
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Herein, we describe the synthesis and spectroscopic properties of five novel pyrrolodeoxycytidine analogs, and the related 5-(1-pyrenylethynyl)-2’-deoxycytidine analog; as well as fluorescence characterization of 5-(p-methoxyphenylethynyl)-2’-deoxyuridine. Within this series of compounds, rigidification of the structure from 6-phenylpyrrolodeoxycytidine to 5,6-benzopyrroldeoxycytidine made remarkable improvement of the fluorescence quantum yield (Φ ~1, EtOH) and substantially increased the Stokes shift. Exchange of the phenyl group of 6-phenylpyrrolodeoxycytidine for other heterocycles (benzofuryl or indolyl) produced an increase in the extinction coefficient at the excitation wavelength while preserving high quantum yields. The steady-state fluorescence response to the environment was determined by sensitivity of Stokes shift to solvent polarity. The effect of solvent polarity on fluorescence emission intensity was concurrently examined and showed that 5,6-benzopyrrolodeoxycytidine is highly sensitive to the presence of water. On the other hand, the previously synthesized 5-(p-methoxyphenylethynyl)-2’-deoxyuridine was found to be sensitive to solvent viscosity indicating molecular rotor behavior.
Introduction
The design and synthesis of novel fluorescent probes capable of incorporation into DNA has experienced tremendous growth within the last two decades.Citation1-Citation5 Naturally occurring nucleobases are virtually non-fluorescent under ambient conditionsCitation6 and thus require chemical modification to be endowed with desirable fluorescence properties. Such fluorescent properties may come from attachment of a pendent fluorophore (e.g. pyrene, anthracene).Citation2 Well-established synthetic protocols based on transition metal catalyzed cross-coupling reactionsCitation7 are oftentimes used for this purpose. Unfortunately, the introduction of a fluorophore extrinsic to the nucleobase often results in constructs possessing fluorescence properties insensitive to the change in the microenvironment of the nucleobase itself.Citation1-Citation3 Taking this into consideration, the development of fluorescent nucleobase analogs that function as reporters of microenvironmental change is of great interest.
An elegant solution to the problem outlined above would be the incorporation of fluorescent properties into the core of the nucleobase itself.Citation1-Citation3 Such structural modifications should afford nucleobase analogs capable of reporting duplex formation or mismatch discrimination, preferably via changes in Stokes shift or fluorescence intensity. The term intrinsic fluorophoresCitation1 has been coined to describe this group of nucleobase analogs. One goal of the design of intrinsically fluorescent nucleobase analogs is to make only modest structural changes when compared with their natural counterparts.Citation1 Another crucial design feature is to retain the Watson-Crick hydrogen bonding face so that canonical base pairing is not disturbed. When both of these design criteria are met, the analog is referred to as an isomorphous nucleobase analog.Citation2 Such analogs are useful for probing nucleic acid structures, dynamics and interactions.Citation2 Despite the ever growing number of fluorescent nucleobase/nucleoside analogs reported, successful de novo design of nucleobase fluorophores is still very challenging and many analogs are produced by the familiar design-synthesize-evaluate (and repeat) paradigm.
As a result of a long standing interest in the development of intrinsically fluorescent nucleobases, we have investigated structural modifications of deoxycytidine and deoxyuridine.Citation1 It was found that the modest fluorescence associated with deoxypyrrolocytidine moietyCitation8-Citation11 (, 1a) may be dramatically improved upon substitution of the position 6 of the parent heterocycle 1 with various substituted phenyl groups.Citation12,Citation13 Among them 6-phenylpyrrolocytidineCitation14 (, 1b) and some of its derivatives have been particularly well studied.Citation15,Citation16 These modified nucleobases were found to stabilize the DNA (RNA)-PNA duplexesCitation16 and the fluorescence associated with them was found to be responsive to match duplex formation.Citation15
Figure 1. Nucleoside analogs 1a–e, 2a, 2b, 3a, and 3b.
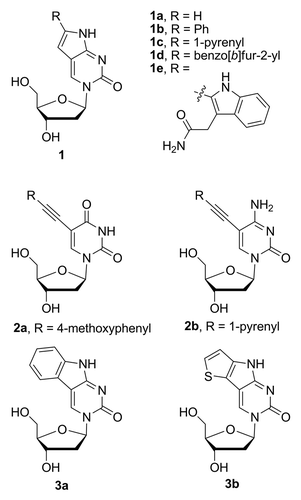
To further investigate the effect of substitution at position 6 on the fluorescence properties of pyrrolocytidine, we have prepared several analogs including the incorporation of the large hydrocarbon-based fluorophore pyrene (, 1c) and the heteroaromatics benzo[b]furan (, 1d) and indole (, 1e). Benzo[b]furan-modified nucleobases were shown to possess interesting fluorescence properties,Citation17 while tryptophan residues within proteins and other indole derivatives are known to exhibit strong solvatochromism.Citation18-Citation20 It is also worth noting, that another heteroaromatic 6-pyrrolocytosine nucleobase analog modified with thiophene (, 1f) has been recently prepared and studied in Tor’s laboratory.Citation21
In order to investigate the role of the rotatable biaryl bond at the site of substitution on the fluorescence properties, we have prepared the 5,6-benzo-fused pyrrolocytidine analog (, 1a) to compare with the properties of 3b. A related thienyl-fused nucleobase analog (, 3b) has recently been reported by Tor.Citation21 Much earlier, Matteucci and von Krosigk synthesized 3a, via a different synthetic route; however, its fluorescent properties were not reported.Citation22
In related studies, we have prepared the uncyclized C5-substituted 1-pyrenylethynyl-2’-deoxycytidine analog 2b to compare with the pyrrolocytidine analog 1c (). This analog has been prepared previously by semi-automated synthetic strategy using on-resin Sonogashira- cross-coupling,Citation23 but has not been characterized as the nucleoside. We have also included new characterization of the 4-methoxyphenylethynyl-substituted deoxyuridine analog 2a () which has been investigated previously and found to possess useful fluorescence properties as a hybridization probe.Citation24
Overall, we report the synthesis and spectral characterization of five pyrrolocytidine deoxynucleosides and a C5-ethynyl derivative of deoxycytidine (2b) and of deoxyuridine (2a). In particular, we have examined the effects of solvent polarityCitation25 and solvent viscosityCitation26 on the fluorescence associated these analogs.
Results and Discussion
Chemistry
The nucleoside analogs 1b–e () were obtained via a one-pot reaction cascade involving a Sonogashira cross-coupling between an appropriately hydroxyl-protected 5-iodo-2’-deoxycytidine and terminal alkyne followed by an intramolecular 5-endo-dig cyclization.Citation12 As previously reported, when modified reaction conditions were used, the products of the Sonogashira cross-coupling alone (, 2a and b) were produced in high yields.Citation13
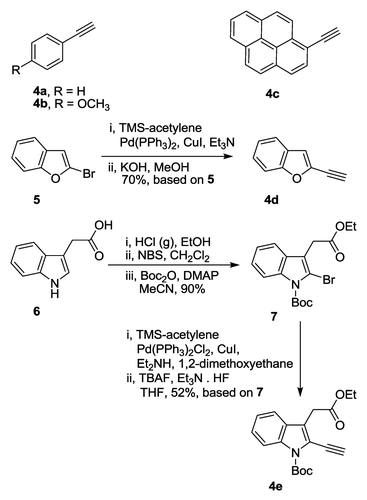
The structures and preparation of the required alkynes are shown in Scheme 1. Phenylacetylene (4a), 4-methoxyphenylacetylene (4b), and 1-ethynylpyrene (4c) were commercially available, while 2-ethynylbenzo[b]furan (4d) and ethyl 2-ethynyl-1-Boc-indole-3-yl acetate (4e) were synthesized. A slight modification of the recently described protocolCitation27 starting with Sonogashira cross-coupling of 2-bromobenzo[b]furan (5) with trimethylsilylacetylene (TMS-acetylene), followed by the removal of the TMS protecting group under basic conditions furnished the alkyne 4d in 70% overall yield (Scheme 1). Indole-3-acetic acid (6) was esterified,Citation28 followed by the bromination at the 2 positionCitation29 and Boc-protection of indole nitrogen. Ethyl 1-Boc-2-bromoindole-3-yl acetate (Scheme 1, 7) was obtained in 90% yield, based on ethyl 2-bromoindole-3-yl acetate. Subsequent Sonogashira cross-coupling with TMS-acetylene followed by the removal of TMS protecting groupCitation30 afforded the desired alkyne 4e in 52% based on 7 (Scheme 1). Procedures describing the preparation of alkynes 4d and 4e can be found in Materials and Methods section.
Scheme 1. Chemical structures and syntheses of alkynes 4a–e.
With alkynes 4a–e in hand, the protected 5-iododeoxycytidine was subjected to Sonogashira cross-coupling (Scheme 2). The products of cyclization were obtained in serviceable yields (50–75%, based on 8a–c) after purification by flash column chromatography (FCC). The acetyl groups in 1b–d were removed in good yields (>75%) by treatment with K2CO3 in alcoholic solvents (MeOH or EtOH), and the products were purified once again by FCC. To obtain the desired indole-modified pyrrolocytidine analog 1e, the product of cyclization was first treated with saturated solution of NH3 in EtOH to effect amidation of the ethyl ester, followed by the removal of protecting tert-butyldimethylsilyl (TBDMS) groups by treatment with Et3N · 3HF in THF thus providing 1e in 68% yield (Scheme 2) after FCC purification.
Scheme 2. Syntheses of nucleobase analogs 1b–e and 2b.
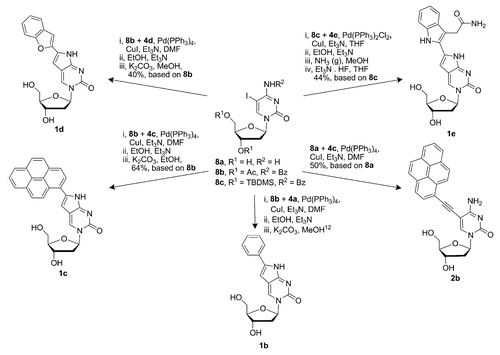
When 5-iododeoxycytidineCitation31,Citation32 (8a) and 1-ethynylpyrene (4c) were subjected to the conditions of Sonogashira cross-coupling, the deoxycytidine analog 2b was obtained in 50% yield (Scheme 2). The 4-methoxyphenyl-modified deoxyuridine analog 2a was obtained in the same manner from deoxyuridine (9) and 4-methoxyphenylacetylene (4b) as described previously by our groupCitation24 (Scheme 3).
Scheme 3. Synthesis of the nucleobase analogs 2a and 3a.
