
Abstract
PNA is hybrid molecule ideally suited for bridging the functional landscape of polypeptides with the structural diversity that can be engineered with DNA nanostructures. However, PNA can be more challenging to work with in aqueous solvents due to its hydrophobic nature. A solution phase method using strain promoted, copper free click chemistry was developed to conjugate the fluorescent dye Cy5 to 2 bifunctional PNA strands as a first step toward building cyclic PNA-polypeptides that can be arranged within 3D DNA nanoscaffolds. A 3D DNA nanocage was designed with binding sites for the 2 fluorescently labeled PNA strands in close proximity to mimic protein active sites. Denaturing polyacrylamide gel electrophoresis (PAGE) is introduced as an efficient method for purifying charged, dye-labeled PNA conjugates from large excesses of unreacted dye and unreacted, neutral PNA. Elution from the gel in water was monitored by fluorescence and found to be more efficient for the more soluble PNA strand. Native PAGE shows that both PNA strands hybridize to their intended binding sites within the DNA nanocage. Förster resonance energy transfer (FRET) with a Cy3 labeled DNA nanocage was used to determine the dissociation temperature of one PNA-Cy5 conjugate to be near 50°C. Steady-state and time resolved fluorescence was used to investigate the dye orientation and interactions within the various complexes. Bifunctional, thermostable PNA molecules are intriguing candidates for controlling the assembly and orientation of peptides within small DNA nanocages for mimicking protein catalytic sites.
Abbreviations:
- PNA, peptide nucleic acid
- DBCO, dibenzocyclooctyl
- PAGE, polyacrylamide gel electrophoresis
- RP-HPLC, reverse-phase high pressure liquid chromatography
- MALDI-MS, matrix assisted laser desorption ionization mass spectrometry
- IEX-FPLC, ion-exchange fast protein liquid chromatography
- EtBr, ethidium bromide
- DTNB, 5, 5′-dithiobis-(2-nitrobenzoic acid)
- TCEP, tris(2-carboxyethyl)phosphine
Introduction
DNA nanostructures are an intriguing platform for manipulating molecules and materials on the nanometer scale,Citation1-3 including proteins and peptides.Citation4,5 DNA nanostructure assembly is governed mostly by Watson-Crick base pairingCitation6 and therefore is more straightforward to engineer than proteins or peptides.Citation7,8 Chemical strategies are routinely used for conjugating nucleic acids to peptides or proteins in order to enhance the functionality of unmodified DNA. However, the number of efficient bioorthogonal coupling strategies can become limiting for creating more highly interconnected peptide complexes that could be used to mimic protein catalytic sites. Alternatively, nucleobase hybridization can be used to leverage a few robust chemical coupling methods for conjugating a number of polypeptides, each to a unique nucleic acid strand, which hybridize to specific binding sites within the DNA scaffold.
PNACitation9 (peptide nucleic acid) is an intriguing synthetic polymer for bridging the diverse functional landscape of polypeptides with the straight-forward construction of nucleic acid nanostructures. Like peptides, PNAs are linked by amide bonds and can be synthesized using similar solid phase protocols,Citation10 thus allowing for seamless synthesis of PNA-peptide conjugates.Citation11 Applications have been recently reviewedCitation12 including a review focused on chemical modifications to PNA.Citation13 Its synthetic backbone makes it highly resistant to nucleases and proteasesCitation14 and useful for targeting DNACitation15 and RNA,Citation16 as well as using it as a molecular probeCitation17 and biosensor.Citation18,19 PNA has also been assembled within DNA scaffolds to investigate applications in bionanotechnology.Citation20-22 Previously we showed the versatility of using PNA linkers for assembling peptidesCitation22 and proteinsCitation23 into a 3D DNA nanocage. The assembly process was rapid and could be performed at or below room temperature, providing mild conditions that preserved the function of the attached polypeptides. Substitutions to the gamma position on the PNA backbone (see Figure S2) have been shown to increase the solubility and thermal stability of the PNA strand,Citation24 as well as provide a handle to introduce functional groups without affecting its binding to a complementary strand.Citation25 Functionalizing PNA at the gamma position would also allow a peptide to be connected anywhere along the backbone or even at multiple locations to form stable cyclic PNA-peptides.Citation21,26 Although conjugating fluorescent dyes and other small molecules to peptides or PNA is greatly simplified when performed after synthesis while the product is still on the solid phase, the conjugation efficiency decreases for larger molecules and the cleavage and purification conditions can be detrimental to some molecules, such as metallopeptidesCitation27 or folded peptides and proteins.
Here we develop a solution phase orthogonal conjugation method using strain-promoted, copper free click chemistry to selectively attach the fluorescent dye Cy5 to the N terminus of 2 bifunctional PNAs containing an azide at the N terminus and a thiol along the backbone at the gamma position. Denaturing PAGE was used to purify the Cy5 labeled conjugates and native PAGE was used to characterize the DNA nanocage when populated with either of 2 Cy5 labeled PNAs. Steady state and time resolved fluorescence spectroscopy was used to characterize the dye labeled constructs.
