest in defining the genetic risk factors for the disease. Several genetic studies have separately identified an association between extracellular superoxide dismutase (ECSOD or SOD3) polymorphisms and altered risk of COPD (Citation[2], Citation[3]). ECSOD is an antioxidant and anti-inflammatory protein found in high concentrations in both lung tissue and in the lung lining fluids (Citation[4]). It has been hypothesized by many that COPD progression involves both the release of free radicals and redox sensitive proteases that result in small airway inflammation, fibrosis and alveolar wall destruction.
Pathophysiology of COPD
COPD is a clinical syndrome defined by chronic expiratory airflow obstruction leading to exercise intolerance and dyspnea. However, a comprehensive description of the disease remains a work in progress. Classic descriptions of chronic bronchitis and emphysema define polar aspects of COPD disease phenotypes; however, the majority of COPD patients display mixed patterns of airway disease and emphysematous loss of lung parenchyma. High resolution chest CT scans of COPD subjects demonstrate various combinations of large and small airway disease leading to air trapping and variable patterns and degrees of emphysema.
The time course and progression of COPD includes: cigarette smoke or environmentally induced airway inflammatory disease that leads to chronic cough and sputum production. There is a significant gap of years between smoking exposure and the development of clinical disease. Ultimately, immune defenses are activated in response to chronic inflammatory cytokines and recurrent infections. Recurrent infections in COPD patients are associated with alterations in immune mechanisms and a role for both macrophages and neutrophils has been defined in COPD pathogenesis (Citation[5]). The unique characteristic of patients with COPD is that they develop persistent chronic inflammation that is not successfully controlled. These inflammatory events break down lung collagen by chronic activation of local proteases. The results of these chronic inflammatory events are changes in alveolar size and lung compliance producing emphysema (Citation[6]), and chronic airway inflammation.
Cigarette smoking, COPD and free radicals in the lung
The lung has an extensive surface area that is exposed to environmental irritants, such as cigarette smoke, which causes free radical production. Each puff of cigarette smoke has been reported to contain up to 1014 free radical molecules as well as 4700 chemicals (Citation[7]). Some of these chemicals are short term free radicals such as superoxide (O2· −) and nitric oxide (NO) and others are long acting free radicals such as semiquinones (Citation[8]). Cigarette smoke enhances recruitment of inflammatory cells to the lung (Citation[9]). Inflammatory cells, such as activated neutrophils and macrophages, can produce large amounts of reactive oxygen species (ROS), mainly through the NADPH oxidase system (Citation[10]). It has been hypothesized that the ROS released by inflammatory cells recruited to sites of injury, cause extensive tissue damage which leads to chronic inflammation (Citation[11]). ROS release by neutrophils and macrophages not only damages surrounding tissues, it can also directly damage/inactivate antioxidant enzymes (Citation[11]). ROS from activated neutrophils can cause proteolysis of the antioxidant, ECSOD, rendering it inactive (Citation[11]). Thus, chronic free radical production can inhibit the activity of the very enzymes released to protect the body from free radical damage.
Free radicals and reactive species that generate oxidative stress are short lived; thus, oxidative stress is identified by measuring end products of free radical reactions that have already occurred. These oxidative stress markers include lipid peroxidation (isoprotanes), protein carbonyls, nitrotyrosine formation, glutathione levels and DNA damage. COPD has been associated with increased isoprostanes and lipid peroxidation (12–15), and elevated nitric oxide production and nitrotyrosine formation (Citation[16], Citation[17]), supporting a role for oxidative stress in the disease. Moreover, these markers are elevated even in mild COPD (Citation[18], Citation[19]). Leukocytes from COPD subjects have increased generation of superoxide, increased SOD activity, increased protein carbonyls and increased glutathione levels as compared to controls (Citation[20]).
Lung inflammation persists even after a person stops smoking (Citation[21]). One study found that for an ex-smoker, it takes up to 3 years of not smoking for lung macrophage numbers to decrease to the levels of a never smoker (Citation[9]). A recent prospective study found that oxidative stress persists in the lungs for months after smoking cessation (Citation[22]). Another study demonstrated that respiratory bronchiolitis, a common inflammatory lesion of the respiratory bronchioles associated with smoking, can occur or persist well after a person has stopped smoking. Respiratory bronchiolitis was found in 42% of patients who had quit smoking for 3 years and 33% who had quit for 5 years (Citation[21]). Hogg et al. found increased lymphocyte numbers in airways of patients with severe COPD that had not smoked for an average of 9 years (Citation[23]). Taken together these data indicate that smoking causes chronic inflammation and oxidative stress in the lung and that both persist well after a person has stopped smoking.