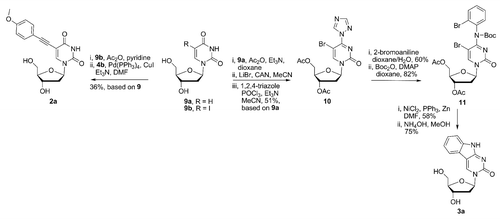
The synthesis of 5,6-benzo-fused cytidine analog 3a was performed similarly as for the related PNA monomer.Citation33 Deoxyuridine (9) was protected by acetylation of the hydroxyl groups, followed by bromination using a mixture of LiBr and cerium ammonium nitrate (CAN).Citation34 Subsequent reaction of the crude intermediate with POCl3 and 1,2,4-triazoleCitation33 afforded the deoxycytidine analog 10 (Scheme 3) in 51% overall based on 9, after chromatographic purification. With intermediate 10 in hand, substitution at C4 with 2-bromoaniline was performed in wet dioxane. Although this reaction is well known, and usually performed under anhydrous conditions, we found that without some small amount of water present the reaction did not proceed well. After installation of the N4 group, the exocyclic nitrogen was masked with a Boc groupCitation33 which provided the cyclization substrate 11 (49% overall yield, based on 10, Scheme 3). The key step in the synthesis of the 5,6-benzo-fused cytosine analog 3a was the reductive Ni-mediated cyclization,Citation35 performed in the same manner as described recently.Citation33 The protected 5,6-benzo-fused cytosine analog was obtained in 58% yield after chromatographic purification. It is interesting to point out, that the current synthetic yield represents ca. 2-fold improvement, compared with the synthesis of related 5,6-benzo-fused cytosine PNA monomer.Citation33 The removal of acetyl groups was effected by treatment with NH4OH in MeOH, affording the desired monomer 3a in 75% yield. The synthesis of the heterocyclic skeleton (pyrimido[4,5-b]indole) present in 3a has been achieved previously in 6 steps and 15% overall yield starting from 5-iodo-deoxyuridine.Citation22 Stille coupling of an appropriately substituted organotin moiety containing aniline with 5-iododeoxyuridine and subsequent DBU-mediated cyclization of the resulting heterobiaryl intermediate were the key steps in the previously described synthesis.Citation22 The current synthesis of 3a provided the desired product in 11% overall yield over 7 steps. Although proceeding in a marginally lower yield, our synthesis has the advantages of avoiding toxic organotin reagents and only involves one protection/deprotection step whereas two are involved in the Matteucci synthesis.Citation22 Moreover, intermediates 10 and 11 can also be utilized to prepare the nucleosides bearing various unusual heterocyclic skeletons, as described recently.Citation33
Fluorescence properties
The spectral properties of compounds 1b–e, 2a, 2b, and 3a have been studied in detail and are reported in . UV-Vis and fluorescence spectra were measured in EtOH, dioxane and water, in order to evaluate the influence of the polarity of the medium. Likewise, fluorescence quantum yields (Φf) and molar extinction coefficients (ε) have been determined for all of the compounds in each solvent, except for the molar extinction coefficients of 1c and 2b in pure water due to insufficient solubility.
Table 1. Fluorescence properties of nucleobase analogs 1b-1e, 2a, 2b, and 3a
In general, with comparison to the parent cytidine analog 1b previously studied in our laboratory,Citation12 substitution at position 6 of pyrrolodeoxycytidine with larger aromatic groups has a positive effect on both the quantum yield and extinction coefficients in organic solvents. Whereas the quantum yield of 6-phenylpyrrolodeoxycytidine is good in organic solvent (Φf = 0.28, dioxane), its ε value at the longest wavelength absorption band used for excitation is relatively small. The increased conjugation provided by the larger aromatic substituents makes a greater impact on the ε value and correspondingly leads to greater fluorophore brightness (defined as: ε × Φf). Groups larger than phenyl (i.e., pyrenyl, benzo[b]furanyl, indolyl) also produce a bathochromic shift of the lowest energy absorption band. However, the absorption shift is relatively small (≤20 nm in all solvents examined) and results in the fluorophore excitation wavelength being in the near UV and the emission being in the visible blue region. All of the pyrrolcytidine analogs showed a response to the nature of the solvent with greater Stokes shift for the more polar solvent (H2O) vs. ethanol or dioxane. The intensity of emission varied for the pyrrolcytidine analogs within the dioxane, ethanol, and water series examined, but not in a uniform manner. Whereas 6-phenylpyrrolodeoxycytidine 1b showed near constant quantum yield in dioxane and water, the benzo[b]furanyl analog 1d displayed an increased quantum yield while the 1-pyrenyl (1c) and indolyl (1e) exhibited dramatic quenching (). Although it was known that incorporation of 1b into oligonucleotides yield constructs that maintain fluorescence in aqueous media,Citation14,Citation36 these initial results indicate that nucleoside analog 1d may behave in a similar manner, yet be more emissive. In order to examine the effect of the medium polarity on fluorescence emission, further studies were undertaken, vide infra (see “Solvent polarity effects”).
The 6-pyrenyl, benzo[b]furanyl, and indolyl pyrrolocytidine analogs are new compounds and, due to the lack of comparators, will be examined in relation to similar 5-substituted cytidine compounds. Firstly, 5-(pyren-1-yl)deoxycytidine (PydC) has been prepared by Wagenknecht and used to study electron transport in DNA.Citation37,Citation38 PydC demonstrated steady-state fluorescence in acetonitrile (λexcit. = 340 nm) with emission maximum at approximately 380 nm and showing some vibronic structure. It was posited that this emission resulted from a locally excited state, i.e. on the pyrene moiety. In water, the fluorescence was dramatically quenched and this was ascribed to fast electron transfer to cytosine nucleobase followed by rapid protonation of the base radical. The analogous pyrenylpyrrolodeoxycytidine (1c) possesses a solvent dependent red-shifted absorbance (λexcit. = 377 - 396 nm) and emission (λemiss. = 457 - 485 nm) with a markedly larger Stokes shift and dramatic quenching in water vs. ethanol or dioxane. The reduction potential of pyrrolocytidine is not known; however, these results suggest that it is the electron acceptor from a photo-excited pyrene. Thusly, the large Stokes shift in water, nearly twice the magnitude as observed in ethanol or dioxane, argues for an excited state species with much greater polar character (or charge separated species) than the ground state. As well, the dramatic fluorescence quenching can be understood if the excited state was rapidly protonated, as for cytidine.
Next, 5-benzo[b]furanylcytidine has recently been reported as a member of the complete ribonucleoside family of benzo[b]furan conjugates.Citation39 Interestingly, this analog possessed rather large Stokes shifts in water, dioxane, methanol and acetonitrile; when excited at 320 nm and it displayed a broad emission centered near 450 nm for each solvent. This analog produced rather weak fluorescence (Φf < 0.06) in a variety of solvents except for glycerol where the quantum yield was improved to 0.127. The pyrrolodeoxycytidine analog 1d offers much improved fluorescence. In the three directly comparable solvents: dioxane, ethanol/methanol and water, 1d possesses quantum yields > 0.4 and a red-shifted absorbance (λ ~375 nm).
Finally, 5-indolylcytidine is unknown but a 5-tryptophanylcytidine derivative has been prepared by the photochemical reaction of 5-bromo-2’-deoxycytidine with N-acetyltryptophan ethyl amide.Citation40 While it was noted that this derivative was fluorescent in water (λexcit. = 350 nm; λemiss. = 445 nm), the quantum yield was not reported. The pyrrolocytidine analog 1e exhibits a bathochromic shift relative to the aforementioned analog (λexcit. = 380 nm; λemiss. = 489 nm) likely as a result of the pyrrolocytosine nucleus vs. the cytosine. Compound 1e shows fluorescence which is strongly dependent on the solvent. It has a high Φf in dioxane and ethanol, 0.32 and 0.48 respectively, while dramatic quenching (occurs in water (Φf < 0.01). The quenching may be a consequence of excited state electron transfer from the indole moiety to the base, similar to the proposed mechanism for the photoreaction reported,Citation40 followed by rapid protonation in analogy to the pyrene-conjugated nucleoside.
Rigidification of the heterocyclic skeleton present in analogs 1b–1e to eliminate the possibility of free rotation about the biaryl bond produces analog 3a. This strategy, i.e. fusion of rings to produce polycyclic heteroaromatic analogs of cytidine, has been previously employed for the preparation of fluorescent nucleosides. Notably, Matteucci’s examples of phenothiazine and phenoxazine derviativesCitation41 which were later described as highly emissive and environmentally insensitive fluorophores useful for fluorescence resonance energy transfer (FRET) based studies.Citation42,Citation43 Saito’s group has reported fluorescently responsive cytidine analogs based on the pyridopyrimidine scaffold which can discriminate between the different environments presented by an opposing nucleoside in the context of hybridized oligonucleotides and has potential use in single-nucleotide polymorphism detection.Citation44,Citation45 A structurally related example has been reported by Sekine for which geometrically locking a 4-N-carbamoyldeoxycytidine transformed the nonfluorescent precursor into a fluorescent analog that experienced quenching when paired to guanosine, but not adenosine, in the context of oligomers.Citation46
The proceeding examples are either bright but relatively insensitive to their environment or environmentally responsive but with low quantum yields (≤ 0.1) or small Stokes shift and peak emission in the UV which are not ideal properties. In the present case, nucleoside 3a possesses remarkably high fluorescence quantum yields and large Stokes shift in the organic solvents of EtOH (Φf > 0.99) and dioxane (Φf = 0.93), while near complete quenching of the fluorescence is observed in water (Φf < 0.01, ). An increase in brightness factor is also observed originating from a larger ε value and greater Φf when compared with 1b. These properties distinguish 3a from known examples except for the structurally similar thiophene-fused analog recently reported by Tor;Citation21 however, 3a shows greater polarity sensitivity, as described below (see “Solvent polarity effects”).
The non-cyclized 5-pyrenylethynyldeoxycytidine analog 2b has been previously prepared via cross-coupling to a 5-iodocytidine already incorporated into an oligonucleotide but not as the free nucleoside.Citation47 Spectral characterization of a duplex containing a single 5-pyrenylethynyldeoxycytidine insert was described as pyrene-like exhibiting vibronic structure in the emission (λemiss. = 408, 430 nm). It was concluded that there was weak electronic coupling between the pyrene moiety and cytosine base. Similarly, we note that the 5-pyrenylethynyldeoxycytidine analog 2b has much different fluorescence spectra than pyrenylpyrrolodeoxycytidine 1c (see Fig. S2–S4). Whereas the qualities of the spectra of 1c resemble other pdC analogs, i.e., a red-shifted absorption relative to the precursor components – in this case cytosine and ethynylpyrene, and a broad, featureless emission > 450 nm; the spectra of 2b is distinctly different retaining vibronic structure and lacking a large bathochromic shift. In our description of nucleobase fluorophores, 1c behaves as an integrated fluorophore and 2b behaves more like a pendant fluorophore.
Solvent polarity effects
Nucleoside probes that respond fluorimetrically to polarity find use in the interrogation of nucleic acid structures. Such responsive nucleosides can be deliberately designed, such as the 6-dimethylamino-2-acylnaphthalene-deoxyuridine conjugate synthesized by Saito and coworkers exploiting the known polarity responsiveness of the substituted naphthalene moiety.Citation48,Citation49 Alternatively, the property of polarity sensitivity may be discovered serendipitously when the primary goal is endowing a nucleoside with fluorescence. During the past few years, Tor and coworkers have established methods to characterize the polarity dependent fluorescence of nucleoside analogs. Recently, they have described the use of a simple binary solvent system (dioxane and water in varying proportions) in order to adjust the polarity of the medium while minimizing changes in other parameters, such as solvent viscosity.Citation21 The variation in Stokes shift is then plotted againt Reichardt’s microscopic solvent polarity parameter to quantify the polarity sensitivity.Citation50 In order to make direct comparisons with previously reported data, we have adopted the Tor method to evaluate the change in fluorescence with the respect to changes in medium polarity. The variation of Stokes shift and emission intensity as the solvent polarity was changed was determined for each of the nucleoside analogs 1b–1e, 2a, 2b, and 3a and is summarized in while the spectral data are presented in and and the Supplemental Materials.
Figure 2. Photophysical characterization of cytidine analogs. Absorbance in water (dashed black line) and dioxane (solid black line) and emission (100% dioxane, red; 90% dioxane/10% water, blue; 80% dioxane/20% water, pink; 70% dioxane/30% water, brown; 60% dioxane/40% water, light green; 40% dioxane/60% water, dark green; 20% dioxane/80% water, turquoise; 10% dioxane/90% water, orange; 100% water, purple). (A) 1b; (B) 1c; (C) 1d; (D) 1e; (E) 2b; (F) 3a.
