
ABSTRACT
Introduction
The deployment of Artemisinin-based combination therapies and transmission control measures led to a decrease in the global malaria burden over the recent decades. Unfortunately, this trend is now reversing, in part due to resistance against available treatments, calling for the development of new drugs against untapped targets to prevent cross-resistance.
Areas covered
In view of their demonstrated druggability in noninfectious diseases, protein kinases represent attractive targets. Kinase-focussed antimalarial drug discovery is facilitated by the availability of kinase-targeting scaffolds and large libraries of inhibitors, as well as high-throughput phenotypic and biochemical assays. We present an overview of validated Plasmodium kinase targets and their inhibitors, and briefly discuss the potential of host cell kinases as targets for host-directed therapy.
Expert opinion
We propose priority research areas, including (i) diversification of Plasmodium kinase targets (at present most efforts focus on a very small number of targets); (ii) polypharmacology as an avenue to limit resistance (kinase inhibitors are highly suitable in this respect); and (iii) preemptive limitation of resistance through host-directed therapy (targeting host cell kinases that are required for parasite survival) and transmission-blocking through targeting sexual stage-specific kinases as a strategy to protect curative drugs from the spread of resistance.
1. Introduction
Malaria is a devastating infectious disease causing 241 million infections and 627,000 deaths worldwide in 2020 [Citation1]. The causative organisms are unicellular eukaryotic parasites of the Plasmodium genus. The Global Technical Strategy (GTS) for malaria aims to achieve a 90% reduction in global malaria mortality rate and case incidence by 2030 as compared to 2015 [Citation2]. Unfortunately, it is recognized that progress toward GTS milestones has stalled in recent years [Citation1,Citation2]. This may be attributed to a convergence of various biological threats, including pfhrp2/3 gene deletion hindering detection and diagnosis, spread of insecticide-resistant Anopheles vectors, and, of particular concern, the rise of drug resistance toward frontline antimalarials amongst Plasmodium populations [Citation1]. Artemisinin-based combination therapy (ACT) remains the frontline therapy for malaria globally. The first report of artemisinin resistance was amongst P. falciparum strains isolated from Cambodia in 2009 [Citation3] and has since spread throughout the Greater Mekong Subregion [Citation1]. More recently, there have been reports of independent emergence of artemisinin resistance in clinical isolates of parasites from Rwanda [Citation4,Citation5] and Uganda [Citation6], which may be the beginning of a worrying trend in Africa. This demonstrates the need to expand our arsenal of antimalarial treatments by the development of new drugs with novel mechanisms of action. We believe that inhibition of Plasmodium and host kinases could answer this call for action.
In this review, we highlight promising Plasmodium and human kinases as targets to develop new antimalarial agents for, and progress made by the medicinal chemistry community toward targeting these kinases. For a comprehensive survey of all inhibitors available for Plasmodium kinases, readers are advised to consult excellent reviews by Moolman et al. and Mustière et al. [Citation7,Citation8].
1.1. Human and plasmodium kinome
A comparative study of the kinomes of Homo sapiens, Plasmodium falciparum and Plasmodium vivax has been published recently [Citation9]; the reader is directed to this paper for a comprehensive discussion of the topic. Briefly, this showed that there are minimal differences between the kinomes of the two Plasmodium species, that both show significant divergence from the human kinome (). Notably, the Tyrosine Kinase (TK) group is absent in malaria parasites (whereas the human kinome comprises approximately 47 TKs). Furthermore, Plasmodium spp. also possess families such as the Calcium-dependent Protein Kinases (CDPK) and the FIKKs (named after a shared Phe-Ile-Lys-Lys motif) that are absent from the human kinome. The parasite kinomes includes members of most eukaryotic kinase groups, but in many cases these enzymes are widely divergent from their human homologues, suggesting selective inhibition can be achieved [Citation9].
Figure 1. Phylogenetic tree of the Plasmodium falciparum kinome, highlighting kinases that are targets of ongoing drug discovery. Highlighted in blue are the parasite kinases discussed as ongoing targets for drug discovery. The typical protein group families were annotated using Adobe illustrator along with the Aurora kinase family (ARK) and the Apicomplexan-specific kinase family FIKK. Bootstraps values ≥ 30 are represented and annotated on the respective branches; these values relate to the full tree developed using the kinomes of P. falciparum, P. vivax and H. sapiens published in [Citation9,Citation10], which should be consulted for details.
![Figure 1. Phylogenetic tree of the Plasmodium falciparum kinome, highlighting kinases that are targets of ongoing drug discovery. Highlighted in blue are the parasite kinases discussed as ongoing targets for drug discovery. The typical protein group families were annotated using Adobe illustrator along with the Aurora kinase family (ARK) and the Apicomplexan-specific kinase family FIKK. Bootstraps values ≥ 30 are represented and annotated on the respective branches; these values relate to the full tree developed using the kinomes of P. falciparum, P. vivax and H. sapiens published in [Citation9,Citation10], which should be consulted for details.](/cms/asset/5a6f1ca0-dc56-4a28-b7f2-abe8c027a3d5/iett_a_2185511_f0001_oc.jpg)
1.2. A Target-Based Approach
Historically, due to limited number of validated targets in Plasmodium, phenotypic screening has been a popular method to discover novel antimalarials. One major advantage of phenotypic screening is that the starting point has already demonstrated efficacy against the parasite, thereby overcoming hurdles in physiochemical properties that may limit target exposure. However, target-based drug discovery offers numerous key advantages over traditional phenotypic screening. Firstly, an identified target facilitates structure-guided rational compound optimization efforts, especially to gain selectivity over undesired off-targets (such as a human orthologue of an essential Plasmodium kinase). Target-based drug discovery also enables (i) the development of in vitro biochemical screens, which may identify hits that may lack sufficient potency to be identified in a phenotypic screen, and (ii) the utilization of novel screening technologies such as virtual screening, fragment-based screening, and DNA-encoded library screening technologies, which cover a larger proportion of chemical space to increase the likelihood of discovery of novel chemical matter. From a clinical development perspective, knowledge of the target is also crucial for developing combination therapies and for resistance monitoring. For an in-depth discussion of target-based antimalarial drug discovery, readers are encouraged to consult publications by the Malaria Drug Accelerator (MalDA), a consortium aiming to improve and accelerate early antimalarial drug discovery process by identifying novel targets [Citation11,Citation12].
Target identification is crucial toward realizing the benefits of target-based drug discovery. Two major target identification techniques have been used to identify antiplasmodial kinase targets. In vitro resistance generation with follow-up whole-genome sequencing is one such technique used frequently for identification of targets of kinase inhibitors. This has been successfully implemented for the imidazopyrazine KDU691 (PfPI4Kβ) [Citation13], the 2-aminopyridine MMV390048 (PfPI4Kβ) [Citation14], the 2-aminopyrazine UCT943 (PfPI4Kβ) [Citation15], and 7-azaindole TCMDC-135051 (PfCLK3) [Citation16]. Chemoproteomics is the other broad class of commonly-utilized target-identification technique, as reviewed in [Citation17]. The main benefits of chemoproteomics are that it offers evidence of direct target engagement, and it may be utilized even when the target is refractory to resistance generation. One contributing factor to the popularity of chemoproteomics in kinase target identification is due to the availability of the established Kinobead technology, which uses a selection of promiscuous kinase inhibitors immobilized onto Sepharose beads to pull down kinases from cell lysates [Citation18]. While originally designed for human kinase pulldowns, Kinobeads has been repurposed for pulldowns of kinases from Plasmodium lysate [Citation14,Citation18–22]. This technique has been implemented successfully in the identification of the kinase targets of the 2-aminopyridine MMV390048 (PfPI4Kβ) [Citation14], pyridine-imidazole MMV030084 (PfPKG) [Citation22], aminopyrimidine-thiazole compounds (PfPKG) [Citation20,Citation21], and purfalcamine (PfCDPK1) [Citation23].