Cigarette smoke can overwhelm the capacity of lung antioxidant defenses and lead to chronic oxidative stress and inflammation in the respiratory system (Citation[24], Citation[25]). Oxidative stress in the lung may also be perpetuated by recurrent infections with excess accumulation of inflammatory cells.
ECSOD background
The lung has several lines of defense to protect against oxidants, pollutants, and irritants, such as cigarette smoke. One important line of defense is the production of antioxidant enzymes (Citation[4]). Superoxide dismutases are powerful antioxidant enzymes that reduce the superoxide radical to a less reactive hydrogen peroxide molecule (Citation[26]). There are two superoxide dismutases localized in lung cells (MnSOD and CuZnSOD) and one superoxide dismutase primarily localized in the extracellular space of the lung (ECSOD, ) (26–28). Other lung antioxidant enzymes such as catalase, glutathione peroxidases, and the thioredoxin/peroxiredoxin and glutaredoxin families of enzymes further contribute to the scavenging of hydrogen peroxide to water (Citation[4]). Together these antioxidant enzymes work to protect the lung from oxidatative stress.
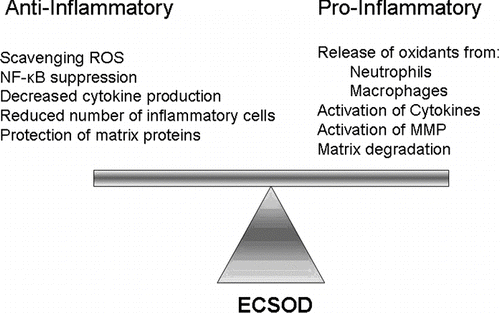
Figure 1 ECSOD modulates oxidative damage and inflammation. ECSOD creates a less oxidizing environment by reducing the highly reactive superoxide radical (O2· −) to the less reactive hydrogen peroxide molecule (H2O2). Other lung antioxidants, such as catalase and peroxidases, further reduce H2O2 to water. Thus, in the presence of ECSOD and other antioxidants there are fewer reactive oxygen species, which creates an anti-inflammatory environment. However, when ECSOD is not present, a more oxidizing environment occurs because O2· − quickly forms other reactive oxygen species, i.e., hydroxyl radical (OH·), peroxynitrite (ONOO−), and bleach (HOCl). A more oxidizing environment promotes pro-inflammatory signaling that can lead to matrix degradation.
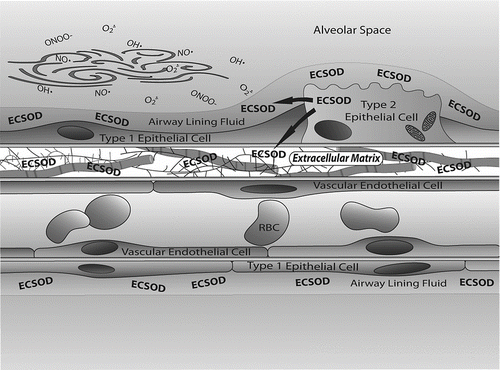
The major SOD in extracellular fluids is ECSOD (Citation[27]). ECSOD is found in high abundance in the lung, the fluid lining the lung and in the vasculature (Citation[29], Citation[30]). In particular, ECSOD is located in the lung extracellular matrix, at airway epithelial cell junctions, in the lining of vessels and at the surface of airway smooth muscles (Citation[29], Citation[30]). ECSOD is a 135,000 mw tetrameric glycoprotein that has a N-terminal signal peptide for secretion out of the cell, a copper/zinc containing activity domain and a C-terminal heparin binding tail (Citation[31]). The positively charged heparin binding tail allows ECSOD to bind to negatively charged extracellular matrix elements and to endothelial cells (Citation[32]). The distribution of ECSOD in extracellular compartments indicates that the enzyme plays a critical role in protecting extracellular matrix proteins from free radical damage and potentially protecting this tissue from the progression of chronic inflammation ().