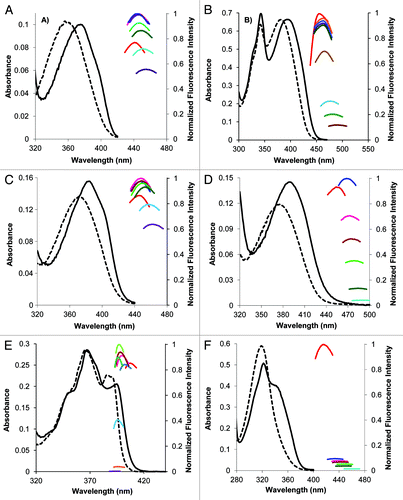
Figure 3. Photophysical characterization of uridine analog 2a. Top panel: fluorescence changes in response to changes in polarity. Absorbance in water (dashed black line) and dioxane (solid black line) and emission in 9:1 dioxane/water, blue; 8:2 dioxane/water, pink; 7:3 dioxane/water, brown; 6:4 dioxane/water, light green; 4:6 dioxane/water, dark green; 2:8 dioxane/water, turquoise; 1:9 dioxane/water, orange. Bottom panel: fluorescence response to changes in medium viscosity, absorbance in MeOH (solid black line) and emission in MeOH, blue; 7:3 MeOH/glycerol, pink; 1:1 MeOH/glycerol, orange; 4:6 MeOH/glycerol, turquoise; 2:8 MeOH/glycerol, purple; 15:85 MeOH/glycerol, brown; 1:9 MeOH/glycerol, green.
From the Stokes shifts dependence on the microenvironmental polarity parameter, the parent nucleoside analog 1b () displays moderate polarity sensitivity (62 cm−1/[kcal · mol−1], ) and is comparable to that associated with the benzofuranyl-containing analog 1d (). Nucleoside 1d not only possesses a much improved quantum yield over the previously reported 5-benzofuranylcytidine, the polarity sensitivity is also much larger 72 cm−1/(kcal mol−1) vs. ~26 cm−1/(kcal · mol−1) estimated from data presented by Srivatsan.Citation39
The indole-modified analog 1e possesses greater polarity sensitivity [93 cm−1/(kcal · mol−1)] likely reflecting the properties of the indole group (). The variation of tryptophan fluorescence with the environment is a known feature exploited in studying the protein conformation.Citation18,Citation19
The Stokes shifts of the pyrene-containing derivatives 1c () and 2b () are less sensitive to changes in the solvent polarity (), and show no evidence of aggregation or excimer formation. The lack of Stokes shift response to polarity change is not unexpected based on the well known insensitivity of pyrene itself. However, it is notable that the pyrenylpyrrolocytosine derivative 1c exhibits a weak positive effect consonant with the other pyrrolocytosine derivatives, and different than the pyrenylethynyl-dC 2b, which also supports the notion that 1c is an integrated fluorophore with substantial electronic communication between the pyrene and pC moieties.
The 5,6-benzo-fused analog 3a (105 cm−1/[kcal · mol−1], , ) possesses greater sensitivity of Stokes shift to solvent polarity than the unfused congener 1b. Interestingly, 3a is extremely sensitive to the presence of water; the addition of 10% water results in ca. 94% reduction in fluorescence intensity. This behavior is obviously different than the overall trend observed for analogs 1b-1e, 2a, and 2b which display a non-unidirectional effect on emission intensity with the increasing polarity, providing the maximum fluorescence emission in dioxane solutions containing ca. 10–40% of water. The extraordinary sensitivity to the presence of water may be useful property for examining nucleoside binding proteins/transporters or after the incorporation into oligonucleotides to report on ligand/protein/enzyme binding that excludes access of water to the fluorophore.
Solvent viscosity effects
It has been well documented that fluorescent nucleosides that feature a rotatable bond as part of the chromophore can display an increase of fluorescence intensity in the solvents with increased viscosity.Citation26 With the exception of analog 3a, all analogs described in this study possess a potentially rotatable C-C biaryl (or alkyne-aryl) bond as part of the fluorophore. We have, therefore also decided to investigate the relationship between the emission intensity and viscosity of the solvents.
Our studies began with examining the emission intensity of parent analog 6-phenylpyrrolodeoxycytidine 1b in mixtures of MeOH and glycerol that vary in viscosity. The emission intensity associated with 1b was found to be independent of solvent viscosity; therefore none of remaining pyrrolocytidine analogs 1c-1e were studied. However, it was observed previously that the phenylethynyldeoxyuridine 2a exhibited strong “turn on” of fluorescence in a duplex vs. single-stranded state. It was suspected that 2a may exhibit molecular rotor properties based on this observation of increased fluorescence in a more restricted environment (duplex) than one that would permit free rotation about the alkyne-aryl bonds (single strand). The nucleoside 2a has been evaluated as shown in .
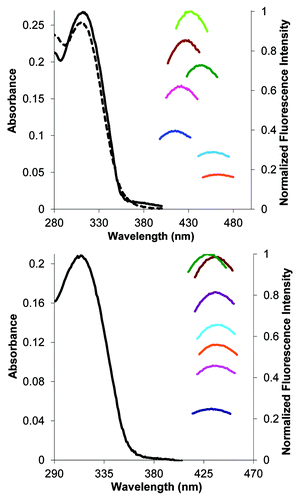
The phenylethynyldeoxyuridine 2a displays a substantial response to changes in solvent polarity (, top panel), possessing the highest value yet reported (measured under comparable conditions) for neutrally-charged nucleosides.Citation21,Citation51 It also displays a trend of increased fluorescence in solvents mixtures of higher viscosity, at the near constant polarity of methanol/gycerol solvent mixtures (, bottom panel), which indicates molecular rotor behavior.Citation26
In contrast, to the properties of 2a, the pyrenylethynyldeoxycytidine 2b showed no clear dependence on solvent viscosity (data not shown). This behavior can be understood in the following way. The phenylethynyluracil moiety is entirely part of the fluorophore and the rotatable bonds are integral to the fluorophore structure whereas the pyrene moiety is the fluorophore and the rotatable bonds are external to it, thus having little effect on the fluorescence. This notion is supported by the observation that the fluorescence emission of 2b shows vibronic structure not dissimilar to pyrene (Fig. S3) and is very similar to that reported for 1-phenylethynylpyrene.Citation52 In addition, the lack of response of the Stokes shift to changes in solvent polarity parallels pyrene itself which shows approximately constant emission wavelength but changes in relative intensities of the so-called I1 and I3 bands which form the basis of the Py scale.Citation53
Materials and Methods
General remarks
Reagents were commercially available unless otherwise stated and all solvents were reagent grade except for water (18.2 MΩ· cm millipore water). Dry solvents (dioxane, DMF) for chemical synthesis were obtained by drying on activated Al2O3 columns in a solvent purification system. Spectroscopic grade solvents (dioxane, DMSO, glycerol, EtOH, MeOH) have been used to perform spectroscopic studies. Solvents were removed under reduced pressure in a rotary evaporator and organic extracts were dried over Na2SO4. Reaction mixtures involving air sensitive reagents (Pd[PPh3]4, CuI) have been degassed by passing the stream of N2 gas into the reaction mixture cooled to –78 °C. The flask was then evacuated and the passing of N2 was repeated twice more. FCC was performed using silica gel (SiO2; mesh size 230–400 Å). Thin-layer chromatography (TLC) was performed on an Al backed silica gel plate with compounds visualized by I2 vapors, 5% ninhydrin stain, phosphomolybdic acid stain, and UV light. 1H and 13C NMR spectra were recorded on a 400 MHz spectrometer. Chemical shifts (δ) are reported in parts per million, and are referenced as follows: CDCl3 (7.26 ppm), DMSO-D6 (2.49 ppm) for 1H NMR and CDCl3 (77.0 ppm), DMSO-D6 (39.5 ppm) for 13C NMR (100 MHz). Mass spectra (MS) were obtained on a mass spectrometer using electron impact (EI), chemical ionisation (CI), or electrospray ionisation (ESI). UV-VIS spectra were acquired using an UV-VIS spectrofotometer. Steady-state fluorescence spectra were acquired using a fluorimeter. All fluorescence measurements were performed using a 1 cm wide four-sided quartz cuvette. UV-VIS measurements were performed using a 1 cm two-sided quartz glass cuvette.
Synthesis of 2-ethynylbenzo[b]furan (4d)
2-Bromobenzo[b]furan (5591 mg, 3 mmol) was dissolved in Et3N (10 mL), followed by the addition of Pd(PPh3)2Cl2 (42 mg, 0.06 mmol) and CuI (6 mg, 0.03 mmol). The mixture was degassed followed by the addition of TMS-acetylene (640 μL, 4.5 mmol). The mixture was then stirred (in the atmosphere of N2) for 2 h at 60 °C, was cooled to room temperature (RT), the solid residue formed during the reaction was filtered off with suction and the filter was washed with hexanes. The filtrate was concentrated; the residue was dissolved in MeOH (6 mL) containing KOH (168 mg, 3 mmol). The mixture was stirred for 30 min at RT, the solvent was evaporated and the residue was subjected to FCC on 50 g SiO2 eluted with light petroleum ether. The eluate was concentrated to afford 2-ethynylbenzo[b]furan (4d, 300 mg, 70%, based on 5) as colorless oil. The spectral data for 4d were in agreement with those previously reported.Citation27
Synthesis of indole containing alkyne 4e
Ethyl 1-Boc-2-bromoindole-3-yl acetate (7) was prepared from 2-bromoindole-3-yl acetic acid (6) according to literature procedures.Citation28,Citation29 Subsequently the alkyne functionality was installed in two steps. Ethyl 1-Boc-2-bromoindole-3-yl acetate (7, 3.8 g, 10 mmol) and TMS-acetylene (4.1 mL, 29.5 mmol) were mixed with diethylamine (6 mL) and dimethoxyethane (6 mL). The mixture was degassed followed by the addition of Pd(PPh3)2Cl2 (350 mg, 0.5 mmol) and CuI (300 mg, 1.5 mmol). The reaction mixture was then stirred (in the atmosphere of N2) for 12 h at 55 °C. The solvent was evaporated and the residue was subjected to FCC on 100 g SiO2 eluted with hexanes/EtOAc (98:2) to afford ethyl 1-Boc-2-(2-TMS-ethynyl)indole-3-yl acetate (2.6 g, 65%). 1H NMR (CDCl3): δ = 7.92 (d, 1H, J = 8.6), 7.23 (d, 1H, J = 7.8), 7.07 (t, 1H, J = 7.8), 6.96 (m, 1H), 3.87 (q, 2H, J = 7.0), 3.54 (s, 2H), 1.40 (s, 9H), 0.97 (t, 3H, J = 7.0), 0.0 (s, 9H). 13C NMR (CDCl3): δ = 169.8, 149.0, 135.5, 128.1, 125.8, 122.9, 122.7, 118.9, 115.4, 104.4, 95.8, 84.1, 60.6, 31.0, 27.9, 13.96, –0.33. HRMS (EI): calcd. for C22H29NO4Si [M+] 399.1866, found 399.1873.