Results and Discussion
DNA-PNA complex design
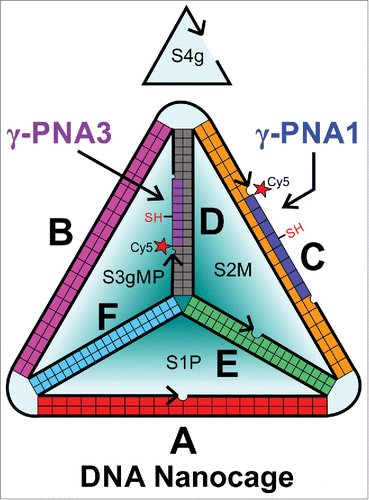
Previously we incorporated 2 fluorescently labeled peptidesCitation22 or proteinsCitation23 within a 3D DNA nanocage using PNA linkers. Briefly, we modified an existing DNA tetrahedron design by introducing 8nt single stranded domains complementary to a PNA strand functionalized on the N terminus with a short peptide or small protein oriented toward the center of the DNA nanocage. This design allowed the DNA nanocage to be preassembled, purified and then subsequently populated with one or 2 PNA-polypeptides under mild conditions. Here we introduce a third 8nt PNA binding site (PNA3), to be used in conjunction with the PNA1 binding site for introducing guest molecules, as shown in . The PNA3 binding site is closer in proximity to the PNA1 binding site (3 nm) than a similar PNA binding site on edge F used in the previous design (5–6 nm),Citation22 which is better suited for assembling protein active sites. Furthermore, both PNA1 and PNA3 sequences were modified to increase the predicted thermal stabilityCitation28 from 37°C to near 50°C by increasing the purine content. Throughout this report we use the nomenclature defined in Figure S1, Table S1 and Table S2 when we describe the various strands and constructs used in each experiment.
Figure 1. Schematic showing the arrangement of DNA and PNA sequences in the complex, written in the 5′ (N) to 3′ (C) direction. γ-PNA1 and γ-PNA3 are labeled with Cy5, thiol functionalized at the gamma position of 4th PNA base, and attached to edge C and D respectively. This schematic was redrawn fromCitation39 with changes to incorporate the gamma modified PNAs.
Fluorescent labeling and purification of γ-PNA
As a proof of concept to form cyclic PNA-peptides for introducing into 3D DNA nanocages, a solution phase conjugation method was developed for labeling PNA with Cy5 that is compatible with bifunctional PNA strands. Cy5 functionalized with the strained alkyne dibenzocyclooctyl (DBCO) was used to label each γ-PNA at the N terminal azide position, as shown in Figure S3 and described in the methods section. The chemical structures of PNA1-Cy5 and PNA3-Cy5 are shown in Figure S4. Strained alkynes can be efficiently conjugated to azides without adding any copper catalyst,Citation29,30 which could interfere with assembling metallopeptide complexes and can be difficult to completely remove.Citation30,31 We were unable to separate the unreacted Cy5 from the γ-PNA-Cy5 conjugate using reverse-phase high pressure liquid chromatography (RP-HPLC). We had similar issues trying remove unreacted fluorescein (FAM) from γ-PNA-FAM, which suggests the dye labeled γ-PNA sequences may strongly interact with the unreacted dyesCitation32,33 or they have similar levels of hydrophobicity. Denaturing PAGE has been shown as an alternative to RP-HPLC for purifying unmodified PNA under acidic conditions (pH 3) with single-base resolution.Citation34 We adapted this technique to work under slightly alkaline conditions (pH 8.5) where natural PNA is uncharged, and the γ-PNA-Cy5 conjugates are negatively charged and of significantly greater size (∼3.5 kDa) than the unreacted Cy5 (1 kDa). shows the denaturing PAGE gel after the reaction of Cy5 with γ-PNA3. The unreacted Cy5 migrates significantly faster than the γ-PNA-Cy5 conjugate. Matrix assisted laser desorption ionization mass spectrometry (MALDI-MS) of the other weakly fluorescent bands were found to contain impurities.
Figure 2. Purification of Cy5 labeled γ-PNAs using denaturing PAGE and time course elution from gel blocks. (A) Cy5 fluorescence image of a 10% denaturing PAGE gel containing a control with Cy5 only (Lane 1) and the γ-PNA3 and Cy5 reaction cocktail (Lanes 3–11). (B) Cy5 fluorescence image of the of γ-PNA1-Cy5 bands cut from a 7.5% gel, similar to that shown in [A], when eluted with water and centrifuged in Spin-X™ tubes up to 4 times to remove the product eluted from the gel blocks. (C) The fluorescence intensity of the gel blocks after each elution in (B) was used to determine the fraction of PNA remaining in the gel blocks for γ-PNA1-Cy5 and γ-PNA3-Cy5. Each curve was fit with an exponential decay function as described in the methods section. The parameters for the fitting functions are shown in Table S4.
