
Figures & data
Table 1. Case profiles.
Figure 1. Workflow of protein extraction from the formalin-fixed paraffin-embedded (FFPE) samples. The FFPE samples are sectioned into two tubes, one for proteinase K (PK) treatment (d) and the other without PK treatment (e), and the proteins are then extracted.
Figure 2. Western blot analyses of the formalin-fixed paraffin-embedded (FFPE) samples with the 3F4 anti-prion protein (PrP) antibody. (a) in control cases, proteinase K-untreated (PK [−]) samples show strong PrP signals at approximately 25–40kDa. Proteinase K-treated (PK [+]) samples show no signal. (b, c, d) in the sporadic Creutzfeldt–Jakob disease (sCJD) cases, PK-resistant PrP (PrPres) signals are observed with varied densities per case. In sCJD 1, 2, and 3, three PrPres bands are detected at 21–30 kDa (white arrowheads). sCJD 4, 6 and 7 also show three PrPres bands, but the unglycosylated PrPres are slightly wider at 19–21 kDa (black arrowheads). In sCJD 5, faint PrPres is detected at 21–30 kDa. In all sCJD cases, high molecular smear PrPres signals are observed with varied densities per case. (e) PK digestion reveals three bands of PrPres at 21–30 kDa (white arrowheads) in Gerstmann–Sträussler–Scheinker disease (GSS) 1 and 3, as well as low molecular weight bands at approximately 8 kDa (arrows) and strong high molecular weight smear bands. In GSS 2, a high molecular weight smear band and a band at approximately 8 kDa (arrow) are present. In glycosylphosphatidylinositol-anchorless prion disease (GPIALP) cases, PK digestion reveals smear bands from low to high molecular weight and low molecular bands at approximately 9 kDa (arrows). In V180I CJD cases, weak high molecular PrPres smear signals are observed in V180I CJD 1, PrPres signal are unclear in V180I CJD 2.
![Figure 2. Western blot analyses of the formalin-fixed paraffin-embedded (FFPE) samples with the 3F4 anti-prion protein (PrP) antibody. (a) in control cases, proteinase K-untreated (PK [−]) samples show strong PrP signals at approximately 25–40kDa. Proteinase K-treated (PK [+]) samples show no signal. (b, c, d) in the sporadic Creutzfeldt–Jakob disease (sCJD) cases, PK-resistant PrP (PrPres) signals are observed with varied densities per case. In sCJD 1, 2, and 3, three PrPres bands are detected at 21–30 kDa (white arrowheads). sCJD 4, 6 and 7 also show three PrPres bands, but the unglycosylated PrPres are slightly wider at 19–21 kDa (black arrowheads). In sCJD 5, faint PrPres is detected at 21–30 kDa. In all sCJD cases, high molecular smear PrPres signals are observed with varied densities per case. (e) PK digestion reveals three bands of PrPres at 21–30 kDa (white arrowheads) in Gerstmann–Sträussler–Scheinker disease (GSS) 1 and 3, as well as low molecular weight bands at approximately 8 kDa (arrows) and strong high molecular weight smear bands. In GSS 2, a high molecular weight smear band and a band at approximately 8 kDa (arrow) are present. In glycosylphosphatidylinositol-anchorless prion disease (GPIALP) cases, PK digestion reveals smear bands from low to high molecular weight and low molecular bands at approximately 9 kDa (arrows). In V180I CJD cases, weak high molecular PrPres smear signals are observed in V180I CJD 1, PrPres signal are unclear in V180I CJD 2.](/cms/asset/5ed6a7cc-4f00-4da1-ab91-999f14c2d9e2/kprn_a_2337981_f0002_b.gif)