Ethyl 1-Boc-2-(2-TMS-ethynyl)indole-3-yl acetate (1.6 g, 4 mmol) was dissolved in THF (6 mL) and the solution was cooled to -10 °C. Et3N · HF (0.6g, 16 mmol) was added to the stirred mixture followed by a dropwise addition (over the period of 15 min) of 1 M solution of TBAF in THF (8 mL, 8 mmol). The mixture was allowed to warm up to 0 °C and was stirred for 1 h at 0 °C. The precipitate formed during the reaction was filtered off using a short plug of SiO2, the filter was washed with hexanes/EtOAc (1:1) and the filtrate was concentrated. The residue was then subjected to FCC on 50 g SiO2, hexanes/EtOAc (98:2) to afford ethyl 1-Boc-2-ethynylindole-3-yl acetate (4e, 1.06 g, 80%). 1H NMR (CDCl3): δ = 8.12 (d, 1H, J = 7.8), 7.49 (d, 1H, J = 7.4), 7.33 (t, 1H, J = 7.4), 7.23 (t, 1H, J = 8.0), 4.12 (q, 2H, J = 7.0), 3.83 (s, 2H), 3.64 (s, 1H), 1.66 (s, 9H), 1.20 (t, 3H, J = 7.0). 13C NMR (CDCl3): δ = 169.6, 148.8, 135.2, 127.8, 125.6, 127.7, 122.4, 118.9, 118.0, 115.2, 86.7, 84.1, 74.6, 60.5, 30.6, 27.6, 13.7. HRMS (EI): calcd. for C19H21NO4 [M+] 327.1471, found 327.1479.
Spectral data associated with all of the intermediates were in agreement with those published previously,Citation30 see supplemental information.
Synthesis of N4-benzoyl-5′-iodo-5′,3′-di-O-Ac-deoxycytidine (8b)
Deoxycytidine (1.06 g, 4.67 mmol) was dissolved in AcOH (10mL, sonication required), followed by the addition of CHCl3 (10 mL) and acetyl chloride (4 mL). The mixture was then stirred for 18 h at RT; was cooled to 0 °C and excess acetyl chloride was quenched by the addition of MeOH (4 mL). The solvents were evaporated, the residue was coevaporated with toluene (ca. 100 mL), followed by dissolving the residue in saturated NaHCO3 solution (50 mL). The product was extracted with 8 × 50 mL of CH2Cl2/MeOH (95:5), combined organic extract was dried and was concentrated to leave 5′,3′-di-O-Ac-deoxycytidine (1.44 g, 99%) as colorless oil of sufficient purity to be used in the next step. 1H NMR (DMSO-d6): δ = 9.58 (br,s, 1H), 8.62 (br,s,1H), 7.92 (d, J1 = 7.82 Hz, 1H), 6.16 (m, 1H), 6.09 (t, J1 = 6.84 Hz, 1H), 5.18 (m, 1H), 4.23 (br,s,3H), 2.41 (m, 2H), 2.06 (s, 3H), 2.04 (s, 3H). 13C NMR (DMSO-d6): δ = 170.19, 170.06, 160.66, 148.30, 143.78, 94.47, 86.11, 81.88, 73.83, 63.57, 36.58, 20.78, 20.63. HRMS (ESI) m/z calcd. for C13H18N3O6 [MH+] 312.1196, found 312.1198.
Iodine (698 mg, 2.75 mmol) and iodic acid (418 mg, 2.38 mmol) were added to a vigorously stirred mixture containing 5′,3′-di-O-Ac-deoxycytidine (1.44 g, 4.63 mmol) in AcOH (10.5 mL), CCl4 (7 mL) and water (7 mL). Resulting mixture was stirred vigorously for 24 h at 40 °C, the volatiles were evaporated and the residue was partitioned between saturated NaHCO3 solution (50 mL) and EtOAc (50 + 2 × 30 mL). Combined organic extract was dried, was concentrated and was subjected to FCC on 60 g SiO2 eluted with CH2Cl2/MeOH (98:2) later replaced with CH2Cl2/MeOH (95:5). Evaporation of the eluate afforded 5-iodo-5′,3′-di-O-Ac-deoxycytidine (1.12 g, 55%) as pale yellow oil. 1H NMR (DMSO-d6): δ = 9.14 (br, s, 1H), 8.47 (br, s, 1H), 8.17 (s, 1H), 6.06 (t, J1 = 6.84 Hz, 1H), 5.18 (m, 1H), 4.26 (m, 3H), 2.47 (m, 1H), 2.37 (m, 1H), 2.09 (s, 1H), 2.06 (s, 1H). 13C NMR (DMSO-d6): δ = 170.06, 170.01, 161.34, 149.97, 148.52, 86.18, 81.91, 73.81, 63.51, 57.31, 48.61, 36.76, 20.78. LRMS (EI) m/z calcd. for C13H16IN3O6 [M+] 437.0084, found 437.0084.
Benzoic anhydride (748 mg, 3.31 mmol) was added to a solution of 5-iodo-5′,3′-di-O-Ac-deoxycytidine (1.11 g, 2.54 mmol) in dry pyridine (8 mL). The mixture was stirred for 3 h at 85 °C (N2 atmosphere). Pyridine was removed by coevaporation with toluene (3 × 100 mL), the residue was then partitioned between saturated aq. NaHCO3 (50 mL) and EtOAc (50 + 30 mL). Combined organic extract was dried, was concentrated and was subjected to FCC on 40 g SiO2 eluted with hexanes/acetone (2:1). The eluate was concentrated to ca. one quarter of its original volume (crystallization of the product was visible) and was set aside for 1 h at –10 °C. Separated crystals were filtered off, were washed with hexanes and were dried to leave N4-benzoyl-5′-iodo-5′,3′-di-O-Ac-deoxycytidine 8b (883 mg, 64%) as colorless crystals. 1H NMR (DMSO-D6): δ 12.86 (s, D2O exch., 1H); 8.22 (m, 3H); 7.63 (m, 1H); 7.52 (m, 2H); 6.12 (t, J = 6.5 Hz, 1H); 5.21 (m, 1H); 4.28 (m, 3H); 2.54 (m, 1H); 2.39 (m, 1H); 2.13 (s, 3H); 2.07 (s, 3H). 13C NMR (DMSO-D6): δ 178.1, 170.1, 170.0, 156.2, 147.2, 146.5, 136.2, 132.9, 129.5, 128.4, 86.0, 82.0, 73.8, 70.0, 63.5, 36.5, 20.8 (2 × C). HRMS (ESI) m/z: found 542.0404 [M+H]+ (542.0424 calcd. for C20H21IN3O7).
Synthesis of N4-benzoyl-5′-iodo-5′,3′-di-O-TBDMS-deoxycytidine (8c)
Benzoic anhydride (4.1 g, 18 mmol) was added to a suspension of crude 5′-iodo-5′,3′-di-O-TBDMS-deoxycytidine (prepared from 5.5 mmol of 5′-iododeoxycytidine according to the literature procedureCitation44) in MeCN (25 mL) followed by the addition of DMAP (20 mg, 0.17 mmol). Resulting mixture was stirred (atmosphere of N2) for 8 h at 70 °C. The solvent was evaporated; the residue was subjected to FCC on 100 g SiO2, eluted with CH2Cl2/MeOH (98:2) later replaced with CH2Cl2/MeOH (95:5) to afford N4-benzoyl-5′-iodo-5′,3′-di-O-TBDMS-deoxycytidine (8c, 3.2 g, 85%). 1H NMR (CDCl3): δ 13.25 (s, D2O exch., 1H); 8.38 (m, 2H); 8.25 (s, 1 H); 7.55 (m, 1H); 7.47 (d, 2 H); 6.29 (dd, J = 8.0, 5.5 Hz, 1H); 4.41 (m, 1H); 4.03 (dd, J = 4.5, 2.0 Hz, 1H); 3.93 (dd, J = 11.5, 2.5 Hz, 1H); 3.79 (dd, J = 11.5, 2.5 Hz, 1H), 2.39 (ddd, J = 13.0, 8.0, 2.0 Hz, 1H); 2.02 (ddd, J = 13.5, 8.0, 6.0 Hz, 1H); 0.96 (s, 9H); 0.90 (s, 9H); 0.18 (s, 3H); 0.17 (s, 3H); 0.10 (s, 3H); 0.09 (s, 3H). 13C NMR (CDCl3): δ 179.7, 156.9, 147.4, 145.6, 136.6, 132.8, 130.2, 128.2, 88.7, 86.4, 72.6, 69.3, 63.0, 42.2, 26.2, 25.7, 18.5, 18.0, –4.7, –4.9, –5.1, –5.3. HRMS (ESI) m/z: found 708.1786 [M+Na]+ (708.1762 calcd. for C28H44IN3O5Si2Na).
General protocol for the tandem Sonogashira cross-coupling/5-endo-dig cyclization cascade
Compound 1b was prepared as described in the literature.Citation12
A round bottom flask containing N4-benzoyl-5-iodo-5′,3′-di-O-Ac-deoxycytidine (8b, 420 mg, 0.78 mmol) and corresponding alkynes as follows: 2-ethynylpyrene (4c, 265 mg, 1.17 mmol); 2-ethynylbenzo[b]furan (4d, 197 mg, 1.39 mmol) and ethyl 1-Boc-2-ethynylindole-3-yl acetate (4e, 457 mg, 1.4 mmol) was charged with N2 and dry DMF (3 mL, alkynes 4c and 4d) or dry THF (2.5 mL, alkyne 4e) was added. The mixtures were degassed followed by the addition of Pd(PPh3)4 (alkynes 4c and 4d, 90 mg, 0.08 mmol) or Pd(PPh3)2Cl2 (alkyne 4e, 56 mg, 0.08 mmol) and CuI (30 mg, 0.16 mmol). The mixtures were degassed again, Et3N (1.08 mL, 7.76 mmol) was added and the mixtures were stirred (in the dark, under N2 atmosphere) for 18 h at 50 °C (alkyne 4c), for 8 h at rt and 18 h at 60 °C (alkyne 4d) and for 24 h at 50 °C (alkyne 4e). EtOH and Et3N were added (2 mL each) and the stirring continued for 18 h at 50 °C (alkyne 4c), 18 h at 80 °C (alkyne 4d) and for 18 h at 55 °C (alkyne 4e). The mixtures were cooled to RT, were diluted with 4% EDTA solution (50 mL) and were extracted with CH2Cl2 (2 × 25 mL, alkyne 4c), or EtOAc (2 × 30 + 20 mL, alkyne 4d). Combined organic extracts were washed with brine (3 × 50 mL), were dried and were concentrated. The residues were subjected for FCC purification on 50 g SiO2 eluted with toluene later replaced with toluene/MeOH (98:2, alkyne 4c) or 30 g SiO2 eluted with Et2O/acetone (3:1) later replaced with Et2O/acetone (2:1, alkyne 4d). In the case of alkyne 4e no aqueous extractive workup was performed, the solvent was evaporated and the residue was subjected for FCC on 60 g SiO2 eluted with CH2Cl2/MeOH (98:2) later replaced with CH2Cl2/MeOH (95:5). Evaporation of the eluates afforded the desired products derived from alkynes 4d and 4e of sufficient purity. In the case of product derived from the alkyne 4c, the impurities were removed by adding the solution of the cyclization product in CH2Cl2 (ca. 2 mL) into hexanes (ca. 20 mL). The precipitate was separated by filtration, washed with hexanes and was dried to afford 6-(2-pyrenyl)-pyrrolo-5′,3′-di-O-Ac-deoxycytosine (296 mg, 75%) as yellow solid. 1H NMR (DMSO-D6): δ 12.02 (s, D2O exch., 1H); 8.62 (s, 1H); 8.52 (m, 1H); 8.36 (m, 3H); 8.25 (m, 4H); 8.13 (m, 1H); 6.74 (s, 1H); 6.38 (t, J = 6.5 Hz, 1H); 5.27 (d, J = 6.5 Hz, 1H), 4.37 (m, 3H), 2.62 (m, 1H), 2.42 (m, 1H), 2.11 (s, 3H), 2.09 (s, 3H). 13C NMR (DMSO-D6): δ 170.3, 170.1, 159.9, 153.9, 138.8, 136.5, 130.9 (2 × C), 130.4, 128.3, 128.1, 128.0, 127.3, 127.1, 126.6, 126.3, 125.8, 125.4, 124.9, 124.3, 124.2, 123.8, 109.7, 102.0, 87.4, 82.1, 74.2, 63.8, 38.0, 20.8, 20.7. HRMS (ESI) m/z found 536.1816 [M+H]+ (calcd. 536.1822 for C31H26N3O6).