![Figure 2. Purification of Cy5 labeled γ-PNAs using denaturing PAGE and time course elution from gel blocks. (A) Cy5 fluorescence image of a 10% denaturing PAGE gel containing a control with Cy5 only (Lane 1) and the γ-PNA3 and Cy5 reaction cocktail (Lanes 3–11). (B) Cy5 fluorescence image of the of γ-PNA1-Cy5 bands cut from a 7.5% gel, similar to that shown in [A], when eluted with water and centrifuged in Spin-X™ tubes up to 4 times to remove the product eluted from the gel blocks. (C) The fluorescence intensity of the gel blocks after each elution in (B) was used to determine the fraction of PNA remaining in the gel blocks for γ-PNA1-Cy5 and γ-PNA3-Cy5. Each curve was fit with an exponential decay function as described in the methods section. The parameters for the fitting functions are shown in Table S4.](/cms/asset/85ce3eb0-415d-4430-8935-edccaffa7c29/kdpx_a_992181_f0002_oc.gif)
The bands for the γ-PNA-Cy5 conjugates were cut from the gel and eluted with nanopure water up to 4 times to extract the product, as detailed in Table S3. The fluorescent signal remaining in the gel blocks after each elution step, for both γ-PNA3-Cy5 and γ-PNA1-Cy5, was integrated and fit with an exponential function, as shown in , and described in the methods section. Both samples are efficiently eluted after 3–4 elution steps. Despite a lower percent gel with larger pores, γ-PNA1-Cy5 elutes with slightly lower efficiency, which may be due to its reduced solubility compared to γ-PNA3-Cy5. Although the times allowed for elution during each step were significant different (1.25–115 hours), it appears that equilibrium was still achieved after each elution step, which suggests that the elution could be performed in much less time (i.e., 4 elutions over 6 hours). The eluted product was desalted using RP-HPLC. The expected molecular weight of each collected product was detected by MALDI-MS, as shown in Figure S5 for γ-PNA1-Cy5 and Figure S6 for γ-PNA3-Cy5. Both samples show an impurity +70 Da from the expected molecular weights, which was observed to be more prevalent with longer conjugation times. Native PAGE analysis of γ-PNA3-Cy5 shows the expected band as well as a slower moving band, which may be dimerization of γ-PNA3-Cy5 via an intramolecular disulfide between thiols at the gamma position. The dimer may be the result of insufficient disulfide cleavage by tris(2-carboxyethyl)phosphine (TCEP) prior to PAGE analysis.
Although MALDI-MS did not show evidence of any PNAs labeled with Cy5 on both at the azide and thiol positions, the free thiol is the most reactive group remaining on the PNA molecule and is a logical place for side reactions to occur that could lead to the conspicuous +70 Da impurity. To test this hypothesis, the azide and thiol functionalized PNAs were reacted with 5,5′-dithiobis-(2-nitrobenzoic acid (DTNB) prior to conjugation with Cy5, which forms a disulfide with any free thiols. During purification by denaturing PAGE 3 main bands were observed, as shown in Figure S7. The slowest moving band is only observed for PNA1 and is attributed to PNA1-Cy5 disulfide dimers; since the thiols were already oxidized they could not react with DTNB. The next 2 bands are observed for both PNAs and are attributed to the Cy5 conjugated PNA with and without the negatively charged 5-thio-2-nitrobenzoic acid (TNB−) protectant group. Each major band was cut, eluted, reduced with TCEP to remove TNB and desalted using RP- HPLC. Each sample contained the desired product without the +70 Da impurity, as shown in the MALDI-MS for PNA1-Cy5 and PNA3-Cy5 in Figure S8 and Figure S9. These data suggest the impurity was due to a side reaction with the unprotected thiol despite the reaction taking place under acidic conditions (pH 4), but which can be circumvented by protecting the thiol using DTNB or other reversible thiol protectant group.