Figure 3. Western blot analyses of the formalin-fixed paraffin-embedded (FFPE) samples with the EP1802Y anti-prion protein (PrP) antibody. (a) in control cases, proteinase K-untreated (PK [−]) samples show PrP signals at 25–40 kDa. In proteinase K-treated (PK [+]) samples, there is no PrPres signals. (b, c) the PK (+) samples of sporadic Creutzfeldt–Jakob disease (sCJD) 1–7 show unglycosylated PK-resistant PrP (PrPres) signals at 21 kDa (case 1–5 and 7: white arrowheads) and about 19 kDa (case 6: arrowhead). High molecular smear PrPres signals are observed in all sCJD cases (*). In addition, a low molecular weight band at approximately 16 kDa (arrows) are also found in sCJD cases 1–5 and 7. The 16 kDa PrPres signal is not observed in sCJD 6. (D) in the Gerstmann–Sträussler–Scheinker disease (GSS) cases, unglycosylated PrPres at 21 kDa (white arrowheads) and high molecular smear bands are present (*). The low molecular weight band at approximately 8 kDa is not detectable. In glycosylphosphatidylinositol-anchorless prion disease (GPIALP) cases, no PrPres signal is detected. In the PK (+) sample of V180I CJD 1 (pentosan polysulfate [PPS]-treated case), a high molecular smear band (*) is visible, whereas the PrPres signal is substantially weak in V180I CJD 2 (PPS-untreated case).
![Figure 3. Western blot analyses of the formalin-fixed paraffin-embedded (FFPE) samples with the EP1802Y anti-prion protein (PrP) antibody. (a) in control cases, proteinase K-untreated (PK [−]) samples show PrP signals at 25–40 kDa. In proteinase K-treated (PK [+]) samples, there is no PrPres signals. (b, c) the PK (+) samples of sporadic Creutzfeldt–Jakob disease (sCJD) 1–7 show unglycosylated PK-resistant PrP (PrPres) signals at 21 kDa (case 1–5 and 7: white arrowheads) and about 19 kDa (case 6: arrowhead). High molecular smear PrPres signals are observed in all sCJD cases (*). In addition, a low molecular weight band at approximately 16 kDa (arrows) are also found in sCJD cases 1–5 and 7. The 16 kDa PrPres signal is not observed in sCJD 6. (D) in the Gerstmann–Sträussler–Scheinker disease (GSS) cases, unglycosylated PrPres at 21 kDa (white arrowheads) and high molecular smear bands are present (*). The low molecular weight band at approximately 8 kDa is not detectable. In glycosylphosphatidylinositol-anchorless prion disease (GPIALP) cases, no PrPres signal is detected. In the PK (+) sample of V180I CJD 1 (pentosan polysulfate [PPS]-treated case), a high molecular smear band (*) is visible, whereas the PrPres signal is substantially weak in V180I CJD 2 (PPS-untreated case).](/cms/asset/9b14b386-50b5-4037-8717-900317a7757d/kprn_a_2337981_f0003_b.gif)
Table 2. Relative percentage of protease-resistant prion protein by molecular weight.
Figure 4. Western blotting analyses of the formalin-fixed paraffin-embedded (FFPE) samples with the Tohoku 1 (specific for type 1 scrapie prion protein [PrPSc]) and Tohoku 2 (specific for type 2 PrPSc) anti-proteinase K-resistant PrP (PrPres) antibodies. (a, b) Sporadic Creutzfeldt–Jakob disease (sCJD) 1 and 3 show bands of PrPres at 21–30 kDa and high molecular smears with Tohoku 1, whereas Tohoku 2 shows no signal. In sCJD 2, 4, and 5, PrPres signals are observed both with Tohoku 1 and 2. sCJD 6 shows a weak signal with Tohoku 1 but a strong signal with Tohoku 2. (c, d) sCJD 7 shows a smear band with Tohoku 1 and a weak smear band at approximately 20 kDa with Tohoku 2.