2. Plasmodium kinase targets
2.1. Plasmodium falciparum phosphatidylinositol 4-kinase beta (PfPI4Kβ)
PfPI4Kβ (PF3D7_0509800) is the front-runner Plasmodium target in the field of kinase-targeted antimalarials, with one of its inhibitors MMV390048 being the first kinase-targeted antimalarial drug candidate reaching clinical trials [Citation24,Citation25] (see for a summary of the specific Plasmodium kinases discussed in this section and for the structures of their inhibitors). PfPI4Kβ is one of the seven kinases in the P. falciparum phosphoinositide lipid kinase (PIK) family, and is not classified as a protein kinase [Citation26]. Members of the PIK family phosphorylate phosphatidylinositols at specific positions, generating secondary signaling molecules crucial for a variety of cellular functions including vesicular trafficking, endocytic and exocytic processes, lipid distribution and metabolism [Citation27]. Specifically, PfPI4Kβ is important for membrane trafficking, and membrane biogenesis around developing merozoites in the late asexual blood stage [Citation13]. Other functions of PfPI4Kβ include regulating PfCDPK7 localization and activity, thereby regulating phospholipid biosynthesis [Citation28]. PfPI4Kβ has been genetically validated to be essential for the asexual blood stage of P. falciparum [Citation29], and pharmacologically validated to be essential for the liver stage, asexual blood stage, and gametocyte stages of Plasmodium [Citation13].
Figure 2. Inhibitors of specific Plasmodium kinases. Indicated below the structures are their IC50 values against the target kinases and their EC50 values against Plasmodium. Unless otherwise stated, EC50 values refer to activity in parasite viability assays in the asexual blood stage.
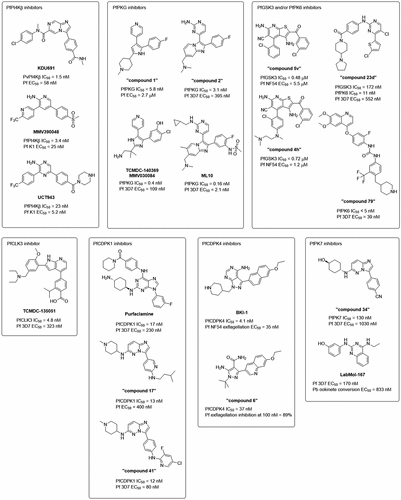
Table 1. Summary of Plasmodium kinase targets discussed, their genetic essentiality to the asexual blood stage of P. falciparum [Citation29,Citation52], biological function, reported inhibitors, availability of commercial screening and crystal structures, and ability to be pulled down from P. falciparum lysate using Kinobeads [Citation14,Citation18–22].
Two key classes of PfPI4Kβ inhibitors have been reported in the literature, the imidazopyrazine/imidazopyridazine scaffold and the 2-aminopyridine/2-aminopyrazine scaffold.
From one of the hits discovered through a large high-throughput cell-based screen of 1.7 million compounds against P. falciparum asexual blood stage parasites [Citation30], a medicinal chemistry campaign led to the discovery of inhibitors with the imidazopyrazine/imidazopyridazine scaffold, exemplified by the imidazopyrazine KDU691, with potent activity against asexual blood stage P. falciparum (EC50 = 58 nM) [Citation31]. Through generation of resistant mutants, PfPI4Kβ was identified to be the target of KDU691, and biochemical assays corroborated the finding (IC50 of PvPI4K = 1.5 nM) [Citation13]. KDU691 also has good potential to be developed into a drug candidate effective against multiple life stages of the parasite due to activity against P. yoelii liver stage schizonts (EC50 = 36 nM), P. cynomolgi hypnozoites (EC50 = 196 nM), P. falciparum gametocytes (EC50 = 220 nM), and P. falciparum oocysts (EC50 = 316 nM) [Citation13]. Unfortunately, inhibition of hypnozoite forms in an in vitro assay did not translate into in vivo radical cure of P. cynomolgi [Citation32]. KDU691 has demonstrated in vivo prophylactic effect against the P. berghei murine model of infection, where a single dose of 7.5 mg/kg rapidly eliminates liver stage parasites post-infection [13]. When dosed at 50 mg/kg twice daily, an 80% cure rate was also achieved against blood stage infection [Citation13]. When used as a monotherapy, the imidazopyrazines target the late stage of the asexual blood stage cycle and are inactive against the other intraerythrocytic stages [Citation13,Citation33], but when used in conjunction with dihydroartemisinin (DHA), KDU691 has also been reported to inhibit the recovery of DHA-pretreated quiescent ring-stage P. falciparum [Citation33], a known resistance mechanism to DHA [Citation34]. This demonstrates potential drug synergy with the current frontline antimalarials and potential to limit resistance development and transmission.
The second key class of PfPI4Kβ inhibitors possesses the 2-aminopyridine/2-aminopyrazine scaffold. This series was discovered through screen of 36,608 compounds from the BioFocus DPI SoftFocus kinase library against the P. falciparum asexual blood stage [Citation35]. Medicinal chemistry optimization for in vitro activity against the P. falciparum asexual blood stage led to the discovery of the 2-aminopyridine MMV390048 (also referred to as MMV048) (EC50 = 25–28 nM) [Citation35]. MMV390048 is also active against P. cynomolgi liver stage schizonts (EC50 = 64 nM) and hypnozoites (61 nM), P. falciparum early and late-stage gametocytes (EC50 = 140–285 nM), and formation of oocysts (EC50 = 111 nM) [Citation14]. MMV390048 demonstrates in vivo efficacy in P. berghei mouse model, eliminating all parasitemia with a single dose of 30 mg/kg [Citation35], and in a P. falciparum severe combined immunodeficient (SCID) mouse model, with an ED90 of 0.57 mg/kg [Citation14]. In vivo, MMV390048 also reduces P. berghei mouse-to-mosquito-to-mouse transmission at 2 mg/kg and possesses prophylactic effect against P. cynomolgi infection in macaques at 20 mg/kg [Citation14]. The target of MMV390048 was identified to be PfPI4Kβ by resistant mutant generation and by chemoproteomics experiments (Kdapp = 100–300 nM), which was corroborated with the biochemical inhibition of P. vivax PI4K (IC50 = 3.4 nM) [Citation14]. In absence of a crystal structure of PfPI4Kβ, the selectivity of MMV390048 over human PI4Ks [Citation14] was explained by homology modeling built from the structure of human PI4Kβ [Citation36]. As one of the most promising compounds available, MMV390048 has completed Phase I clinical development [Citation24,Citation25]. However, the first Phase II clinical trial was terminated due to a strategic business decision by the MMV [Citation37], although rapid and complete clearance of P. vivax asexual blood stage parasites and gametocytes were indeed demonstrated in all eight patients enrolled before study discontinuation with a single 120 mg oral dose, supporting clinical validation of PfPI4Kβ [Citation38]. Nonetheless, MMV390048 has paved the way for future clinical trials of PfPI4K inhibitors.