Assembly of the DNA nanocages with fluorescently labeled γ-PNAs
DNA nanocages, for binding either γ-PNA1 (DNA1γ) or γ-PNA3 (DNA3γ), were assembled and purified by ion-exchange fast protein liquid chromatography (IEX-FPLC), and incubated with increasing molar excess (0–3x or 10x) of the corresponding γ-PNA sequence at room temperature, as described previously.Citation22 The assembled complexes were analyzed directly by native PAGE at 4°C, as shown in . A small gel shift is observed for the DNA nanocage upon binding each γ-PNA, with respect to the empty DNA nanocage, and is attributed to the single stranded γ-PNA binding domain becoming double stranded and more rigid, as observed previously.Citation22 A 2−5x excess of unlabeled γ-PNA is required to achieve quantitative binding to the DNA nanocage. This is similar to the 2x excess of PNA-peptide required for quantitative binding to the same DNA nanocage.Citation22 Some PNA sequences show mild aggregation at concentrations as low as 1 μM,Citation24 which could prevent binding of some PNA to the DNA nanocage. The larger excess required for γ-PNA1 may be due to its reduced solubility compared to γ-PNA3. However, when γ-PNA1 is labeled with the negatively charged Cy5, only 2x excess is required for quantitative binding to the DNA nanocage, as shown in the right panel of , suggesting the Cy5 label may improve the solubility of γ-PNA1. also shows the Cy5 fluorescence of the DNA nanocages containing the Cy5 labeled γ-PNAs, as well as a more diffuse band containing some unhybridized γ-PNA-Cy5. These results indicate the proper assembly of the γ-PNAs into the DNA nanocage before and after Cy5 labeling.
Figure 3. Gel showing the binding of the unlabeled and labeled γ-PNAs to the DNA nanocage. [Left] Ethidium bromide image of the unlabeled DNA1γ and DNA3γ nanocages with increasing amount of unlabeled γ-PNA1 and γ-PNA3, respectively. [Right] The unlabeled DNA1γ and DNA3γ nanocages with increasing amount of Cy5 labeled γ-PNA1 and γ-PNA3, respectively. The ethidium bromide (EtBr) fluorescence (left panel) and Cy5 fluorescence (right panel) are shown, with the corresponding excitation and emission wavelengths (Ex/Em).
![Figure 3. Gel showing the binding of the unlabeled and labeled γ-PNAs to the DNA nanocage. [Left] Ethidium bromide image of the unlabeled DNA1γ and DNA3γ nanocages with increasing amount of unlabeled γ-PNA1 and γ-PNA3, respectively. [Right] The unlabeled DNA1γ and DNA3γ nanocages with increasing amount of Cy5 labeled γ-PNA1 and γ-PNA3, respectively. The ethidium bromide (EtBr) fluorescence (left panel) and Cy5 fluorescence (right panel) are shown, with the corresponding excitation and emission wavelengths (Ex/Em).](/cms/asset/68e5614c-019f-47c6-a237-24c49afdff9f/kdpx_a_992181_f0003_oc.gif)
Fluorescence characterization of the Cy5 fluorescently labeled γ-PNAs
Steady-state fluorescence spectra of the Cy5 labeled γ-PNAs and Cy3 labeled DNA nanocage were recorded to determine the peak wavelength and quantum yield, as shown in Table S5, and described in the supporting information methods section. The quantum yields of Cy5 conjugated to PNA and hybridized into the DNA nanocages (0.38) and PNA1-Cy5 alone (0.42) and Cy5-DBCO (0.28) are similar to another report (0.30),Citation35 but is much lower for PNA3-Cy5 (0.11). Interestingly, the quantum yield for PNA3-Cy5 is restored upon binding to the DNA nanocage. The quantum yield of cyanine dyes is affected by their ability to photoisomerize, where lower quantum yields are observed from more freely moving dyes; the dye mobility was investigated by time resolved fluorescence and is discussed later.Citation36 The quantum yields found for Cy3 for conjugated to the DNA nanocages (0.49) are much higher than that reported for the free dye (0.04),Citation35 but in agreement with values when conjugated to ssDNA (0.4)Citation36 or in glycerol (0.52)Citation35 due to reduced photoisomerization under restricted mobility.Citation36 This is consistent with Cy3 strongly interacting with the slow tumbling of the 80 kDa DNA nanocage.
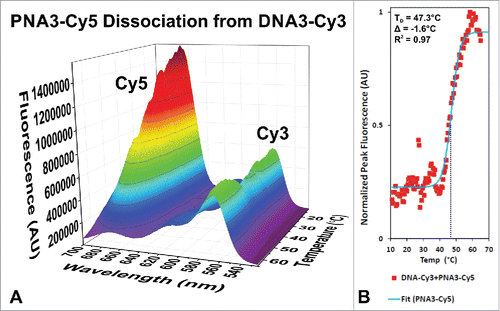
Energy transfer efficiency between a Cy3 labeled DNA nanocage and the Cy5 labeled PNAs were used to investigate the thermal stability of the PNAs bound the DNA nanocage. shows the fluorescence emission intensity when the donor (Cy3) is excited at 475 nm as a function of temperature for the DNA3-Cy3 + PNA3-Cy5 complex. At low temperature the Cy3 fluorescence is quenched by Cy5 and there is significant fluorescence emission from Cy5 due to energy transfer from Cy3 because of the close proximity of the dyes in the fully assembled DNA-PNA complex. As the temperature increases and PNA3-Cy5 dissociates out of the DNA1-Cy3 nanocage, the Cy5 emission is lost and the Cy3 emission is restored. The change in Cy3 fluorescence intensity as a function of temperature is plotted in and fit with a sigmoidal dose response curve, where the inflection point indicates the dissociation temperature (TD). The determined TD for PNA3-Cy5 (47.3°C) is in agreement with theoryCitation28 (48.9°C) and confirms our earlier hypothesis that increasing the purine content could be used to increase the thermal stability of our previous DNA-PNA-peptideCitation22 and DNA-PNA-proteinCitation23 complexes without increasing the PNA sequence length, which may affect the DNA nanocage assembly.