Figure 2 ECSOD protects the extracellular matrix within the lung from oxidant damage. ECSOD has a positively charged binding tail that binds to negatively charged collagen and proteoglycans found in the extracellular matrix. In lung, this results in high levels of ECSOD being associated with extracellular matrix elements found in the thick portions of the alveolar septum. ECSOD is synthesized in alveolar type II cells and is secreted into airway lining fluids. Potential sources of oxidants include inhaled airborne molecules as well as oxidants generated by cellular metabolism and those released by inflammatory cells. Protection of both airway and alveolar septa from oxidative stress is necessary to ensure that the large surface area of the lung and extensive alveolar fibroskeleton remain intact and functional. Cleavage of these structural proteins may play a role in the development of emphysema.
ECSOD function in lung
ECSOD is important in protecting the lung from free radical damage and in controlling inflammation (Citation[33]). ECSOD ameliorates a wide range of lung injuries. ECSOD protects mice from asbestos-induced lung injury (Citation[34]). Asbestos exposed mice lacking ECSOD have a greater inflammatory response, more fibrosis, and more oxidative damage as compared to exposed wild-type mice (Citation[35]). In another lung injury model, hyperoxia, toxicity is reduced by high levels of ECSOD (Citation[36]). In that particular model, animals that overexpress ECSOD had significantly lower mortality rates as compared to wild type animals and had fewer inflammatory cells in their bronchial alveolar lavage fluid as compared to wild-type mice (Citation[36]). Recently, Gongora, et al. found that conditional ECSOD knock-out in mature animals leads to acute lung injury at ambient levels of oxygen (Citation[37]).
ECSOD has also been shown to protect against bleomycin-induced pulmonary fibrosis. Mice that overexpressed ECSOD had less fibrosis and reduced total lung collagen (Citation[38]). Finally, ECSOD inhibits inflammation associated with lipopolysaccharide (LPS) exposure (Citation[39]). ECSOD reduced the number of neutrophils in the lung airways and reduced expression of the inflammatory cytokines, TNF-α and MIP-2 (Citation[39]).
ECSOD has been shown to play an important role in protecting lung extracellular matrix from inflammation in vitro (Citation[33]). ECSOD binds to a variety of human extracellular matrix proteins via its C-terminal binding domain (Citation[4]). ECSOD binds to collagen, hyaluronan, and heparan sulfate, all proteins found in high abundance in the lung extracellular matrix (40–42). Under oxidative conditions, the extracellular matrix becomes damaged by free radical production and the proteins that make up the extracellular matrix fragment. Accumulation of these collagen, hyaluronan and heparan sulfate fragments elicits inflammatory responses in the extracellular matrix (40–42). When present, ECSOD binds to extracellular matrix proteins and protects these proteins from fragmentation (40–42). Thus, ECSOD protects the human extracellular compartment from damage due to oxidative stress.
ECSOD has been shown to inhibit fibrosis. Epithelial cells and macrophages release transforming growth factor-β (TGF-β), which stimulates fibroblast proliferation and leads to fibrotic lesions (Citation[43]). ECSOD inhibits the TGF-β signaling pathway and thus prevents fibrosis in vivo (Citation[44]). COPD is a disease characterized by sites of inflammation and fibrosis and ECSOD is critical in the control of both of these processes.
Genetic studies involving ECSOD in lung function and COPD disease
Significant familial aggregation of both spirometric lung function and COPD has been shown in a number of studies (45–48). Several genetic studies have shown that ECSOD polymorphisms are associated with normal and altered lung function. In mice, SOD3 variants were associated with reduced lung function (Citation[49]). In humans, associations between ECSOD polymorphisms and reduced lung function in children and adults have been observed (Citation[2], Citation[50]). Genome wide association in the Framingham Heart study showed association of a SNP close to the ECSOD gene with percent predicted FEV1 and percent predicted FVC (Citation[51]). Arcaroli et al. have shown that certain ECSOD haplotypes reduce lung inflammation and decrease the severity of acute lung injury and mortality (Citation[52]). Therefore, both animal and human genetic studies have shown that polymorphisms in ECSOD are important in lung function, with specific polymorphisms associated with either an increase or a decrease in lung function.