Further medicinal chemistry optimization was attempted on this series [Citation39–42], which eventually led to the discovery of 2-aminopyrazine UCT943, a next-generation inhibitor in the same series with better solubility and decreased hERG (Ether-à-go-go-Related Gene, a human potassium ion channel of which inhibition is implicated in fatal cardiac arrhythmia) inhibition [Citation41]. UCT943 inhibits PvPI4K (IC50 = 23 nM) [Citation15], is more potent than MMV390048 against the P. falciparum asexual blood stage (EC50 = 5.2–5.4 nM), P. berghei liver stage (EC50 = 0.92 nM), and P. falciparum early and late stage gametocytes (EC50 = 134 nM and 66 nM respectively) [Citation41], while equally potent in the transmission-blocking assay (EC50 = 96 nM) [Citation15]. In vivo, UCT943 demonstrates a complete cure in the P. berghei mouse model and a P. falciparum SCID mouse model at a dose of 4 × 10 mg/kg (ED90 = 0.25–1 mg/kg) [Citation41]. We believe that UCT943 has excellent potential as a drug candidate and look forward to clinical development of this compound.
Apart from the two key classes discussed above, other reported inhibitors of PfPI4Kβ include the bipyridine scaffold [Citation43], the naphthyridine scaffold [Citation44], and the Torin-2 scaffold [Citation45], all three also demonstrating in vivo efficacy in mouse models. This demonstrates that inhibition of PfPI4Kβ is not limited to the two above scaffolds, and further validates PfPI4Kβ as a target worthy of further investigation. As the only clinically validated Plasmodium kinase target, PfPI4Kβ is the most promising target for further development of antimalarials, especially considering its essentiality across multiple life stages of Plasmodium, although we believe that the continued exploration and validation of additional targets is a worthwhile endeavor.
2.2. Plasmodium falciparum cGMP-dependent protein kinase (PfPKG)
PfPKG (PF3D7_1436600) is a serine/threonine protein kinase from the AGC kinase group [Citation46] (). PfPKG contains three functional cGMP-binding domains, one degenerate cGMP-binding domain and a catalytic domain, with activity enhanced by cGMP binding [Citation47]. A crystal structure of the apo form of PfPKG has been solved (PDB: 5DYK) [Citation48]. PfPKG is expressed mainly in the late schizont stages [Citation49], and is involved in direct/indirect phosphorylation of proteins responsible for parasite egress, invasion, calcium signaling and proteolysis [Citation50,Citation51]. Both reverse genetics and saturation mutagenesis approaches have genetically validated PfPKG to be essential for the asexual blood stage of P. falciparum [Citation29,Citation52], and many reports of unsuccessful disruption of the PKG gene supports the classification of its essentiality [Citation53–56]. Inhibition of PfPKG leads to arrest during the schizont stage of blood stage development [Citation54,Citation57], and is responsible for discharge of serine protease PfSUB1 into the parasitophorous vacuole [Citation58,Citation59] to initiate processing of multiple egress-related proteins [Citation60]. Through phosphorylation of PfICM1 [Citation19], PfPKG is also responsible for calcium mobilization from intracellular stores, thus acting upstream of the CDPK signaling pathway for merozoite egress [Citation61]. In addition to PfPKG’s role in asexual blood stage development, PfPKG is also found to be essential for gametogenesis [Citation55]. P. berghei PKG has also been found to be essential for ookinete gliding motility [Citation56], for the release of merozoites from late liver stage schizonts [Citation62,Citation63] and for hepatocyte invasion by sporozoites [Citation63]. Collectively this suggests that inhibition may fulfill both transmission-blocking and prophylactic treatment roles in addition to therapeutic effects against the asexual blood stage.
Both the trisubstituted pyrrole ‘compound 1’ and the aminopyrimidine-imidazopyridine ‘compound 2’ were discovered initially for inhibition of PKG of other apicomplexan parasites Eimeria tenella and Toxoplasma gondii [Citation64–67], but have since been repurposed for functional studies of PfPKG; the discovery of many of the above-discussed functions of PfPKG was enabled by one or both of these compounds [Citation50,Citation54–56,Citation58,Citation59,Citation61,Citation63]. To distinguish on-target effects from potential inhibitor off-targets, they were often used in conjunction with a resistant mutant of PfPKG, in which the small Thr618 gatekeeper has been mutated to a larger Gln residue to preclude inhibitor binding. The IC50 of ‘compound 1’ against PfPKG notably increased from 5.8 nM to 17.8 μM with this single nucleotide substitution [Citation55].
Structurally related to ‘compound 1,’ a pyridine-imidazole compound MMV030084 was reported as a PfPKG inhibitor with activity against the P. falciparum asexual blood stage (EC50 = 109–120 nM), male gamete exflagellation (EC50 = 141 nM) and the P. berghei liver stage (EC50 = 199 nM) [Citation22]. Its on-target PfPKG activity was confirmed by chemoproteomics pulldown experiments as well as conditional knockdown susceptibility experiments [Citation22].
Further medicinal chemistry exploration of the aminopyrimidine-imidazopyridine scaffold of ‘compound 2’ then led to the discovery of ML10 [Citation68]. ML10 demonstrates potent inhibition of PfPKG (IC50 = 0.16 nM), the P. falciparum asexual blood stage (EC50 = 2.1 nM), and reduces oocyst numbers in mosquitoes (EC50 = 41 nM) [Citation68]. In an in vivo P. falciparum SCID mouse model, ML10 demonstrates a complete cure with twice daily oral doses of 100 mg/kg [Citation68]. A crystal structure was obtained with P. vivax PKG (PDB: 5EZR), confirming that the fluorophenyl group occupies a pocket beside the Thr gatekeeper residue, and providing a structural rationale for the drop in potency in this series upon mutation to a larger Gln residue [Citation68]. Further explorations of this aminopyrimidine-imidazopyridine scaffold have also since been described [Citation69,Citation70], and a structurally-related series of aminopyrimidine-thiazoles has been investigated both after a scaffold hop and from separate screening efforts [Citation20,Citation21,Citation71]. During the discovery of the aminopyrimidine-thiazole series, it was noted that PfPKG inhibitors typically exhibit a slow killing rate, but inhibitors that also target other kinases (e.g. SRPK2/CLK2) confer a fast killing rate [Citation20,Citation21]. Recent screening and scaffold hopping approaches have also led to the discovery of related aminopyrimidine-isoxazole [Citation72] and aminopyrimidine-imidazole scaffolds as PfPKG inhibitors [Citation73].
Additionally, compounds from the imidazopyridazine scaffold have also been demonstrated to inhibit PfPKG. They were discovered as dual inhibitors of PfPI4Kβ and PfPKG [Citation74] or PfCDPK1 and PfPKG [Citation75].
For dedicated reviews of PfPKG inhibitors, readers are encouraged to read reviews by Baker et al. [Citation76] and Rotella et al. [Citation77]. We believe that PfPKG is one of the most promising targets for next-generation antimalarials because of its proven essentiality for multiple life stages of Plasmodium, its refractoriness to resistance-conferring mutations [Citation22,Citation68] reflecting its critical role in the Plasmodium life cycle, and the small gatekeeper residue enabling selective inhibitor development.