Figure 4. Fluorescence spectra of the Cy5 labeled PNA3 bound to the Cy3 labeled DNA3 nanocage as a function of temperature. The Cy3 labeled DNA nanocage (DNA3-Cy3) containing Cy5 labeled γ-PNA3 was excited at 475 nm and the emission was monitored as a function of wavelength as the temperature was increased from 11°C to 85°C as shown in (A). The peak donor fluorescence is plotted versus temperature and fitted with a sigmoidal dose response curve (B), where the inflection point corresponds to the dissociation temperature (TD) indicated on the plot. The difference (Δ) from a theoretical value (48.9°C) is also shown, which is greater than the value predicted for a similar DNA sequence (37.5°C).
Interestingly, when we tried the same experiment with the DNA1-Cy3 + PNA1-Cy5 complex, which places Cy5 closer to the vertex of the DNA nanocage containing Cy3, no energy transfer was observed. As shown previously in , the native PAGE of DNA1-PNA1 and DNA3-PNA3 complexes were identical, suggesting that both were assembled properly. However, energy transfer is dependent not only on distance, but also on the orientation between the dyes. Dyes that have limited rotational mobility and are placed at orthogonal orientations will not transfer energy even at very close distances. We then investigated the mobility of the dyes attached to the complexes using time resolved fluorescence anisotropy. Time correlated single photon counting (TCSPC) was used to measure the fluorescence decay kinetics of the Cy3 (Ex. 500, Em. 600 nm) and Cy5 (Ex. 600, Em. 670 nm) containing constructs when excited with vertically polarized light and when the emission compared between vertical (IVV) and horizontal (IVH) polarization with respect to the excitation source.
shows the fluorescence decay kinetics of each sample with Cy5 at both vertical and horizontal emission polarization. The difference was used to calculate the time resolved fluorescence anisotropy as shown in , and described in the supporting information methods section. The Cy5-DBCO anisotropy decays from the maximum anisotropy value of 0.4 (i.e., fully polarized) to 0 (i.e., fully depolarized) within 3 ns due to its rapid rotation when free in solution. The anisotropy of Cy5 conjugated to each PNA strands decays to 0 within 7 ns due to the slower motion of the larger molecule. Interestingly, PNA3-Cy5 decays faster than PNA1-Cy5 within the first 1 ns, and may contribute to the lower observed quantum yield for PNA3-Cy5. When the Cy5 labeled PNAs are hybridized to the large DNA nanocage the anisotropy decays much slower to 0.25–0.27 after 8 ns, suggesting that the dyes are strongly interacting with the DNA nanocage. These values are in agreement with our previous study using tetramethylrhodamine (TMR) labeled DNA nanocages (0.24),Citation22 which also highly interacts with the DNA nanocage.
Figure 5. Fluorescence decay and anisotropy of Cy5 labeled γ-PNA hybridized to a DNA nanocage. (A–E) Plot of the parallel (Ivv) and perpendicular (Ivh) fluorescence decay of Cy5 free in solution (A), when conjugated to γ-PNA1 (B) or γ-PNA3 (C) and when conjugated to PNA and hybridized into DNA1 nanocage (D) or DNA3 nanocage (E), respectively. Panel (F): The anisotropy for each data point of the decays, shown in panels A–E, was calculated as described in the Supporting Information, Materials and Methods section, and the results are plotted here. The central moving average for each dataset is reported in the same plot for clarity.
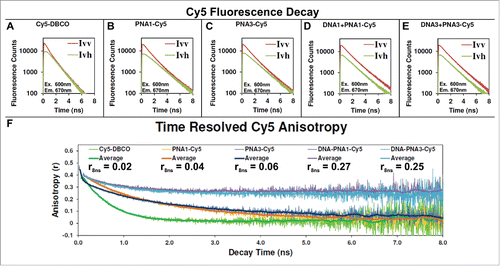
Figure 6. Fluorescence decay and anisotropy of Cy3 labeled DNA nanocages. (A–C) Plot of the parallel (Ivv) and perpendicular (Ivh) fluorescence decay of Cy3 conjugated to a ssDNA (A), when the ssDNA-Cy3 is assembled into either the DNA1-Cy3 (B) or DNA3-Cy3 nanocage, respectively. Panel (D): The anisotropy for each data point of the decays, shown in panels A–C, was calculated as described in the Supporting Information, Materials and Methods section, and the results are plotted here. The central moving average for each data set is reported in the same plot for clarity.