6-(2-Benzo[b]furan-2-yl)-pyrrolo-5′,3′-di-O-Ac-deoxycytosine (186 mg, 53%), yellow solid. 1H NMR (DMSO-D6) δ 11.87 (s, D2O exch., 1H); 8.57 (s, 1H); 7.71 (d, J = 8 Hz, 1H); 7.63 (d, J = 8 Hz, 1H); 7.35 (m, 2H); 7.29 (dd, J = 8, 8 Hz, 1H); 6.79 (s, 1H); 6.29 (t, J = 6.5 Hz, 1H); 5.23 (m, 1H); 4.34 (m, 3H), 2.73 (m, 1H); 2.38 (m, 1H); 2.09 (s, 3H); 2.06 (s, 3H). HRMS (ESI) m/z: found 452.1468 [M + H]+ (calcd. 452.1458 for C23H22N3O7).
Ethyl 6-[(1-Boc-indole-3-yl acetate)-2-yl]-pyrrolo-5′,3′-di-O-TBDMS-deoxycytosine (394 mg, 64%), yellow solid. 1H NMR (CDCl3): δ 9.42 (br s, 1 H); 8.77 (s, 1H); 8.21 (d, J = 8.0, 1H); 7.58 (d, J = 8.0, 1H); 7.39 (dd, J = 8.0, 8.0, 1H); 7.29 (dd, J = 8.0, 8.0, 1H); 6.42 (s, 1H); 6.39 (m, 1H); 4.41 (dd, J = 11.5, 6.0, 1H); 4.17 (q, J = 7.5, 2H);, 4.03 (m, 1H); 4.00 (m, 1H); 3.85 (m, 1H); 3.68 (s, 2H); 2.61 (m, 1H); 2.21 (m, 1H); 1.42 (s, 9H); 1.26 (t, J = 7.0, 1H); 0.95 (s, 9H); 0.89 (s, 9H); 0.17 (s, 3H); 0.14 (s, 3H); 0.07 (2 × s, 6H). 13C NMR (CDCl3): δ 171.1, 158.5, 154.5, 149.5, 136.6, 136.3, 131.0, 128.8, 127.9, 125.7, 123.1, 119.3, 117.1, 115.7, 108.6, 102.4, 87.7, 84.2, 69.8, 61.7, 61.3, 42.6, 31.2, 27.8, 26.0, 25.7, 18.4, 17.9, 14.2, -4.5, -5.0, -5.4 (2 × C). HRMS (ESI) m/z: found 803.3860 [M+Na]+ (calcd. 803.3847 for C40H60N4O8Si2Na).
Removal of the protecting groups from the cyclization products derived from alkynes 4c and 4d
Separate suspensions of 6-(1-pyrenyl)pyrrolo-5′,3′-di-O-Ac-deoxycytosine (100 mg, 0.19 mmol) and K2CO3 (5 mg, 0.04 mmol) in EtOH (20 mL) and 6-(benzo[b]fur-2-yl)pyrrolo-5′,3′-di-O-Ac-deoxycytosine (205 mg, 0.45 mmol) and K2CO3 (157 mg, 1.13 mmol) in MeOH (6 mL) were stirred at RT (to form 1c) or at 0 °C (to form 1d), until the TLC analysis revealed the complete consumption of starting material. The solids were removed by filtration, the filtrates were evaporated and the residues were purified by trituration (CH2Cl2, ca. 10 mL) and subsequent crystallization using EtOH/water (product 1c) or by FCC on 20 g SiO2 eluted with EtOAc/MeOH (95:5) later replaced with EtOAc/MeOH (9:1), followed by the evaporation of the eluate.
6-(1-Pyrenyl)pyrrolodeoxycytosine (1c, 84 mg, 85%) as yellow solid. 1H NMR (DMSO-D6): δ 11.97 (s, D2O exch., 1H); 8.86 (s, 1H); 8.52 (d, 1H); 8.36 (m, 3H); 8.25 (m, 4H); 8.13 (m, 1H); 6.69 (s, 1H); 6.33 (t, J = 6.5 Hz, 1H); 5.32 (d, D2O exch., J = 4.0 Hz, 1H); 5.17 (t, D2O exch., J = 5.0, 1H); 4.30 (m, 1H); 3.94 (m, 1H); 3.69 (m, 2H), 2.42 (ddd, J = 13.5, 6.0, 4.0 Hz, 1H), 2.09 (ddd, J = 13.5, 6.0, 4.0 Hz). 13C NMR (DMSO-D6): δ 159.6, 154.0, 138.9, 136.9, 130.9, 130.8, 130.4, 128.3, 128.1, 128.0, 127.3, 127.1, 126.6, 126.4, 125.8, 125.4, 124.9, 124.2, 124.1, 123.9, 109.3, 101.9, 88.0, 87.2, 70.1, 61.1, 41.6. HRMS (ESI) m/z found 452.1601 [M+H]+ (calcd. 452.1610 for C27H22N3O4).
6-(Benzo[b]furan-2-yl)pyrrolodeoxycytidine (1d, 124 mg, 75%) as yellow solid. 1H NMR (DMSO-D6) δ 12.08 (s, D2O exch., 1H); 8.79 (s, 1H); 7.70 (d, J = 8 Hz, 1H); 7.62 (d, J = 8 Hz, 1H); 7.36 (m, 2H); 7.28 (dd, J = 8, 8 Hz, 1H); 6.73 (s, 1H); 6.24 (t, J = 6.5 Hz, 1H); 5.30 (d, D2O exch., J = 4.5 Hz, 1H); 5.15 (t, D2O exch., J = 5.5 Hz, 1H); 4.25 (m, 1H); 3.92 (m, 1H), 3.67 (m, 2H), 2.39 (m, 1H); 2.05 (m, 1H). 13C NMR (DMSO-D6) δ 159.7, 154.3, 153.8, 148.0, 137.6, 130.0, 128.2, 125.3, 123.6, 121.5, 111.1, 108.6, 103.5, 98.6, 88.0, 87.2, 69.9, 61.0, 41.5. HRMS (ESI) m/z: found 368.1228 [M + H]+ (calcd. 368.1246 for C19H18N3O5).
Removal of the protecting groups from the cyclization product derived from alkyne 4e
Ethyl 6–2-[(1-Boc-indole-3-yl acetate)-2-yl]-pyrrolo-5′,3′-di-O-TBDMS-deoxycytosine (250 mg, 0.32 mmol) was added to a solution of NH4OH (ca. 28%, 1 mL) in MeOH (4 mL) cooled to 0 °C in a sealed vessel. The mixture was then stirred for 12 h at 55 °C, the solvent was evaporated and the residue was purified by FCC on 30 g SiO2 eluting with CH2Cl2/MeOH (98:2) later replaced with CH2Cl2/MeOH (95:5). The eluate was evaporated and the residue was dissolved in THF (3 mL), the solution was cooled to 0 °C, followed by the addition of Et3N · 3HF (155 mg, 1 mmol). The mixture was then stirred at RT for 12 h, the solvent was evaporated, the residue was triturated with Et2O (ca. 20 mL), the product was crystallized from MeOH to afford 6-[(indole-3-yl acetamide)-2-yl]pyrrolodeoxycytosine (1e, 92 mg, 68%), yellow solid. 1H NMR (DMSO-D6): δ 12.47 (s, D2O exch., 1H); 11.37 (s, D2O exch., 1H); 8.77 (s, 1H); 8.06 (s, D2O exch., 1H); 7.82 (m, 1H); 7.45 (s, D2O exch., 1H); 7.37 (m, 1H); 7.15 (m, 1 H); 7.05 (m, 1 H); 6.59 (s, 1H), 6.27 (t, J = 6.0 Hz, 1H); 5.26 (m, D2O exch., 1H); 5.14 (m, D2O exch., 1H); 4.26 (m, 1H); 3.90 (m, 1H); 3.66 (m, 4H); 2.37 (m, 1H); 2.05 (m, 1H). 13C NMR (DMSO-D6): δ 174.5, 159.2, 153.8, 136.4, 136.0, 132.4, 128.4, 127.7, 122.6, 119.3, 118.9, 111.1, 109.1, 107.5, 97.8, 87.9, 87.0, 69.7, 64.9, 60.9, 41.5. HRMS (ESI) m/z: found 424.1622 [M + H]+ (calcd. 424.1621 for C21H22N5O5).
Sonogashira cross-coupling to form the analogs 2a and 2b
Uridine analog 2a was prepared from 4b and 9b according to the literature procedure; spectral data were consistent with those reported previously.Citation24
5-Iododeoxycytidine (8a, 500 mg, 1.41 mmol) and 1-ethynylpyrene (4c, 410 mg, 1.83 mmol) were dissolved in dry DMF (5 mL). The mixture was degassed, then Pd(PPh3)4 (160 mg, 0.14 mmol) and CuI (53 mg, 0.28 mmol) were added and the mixture was degassed again. Et3N (720 μL, 5.64 mmol) was then added and the mixture was stirred (in the dark, under N2 atmosphere) for 18 h at RT. The solvent was evaporated; the residue was subjected to FCC on 30 g SiO2 eluting with CH2Cl2/MeOH (95:5). Evaporation of the eluate afforded 5-(1-pyrenylethyn-2-yl)deoxycytidine (2b, 310 min, 50%), yellow solid. 1H NMR (DMSO-D6): δ 8.63 (s, 1H); 8.58 (m, 1H); 8.43 (m, 1H); 8.34 (m, 4H); 8.23 (m, 2H); 8.13 (m, 1H); 7.92 (s, D2O exch., 1H); 7.26 (s, D2O exch., 1H); 6.19 (t, J = 6.0 Hz, 1H); 5.28 (m, D2O exch., 2H); 4.30 (m, 1H); 3.86 (m, 1H); 3.74 (m, 1H); 3.67 (m, 1H); 2.24 (m, 1H); 2.15 (m, 1H). 13C NMR (DMSO-D6): δ 163.8, 153.4, 145.4, 130.8, 130.7, 130.6, 130.5, 129.8, 128.6, 128.2, 127.2, 126.7, 125.9, 125.8, 125.2, 124.7, 123.6, 123.4, 117.1, 92.6, 57.6, 87.5, 85.6, 69.7, 60.8, 56.0, 40.9. HRMS (ESI) m/z found 452.1590 [M+H]+ (calcd. 452.1610 for C27H22N3O4).