2.3. Plasmodium falciparum glycogen synthase kinase 3 (PfGSK3)
PfGSK3 (PF3D7_0312400) is one of the three members of the GSK3 family within the CMGC group [Citation46] (). PfGSK3 has been genetically validated to be essential for P. falciparum asexual blood stage reproduction [Citation29,Citation52,Citation78], but its exact functions are still currently unclear. PfGSK3 is localized with Maurer’s clefts in the red blood cell cytoplasm [Citation79], membranous structures that are responsible for protein trafficking and host cell remodeling [Citation80]. One of the known substrates of PfGSK3 is PfAMA1 [Citation81], a key protein for merozoite invasion of erythrocytes [Citation82]. Inhibition of either PfGSK3- or PfPKA-mediated phosphorylation of PfAMA1 leads to disruption of ability of merozoites to invade erythrocytes [Citation81].
A series of thienopyridine inhibitors was identified from a high-throughput screen of 10,480 compounds [Citation83]. After medicinal chemistry optimization, the most promising analogue, ‘compound 5v,’ demonstrates selectivity for PfGSK3 (IC50 = 0.48 μM) over human GSK3α/β (IC50 > 100/3.3 μM) and is effective against the P. falciparum asexual blood stage (EC50 = 5.5 μM) [Citation83]. Through the use of docking studies with human and P. falciparum GSK3, the selectivity is rationalized to be due to interactions with the longer Met gatekeeper residue of PfGSK3, which is absent with the shorter Leu gatekeeper residue in the human orthologue, as well as binding of the ortho-chloro substituent in a cavity of the active site of PfGSK3 absent in human GSK3 [Citation83,Citation84]. Further optimization of this scaffold led to the discovery of ‘compound 4h,’ which also demonstrates selectivity for PfGSK3 (IC50 = 0.72 μM) over human GSK3 (IC50 = 40.2 μM) and activity against the P. falciparum asexual blood stage (EC50 = 1.2 μM) [Citation85]. It is noted that this scaffold may exhibit its anti-plasmodial activity through additional targets, since analogues inactive against PfGSK3 still maintained potency against P. falciparum [Citation86]. While in vivo validation has not yet been reported for this series of analogues, these works demonstrated that selectivity between human and P. falciparum GSK3 is possible, and we look forward to further development of this scaffold.
Compounds from a separate series of benzofuran analogues have also been reported as selective PfGSK3 inhibitors, but anti-plasmodial activity was not disclosed despite strong inhibition of PfGSK3 (IC50 = 0.49 nM) [Citation87]. Further biological characterization would be necessary to determine the full potential of this series.
Recently, we have also published a series of dual PfGSK3/PfPK6 inhibitors based on a 2-amino-4-aryl-pyrimidine scaffold discovered through a screen of a focused library of 110 kinase inhibitors [Citation88]. As an exemplar of this series, ‘compound 23d’ potently inhibits both kinases (PfGSK3/PfPK6 IC50 = 172/11 nM) and exhibits anti-plasmodial activity against P. falciparum asexual blood stage (EC50 = 552 nM). Further biological evaluation and optimization for on-target activity would be necessary to develop this series into a novel antimalarial, but dual inhibition of two putatively essential P. falciparum kinases is a promising start.
We believe that PfGSK3 holds potential as an antimalarial target, but further validation, especially in vivo, would be necessary. We note that PfGSK3 is one of the kinases which is commercially screenable on Luceome’s KinaseSeeker platform, facilitating future drug discovery efforts.
2.4. Plasmodium falciparum protein kinase 6 (PfPK6)
PfPK6 (PF3D7_1337100) is a hybrid kinase in the CMGC group of kinases with homology with both CDKs and MAPKs [Citation46,Citation89] (). Both reverse genetics and saturation mutagenesis approaches have confirmed the essentiality of PfPK6 for P. falciparum asexual blood stage reproduction [Citation29,Citation52]. Not much is known about the exact function of PfPK6. Due to its homology with human CDK2 and its expression profile in late ring, trophozoites, and early schizonts, PfPK6 is postulated to be involved in the cell cycle of the Plasmodium parasite in a cyclin-independent manner [Citation89].
In a screen of the Tres Cantos Antimalarial Set (TCAMS, see below) [Citation90] for biochemical inhibition of five kinases including PfPK6, Crowther et al. identified various scaffolds as PfPK6 inhibitors, including eight compounds from a 2,4-diaminopyrimidine series of compounds labeled ‘Scaffold J’ with inhibition of PfPK6 up to 138 nM [Citation91]. Recently, we have disclosed a series of structurally similar quinoline-based inhibitors with a type II pharmacophore as PfPK6 inhibitors, culminating in the discovery of ‘compound 79’ [Citation92]. ‘Compound 79’ demonstrates potent inhibition of PfPK6 (IC50 < 5 nM), P. falciparum in the asexual blood stage (EC50 = 39 nM), and P. berghei liver stage (EC50 = 220 nM). However, profiling of ‘compound 79’ against a panel of human and P. falciparum kinases revealed that it likely exerts anti-plasmodial effects not only via PfPK6 inhibition but via polypharmacology. Although further optimization for selectivity for PfPK6 and desired pharmacokinetic properties is necessary, this scaffold holds great promise for proceeding into further in vivo validation.
As mentioned previously, we have also reported a series of dual PfGSK3/PfPK6 inhibitors based on a 2-amino-4-aryl-pyrimidine scaffold [Citation88]. These compounds are generally more potent against PfPK6 than PfGSK3, suggesting that they act more likely via PfPK6 inhibition, although further biological characterization is required to identify their targets.
PfPK6 has potential to be a novel antimalarial target, but further biological characterization of its functions would be necessary. We note that PfPK6 is screenable commercially on Luceome’s KinaseSeeker platform and believe this will facilitate further drug discovery efforts. In vivo characterization of the inhibitors reported would also lend support for PfPK6 to be a novel target.
2.5. Plasmodium falciparum cyclin-dependent-like kinase 3 (PfCLK3)
PfCLK3 (PF3D7_1114700) is one of the four members of the CLK family, in the CMGC group of kinases [Citation46] (). PfCLK3 lies within a sub-group of CMGC kinases where auto-phosphorylation at a tyrosine residue within the activation loop, which occurs at an intermediate stage of biogenesis, is required for the mature kinase to have full catalytic activity as a serine/threonine protein kinase [Citation52]. The specialized nature of the activation mechanism of kinases within this subgroup, called dual specificity tyrosine regulated protein kinases (DYRK), might point to opportunities for selective targeting [Citation93]. Certainly, human DYRKs are emerging as important targets in a number of human diseases most notably Alzheimer’s disease [Citation94]. Members of the Plasmodium CLK family, PfCLK1, PfCLK2 [Citation95] and PfCLK3 phosphorylate Serine/Arginine-rich (SR) proteins [Citation96]. SR proteins are known to control alternative mRNA splicing and phosphorylation is a key post-translational modification that regulates their activity [Citation97–99]. The human CLKs (of which there are 4) and the closest PfCLK3 human orthologue PRPF4B/PRP4K [Citation100] are essential for mRNA splicing by interacting with splicing factors [Citation101] and phosphorylation of proteins of the spliceosome complex [Citation102], and thus it is believed that PfCLK3 may serve similar functions in Plasmodium. Through both reverse genetics and saturation mutagenesis approaches, PfCLK3 is genetically validated to be essential for P. falciparum asexual blood stage reproduction [Citation29,Citation52]. It has been demonstrated that activity of the PfCLK family of kinases is essential for progression of P. falciparum from the trophozoite to schizont stages, as well as for gametocyte viability and exflagellation [Citation16,Citation96]. Inhibition of PfCLK3 could lead to downregulation of up to 779 gene transcripts [Citation16], supporting its role in transcriptional regulation.