![Figure 4. Western blotting analyses of the formalin-fixed paraffin-embedded (FFPE) samples with the Tohoku 1 (specific for type 1 scrapie prion protein [PrPSc]) and Tohoku 2 (specific for type 2 PrPSc) anti-proteinase K-resistant PrP (PrPres) antibodies. (a, b) Sporadic Creutzfeldt–Jakob disease (sCJD) 1 and 3 show bands of PrPres at 21–30 kDa and high molecular smears with Tohoku 1, whereas Tohoku 2 shows no signal. In sCJD 2, 4, and 5, PrPres signals are observed both with Tohoku 1 and 2. sCJD 6 shows a weak signal with Tohoku 1 but a strong signal with Tohoku 2. (c, d) sCJD 7 shows a smear band with Tohoku 1 and a weak smear band at approximately 20 kDa with Tohoku 2.](/cms/asset/a65bb028-86a5-443f-88c7-c9d06ae3b71f/kprn_a_2337981_f0004_b.gif)
Figure 5. Histological findings of the prion disease cases. (A, C, E, G, I, K, M, O, Q, S, U, W: Hematoxylin and eosin staining (HE), B, D, F, H, J, L, N, P, R, T, V, X: Immunohistochemistry (IHC) with 3F4), (A, B: sporadic Creutzfeldt–Jakob disease case 1 (sCJD 1), C, D: sCJD 2, E, F: sCJD 3, G, H: sCJD 4, I, J: sCJD 5, K, L: sCJD 6, M, N: sCJD 7, O, P: Gerstmann–Sträussler–Scheinker disease case 1 (GSS 1), Q, R: glycosylphosphatidylinositol-anchorless prion disease case 1 (GPIALP 1), S, T: V180I CJD 1 (treated with pentosan polysulfate (PPS)), U, V: V180I CJD 2 without PPS, W, X: non prion case: control 3). (A, B: sCJD 1) HE staining indicating neuronal loss, gliosis and spongiform changes (a). IHC with 3F4 displaying synaptic prion protein (PrP) deposits in the cerebral cortex (b). (C, D: sCJD 2) HE staining shows spongiform changes and some large confluent vacuoles (inset) (c). 3F4 reveals synaptic PrP and perivacuolar PrP deposits (inset) (d). (E, F: sCJD 3) HE staining indicating neuronal loss, gliosis and spongiform changes (e). 3F4 displaying synaptic PrP deposits in the cerebral cortex (f). (G, H: sCJD 4) HE staining shows spongiform changes and some large confluent vacuoles (g). 3F4 reveals synaptic PrP deposits, perivacuolar PrP deposits and perineuronal PrP deposits (inset) (h). (I, J: sCJD 5) HE staining shows spongiform changes and some large confluent vacuoles (I). 3F4 reveals synaptic PrP and perivacuolar PrP deposits (j). (K, L: sCJD 6) HE staining shows spongiform changes (k). 3F4 reveals synaptic, perineuronal (inset), and plaque-type deposits (inset) (l). (M, N: sCJD 7) HE staining shows spongiform changes, gliosis, and large confluent vacuoles (m). 3F4 reveals PrP perivacuolar and diffuse synaptic deposits (n). (O, P: GSS 1) HE staining shows spongiform changes and PrP plaques (inset) (o). 3F4 reveals numerous PrP plaques (inset) and synaptic deposits (p). (Q, R: GPIALP 1) HE staining shows minimal spongiform changes and only mild neuronal loss (q). 3F4 reveals numerous PrP coarse granular deposits in the neuropil and around the vessels (r). (S, T: V180I CJD 1 treated with PPS) HE staining shows spongiform changes and gliosis (s). 3F4 reveals synaptic deposits and many PrP coarse deposits (t). (U, V: V180I CJD 2 without PPS) HE staining shows extensive and severe spongiform changes (u). 3F4 shows virtually no abnormal PrP deposits (v). (W, X: non prion case: control 3) HE shows that the neurons and neuropil are preserved, and there is no obvious neurodegenerative pathology (w). 3F4 shows a weak positive PrP staining in the cortex (x). (Scale bars: A-V: 50 µm, inset: C, D, H, O, P: 25 µm, L: 20 µm).