Preparation of 4-[(1,2,4-triazole)-1-yl]-5-bromo-5′,3′-di-O-Ac-deoxycytidine
Ac2O (4 mL, 42 mmol) was added to a solution of deoxyuridine (9a, 1.14 g, 5 mmol) and Et3N (4 mL, 29 mmol) in dry dioxane (15 mL). The mixture was stirred for 24 h at RT and then the solvent was evaporated. The residue was taken up into water (25 mL) and extracted with EtOAc (3 × 40 ml). The combined organic extracts were dried and then concentrated to afford the desired 5′,3′-di-O-Ac-deoxyuridine (pale yellow oil) of sufficient purity for the subsequent step. 5′,3′-Di-O-Ac-deoxyuridine (1.56 g, 5 mmol), LiBr (521 mg, 6 mmol) and CAN (5.48 g, 10 mmol) were suspended in MeCN (70 mL) and stirred for 90 min at 80 °C. The solvent was evaporated, the residue was taken up into brine (50 ml) followed by the extraction with EtOAc (4 × 50 ml). The combined organic extract was dried and concentrated to afford 5-bromo-5′,3′-di-O-Ac-deoxyuridine (pale yellow oil) of sufficient purity for the subsequent step. 1,2,4-Triazole (3.11 g, 45 mmol) was dissolved in MeCN (190 mL) and the resulting solution was cooled to 0 °C. Et3N (10.5 mL, 75 mmol) was added, followed by the dropwise addition of POCl3 (1 mL, 11 mmol). The mixture was stirred for 30 min at 0 °C at which point a solution of 5-bromo-5′,3′-di-O-Ac-deoxyuridine (2.11 g, 5 mmol) in MeCN (10 mL) was added dropwise over a period of ca. 10 min. The cooling bath was removed and the mixture was heated at 60 °C for 18 h. The solvent was removed by evaporation, the residue was taken up into brine (50 mL), followed by the extraction with EtOAc (4 × 50 mL). The combined organic extract was dried and concentrated. The residue was subjected to FCC on 75 g SiO2 eluting with CH2Cl2/acetone (4:1). Evaporation of the eluate afforded 4-[(1,2,4-triazole)-1-yl]-5-bromo-5′,3′-di-O-Ac-deoxycytidine (10, 1.13 g, 51%, based on 9a), pale yellow oil. 1H NMR (CDCl3): δ 9.16 (s, 1 H); 8.53 (s, 1H); 8.18 (s, 1H); 6.25 (t, J = 6.5 Hz, 1H),; 5.24 (m, 1H); 4.45 (m, 1H); 4.43 (m, 2H); 2.93 (ddd, J = 15.0, 6.5, 3.0 Hz, 1H); 2.23 (m, 1H); 2.16 (s, 3H); 2.13 (s, 3H). 13C NMR (CDCl3): δ 170.3, 170.0, 155.8, 153.5, 152.1, 148.7, 145.2, 88.2, 86.8, 83.7, 73.4, 63.3, 39.2, 20.9, 20.8. HRMS (ESI) m/z: found 442.0366 [M+H]+ (442.0362 calcd. for C15H17BrN5O6).
Preparation of N4-Boc, N4-(2-bromophenyl)-5-bromo-5′,3′-di-O-Ac-deoxycytidine (11)
To install the N4-aryl group, 2-bromoaniline (447 mg, 2.6 mmol) and (1,2,4-triazol-1-yl)-5-bromo-5′,3′-di-O-Ac-deoxycytidine (10, 884 mg, 2 mmol) were dissolved in dioxane (5.4 mL) containing water (600 μL). The mixture was stirred for 48 h at RT. The solvent was evaporated; the residue was taken up into water (40 mL) and was extracted with EtOAc (2 × 50 mL). The combined organic extract was dried and was concentrated. The residue was subjected to FCC on 60 g SiO2 eluting with CH2Cl2/acetone (93:7). Evaporation of the eluate afforded N4-(2-bromophenyl)-5-bromo-5′,3′-di-O-Ac-deoxycytidine (650 mg, 60%), white solid. 1H NMR (CDCl3): δ 8.74 (dd, J = 8.5, 1.5 Hz, 1H); 8.03 (s, D2O exch., 1H); 8.00 (s, 1H), 7.58 (dd, J = 8.0, 1.5 Hz, 1H); 7.38 (m, 1H); 7.03 (m, 1H); 6.31 (dd, J = 7.5, 6.0 Hz, 1H); 5.23 (m, 1H); 4.39 (m, 2H); 4.34 (m, 1H), 2.76 (ddd, J = 14.5, 8.0, 6.0 Hz, 1H); 2.17 (s, 3H); 2.13 (m, 1H); 2.12 (s, 3H). 13C NMR (CDCl3): δ 170.4, 170.2, 157.3, 153.7, 140.4, 135.1, 132.3, 128.5, 125.8, 123.1, 114.7, 89.0, 87.0, 82.8, 74.0, 63.7, 39.0, 20.9 (2 × C). HRMS (ESI) m/z: found 543.9705 [M+H]+ (543.9719 calcd. for C19H20Br2N3O6).
To install the Boc protecting group, a solution of Boc2O (786 mg, 3.6 mmol) in dry dioxane (6 mL) was added to a flask containing N4-(2-bromophenyl)-5-bromo-5′,3′-di-O-Ac-deoxycytidine (650 mg, 1.2 mmol). DMAP (15 mg, 0.12 mmol) was then added and the mixture was stirred at 50 °C for 18 h. The solvent was evaporated and the residue was subjected to FCC on 50 g SiO2 eluted with CH2Cl2/acetone (19:1). Evaporation of the eluate afforded N4-Boc, N4-(2-bromophenyl)-5-bromo-5′,3′-di-O-Ac-deoxycytidine (11, 636 mg, 82%), white solid. 1H NMR (CDCl3): δ 8.21 (s, 1H); 7.61 (m, 1H); 7.31 (m, 2H); 7.19 (m, 1H); 6.20 (dd, J = 7.0, 6.0 Hz, 1H); 5.22 (m, 1H); 4.38 (m, 3H); 2.81 (ddd, J = 14.5, 8.0, 2.5 Hz, 1H); 2.16 (s, 3H); 2.15 (m, 1H); 2.11 (s, 3H). 13C NMR (CDCl3): δ 170.3, 170.2, 164.3, 153.5, 151.2, 143.9, 139.2, 133.4, 130.7, 129.4, 128.1, 95.7, 87.7, 83.8, 83.3, 73.8, 63.5, 39.1, 27.8, 20.9, 20.9 (2 × C). HRMS (ESI) m/z: found 644.0264 [M+H]+ (644.0243 calcd. for C24H28Br2N3O8).
Ni-mediated cyclization and deprotection to form 5,6-benzo-fused analog 3a
Zn dust (392 mg, 6 mmol), NiCl2 (52 mg, 0.4 mmol) and PPh3 (420 mg, 1.6 mmol) were placed in a flask and flushed with N2 for 45 min. Dry DMF (2 mL) was added under a continuous flow of N2 gas and the solution was heated to 80 °C until a dark red color persisted (ca. 20 min). A solution of N4-Boc, N4-(2-bromophenyl)-5-bromo-5′,3′-di-O-Ac-deoxycytidine (11, 258 mg, 0.4 mmol) in dry DMF (2 mL) was added dropwise over a period of ca. 5 min. The reaction was then stirred for 1 h at 80 °C (N2 atmosphere), cooled to RT, and then quenched with a 4% solution of EDTA (50 mL). The aqueous phase was extracted with EtOAc (4 × 50 ml), the combined organic extract was dried and then concentrated. The residue was subjected to FCC on 35 g SiO2 eluted with CH2Cl2/MeOH (98:2) which was later replaced with CH2Cl2/MeOH (95:5) to afford the diacetylated precursor of the analog 3a (90 mg, 58%), white solid. 1H NMR (DMSO-D6): δ 11.63 (s, D2O exch., 1H); 8.83 (s, 1H); 7.87 (d, J = 8.0 Hz, 1H); 7.34 (ddd, J = 8.0, 8.0, 1.0 Hz, 1H); 7.27 (d, J = 8.0 Hz, 1H); 7.19 (ddd, J = 8.0, 8.0, 1.0 Hz, 1H); 6.33 (dd, J = 8.0, 6.0 Hz, 1H); 5.27 (m, 1 H); 4.38 (m, 2 H); 4.35 (m, 1 H); 2.56 (ddd, J = 14.5, 8.5, 2.5 Hz, 1H); 2.42 (m, 1H); 2.10 (s, 3H); 2.04 (s, 3H). 13C NMR (DMSO-D6): δ 170.2, 170.1, 161.8, 154.3, 140.1, 136.5, 126.8, 121.4, 120.2, 111.3, 109.5, 104.1, 87.2, 82.1, 74.3, 63.7, 37.6, 20.8, 20.6. HRMS (ESI) m/z: found 386.1336 [M+H]+ (386.1352 calcd. for C19H20N3O6).
The free nucleoside was accessed by: NH4OH (ca. 28%, 4.5 mL) was added to a solution of the diacetylated precursor of the analog 3a (90 mg, 0.23 mmol) in MeOH (4.5 mL) and the mixture was stirred for 2 h at 55 °C. Volatiles were evaporated and the desired analog 3a (52 mg, 75%) was purifed by crystallization from MeOH. 1H NMR (DMSO-D6): δ 11.64 (s, D2O exch., 1H); 9.14 (s, 1H); 7.79 (d, J = 8.0 Hz, 1H); 7.32 (dd, J = 8.0, 8.0 Hz, 1H); 7.26 (d, J = 8.0 Hz, 1H); 7.17 (dd, J = 8.0, 8.0 Hz, 1H); 6.26 (t, J = 6.0 Hz, 1H); 5.29 (m, D2O exch., 1H); 4.28 (m, 1H); 3.90 (m, 1H); 3.72 (m, D2O exch., 2H); 2.35 (ddd, J = 13.5, 10.5, 4.5 Hz, 1H); 2.10 (m, 1H). 13C NMR (DMSO-D6): δ 161.6, 154.4, 140.0, 136.7, 126.5, 121.2, 120.6, 119.9, 111.3, 103.6, 87.7, 86.7, 69.5, 60.7, 41.3. HRMS (ESI) m/z: found 302.1119 [M+H]+ (302.1141 calcd. for C15H16N3O4).
Quantum yields determination
Quantum yields (Φ) associated with compounds 1b–e, 2a, 2b, and 3a were determined in dioxane, EtOH and water. All quantum yields (Φ) measured in water were determined using quinine bisulfate in 0.5 M H2SO4 (Φ 0.55) as a standard.Citation54 Quantum yields (Φ) measured in organic solvents for 1c, 1e, and 2b were determined using 9,10-dichloro-anthracene (Φ 0.55) as a standard,Citation55 for 2a and 3a pyrene (Φ 0.53) was used as a standardCitation55 and for 1b and 1d 9,10-diphenyl-anthracene (Φ 0.90) was used as a standard.Citation54 In each case the following equation was used to calculate the relative fluorescence quantum yield using seven samples of varying concentrations.
Φ (Fluorescence quantum yield), I (integrated fluorescence intensity) and Abs (absorbance at λex). The standard is denoted as (ref) while the sample is denoted as (s).
Molar extinction coefficients
Molar extinction coefficients (ε) associated with compounds 1b–e, 2a, 2b, and 3a were estimated by constructing Beer-Lambert plots (A = ε × c × l), wherein A (absorbance), ε (molar extinction coefficient), c (concentration), l (light path length ~1 cm). Molar extinction coefficients (ε) associated with compounds 1b–e, 2a, 2b, and 3a were determined in dioxane, EtOH, and water with the exception of 1c and 2b, wherein an insufficient solubility prevented the determination of molar extinction coefficients (ε) in water.