Five genetic studies have evaluated the relationships of ECSOD and risk of COPD (, (Citation[53])). In two independent studies, using two different populations, ECSOD polymorphisms have been correlated with a reduced risk of developing COPD (Citation[3], Citation[54]). In particular, the ECSOD polymorphism, R213G, has been shown to reduce the risk of smokers to develop COPD (Citation[3], Citation[54]). This polymorphism causes a substitution of an arginine for a glycine at amino acid 213 in the heparin binding tail of ECSOD (). The normal heparin binding tail of ECSOD has a cluster of 6 positively charged amino acids, which allows ECSOD to bind to the negatively charged extracellular matrix (, (Citation[3], Citation[54])). The R213G mutation reduces the positive charge on the binding tail, and markedly alters the protein's affinity for binding to tissues or extracellular matrix. A result of this is that high circulating levels occur for the R231G ECSOD protein- and it is presumed that this is associated with high amounts in the airway lining fluids where it would be in an ideal position to protect the lung from antioxidant injury induced by inhaled oxidants, such as cigarette smoking. Thus, individuals who smoke and are carriers of this ECSOD polymorphism are at a reduced risk of developing COPD (, ).
Figure 3 ECSOD functional mutation (R213G) in binding tail. ECSOD has a described mutation at amino acid number 213 in its binding tail where a C to G nucleotide substitution changes the amino acid from an arginine to a glycine. This arginine is one of a cluster of 6 positively charged amino acids in the carboxy terminus of the protein that creates strong binding affinity to heparin and other negatively charged matrix elements. The result of replacing this one arginine with glycine is that the protein has a marked decrease in affinity for the negatively charged extracellular matrix and a marked increase in its circulating levels. This mutation occurs in 4–6% of northern European populations.
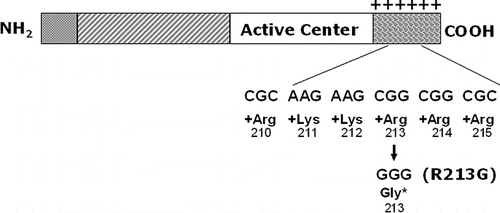
Figure 4 Effect of ECSOD binding tail mutation on COPD risk in smokers. A. This Figure is adapted from Young et al. (Citation[54]) and shows smokers who have not developed COPD have a significantly higher incidence of the G allele at the R213G locus as compared to smokers who developed COPD. This protection from the effects of smoking is presumed to be related to increased levels of ECSOD secreted into the alveolar lining fluid, and thereby, more effective antioxidant protection from airborne oxidants found in cigarette smoke. B. This figure is adapted from Juul et al. (Citation[3]). This prospective study followed individuals for an average of 24 years and found that individuals heterozygous for the ECSOD R213G mutation had significantly lower COPD morbidity and morality rates as compared to individuals that were noncarriers for this ECSOD mutation.
![Figure 4 Effect of ECSOD binding tail mutation on COPD risk in smokers. A. This Figure is adapted from Young et al. (Citation[54]) and shows smokers who have not developed COPD have a significantly higher incidence of the G allele at the R213G locus as compared to smokers who developed COPD. This protection from the effects of smoking is presumed to be related to increased levels of ECSOD secreted into the alveolar lining fluid, and thereby, more effective antioxidant protection from airborne oxidants found in cigarette smoke. B. This figure is adapted from Juul et al. (Citation[3]). This prospective study followed individuals for an average of 24 years and found that individuals heterozygous for the ECSOD R213G mutation had significantly lower COPD morbidity and morality rates as compared to individuals that were noncarriers for this ECSOD mutation.](/cms/asset/5bbb9dd3-1deb-425c-9757-8dbe3a973da0/icop_a_408692_uf0004_b.gif)
Table 1 Summary of genetic association studies of COPD with ECSOD
SUMMARY
Most people who develop COPD are smokers or former smokers. Smoking increases lung oxidative stress, increases inflammatory cytokines, and causes recruitment of inflammatory cells to the lung. When not controlled, this chronic inflammatory process leads to chronic airway disease, destruction of lung tissue and COPD. COPD is found in family clusters, which suggests that genetics plays an important role in the progression of COPD. Several genetic studies have indicated that the R213G mutation in ECSOD is associated with protection from the development of COPD. The R213G mutation results in high circulating levels of ECSOD and likely high levels in alveolar and airway linking fluids where it could protect the lung interface from the effects of inhaled free radicals in tobacco smoke. ECSOD both scavenges free radicals and controls inflammation associated with COPD.