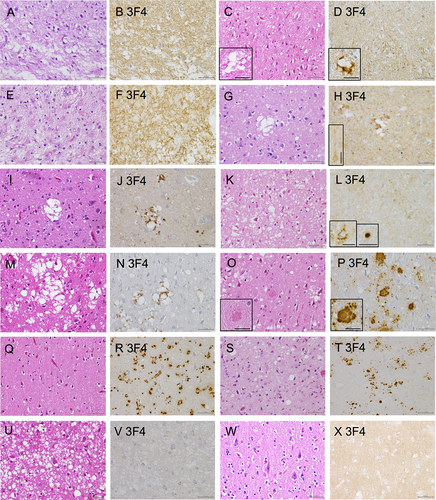
Figure 6. Schema for classing prion diseases using western blotting (WB) from formalin-fixed paraffin-embedded (FFPE) samples. In proteinase K-treated FFPE samples by western blotting with 3F4 antibody, the presence of 21–30 kDa three PrPres bands, high molecular smear PrPres signals, or low molecular PrPres signals strongly suggests prion disease. In 3F4, three PrPres bands + moderate high molecular smear PrPres signals suggest sCJD, strong high molecular smear PrPres signals + moderate three PrPres bands + moderate low molecular PrPres signals suggests GSS or GPIALP. In V180I CJD, it is difficult to detect, and only V180I CJD 1 PPS-treated case show weak high molecular smear PrPres signals. In sCJD cases, western blotting using Tohoku 1 (TH1) and Tohoku 2 (TH2) antibodies specific for type 1 and type 2 PrPres, respectively, can also distinguish between type 1 and type 2 PrPres with this method. In the case of V180I CJD, western blotting with EP1802Y antibody enhanced the high molecular smear PrPres signal, which was weak in 3F4. However, this was also only in PPS-treated case (V180I CJD 1), and it was difficult to identify the PrPres signal in untreated V180I CJD (V180I CJD 2).
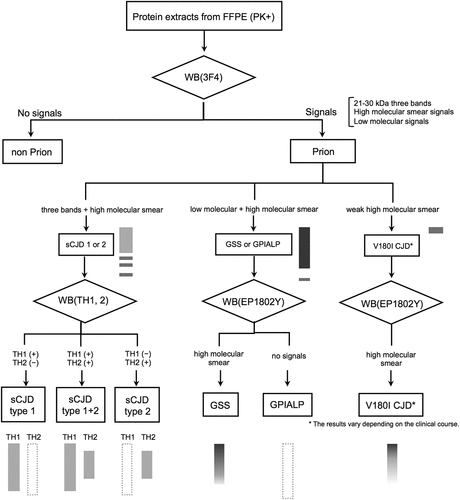
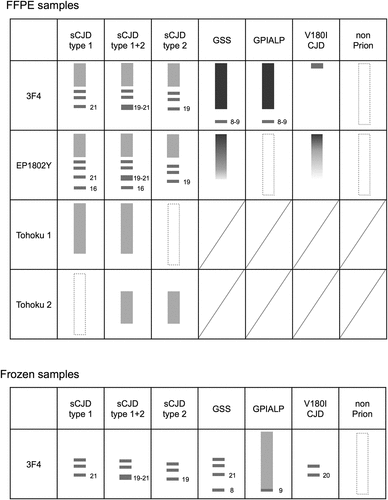
Figure 7. Representative band patterns of western blotting (WB) of formalin-fixed paraffin-embedded (FFPE) samples using multiple antibodies specific for different epitopes. Band patterns for the conventional method using frozen samples are displayed at the bottom of the table.
Data availability statement
Data supporting the findings of this study are available from the corresponding author upon reasonable request.