A screen of 24,619 compounds against PfCLK3 led to the discovery of 7-azaindole TCMDC-135051, a selective inhibitor of PfCLK3 (IC50 = 4.8 nM) [Citation16]. As part of the Tres Cantos Antimalarial Set (TCAMS, see below), TCMDC-135051 demonstrated activity against the P. falciparum asexual blood stage (EC50 = 323 nM) [Citation90], as well as activity against P. berghei liver stage (EC50 = 400 nM), P. falciparum early and late stage gametocytes (EC50 = 800–910 nM), and exflagellation (EC50 = 200 nM) [Citation16]. In vivo, TCMDC-135051 resulted in a near-complete clearance of parasites in a P. berghei mouse model with a twice daily dose of 50 mg/kg [Citation16]. Further medicinal chemistry optimization has since been reported, changing its carboxylic acid to the bioisosteric tetrazole [Citation103]. In particular, TCMDC-135051 has shown to be selective for PfCLK3 over the human ortholog PRPF4B [Citation16], showing great promise for development of a selective inhibitor. While the azaindole scaffold of TCMDC-135051 represents the only known PfCLK3 inhibitor scaffold so far, we look forward to further development of this series and believe that PfCLK3 holds great promise as a novel antimalarial target.
In a recent development, Altiratinib was defined as an inhibitor of the Toxoplasma gondii PfCLK3 orthologue TgPRP4K – with parasiticidal activity that prevents the development of intracellular zoites in the nanomolar range [Citation104]. The significance of this finding is that T. gondii and species of Plasmodium that includes P. falciparum are all members of the apicomplexan phylum of parasitic alveolates which are characterized by possessing an organelle called an apicoplast. Evaluation of the binding mode of TCMDC-135051 to PfCLK3, and Altiratinib to TgPRP4K, has led to the suggestion that a pan-apicomplexan inhibitor to CLK3 orthologues across apicomplexan species might be possible [Citation104].
2.6. Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1)
PfCDPK1 (PF3D7_0217500) is one of the seven kinases from the CDPK family within the CAMK group of kinases () [Citation46,Citation105,Citation106]. Since there are no mammalian orthologues of CDPKs, CDPKs are an enticing target for selective inhibition of Plasmodium kinases over human kinases. We note that PfCDPK1 is commercially screenable by the DiscoverX platform, and mining KINOMEscan data from literature might be fruitful in finding PfCDPK1 inhibitors. While a crystal structure of PfCDPK1 is not available, a crystal structure of P. berghei CDPK1 has been solved (PDB: 3Q5I), which may aid structure-based drug discovery efforts.
Like most CDPKs, PfCDPK1 has a kinase domain and a calmodulin-like domain with four calcium-binding EF-hands, and its activity is upregulated by Ca2+-binding [Citation107]. PfCDPK1 is genetically validated to be essential for the asexual blood stage of P. falciparum [Citation29,Citation52]. Early studies reveal that PfCDPK1 phosphorylates erythrocytic membrane proteins [Citation108] and is proposed to control membrane biogenesis processes by virtue of its membranous localization at the parasitophorous vacuolar membrane (PVM) [Citation109]. PfCDPK1 has been demonstrated to be essential for early schizogony [Citation110] as well as for microneme secretion and invasion in merozoites [Citation111]. In schizonts and merozoites, PfCDPK1 localizes to the plasma membrane [Citation110–112], and is also known to phosphorylate PfMTIP and PfGAP45 [Citation112–114], components of the glideosome, a key motor complex required for merozoite egress and invasion [Citation115]. Phosphoproteomics analysis also confirms PfCDPK1 is involved in phosphorylation of proteins involved in invasion and motility [Citation116]. PfCDPK1 also regulates PfPKA activity, with implications on microneme secretion and invasion [Citation116]. Accordingly, inhibition of PfCDPK1 by K252a inhibits merozoite invasion, and egress at higher concentrations [Citation112]. Additionally, PfCDPK1 has also been demonstrated to be essential for both male and female gametogenesis and transmission to mosquitoes [Citation117]. In P. berghei, CDPK1 has been identified to be essential for ookinete development through translational activation of stored mRNAs in the female gametocyte [Citation118]. In liver stages, PbCDPK1 has been shown to be essential for sporozoite motility and invasion of hepatocytes [Citation119].
While knockout of PfCDPK1 has been unsuccessful [Citation23,Citation29,Citation52,Citation117], knockout of mutant PfCDPK1 T145M is possible [Citation117], and there have been contrasting reports on the ability of PbCDPK1 to be knocked out [Citation53,Citation120], thereby disputing its essentiality and calling into question if CDPK1 is a good cross-species target. This also brings into question the suitability of using P. berghei mouse models for evaluation of PfCDPK1 inhibitors. With that said, target vulnerability for PfCDPK1 is also equivocal. Despite unsuccessful attempts at PfCDPK1 knockout, it has been reported that partial knockdown of PfCDPK1 and PbCDPK1 have shown no effect on normal cellular reproduction in the asexual blood stages [Citation110,Citation118], although another study showed partial knockdown of PfCDPK1 leads to decrease in invasion and parasitemia [Citation116]. The main implication for drug discovery efforts is that chemical inhibition of PfCDPK1 seems required to be extensive to manifest therapeutic effects.
A screen of approximately 20,000 compounds against PfCDPK1 led to the discovery of 2,6,9-trisubstituted purines as PfCDPK1 inhibitors, culminating in the discovery of purfalcamine [Citation23]. Purfalcamine is a potent inhibitor of PfCDPK1 (IC50 = 17 nM) active against the P. falciparum asexual blood stage (EC50 = 230 nM), arresting parasites at the schizont stage [Citation23], inhibits microneme discharge and inhibits erythrocyte invasion [Citation111]. Unfortunately, in vivo, purfalcamine did not demonstrate efficacy beyond a delay in onset of parasitemia [Citation23], demonstrating that further optimization is necessary.
The imidazopyridazine series have also been reported as PfCDPK1 inhibitors. A screen of 54,733 compounds yielded 11 imidazopyridazines with IC50 values <100 nM [Citation121]. Separately, the imidazopyridazine scaffold was discovered in another screen of 35,422 compounds [Citation122,Citation123]. Optimization led to the discovery of ‘compound 17,’ which was active against PfCDPK1 (IC50 = 13 nM), and P. falciparum asexual blood stage (EC50 = 400 nM) [Citation122]. ‘Compound 17’ demonstrated a modest effect in vivo, with a 46% parasitemia reduction in a P. berghei mouse model with an daily oral dose of 50 mg/kg [Citation122]. Further medicinal chemistry optimization has been reported [Citation124,Citation125], which led to the discovery of ‘compound 41’ [Citation125]. ‘Compound 41’ demonstrated equal potency against PfCDPK1 (IC50 = 12 nM), an improved P. falciparum asexual blood stage potency (EC50 = 80 nM), but only slight improvement in an in vivo P. berghei mouse model (51% parasitemia reduction with a daily oral dose of 50 mg/kg) [Citation125]. The disappointing in vivo results were possibly attributed to insufficient extent of target inhibition or differences in P. berghei versus P. falciparum biology [Citation125].