Both animal models and human genetic studies have demonstrated associations of ECSOD with lung function and development of smoking related diseases. A common ECSOD polymorphism has been shown to reduce the risk of developing COPD. While this polymorphism could account for only a small portion of the smokers who are resistant to developing COPD, it demonstrates the potential importance of ECSOD in protecting the lung from oxidative stress. A relative deficiency or inadequate up-regulation of ECSOD in response to stress may be an important component in enhancing risk for development of COPD.
Declaration of interest:
The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
ACKNOWLEDGMENTS
We would like to thank Michael Weaver for his graphic design work presented in this manuscript.
The project described is being supported by Award Numbers U01HL089897 and U01HL089856 from the National Heart, Lung, And Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, And Blood Institute or the National Institutes of Health. We would also like to acknowledge the Monfort Family Foundation and the Department of Defense (W81XW-07-1-0550) for supporting this work.
REFERENCES
- Lokke A, Lange P, Scharling H, Fabricius P, Vestbo J. Developing COPD: a 25 year follow up study of the general population. Thorax 2006; 61: 935–939
- Dahl M, Bowler R P, Juul K, Crapo J D, Levy S, Nordestgaard B G. Superoxide dismutase 3 polymorphism associated with reduced lung function in two large populations. Amer J Respir Crit Care Med 2008; 178: 906–912
- Juul K, Tybjaerg-Hansen A, Marklund S, Lange P, Nordestgaard B G. Genetically increased antioxidative protection and decreased chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 2006; 173: 858–864
- Kinnula V L, Crapo J D. Superoxide dismutases in the lung and human lung diseases. Amer J Respir Crit Care Med 2003; 167: 1600–1619
- Tetley T D. Inflammatory cells and chronic obstructive pulmonary disease. Curr Drug Targets 2005; 4: 607–618
- Yoshida T, Tuder R M. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev 2007; 87: 1047–1082
- Church D F, Pryor W A. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect 1985; 64: 111–126
- Kirkham P, Rahman I. Oxidative stress in asthma and COPD: antioxidants as a therapeutic strategy. Pharmacol Therapeut 2006; 111: 476–494
- Agius R M, Rutman A, Knight R K, Cole P J. Human pulmonary alveolar macrophages with smokers' inclusions: their relation to the cessation of cigarette smoking. Br J Exper Pathol 1986; 67: 407–413
- Iles K E, Forman H J. Macrophage signaling and respiratory burst. Immunol Res 2002; 26: 95–105
- McCord J M, Gao B, Leff J, Flores S C. Neutrophil-generated free radicals: possible mechanisms of injury in adult respiratory distress syndrome. Environ Health Perspect 1994; 102(Suppl 10)57–60
- Biernacki W A, Kharitonov S A, Barnes P J. Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax 2003; 58: 294–298
- Paredi P, Kharitonov S A, Leak D, Ward S, Cramer D, Barnes P J. Exhaled ethane, a marker of lipid peroxidation, is elevated in chronic obstructive pulmonary disease. Amer J Respirat Crit Care Med 2000; 162: 369–373
- Pratico D, Basili S, Vieri M, Cordova C, Violi F, Fitzgerald G A. Chronic obstructive pulmonary disease is associated with an increase in urinary levels of isoprostane F2alpha-III, an index of oxidant stress. Amer J Respir Crit Care Med 1998; 158: 1709–1714
- Rahman I, van Schadewijk A A, Crowther A J, Hiemstra P S, Stolk J, MacNee W, De Boer W I. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Amer J Respirat Crit Care Med 2002; 166: 490–495
- Corradi M, Pesci A, Casana R, Alinovi R, Goldoni M, Vettori M V, Cuomo A. Nitrate in exhaled breath condensate of patients with different airway diseases. Nitric Oxide 2003; 8: 26–30