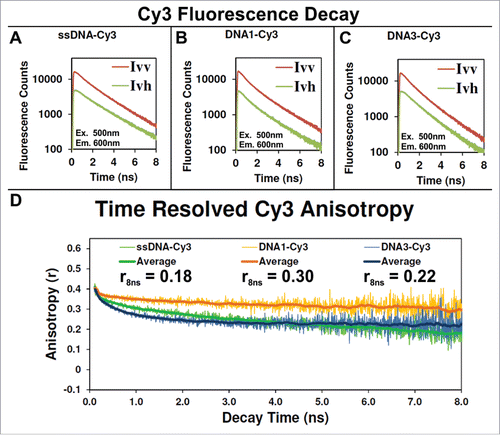
shows the fluorescence decay kinetics of Cy3 for horizontal and vertical emission polarizations, first when conjugated to single-stranded DNA (A), then when the same ssDNA is annealed into either DNA1-Cy3 or DNA3-Cy3 nanocages. The calculated anisotropy for DNA3-Cy3 decays to 0.22 within 8 ns, suggesting that it is also highly interacting with the DNA nanocage. Interestingly, the calculated anisotropy of DNA1-Cy3 decays only to 0.3 within 8 ns, suggesting that its rotation may be restricted in a mostly static orientation relative to the DNA nanocage where the anisotropy only decays due to the slower diffusion of the DNA nanocage. The lack of energy transfer suggests that this static orientation is orthogonal to the donor. This type of orientation dependence was observed because of strong interaction between Cy3 and Cy5 with the nucleobases of a linear DNA helix as the length was extended due to the rotation of DNA nucleobases along the helix.Citation37 This effect could be more dramatic for helices joined at more orthogonal angels, such as the 60⁰ angle between adjoining edges of our 3D DNA tetrahedron. Finally, TCSPC was also used to measure the fluorescence lifetimes of each Cy3 and Cy5 labeled complex, as shown in Figure S10 and listed in Table S5. The lifetimes of DNA1-Cy3 and DNA3-Cy3 (1.3 ns) are in agreement with other reports that show that the natural lifetime of the free Cy3 dye (0.18 ns) can be extended up to 2.0 ns when its rotation is restricted to minimize photoisomerization.Citation36,37 The observed Cy3 lifetimes are consistent with the high anisotropy due to rotational restriction by the DNA nanocage. The lifetime of the free Cy5-DBCO (1.1 ns) is in excellent agreement with another report (1.0 ns).Citation38 The observed lifetime of Cy5 conjugated to PNA (1.4–1.5 ns) is consistent with the motion of Cy5 being restricted to minimize photoisomerization.Citation38
Conclusion
We introduced a bifunctional PNA design using copper free click chemistry to label the PNA with the dye Cy5 in solution while protecting a thiol group for future conjugation reactions. Denaturing PAGE is introduced as a method to efficiently purify PNA fluorescently labeled with Cy5. The fluorescence of the product was used to monitor its elution from the gel blocks, which allows the method to be extended to other charged molecules that may not be so easily detected. The Cy5 labeled γ-PNAs were assembled into DNA nanocages under benign conditions that were previously shown to preserve polypeptide function.Citation23 Energy transfer between PNA3-Cy5 and DNA1-Cy3 was used to confirm the high thermal stability of the PNA sequence. Interestingly, no energy transfer is observed for PNA1-Cy5 that may be due to the high anisotropy of Cy3 in that complex keeping the dyes at orthogonal orientations. Bifunctional, thermostable PNA molecules are intriguing candidates for controlling the assembly and orientation of peptides within small DNA nanocages for mimicking protein catalytic sites.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
992181_Supplementary_Materials.zip
Download Zip (1.8 MB)Acknowledgments
We want to thank Drs. Hao Yan and Yan Liu for helping to conceive of and develop the proposed application for using DNA nanostructures to assemble artificial catalytic centers. We also want to than John Lopez and Zach Laughrey for their consultation on MALDI-MS.
Funding
This work was supported by the Center for Bio-Inspired Solar Fuel Production, an Energy Frontier Research Center funded by the US. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001016.