In the above studies, the authors noted poor correlations between PfCDPK1 inhibition and anti-plasmodial activity [Citation121,Citation123], suggesting that other targets may be involved for the imidazopyridazines. Indeed, a later study showed that the imidazopyridazines could be classified into two classes; the first class of compounds have their primary target as PfPKG and act via prevention of egress while the second class of compounds inhibits the parasite growth at the trophozoite stage and may act via binding to HSP90 [Citation75]. Indeed, reduction in PfCDPK1 activity can be compensated by PfPKG [Citation126], supporting the notion that inhibitors of PfCDPK1 also require inhibition of PfPKG for maximal effectiveness. Additionally, bumped kinase inhibitors capable of selectively inhibiting the PfCDPK1 T145G mutant in vitro did not have any differential effect between parasite lines expressing the T145G mutant and wild-type parasites, further suggesting that inhibition of PfCDPK1 may not impact parasite viability [Citation75].
In summary, after extensive biological and chemical biology investigations, PfCDPK1 does not seem as promising a target as other enzymes such as PfPI4Kβ or PfPKG. With challenges against its essentiality and target vulnerability, PfCDPK1 may be a questionable target against the asexual blood stage, although PfCDPK1 still remains a potential target against sexual stages of the parasite.
2.7. Plasmodium falciparum calcium-dependent protein kinase 4 (PfCDPK4)
Like PfCDPK1, PfCDPK4 (PF3D7_0717500) is another member of the seven kinases from the CDPK family within the CAMK group of kinases () [Citation46,Citation105,Citation106]. Unlike PfCDPK1, PfCDPK4 has been genetically validated to be dispensable for the asexual blood stage of P. falciparum [Citation29,Citation52,Citation127]. PfCDPK4 is expressed in gametocytes and localizes to its periphery, and its activity is upregulated upon Ca2+ binding [Citation128,Citation129]. While PfCDPK4 is not essential for gametocytogenesis, it is essential for male gamete exflagellation by mediating phosphorylation of proteins involved in replication, transcription, mRNA processing, translation, motility, as well as other kinases [Citation127]. Analogously, P. berghei CDPK4 has also been established to be essential for both male gametocyte exflagellation and ookinete infection of mosquitoes [Citation130]. During exflagellation, it is vital for the initiation of DNA replication and mitotic spindle assembly, as well as activation of axoneme motility [Citation131]. Additionally, PbCDPK4 has been shown to be essential for sporozoite motility and invasion of hepatocytes [Citation63,Citation119]. Interestingly, chemical inhibition, but not conditional knockout, of PbCDPK4 has a detrimental effect on merosome formation in pre-erythrocytic stages [Citation119]. It has also been found that PbCDPK4 interacts with PbPKG to control merozoite invasion in the erythrocytic stage despite its dispensability [Citation132], suggesting possible synergy of development of dual PKG and CDPK4 inhibitors for symptomatic treatment of malaria.
The major class of PfCDPK4 inhibitors reported are bumped kinase inhibitors (BKIs). One early example with a pyrrolopyrimidine core is BKI-1, a potent inhibitor of PfCDPK4 (IC50 = 4.1 nM) and P. falciparum exflagellation (EC50 = 35 nM) [Citation133]. In vivo, BKI-1 completely blocks formation of oocysts when dosed i.p. for 10 mg/kg [Citation133]. Further medicinal chemistry efforts have been reported [Citation134–136], culminating in a scaffold hop to the 5-aminopyrazole-4-carboxamide core, as exemplified by ‘compound 6,’ which inhibits PfCDPK4 (IC50 = 37 nM) and P. falciparum exflagellation (89% inhibition at 100 nM) [Citation136]. BKIs generally achieve high selectivity for PfCDPK4 (and other apicomplexan orthologues) over most other kinases, including human kinases, by binding of the bulky naphthyl group in the pocket adjacent to the serine gatekeeper residue, a smaller residue than most human kinases [Citation137]. Mutation of the Ser gatekeeper to a Met residue abolishes binding to BKIs [Citation134,Citation135]. One challenge noted with BKIs, like all non-gametocyte-killing transmission-blocking drug candidates, is to maintain a prolonged exposure with sufficient concentration in the bloodstream until gametocytes are cleared after injection [Citation135].
We believe that PfCDPK4 is a potential target for the development of transmission-blocking antimalarials. To facilitate inhibitor development, crystal structures of PfCDPK4 have been solved (PDB: 4QOX and 4RGJ). As have been demonstrated by the BKIs, the small serine gatekeeper residue would likely be the key for developing selective inhibitors.
The fact that selective CDPK4 inhibitors will not affect asexual growth is actually an asset: compounds that target a kinase that is required for both asexual proliferation and sexual differentiation will initially have both curative and transmission-blocking activities; however, acting on asexual proliferation will, potentially rapidly, select for target mutations that render the kinase less susceptible to the inhibitor. This will also impair the transmission-blocking activity of the compound since the target is the same at both stages. Therefore, the development of compounds targeting transmission exclusively is a powerful strategy in the fight against resistance; since gametocytes do not proliferate, the selection of resistant targets is predicted to be much slower for such compounds, which could be used to protect curative drugs against the spread of resistance. In addition to CDPK4, several other kinases that are dispensable for asexual proliferation are essential for sexual development (e.g. Pfnek-2 [Citation138] and Pfnek-4 [Citation139]), and thus represent prime target for transmission-blocking intervention. See [Citation53] for a kinome-wide study of kinases implicated in sexual development in the murine model P. berghei.
2.8. Plasmodium falciparum protein kinase 7 (PfPK7)
PfPK7 (PF3D7_0213400) was initially classified as an orphan kinase [Citation46] but recently reclassified under the CAMK group () [Citation9]. PfPK7 is a hybrid kinase, displaying homology to both MAPKK/MEK and fungal PKA subfamilies [Citation140]. Even though PfPK7 has been shown to be non-essential for P. falciparum blood stage reproduction through reverse genetics and saturation mutagenesis approaches [Citation29,Citation52], it was reported that disruption of PfPK7 halves the growth rate of P. falciparum in the erythrocytic stages, and decreases the number of merozoites generated per schizont [Citation141,Citation142]. Through phosphoproteomics studies, PfPK7 was proposed to act upstream of PfCK1, PfCK2 and PfPKB in signaling cascades [Citation142]. PfPK7 has also been implicated in melatonin signaling and melatonin-induced regulation of the cell cycle [Citation143,Citation144]. Beyond the asexual blood stage, PfPK7 has been shown to be essential for infection of mosquitoes and formation of oocysts [Citation141].
A screen of a kinase-targeted library of 568 compounds led to the discovery of imidazopyridazine compounds as micromolar inhibitors of PfPK7 [Citation145]. With these inhibitors, the structure of PfPK7 has been solved (PDB: 2PMN and 2PMO) [Citation145]. Separately, a high-throughput screen against PfPK7 identified another series of imidazopyridazines as PfPK7 inhibitors [Citation146]. Subsequent medicinal chemistry optimization cumulated in the discovery of ‘compound 34,’ which inhibits PfPK7 (IC50 = 130 nM) and is active against P. falciparum blood stage proliferation (EC50 = 1.03 μM), although the authors noted that this compound may act promiscuously [Citation146].
Recently, a virtual screening approach has been applied to PfPK7, using shape-based models to screen for PfPK7 inhibitors and machine learning models to select compounds active against the asexual blood stage [Citation147]. Eight hits were experimentally validated, and the 2,4-diaminoquinazoline LabMol-167 was found to be active against the asexual blood stage of P. falciparum (EC50 = 170 nM) and inhibits P. berghei ookinete conversion (EC50 = 855 nM) [Citation147]. While experimental conformation of PfPK7 inhibition is lacking, docking suggests that LabMol-167 is capable of binding with PfPK7, as well as PfPK5 putatively [Citation147].