- Ichinose M, Sugiura H, Yamagata S, Koarai A, Shirato K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Amer J Respir Critical Care Med 2000; 162: 701–706
- Kinnula V L, Ilumets H, Myllarniemi M, Sovijarvi A, Rytila P. 8-Isoprostane as a marker of oxidative stress in nonsymptomatic cigarette smokers and COPD. Eur Respir J 2007; 29: 51–55
- Rytila P, Rehn T, Ilumets H, Rouhos A, Sovijarvi A, Myllarniemi M, Kinnula V L. Increased oxidative stress in asymptomatic current chronic smokers and GOLD stage 0 COPD. Resp Res 2006; 7: 69
- Nadeem A, Raj H G, Chhabra S K. Increased oxidative stress and altered levels of antioxidants in chronic obstructive pulmonary disease. Inflammation 2005; 29: 23–32
- Fraig M, Shreesha U, Savici D, Katzenstein A L. Respiratory bronchiolitis: a clinicopathologic study in current smokers, ex-smokers, and never-smokers. Amer J Surg Pathol 2002; 26: 647–653
- Louhelainen N, Rytila P, Haahtela T, Kinnula V L, Djukanovic R. Persistence of oxidant and protease burden in the airways after smoking cessation. BMC Pulmon Med 2009; 9: 25
- Hogg J C, Chu F, Utokaparch S, Woods R, Elliott W M, Buzatu L, Cherniack R M, Rogers R M, Sciurba F C, Coxson H O, Pare P D. The nature of small-airway obstruction in chronic obstructive pulmonary disease. New Engl J Med 2004; 350: 2645–2653
- Bowler R P, Barnes P J, Crapo J D. The role of oxidative stress in chronic obstructive pulmonary disease. COPD 2004; 1: 255–277
- Rahman I. The role of oxidative stress in the pathogenesis of COPD: implications for therapy. Treat Respir Med 2005; 4: 175–200
- McCord J M, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 1969; 244: 6049–6055
- Marklund S L. Human copper-containing superoxide dismutase of high molecular weight. Proceedings of the National Academy of Sciences of the United States of America. 1982; 79: 7634–7638
- Weisiger R A, Fridovich I. Mitochondrial superoxide dimutase. Site of synthesis and intramitochondrial localization. J Biol Chem 1973; 248: 4793–4796
- Oury T D, Chang L Y, Marklund S L, Day B J, Crapo J D. Immunocytochemical localization of extracellular superoxide dismutase in human lung. Lab Invest J Tech Meth Pathol 1994; 70: 889–898
- Oury T D, Day B J, Crapo J D. Extracellular superoxide dismutase in vessels and airways of humans and baboons. Free Rad Biol Med 1996; 20: 957–965
- Folz R J, Crapo J D. Extracellular superoxide dismutase (SOD3): tissue-specific expression, genomic characterization, and computer-assisted sequence analysis of the human EC SOD gene. Genomics 1994; 22: 162–171
- Inoue M, Watanabe N, Matsuno K, Sasaki J, Tanaka Y, Hatanaka H, Amachi T. Expression of a hybrid Cu/Zn-type superoxide dismutase which has high affinity for heparin-like proteoglycans on vascular endothelial cells. J Biol Chem 1991; 266: 16409–16414
- Gao F, Kinnula V L, Myllarniemi M, Oury T D. Extracellular superoxide dismutase in pulmonary fibrosis. Antioxidants Redox Signal 2008; 10: 343–354
- Tan R J, Fattman C L, Watkins S C, Oury T D. Redistribution of pulmonary EC-SOD after exposure to asbestos. J Appl Physiol 2004; 97: 2006–2013
- Fattman C L, Tan R J, Tobolewski J M, Oury T D. Increased sensitivity to asbestos-induced lung injury in mice lacking extracellular superoxide dismutase. Free Rad Biol Med 2006; 40: 601–607
- Folz R J, Abushamaa A M, Suliman H B. Extracellular superoxide dismutase in the airways of transgenic mice reduces inflammation and attenuates lung toxicity following hyperoxia. J Clin Invest 1999; 103: 1055–1066
- Gongora M C, Lob H E, Landmesser U, Guzik T J, Martin W D, Ozumi K, Wall S M, Wilson D S, Murthy N, Gravanis M, Fukai T, Harrison D G. Loss of extracellular superoxide dismutase leads to acute lung damage in the presence of ambient air: a potential mechanism underlying adult respiratory distress syndrome. Amer J Pathol 2008; 173: 915–926