Solvent polarity study
Stock solutions of compounds 1b–e, 2a, 2b, and 3a (on the order of 10−3 M) were prepared in DMSO. These stock solutions were diluted (~10× , depending on fluorophore brightness) in dioxane. These solutions were then further diluted (25×) into volumetric flasks with appropriate volumes of water and dioxane to prepare working solutions containing 100%, 90% 80%, 70%, 60%, 40%, 20%, and 10% dioxane in water. A 100% water sample was prepared in similar fashion using an equivalent amount of the DMSO solution. The ET(30) values were obtained from the literature.Citation56 Excitation and emission spectra for each sample were then obtained with the measurement of the maxima over a narrow window (approx. 50 nm, average of ten independent measurements). Plots of solvent polarity (ET[30]) vs. Stokes shift (cm−1) were then constructed (see Supplemental Materials) and the slope was calculated to provide a numerical representation of solvent polarity sensitivity in cm−1/(kcal mol−1). This procedure is similar to the method reported by Tor and colleagues.Citation21 Points were excluded if the fluorescence emission was too low to reliably find a maximum in either the excitation, or the emission spectra. The points obtained for 100% water and 100% dioxane solutions associated with compound 2a were not taken into consideration due to excessive changes in the electronic structure between the neat and binary solutions preventing the selection of a constant electronic transition to monitor. For the compounds 1c and 2b a significant shift in the linear trend can be observed. As such the largest continuously linear segments were used to represent the polarity sensitivity of the compound.
Solvent viscosity study
Solutions of 70%, 50%, 40%, 20%, 15%, and 10% methanol in glycerol (5 mL each) were prepared by stirring the solvent mixtures overnight (RT) in sealed vials to ensure homogeneity. A known amount of compound 2a was then dissolved in DMSO to obtain a stock solution with the concentration 2.79 × 10−3 M. Then, 20 µL of the stock solution was added to the solutions of glycerol in MeOH prepared previously. UV-Vis absorbance, excitation and emission spectra were then obtained with the emission maximum being averaged over a small window (50 nm). Integrated emission intensities were then normalized using Equation 1. The log of the normalized emission intensities were then plotted () vs. the log of the solvent viscosity, obtained using equation (2).
(1)
(2)
I (Integrated fluorescence intensity), Abs (absorbance at λex), η (viscosity), and w (weight fraction). The normalized data are denoted as (norm) and the data for mixtures are denoted as (mix) respec. Values of 1317 cp (glycerol) and 0.583 cp (MeOH) have been used for the viscosity of the solvents.Citation26 Values of 1.25 kg/L (glycerol) and 0.79 kg/L (MeOH) have been used for the density of the solvents.
Conclusions
The syntheses of five novel pyrrolocytidine analogs (1c–e, 3a) and a 5-ethynylcytidine (2b) analog have been achieved that usefully expand the repertoire of environmentally responsive fluorescent nucleosides. The photophysical characterization of the new analogs has been done along with previously reported nucleosides 1b and 2a for comparison.Citation12,Citation24
The pyrrolocdeoxycytidine analogs 1c–e display fluorophore behavior that is congruent with 6-phenylpyrrolodeoxycytidine 1b, that is, they have the property of being an intrinsic fluorophore, even for the large pyrene substituent. The similarity in fluorescence emission, a broad featureless band at λ > 450 nm, argues for substantial electronic communication between the pC nucleus and the 6-substituent. This is in contrast to the emission of the related 5-pyrenylethynyldeoxycytidine which resembles more closely an isolated pyrene, as previously noted.Citation47 The benefit to variation of the 6-substituent is shown by improvement of fluorophore brightness, primarily by increasing the extinction coefficient at the longest wavelength absorption band used for excitation. As well, variation of the nature of the heteroaromatic substituent at position 6 produces compounds with varying sensitivity to the polarity of the environment, as judged by the emission Stokes shift and emission intensity. For example the benzo[b]furan-2-yl-modified analog 1d is approximately six times brighter than the parent analog 1b, yet possesses similar polarity sensitivity and high emission intensity in water. The indole-containing analog 1e displays not only greater polarity sensitivity, in terms of Stokes shift, but also shows a dramatic change in quantum yield and emission intensity in water. These limited examples tantalizingly suggest: (1) the pyrrolocytosine nucleus is a suitable scaffold for intrinsic nucleobase fluorophore development; and (2) that it may be possible to discover application specific analogs with a desirable combination of responsiveness to water, medium polarity and also possessing high brightness.
For 6-phenylpyrrolodeoxycytidine, the archetypal fluorophore possessing a biaryl bond, solvent viscosity showed no systematic effect on the emission intensity indicating that rotation about this bond is not a significant influence on the radiationless decay of the excited state. Similarly, 5-pyrenylethynyldeoxcytidine (2b) exhibited no dependence of the fluorescence intensity on medium viscosity; however, the spectral properties of 2b indicate that the origin of this effect is different than for 1b. Whereas 1b exhibits spectral properties of an intrinsic nucleobase fluorophore wherein the nucleobase and the 6-substituent (phenyl) form an integrated fluorophore, the pyrene group in 2b appears more independent of the nucleobase. Thus, the rotatable linkage between the nucleobase and the pyrene is not within the core structure of the fluorophore and consequently has little influence on the radiationless relaxation of the excited state. For comparison, we turned to a fluorophore with an integrated acetylenic linkage in the core of the structure: 5-(p-methoxyphenylethynyl)deoxyuridine (2a). For this compound, a strong dependence on solvent viscosity was found resembling that observed for known molecular rotors.Citation26
Although 1b did not show molecular rotor behavior, rigidification of pyrrolocytosine moiety (elimination of the rotatable C-C biaryl bond) to furnish the 5,6-benzo-fused analog 3a produced a large positive impact on the quantum yield in organic solvents (Φ ~1.0) and brightness (compared with parent 1b). Further characterization of the 5,6-benzo-fused pyrrolocytidine analog 3a showed that it is extraordinarily sensitive to the presence of water; combined with a high quantum yield and large Stokes shift, makes this a promising nucleoside probe. The sensitivity of fluorophore 3a may be exploited in studies involving environments with strict exclusion of water, such as DNA-protein interactions.Citation57
| Abbreviations: | ||
| Ac2O | = | acetic anhydride |
| Boc | = | tert-butyloxycarbonyl |
| Boc2O | = | Boc anhydride |
| CAN | = | cerium ammonium nitrate |
| CI | = | chemical ionization |
| DMAP | = | 4-N,N-dimethylaminopyridine |
| DMF | = | N,N-dimethylformamide |
| DMSO | = | dimethylsulfoxide |
| DNA | = | deoxyribonucleic acid |
| EDTA | = | ethylenediamine tetraacetic acid disodium salt |
| EI | = | electron impact |
| ESI | = | electron spray ionization |
| Et3N | = | triethylamine |
| EtOH | = | ehtanol |
| FCC | = | flash column chromatography |
| FRET | = | fluorescence resonance energy transfer |
| HRMS | = | high resolution mass spectrometry |
| MeCN | = | acetonitrile |
| MeOH | = | methanol |
| NBS | = | N-bromosuccimimide |
| NMR | = | nuclear magnetic resonance |
| PNA | = | peptide nucleic acid |
| pC | = | pyrrolocytosine |
| pdC | = | pyrrolodeoxycytidine |
| PydC | = | 5-(pyren-1-yl)deoxycytidine |
| RNA | = | ribonucleic acid |
| RT | = | room temperature |
| TBAF | = | tetrabutylammonium fluoride |
| TBDMS | = | tert-butyldimethylsilyl |
| THF | = | tetrahydrofuran |
| TLC | = | thin layer chromatography |
| TMS | = | trimethylsilyl |
| UV-VIS | = | ultraviolet-visible |
Additional material
Download Zip (1.7 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The Natural Sciences and Engineering Research Council (NSERC) of Canada is gratefully acknowledged for the financial support of this work.
References
- Dodd DW, Hudson RHE. Intrinsically fluorescent base-discriminating nucleoside analogs. Minirew Org Chem 2009; 6:378 - 91; http://dx.doi.org/10.2174/157019309789371659
- Sinkeldam RW, Greco NJ, Tor Y. Fluorescent analogs of biomolecular building blocks: design, properties, and applications. Chem Rev 2010; 110:2579 - 619; http://dx.doi.org/10.1021/cr900301e; PMID: 20205430
- Wilhelmsson LM. Fluorescent nucleic acid base analogues. Q Rev Biophys 2010; 43:159 - 83; http://dx.doi.org/10.1017/S0033583510000090; PMID: 20478079
- Okamoto A, Saito Y, Saito I. Design of base-discriminating fluorescent nucleosides. J Photochem Photobiol Chem 2005; 6:108 - 22; http://dx.doi.org/10.1016/j.jphotochemrev.2005.07.002
- Asseline U. Development and applications of fluorescent oligonucleotides. Curr Org Chem 2006; 10:491 - 518; http://dx.doi.org/10.2174/138527206776055349
- Callis PR. Electronic states and luminescence of nucleic acid systems. Annu Rev Phys Chem 1983; 34:329 - 57; http://dx.doi.org/10.1146/annurev.pc.34.100183.001553
- Johansson Seechurn CC, Kitching MO, Colacot TJ, Snieckus V. Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel Prize. Angew Chem Int Ed Engl 2012; 51:5062 - 85; http://dx.doi.org/10.1002/anie.201107017; PMID: 22573393
- Inoue H, Imura A, Ohtsuka E. Synthesis of dodecadeoxyribonucleotides containing a pyrrolo[2,3-d]pyrimidine nucleoside and their base-pairing ability. Nippon Kagaku Kaishi 1987; 7:1214 - 20
- Woo J, Meyer RB Jr., Gamper HB. G/C-modified oligodeoxynucleotides with selective complementarity: synthesis and hybridization properties. Nucleic Acids Res 1996; 24:2470 - 5; http://dx.doi.org/10.1093/nar/24.13.2470; PMID: 8692683
- Berry DA, Jung K-Y, Wise DS, Sercel AD, Pearson WH, Mackie H, Randolph JB, Somers RL. Tetrahedron Lett 2004; 45:2457 - 61; http://dx.doi.org/10.1016/j.tetlet.2004.01.108