In summary, we believe that PfPK7 is a potential target for the development of new antimalarials. The exact function of PfPK7 remains to be elucidated, but the existence of crystal structures should aid further discovery efforts to develop lead compounds and probes to facilitate study of PfPK7.
3. Kinase inhibitor scaffolds without a known target
As discussed earlier, target-based drug discovery offers several advantages over traditional phenotypic screening methods, and we highlighted several target identification techniques. We note that compounds belonging to known scaffolds of kinase inhibitors are commonly found amongst phenotypic screening hits, but efforts to identify their targets remain limited. Nevertheless, it is crucial to identify the targets of these compounds to facilitate further drug discovery efforts. In this section, we review reports of compounds belonging to a few known kinase inhibitor scaffolds for which target identification is still indeterminate, and hope that efforts may be put forth in the future to illuminate the target(s).
The largest sources of such kinase inhibitor scaffolds may be found by scaffold analysis and target identification of high-throughput phenotypic screening hits. In 2008, Novartis reported a screen of 1.7 million compounds and found 5973 hits active against the P. falciparum blood stage with EC50 values < 1.25 μM [Citation30]. 530 different chemical clusters were identified, amongst them a cluster of compounds similar to staurosporine, a promiscuous pan-kinome inhibitor, was highlighted [Citation30]. Notably, amongst these screening hits are imidazopyridazines which was eventually developed into the PfPI4Kβ inhibitor KDU691 [Citation31]. A follow-up target identification effort using nine target-based biochemical screens was conducted, and identified several inhibitors of the metabolic kinases choline kinase and guanylate kinase [Citation148], but no other kinases were screened. In 2010, St Jude Children’s Research Hospital reported a screen of 309,474 compounds and found 561 hits with EC50 values < 2 μM against the P. falciparum asexual blood stage [Citation149]. Target identification efforts on this set of compounds have been limited, but at least one compound has been discovered to be a weak binder of PfGSK3 (SJ000035864, Kd = 6.33 μM) [Citation149]. Also in 2010, from a screen of almost 2 million compounds, GlaxoSmithKline reported the identification of 13,533 hits with an estimated EC50 values < 2 μM against the asexual blood stage of P. falciparum [Citation90]. This set of hits was thereafter named the Tres Cantos Antimalarial Set (TCAMS) [Citation90]. The authors noted that kinase inhibitors are particularly enriched within the TCAMS [Citation90]. A screen of the TCAMS against PfCDPK1, PfCDPK4, PfMAP2, PfPK6 and PfPK7 in biochemical assays was later conducted, which identified twelve scaffolds that possess different inhibition profiles across the five kinases [Citation91]. However, poor correlations were observed between the biochemical IC50 values and the antiplasmodial EC50 values, suggesting that these scaffolds still have other targets yet to be identified [Citation91]. A 2014 screen by the Medicines by Malaria Venture of 256,263 compounds afforded 178 hits with EC50 values < 1 μM and a selectivity window between antiplasmodial activity and cytotoxicity against HEK-293 cells of at least 10-fold [Citation150]. After subsequent compound filtering, ten clusters of compounds based on chemotypes were reported but further target identification was not performed [Citation150]. Recently in 2021, the National Center for Advancing Translational Sciences reported a screen of 456,817 compounds against the P. falciparum asexual blood stage, yielding 994 active compounds with EC50 values < 2 μM and demonstrated no cytotoxicity against HepG2 cells, which may be organized into 70 compound clusters [Citation151]. These were subsequently also screened against P. berghei in liver stage viability assays [Citation151]. As far as we are aware, the targets for most of these compounds remain unidentified. In a more directed screening approach, in 2017, the University of Dundee reported a screen of 4731 kinase inhibitors, which led to the identification of nine scaffolds with sub-micromolar potency against the P. falciparum asexual blood stage [Citation152]. Of the nine scaffolds, three novel series demonstrated chemical tractability and the authors report early SAR studies [Citation152]. Altogether, these large-scale phenotypic screens provide a rich source of data for scaffold mining and provide active compounds on which the community should focus further target identification efforts.
Apart from large phenotypic screens, we would like to highlight focused SAR studies on several individual kinase inhibitor scaffolds of which further target identification may prove fruitful. See for the structures of some of the scaffolds discussed in this section. One such scaffold is the 2,6,9-trisubstituted purine scaffold. This scaffold is a common CDK inhibitor scaffold. During the discovery of purfalcamine, various other 2,6,9-trisubstituted purines have been identified with antiparasitic activity but with lower PfCDPK1 inhibition [Citation23]. The targets for these compounds were not identified. In fact, the most potent compound against the P. falciparum blood stage discovered in the study was ‘compound 14’ (EC50 = 101 nM), which is a poor inhibitor of PfCDPK1 (IC50 = 3846 nM) [Citation23]. The authors postulated that the quinoline ring constituted the pharmacophore for other antimalarials such as chloroquine, which may explain its antiparasitic activity [Citation23], but there remains the possibility of other kinases being involved. There have been other reports of 2,6,9-trisubstituted purines as antimalarials without any target identification [Citation153–155]. One of the most potent compounds in an early study by Harmse et al. was ‘compound 59,’ which is moderately active against P. falciparum asexual blood stage (EC50 = 530 nM) [Citation153]. Other less potent compounds with EC50 values in the μM range have been reported by Mallari et al. [Citation154] and Houzé et al. [Citation155]. While the targets of these inhibitors have not been identified, other members of this scaffold, e.g. roscovitine, olomoucine and purvalanol A, have been reported as micromolar-potency inhibitors of CMGC group kinases PfPK5 [Citation156] and PfPK6 (ref [Citation89]. and our unpublished observations). We believe that this scaffold holds potential to be developed into an antimalarial drug, but target identification and in vivo characterization are paramount before advancing members of this scaffold forward.
Figure 3. Scaffolds of small molecule kinase inhibitor series with antimalarial activity. Indicated below the structures are their EC50 values determined in asexual blood stage viability assays on various parasite clones.
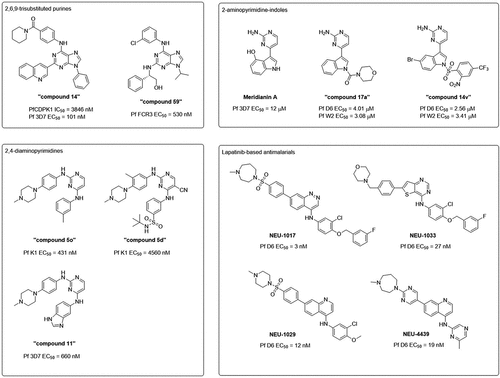
Another kinase inhibitor scaffold investigated for antimalarial drug discovery is the 2,4-diaminopyrimidine scaffold. This scaffold is represented in the TCAMS [Citation90], and a screen of the TCAMS against five Plasmodium kinases found members of the scaffold (‘Scaffold H’ and ‘Scaffold I’) to inhibit PfCDPK1, PfCDPK4, and PfPK6 [Citation91]. Within a SAR campaign of 2,4-diaminopyrimidines inspired by hits from the TCAMS, ‘compound 5o’ was found to be the most potent inhibitor of the P. falciparum asexual blood stage (EC50 = 431 nM), although this compound also inhibits human JAK2, JAK3, and EGFR and is moderately cytotoxic against Vero and A549 cells [Citation157]. As this scaffold is a common kinase inhibitor scaffold, one concern about this series is potential promiscuity and thus cytotoxicity, but analogues such as ‘compound 5d’ have proven to have a lower extent of human kinase inhibition and cytotoxicity at a cost of a moderate drop in antiplasmodial activity (EC50 = 4.56 μM) [Citation157]. This demonstrates that the selectivity window of this series may be optimized. Indeed, the selectivity window was improved with a recent medicinal chemistry study culminating in the discovery of ‘compound 11,’ which inhibits the P. falciparum asexual blood stage (EC50 = 660 nM) with minimal cytotoxicity against HepG2 cells (EC50 > 25 μM) [Citation158]. Using chemo- and bioinformatic approaches, the authors suggested that this series may target the CDPK and NEK family of compounds [Citation158], although this requires further experimental validation. Since the 2,4-diaminopyrimidine scaffold is common kinase inhibitor scaffold, we believe that further target identification using inhibitors discovered in these studies might reveal and validate interesting novel targets as antimalarials.