- Bowler R P, Nicks M, Warnick K, Crapo J D. Role of extracellular superoxide dismutase in bleomycin-induced pulmonary fibrosis. Amer J Physiol 2002; 282: L719–726
- Bowler R P, Nicks M, Tran K, Tanner G, Chang L Y, Young S K, Worthen G S. Extracellular superoxide dismutase attenuates lipopolysaccharide-induced neutrophilic inflammation. Amer J Respir Cell Mol Biol 2004; 31: 432–439
- Gao F, Koenitzer J R, Tobolewski J M, Jiang D, Liang J, Noble P W, Oury T D. Extracellular superoxide dismutase inhibits inflammation by preventing oxidative fragmentation of hyaluronan. J Biol Chem 2008; 283: 6058–6066
- Kliment C R, Tobolewski J M, Manni M L, Tan R J, Enghild J, Oury T D. Extracellular superoxide dismutase protects against matrix degradation of heparan sulfate in the lung. Antioxidants Redox Signal 2008; 10: 261–268
- Petersen S V, Oury T D, Ostergaard L, Valnickova Z, Wegrzyn J, Thogersen I B, Jacobsen C, Bowler R P, Fattman C L, Crapo J D, Enghild J J. Extracellular superoxide dismutase (EC-SOD) binds to type I collagen and protects against oxidative fragmentation. J Biol Chem 2004; 279: 13705–13710
- Cutroneo K R. TGF-beta-induced fibrosis and SMAD signaling: oligo decoys as natural therapeutics for inhibition of tissue fibrosis and scarring. Wound Repair Regen 2007; 15(Suppl 1)S54–60
- Rabbani Z N, Anscher M S, Folz R J, Archer E, Huang H, Chen L, Golson M L, Samulski T S, Dewhirst M W, Vujaskovic Z. Overexpression of extracellular superoxide dismutase reduces acute radiation induced lung toxicity. BMC Cancer 2005; 5: 59
- Cotch M F, Beaty T H, Cohen B H. Path analysis of familial resemblance of pulmonary function and cigarette smoking. Amer Rev Respir Dis 1990; 142: 1337–1343
- Cotch M F, Beaty T H, Munoz A, Cohen B H. Estimating familial aggregation while adjusting for covariates. Application to pulmonary function data from black and white sibships. Ann Epidemiol 1992; 2: 317–324
- Rybicki B A, Beaty T H, Cohen B H. Major genetic mechanisms in pulmonary function. J Clin Epidemiol 1990; 43: 667–675
- Silverman E K, Chapman H A, Drazen J M, Weiss S T, Rosner B, Campbell E J, O'Donnell W J, Reilly J J, Ginns L, Mentzer S, Wain J, Speizer F E. Genetic epidemiology of severe, early-onset chronic obstructive pulmonary disease. Risk to relatives for airflow obstruction and chronic bronchitis. Amer J Respir Crit Care Med 1998; 157: 1770–1778
- Reinhard C, Meyer B, Fuchs H, Stoeger T, Eder G, Ruschendorf F, Heyder J, Nurnberg P, de Angelis M H, Schulz H. Genomewide linkage analysis identifies novel genetic Loci for lung function in mice. Amer J Respir Crit Care Med 2005; 171: 880–888
- Ganguly K, Stoeger T, Wesselkamper S C, Reinhard C, Sartor M A, Medvedovic M, Tomlinson C R, Bolle I, Mason J M, Leikauf G D, Schulz H. Candidate genes controlling pulmonary function in mice: transcript profiling and predicted protein structure. Physiol Genom 2007; 31: 410–421
- Wilk J B, Walter R E, Laramie J M, Gottlieb D J, O'Connor G T. Framingham Heart Study genome-wide association: results for pulmonary function measures. BMC Med Genetics 2007; 8(Suppl 1)S8
- Arcaroli J J, Hokanson J E, Abraham E, Geraci M, Murphy J R, Bowler R P, Dinarello C A, Silveira L, Sankoff J, Heyland D, Wischmeyer P, Crapo J D. Extracellular superoxide dismutase haplotypes are associated with acute lung injury and mortality. Amer J Respir Crit Care Med 2009; 179: 105–112
- Bentley A R, Emrani P, Cassano P A. Genetic variation and gene expression in antioxidant related enzymes and risk of COPD: a systematic review. Thorax 2008; 63: 956–961
- Young R P, Hopkins R, Black P N, Eddy C, Wu L, Gamble G D, Mills G D, Garrett J E, Eaton T E, Rees M I. Functional variants of antioxidant genes in smokers with COPD and in those with normal lung function. Thorax 2006; 61: 394–399