References
- Pinheiro AV., Han D, Shih WM, Yan H. Challenges and opportunities for structural DNA nanotechnology. Nat Nanotechnol 2011; 6:763-72; PMID:22056726; http://dx.doi.org/10.1038/nnano.2011.187
- Linko V, Dietz H. The enabled state of DNA nanotechnology. Curr Opin Biotechnol 2013; 24:555-61; PMID:23566376; http://dx.doi.org/10.1016/j.copbio.2013.02.001
- Seeman NC. Structural DNA nanotechnology: growing along with nano letters. Nano Lett 2010; 10:1971-8; PMID:20486672; http://dx.doi.org/10.1021/nl101262u
- Saccà B, Niemeyer CM. Functionalization of DNA nanostructures with proteins. Chem Soc Rev 2011; 40:5910-21; PMID:21975573; http://dx.doi.org/10.1039/c1cs15212b
- Diezmann F, Seitz O. DNA-guided display of proteins and protein ligands for the interrogation of biology. Chem Soc Rev 2011; 40:5789-801; PMID:21589953; http://dx.doi.org/10.1039/c1cs15054e
- Seeman NC. Nucleic acid junctions and lattices. J Theor Biol 1982; 99:237-47; PMID:6188926
- Sawyer N, Speltz EB, Regan L. NextGen protein design. Biochem Soc Trans 2013; 41:1131-6; PMID:24059497; http://dx.doi.org/10.1042/BST20130112
- Hollfelder F, Lutz S. Just (protein) engineering? Curr Opin Struct Biol 2013; 23:569-70; PMID:23886559; http://dx.doi.org/10.1016/j.sbi.2013.07.003
- Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991; 254:1497-500; PMID:1962210; http://dx.doi.org/10.1126/science.1962210
- Braasch DA, Nulf CJ, Corey DR. Synthesis and purification of peptide nucleic acids. In: Current protocols in nucleic acid chemistry. New York, NY: John Wiley & Sons; 2002. page 4.11.1-4.11.18.
- Kaihatsu K, Corey DR. Synthesis of peptide nucleic acid-peptide conjugates. Methods Mol Biol 2004; 283:207-16; PMID:15197312
- Corradini R, Sforza S, Tedeschi T, Totsingan F, Manicardi A, Marchelli R. Peptide nucleic acids with a structurally biased backbone. Updated review and emerging challenges. Curr Top Med Chem 2011; 11:1535-54; PMID:21510833; http://dx.doi.org/10.2174/156802611795860979
- Rozners E. Recent advances in chemical modification of Peptide nucleic acids. J Nucleic Acids 2012; 2012:518162; PMID:22991652
- Demidov VV, Potaman VN, Frank-Kamenetskii MD, Egholm M, Buchard O, Sönnichsen SH, Nielsen PE. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem Pharmacol 1994; 48:1310-3; PMID:7945427; http://dx.doi.org/10.1016/0006-2952(94)90171-6
- Lansdorp PM, Verwoerd NP, van de Rijke FM, Dragowska V, Little MT, Dirks RW, Raap a K, Tanke HJ. Heterogeneity in telomere length of human chromosomes. Hum Mol Genet 1996; 5:685-91; PMID:8733138; http://dx.doi.org/10.1093/hmg/5.5.685
- Braasch D, Corey D. Novel antisense and peptide nucleic acid strategies for controlling gene expression. Biochemistry 2002; 41:4503-10; PMID:11926811
- Kuhn H, Demidov VV, Coull JM, Fiandaca MJ, Gildea BD, Frank-Kamenetskii MD. Hybridization of DNA and PNA molecular beacons to single-stranded and double-stranded DNA targets. J Am Chem Soc 2002; 124:1097-103; PMID:11829619; http://dx.doi.org/10.1021/ja0041324
- Corradini R, Feriotto G, Sforza S, Marchelli R, Gambari R. Enhanced recognition of cystic fibrosis W1282X DNA point mutation by chiral peptide nucleic acid probes by a surface plasmon resonance biosensor. J Mol Recognit 2004; 17:76-84; PMID:14872540
- Wang J, Palecek E, Nielsen P. Peptide nucleic acid probes for sequence-specific DNA biosensors. J Am Chem Soc 1996; 7863:7667-70; http://dx.doi.org/10.1021/ja9608050
- Lukeman PS, Mittal AC, Seeman NC. Two dimensional PNA/DNA arrays: estimating the helicity of unusual nucleic acid polymers. Chem Commun 2004; 20:1694-5; PMID:15278141; http://dx.doi.org/10.1039/b401103a
- Englund EA, Wang D, Fujigaki H, Sakai H, Micklitsch CM, Ghirlando R, Martin-Manso G, Pendrak ML, Roberts DD, Durell SR, et al. Programmable multivalent display of receptor ligands using peptide nucleic acid nanoscaffolds. Nat Commun 2012; 3:614; PMID:22233624; http://dx.doi.org/10.1038/ncomms1629
- Flory JD, Shinde S, Lin S, Liu Y, Yan H, Ghirlanda G, Fromme P. PNA-peptide assembly in a 3D DNA nanocage at room temperature. J Am Chem Soc 2013; 135:6985-93; PMID:23521013; http://dx.doi.org/10.1021/ja400762c
- Flory JD, Simmons CR, Lin S, Johnson T, Andreoni A, Zook J, Ghirlanda G, Liu Y, Yan H, Fromme P. Low temperature assembly of functional 3D DNA-PNA-protein complexes. J Am Chem Soc 2014; 136:8283-95; PMID:24871902; http://dx.doi.org/10.1021/ja501228c
- Sahu B, Sacui I, Rapireddy S, Zanotti KJ, Bahal R, Armitage BA, Ly DH. Synthesis and characterization of conformationally preorganized, (R)-diethylene glycol-containing γ-peptide nucleic acids with superior hybridization properties and water solubility. J Org Chem 2011; 76:5614-27; PMID:21619025; http://dx.doi.org/10.1021/jo200482d
- Englund EA, Appella DH. Synthesis of gamma-substituted peptide nucleic acids: a new place to attach fluorophores without affecting DNA binding. Org Lett 2005; 7:3465-7; PMID:16048318; http://dx.doi.org/10.1021/ol051143z
- Englund EA, Appella DH. Gamma-substituted peptide nucleic acids constructed from L-lysine are a versatile scaffold for multifunctional display. Angew Chemie Int Ed 2007; 46:1414-8; PMID:17133633; http://dx.doi.org/10.1002/anie.200603483
- Metzler-Nolte N. Biomedical applications of organometal–peptide conjugates. In: Jaouen G, Metzler-Nolte N, editors. Medicinal organometallic chemistry. Berlin, Heidelberg: Springer Berlin Heidelberg; 2010. page 195-217.
