
Figures & data

Figure 1. Schematic of the flavivral lifecycle. The Flaviviral lifecycle starts with the (1) internalization of the virion by attachment and receptor-mediated endocytosis. (2) Under the mildly acidic environment of the endosome, the viral membrane fuses with that of the endosomal membrane, uncoats and the nucleocapsid is released into the cytoplasm. (3) In the cytoplasm, vRNA is translated into a polyprotein by the host translational machinery and inserted into the ER membrane. The polyprotein is subsequently cleaved by host and viral proteins to generate 3 structural proteins and at least 7 non-structural proteins. (4) These non-structural proteins form the viral replication complex. After initial viral replication, the viral proteins assist in generating Replication Organelles (RO) and vesicle packets (VP) in the ER shielding the virus from the cytoplasm. In the RO, +ssRNA is generated and transported to another invagination in the ER, the assembly site, the VP. (5) For encapsidation of the +ssRNA, capsid protein accumulates and complexes on lipid droplets (LD) and is recruited to the VP alongside the +ssRNA. Heterodimers of prM and E are recruited to the VP and trimerize on the membrane. Subsequently the VP can bud into the ER forming an immature virion. (6) Virions accumulate in ER cisternae before transport for maturation through the GBF1-dependent host secretory pathway or through secretory autophagy. (7) The virus can be released as free virions or additionally, in membrane enclosed forms derived from autophagosomes, in a host-derived lipid bilayer as quasispecies
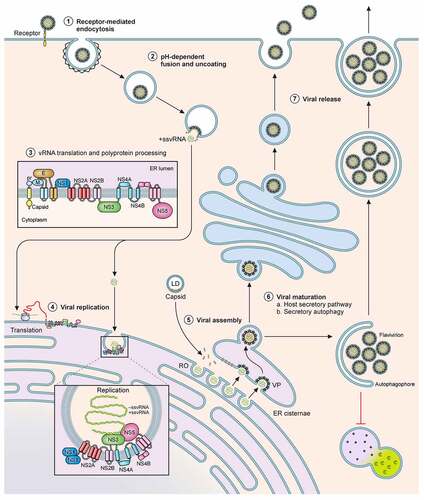
Figure 2. Schematic of the host innate immune response and flaviviral antagonism. Innate immune activation starts by recognition of flaviviral ds- or ssRNA either in the cytoplasm by RLRs; MDA5, RIG-I, PKR; or in the endosomes by TLRs; TLR3, TLR7 and TLR8. Subsequently the NFκB pathway and IRF pathways can be activated to initiate the production of Type I IFN and a range of inflammatory cytokines. The secreted IFNα and IFNβ can subsequently initiate the JAK-STAT signalling cascade to induce ISGs. Flaviviruses have developed multiple routes of intervention in both pathways. The viral non-structural protein NS2A can interact with PKR (YFV) and TBK (ZIKV/DENV) and block signalling. Similarly, NS4B of ZIKV, WNV and DENV serotypes 1,2,4 can prevent phosphorylation of TBK. NS4A of ZIKV is able to antagonize the MAVS signalling pathway by binding MAVS. NS2B/NS3 cleaves STING to prevent mitochondrial DNA-induced Type I IFN production. In the same pathway NS2B of DENV can interact with cGAS and prevent mtDNA recognition. The protease NS5 intervenes in JAK-STAT signalling by either preventing ISGylation of STAT2 (YFV); by inducing degradation of STAT2 (DENV/ZIKV); or by suppressing its maturation (WNV/TBEV). Additionally, secreted hexameric NS1 of WNV, YFV, DENV, can induce Complement factor C4 degradation and thus complement activation
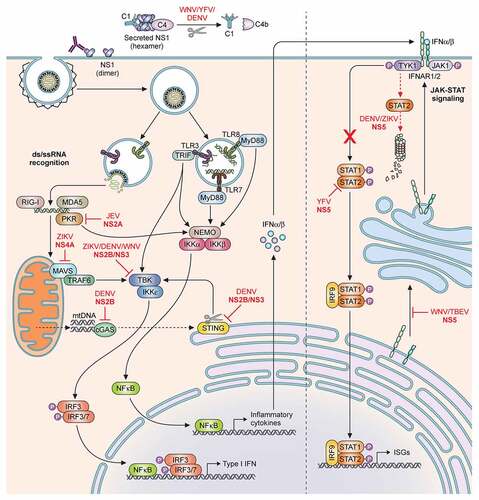
Figure 3. Adaptive immune responses to homologous and heterologous flavivirus infections. Severity of disease outcomes in dengue and Zika infection can be driven by pre-existing immunity against one of them. Primary infection results in production of high affinity neutralising antibodies against the infecting strain. When followed by a secondary infection with the same virus as that of the primary infection, existing memory B and T cells may lead to effective neutralisation of the virus, thereby providing protection. However, in the event of a secondary infection with a heterologous virus (either a different serotype of DENV or with ZIKV), cross-reactive antibodies at sub-neutralising concentrations result in binding and opsonization of the infecting viral particles. These antibody-virus complexes can be taken up by FcR-expressing cells such as monocytes, macrophages and mast cells, causing increased viral replication and hyperinflammatory responses. The pre-existing antibodies can also bind to infected cell surfaces causing NK cell mediated lysis via antibody-dependent cellular cytotoxicity
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analysed in the current study. All figures have been deposited in Figshare with DOI 10.6084/m9.figshare.17004592