- Hardman SJO, Botchway SW, Thompson KC. Evidence for a nonbase stacking effect for the environment-sensitive fluorescent base pyrrolocytosine--comparison with 2-aminopurine. Photochem Photobiol 2008; 84:1473 - 9; http://dx.doi.org/10.1111/j.1751-1097.2008.00368.x; PMID: 18513237
- Hudson RHE, Dambenieks AK, Viirre RD. Fluorescent 7-deazapurine derivatives from 5-iodocytosine via a tandem cross-coupling-annulation reaction with terminal alkynes. Synlett 2004; 13:2400 - 2
- Hudson RHE, Dambenieks AK. Synthesis of N1-unsubstituted 5-alkynylcytosine and derivatives thereof. Heterocycles 2006; 68:1325 - 8; http://dx.doi.org/10.3987/COM-06-10727
- Hudson RHE, Ghorbani-Choghamarani A. Selective fluorometric detection of guanosine-containing sequences by 6-phenylpyrrolocytidine in DNA. Synlett 2007; 870 - 3
- Wojciechowski F, Hudson RHE. Fluorescence and hybridization properties of peptide nucleic acid containing a substituted phenylpyrrolocytosine designed to engage Guanine with an additional H-bond. J Am Chem Soc 2008; 130:12574 - 5; http://dx.doi.org/10.1021/ja804233g; PMID: 18761442
- Wojciechowski F, Hudson RHE. Peptide nucleic acid containing a meta-substituted phenylpyrrolocytosine exhibits a fluorescence response and increased binding affinity toward RNA. Org Lett 2009; 11:4878 - 81; http://dx.doi.org/10.1021/ol9019474; PMID: 19788268
- Tanpure AA, Srivatsan SG. Synthesis and photophysical characterisation of a fluorescent nucleoside analogue that signals the presence of an abasic site in RNA. Chembiochem 2012; 13:2392 - 9; http://dx.doi.org/10.1002/cbic.201200408; PMID: 23070860
- Burstein EA, Vedenkina NS, Ivkova MN. Fluorescence and the location of tryptophan residues in protein molecules. Photochem Photobiol 1973; 18:263 - 79; http://dx.doi.org/10.1111/j.1751-1097.1973.tb06422.x; PMID: 4583619
- Chen Y, Barkley MD. Toward understanding tryptophan fluorescence in proteins. Biochemistry 1998; 37:9976 - 82; http://dx.doi.org/10.1021/bi980274n; PMID: 9665702
- Kirby EP, Steiner RF. The influence of solvent and temperature upon the fluorescence of indole derivatives. J Phys Chem 1970; 74:4480 - 90; http://dx.doi.org/10.1021/j100720a004
- Noé MS, Ríos AC, Tor Y. Design, synthesis, and spectroscopic properties of extended and fused pyrrolo-dC and pyrrolo-C analogs. Org Lett 2012; 14:3150 - 3; http://dx.doi.org/10.1021/ol3012327; PMID: 22646728
- Matteucci MD, von Krosigk U. Hybridization properties of oligonucleotides bearing a tricyclic 2’-deoxycytidine analog based on a carbazole ring system. Tetrahedron Lett 1996; 37:5057 - 60; http://dx.doi.org/10.1016/0040-4039(96)01016-7
- Wagner C, Rist M, Mayer-Enthart E, Wagenknecht H-A. 1-ethynylpyrene-modified guanine and cytosine as optical labels for DNA hybridization. Org Biomol Chem 2005; 3:2062 - 3; http://dx.doi.org/10.1039/b504079e; PMID: 15917887
- Hudson RHE, Ghorbani-Choghamarani A. Oligodeoxynucleotides incorporating structurally simple 5-alkynyl-2′-deoxyuridines fluorometrically respond to hybridization. Org Biomol Chem 2007; 5:1845 - 8; http://dx.doi.org/10.1039/b705805e; PMID: 17551631
- Sinkeldam RW, Greco NJ, Tor Y. Polarity of major grooves explored by using an isosteric emissive nucleoside. Chembiochem 2008; 9:706 - 9; http://dx.doi.org/10.1002/cbic.200700714; PMID: 18286575
- Sinkeldam RW, Wheat AJ, Boyaci H, Tor Y. Emissive nucleosides as molecular rotors. Chemphyschem 2011; 12:567 - 70; http://dx.doi.org/10.1002/cphc.201001002; PMID: 21344595
- Zhang H, Larock RC. Synthesis of β- and γ-carbolines by the palladium/copper-catalyzed coupling and cyclization of terminal acetylenes. J Org Chem 2002; 67:7048 - 56; http://dx.doi.org/10.1021/jo0258857; PMID: 12353999
- Menciu C, Duflos M, Fouchard F, Le Baut G, Emig P, Achterrath U, Szelenyi I, Nickel B, Schmidt J, Kutscher B, et al. New N-(pyridin-4-yl)-(indol-3-yl)acetamides and propanamides as antiallergic agents. J Med Chem 1999; 42:638 - 48; http://dx.doi.org/10.1021/jm981079+; PMID: 10052971
- Mejía-Oneto JM, Padwa A. Intramolecular [3 + 2]-cycloaddition reaction of push-pull dipoles across heteroaromatic π-systems. Org Lett 2004; 6:3241 - 4; http://dx.doi.org/10.1021/ol048915w; PMID: 15355022
- Sato S, Shibuya M, Kanoh N, Iwabuchi Y. Highly enantioselective intramolecular aza-spiroannulation onto indoles using chiral rhodium catalysis: asymmetric entry to the spiro-β-lactam core of chartellines. Chem Commun (Camb) 2009; 6264-6:6264 - 6; http://dx.doi.org/10.1039/b913770j; PMID: 19826689
- Watanabe KA, Su TL, Klein RS, Chu CK, Matsuda A, Chun MW, Lopez C, Fox JJ. Nucleosides. 123. Synthesis of antiviral nucleosides: 5-substituted 1-(2-deoxy-2-halogeno-β-D-arabinofuranosyl)cytosines and -uracils. Some structure-activity relationships. J Med Chem 1983; 26:152 - 6; http://dx.doi.org/10.1021/jm00356a007; PMID: 6298422
- Dodd DW, Swanick KN, Price JT, Brazeau AL, Ferguson MJ, Jones ND, Hudson RHE. Blue fluorescent deoxycytidine analogues: convergent synthesis, solid-state and electronic structure, and solvatochromism. Org Biomol Chem 2010; 8:663 - 6; http://dx.doi.org/10.1039/b919921g; PMID: 20090985
- Suchý M, Hudson RHE. Pyrimidine-fused heterocyclic frameworks based on a 4-arylaminocytosine scaffold: Synthesis, characterization and PNA oligomerization of the fluorescent cytosine analog 5,6-benzopC. J Org Chem 2014; 79:3336 - 47; http://dx.doi.org/10.1021/jo402873e; PMID: 24666330
- Asakura J, Robins MJ. Cerium(IV)-mediated halogenation of C-5 of uracil derivatives. J Org Chem 1990; 55:4928 - 33; http://dx.doi.org/10.1021/jo00303a033
- Colon I, Kelsey DR. Coupling of aryl chlorides by nickel and reducing metals. J Org Chem 1986; 51:2627 - 37; http://dx.doi.org/10.1021/jo00364a002
- Wahba AS, Esmaeili A, Damha MJ, Hudson RHE. A single-label phenylpyrrolocytidine provides a molecular beacon-like response reporting HIV-1 RT RNase H activity. Nucleic Acids Res 2010; 38:1048 - 56; http://dx.doi.org/10.1093/nar/gkp1022; PMID: 19933258
- Huber R, Fiebig T, Wagenknecht H-A. Pyrene as a fluorescent probe for DNA base radicals. Chem Commun (Camb) 2003; 1878-79:1878 - 9; http://dx.doi.org/10.1039/b305732a; PMID: 12932012
- Raytchev M, Mayer E, Amann N, Wagenknecht H-A, Fiebig T. Ultrafast proton-coupled electron-transfer dynamics in pyrene-modified pyrimidine nucleosides: model studies towards an understanding of reductive electron transport in DNA. Chemphyschem 2004; 5:706 - 12; http://dx.doi.org/10.1002/cphc.200301205; PMID: 15179723
- Sabale PM, Nuthanakanti A, Srivatsan SG. Synthesis and fluorescence properties of a full set of extended RNA base analogues. Indian J Chem 2013; 52A:1004 - 13
- Celewicz L. Photochemical reactions of 5-bromocytosine and its N-1-substituted derivatives with Nα-acetyl-L-tryptophan N-ethylamide in aqueous solution. Photochem Photobiol B 1995; 30:129 - 37; http://dx.doi.org/10.1016/1011-1344(95)07176-3
- Lin K-Y, Jones RJ, Matteucci M. Tricyclic 2’-deoxycytidine analogs: synthesis and incorporation into oligonucleotides which have enhanced binding to complementary RNA. J Am Chem Soc 1995; 117:3873 - 4; http://dx.doi.org/10.1021/ja00118a026
- Wilhelmsson LM, Holmén A, Lincoln P, Nielsen PE, Nordén B. A highly fluorescent DNA base analogue that forms Watson-Crick base pairs with guanine. J Am Chem Soc 2001; 123:2434 - 5; http://dx.doi.org/10.1021/ja0025797; PMID: 11456897
- Sandin P, Börjesson K, Li H, Mårtensson J, Brown T, Wilhelmsson LM, Albinsson B. Characterization and use of an unprecedentedly bright and structurally non-perturbing fluorescent DNA base analogue. Nucleic Acids Res 2008; 36:157 - 67; http://dx.doi.org/10.1093/nar/gkm1006; PMID: 18003656
- Okamoto A, Tainaka K, Saito I. Clear distinction of purine bases on the complementary strand by a fluorescence change of a novel fluorescent nucleoside. J Am Chem Soc 2003; 125:4972 - 3; http://dx.doi.org/10.1021/ja034090u; PMID: 12708835
- Okamoto A, Tainaka K, Saito I. Synthesis and properties of a novel fluorescent nucleobase, naphthopyridopyrimidine. Tetrahedron Lett 2003; 44:6871 - 4; http://dx.doi.org/10.1016/S0040-4039(03)01740-4
- Miyata K, Tamamushi R, Ohkubo A, Taguchi H, Seio K, Santa T, Sekine M. Synthesis and properties of a new fluorescent bicyclic 4-N-carbamoyldeoxycytidine derivative. Org Lett 2006; 8:1545 - 8; http://dx.doi.org/10.1021/ol053125n; PMID: 16597106
- Mayer E, Valis L, Wagner C, Rist M, Amann N, Wagenknecht H-A. 1-Ethynylpyrene as a tunable and versatile molecular beacon for DNA. Chembiochem 2004; 5:865 - 8; http://dx.doi.org/10.1002/cbic.200300845; PMID: 15174171
- Tainaka K, Tanaka K, Ikeda S, Nishiza K, Unzai T, Fujiwara Y, Saito I, Okamoto A. PRODAN-conjugated DNA: synthesis and photochemical properties. J Am Chem Soc 2007; 129:4776 - 84; http://dx.doi.org/10.1021/ja069156a; PMID: 17378568
- Okamoto A, Tainaka K, Unzai T, Saito I. Synthesis and fluorescence properties of dimethylaminonaphthalene-deoxyuridine conjugates as polarity-sensitive probes. Tetrahedron 2007; 63:3465 - 70; http://dx.doi.org/10.1016/j.tet.2006.09.113
- Reichardt C. Solvatochromic Dyes as Solvent Polarity Indicators. Chem Rev 1994; 94:2319 - 58; http://dx.doi.org/10.1021/cr00032a005
- Shin D, Sinkeldam RW, Tor Y. Emissive RNA alphabet. J Am Chem Soc 2011; 133:14912 - 5; http://dx.doi.org/10.1021/ja206095a; PMID: 21866967
- Malakhov AD, Malakhova EV, Kyznitsova SV, Grechishnikova IV, Prokhorenko IA, Skorobogatyĭ MV, Korshun VA, Berlin IuA. [Synthesis and fluorescent properties of 5-(1-pyrenylethynyl)-2′- deoxyuridine-containing oligodeoxynucleotides]. Bioorg Khim 2000; 26:39 - 50; PMID: 10806551
- Dong DC, Winnik MA. The Py scale of solvent polarities. Solvent effects on the vibronic fine structure of pyrene fluorescence and empirical correlations with the ET and Y values. Photochem Photobiol 1982; 35:17 - 21; http://dx.doi.org/10.1111/j.1751-1097.1982.tb03805.x
- Eaton DF. Reference materials for fluorescence measurements. Pure Appl Chem 1988; 60:1107 - 14; http://dx.doi.org/10.1351/pac198860071107
- Dawson WR, Windsor MW. Fluorescence Yields of Aromatic Compounds. J Phys Chem 1968; 72:3251 - 60; http://dx.doi.org/10.1021/j100855a027
- Casassas E, Fondrodona G, de Juan A. Solvatochromic parameters for binary mixtures and a correlation with equilibrium constants. Part I. Dioxane-water mixtures. J Solution Chem 1992; 21:147 - 62; http://dx.doi.org/10.1007/BF00647004
- Von Hippel PH, McGhee JD. DNA-protein interactions. Annu Rev Biochem 1972; 41:231 - 300; http://dx.doi.org/10.1146/annurev.bi.41.070172.001311; PMID: 4570958