- Giesen U, Kleider W, Berding C, Geiger A, Ørum H, Nielsen PE. A formula for thermal stability (Tm) prediction of PNA/DNA duplexes. Nucleic Acids Res 1998; 26:5004-6; PMID:9776766; http://dx.doi.org/10.1093/nar/26.21.5004
- Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang P V, Miller IA, Lo A, Codelli JA, Bertozzi CR. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci U S A 2007; 104:16793-7; PMID:17942682; http://dx.doi.org/10.1073/pnas.0707090104
- Campbell-Verduyn LS, Mirfeizi L, Schoonen AK, Dierckx R a, Elsinga PH, Feringa BL. Strain-promoted copper-free “click” chemistry for 18F radiolabeling of bombesin. Angew Chem Int Ed Engl 2011; 50:11117-20; PMID:21956935
- Becer CR, Hoogenboom R, Schubert US. Click chemistry beyond metal-catalyzed cycloaddition. Angew Chem Int Ed Engl 2009; 48:4900-8; PMID:19475588; http://dx.doi.org/10.1002/anie.200900755
- Armitage B. Cyanine dye–DNA interactions: intercalation, groove binding, and aggregation. In: Waring MJ, Chaires JB, editors. DNA binders and related subjects. Berlin Heidelberg: Springer; 2005. page 55-76.
- Smith J, Olson D, Armitage B. Molecular recognition of PNA-containing hybrids: spontaneous assembly of helical cyanine dye aggregates on PNA templates. J Am Chem Soc 1999; 121:2686-95; http://dx.doi.org/10.1021/ja9837553
- Dodd DW, Hudson RHE. Analysis and purification of peptide nucleic acids by denaturing PAGE. Electrophoresis 2007; 28:3884-9; PMID:17922504; http://dx.doi.org/10.1002/elps.200700192
- Mujumdar RB, Ernst LA, Mujumdar SR, Lewis CJ, Waggoner AS. Cyanine dye labeling reagents: sulfoindocyanine succinimidyl esters. Bioconjug Chem 1993; 4:105-11; PMID:7873641; http://dx.doi.org/10.1021/bc00020a001
- Sanborn ME, Connolly BK, Gurunathan K, Levitus M. Fluorescence properties and photophysics of the sulfoindocyanine Cy3 linked covalently to DNA. J Phys Chem B 2007; 111:11064-74; PMID:17718469; http://dx.doi.org/10.1021/jp072912u
- Iqbal A, Arslan S, Okumus B, Wilson TJ, Giraud G, Norman DG, Ha T, Lilley DMJ. Orientation dependence in fluorescent energy transfer between Cy3 and Cy5 terminally attached to double-stranded nucleic acids. Proc Natl Acad Sci U S A 2008; 105:11176-81; PMID:18676615; http://dx.doi.org/10.1073/pnas.0801707105
- Widengren J, Schwille P. Characterization of photoinduced isomerization and back-isomerization of the cyanine dye Cy5 by fluorescence correlation spectroscopy. J Phys Chem A 2000; 104:6416-28; http://dx.doi.org/10.1021/jp000059s
- Goodman RP, Schaap IAT, Tardin CF, Erben CM, Berry RM, Schmidt CF, Turberfield AJ. Rapid chiral assembly of rigid DNA building blocks for molecular nanofabrication. Science 2005; 310:1661-5; PMID:16339440; http://dx.doi.org/10.1126/science.1120367