Related to the 2,4-diaminopyrimidine scaffold is the 2-aminopyrimidine-indole scaffold of the meridianin family of natural products. Meridianin A has been reported to inhibit P. falciparum asexual blood stage reproduction (EC50 = 12 μM) [Citation159]. This has been improved following a medicinal chemistry campaign, affording ‘compound 17a,’ which is more potent against the P. falciparum asexual blood stage (EC50 = 3.08–4.01 μM) [Citation160]. A subsequent study led to the discovery of ‘compound 14v,’ which maintained potency against the P. falciparum asexual blood stage (EC50 = 2.56–3.41 μM) [Citation161]. During the SAR analyses, it was noted that the aminopyrimidine ring is essential for antiplasmodial activity [Citation160]. Since the aminopyrimidine ring is potentially a hinge-binding group for kinase inhibition, this finding suggests that these compounds may act via a kinase target. Indeed, meridianins have been reported as kinase inhibitors of various human kinases [Citation162], although the target(s) responsible for their antiplasmodial activity remains to be elucidated. Interestingly, the 2-amino-4-arylpyrimidine putative hinge-binding group is reminiscent of PfPKG inhibitors ‘compound 2’ and ML10 and is similar to the dual PfGSK3/PfPK6 inhibitor ‘compound 23d.’ Despite the lower potencies of these natural product analogues, target identification may provide valuable insight for the field of natural products repurposing for kinase inhibition and for antimalarial activity.
Another scaffold extensively studied by the Pollastri group is the lapatinib scaffold. Lapatinib is a FDA-approved drug for breast cancer, targeting EGFR and HER2 [Citation163]. It was first repurposed for inhibition of Trypanosoma brucei proliferation [Citation164], but follow-up screening revealed additional activity of the lapatinib analogues against other protozoan parasites including P. falciparum [Citation165]. By substituting the quinazoline core of lapatinib with alternative heterocycles, potent inhibitors against the asexual blood stage of P. falciparum have been discovered [Citation165]. The most potent example was the cinnoline NEU-1017 (Cmpd 68), which is extremely potent against the P. falciparum blood stage (EC50 = 3 nM) [Citation165]. The focus of medicinal chemistry campaigns of compounds of the quinazoline and thienopyrimidine cores have been for T. brucei proliferation inhibition, but compounds are generally active against P. falciparum in the sub-micromolar or low micromolar range of potencies [Citation166–168]. The most potent example from the thienopyrimidine scaffold against the P. falciparum asexual blood stage was NEU-1033 (Cmpd 11j) (EC50 = 27 nM) [Citation166]. Further optimization of compounds with the quinoline core for various antiparasitic activity have been reported, and these analogues are generally more potent against P. falciparum as compared to the quinazolines and thienopyrimidines [Citation169–173]. Quinoline NEU-1029 (Cmpd 7) exhibited excellent potency against the P. falciparum blood stage (EC50 = 12 nM) and P. berghei liver stage (EC50 = 1.8 μM) [Citation170]. However, in vivo NEU-1029 only led to a suppression of parasitemia and life extension of 5 days in a P. berghei mouse model instead of a complete cure, which was attributed to rapid clearance of the compound [Citation170]. Subsequent optimization led to the discovery of NEU-4439 (Cmpd 21), which was potent against the P. falciparum asexual blood stage (EC50 = 19 nM) and demonstrates favorable ADME properties [Citation173]. As far as we are aware, target identification for any of these inhibitors in P. falciparum have not been reported. In T. brucei, lapatinib has been shown to bind to several kinases through competitive elution from affinity chromatography experiments [Citation174], and we believe similar experiments would help to identify the targets in P. falciparum as well. Given the excellent potencies reported by these inhibitors, we believe that target identification might lead to the discovery of high-quality targets. We look forward to further in vivo development of these inhibitors as they hold excellent potential to be developed into novel antimalarials.
In this section, we have reviewed several large phenotypic screens for which kinase inhibitor scaffolds may be identified from, as well as focused SAR studies on selected kinase inhibitor scaffolds. We believe that target identification of exemplars from these scaffolds would not only shed light onto alternative viable antiplasmodial targets, but also facilitate further drug discovery efforts of these chemical series involved.
4. Human kinase targets
When attempting to uncover the mode of action of kinase inhibitors scaffolds that display parasiticidal activity in vitro but whose target(s) is/are unknown, one must keep in mind that targets may be present in both the parasite and the host erythrocyte. Several host kinases have been implicated in infection at both the blood and the liver stages, and pharmacological [Citation175] or reverse genetics [Citation176] interference with several host kinases does impair parasite survival. A promising aspect of host-directed therapy (HDT) is that drugs that target an enzyme of the host cell (rather than the pathogen) are less susceptible to the emergence of resistance because mutations in the target that confer resistance cannot be selected by drug pressure if the target is not under the pathogen’s genetic control. Indeed, preliminary experiments (Adderley and Doerig, unpublished) suggest that highly selective inhibitors of host erythrocyte kinases are largely refractory to the emergence of resistance in in vitro selection experiments. Of particular interest are kinases belonging to groups that are absent from the parasite’s kinome, such as Tyrosine Kinases. To illustrate the avenues that are being opened by such a strategy, it is of interest to cite a recent study showing that co-administering Imatinib (a well-tolerated Tyrosine Kinase inhibitor initially developed for the treatment of leukemia) with standard artemisinin combinations led to a significantly accelerated decline in parasite density in treated patients without added toxicity [Citation177].
This topic, including a full list of host targets and compounds and a full discussion of the salient features of host-directed therapy, is the subject of recent comprehensive reviews [Citation178,Citation179], and therefore will not be covered in detail here.
5. Conclusion
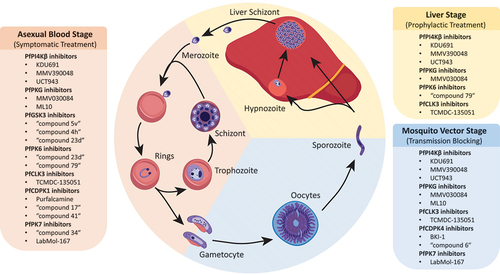
Drug resistance poses a significant threat against humanity’s continued progress toward global elimination of malaria. In search for novel antimalarials, targeting Plasmodium and human kinases have emerged as a promising field of research. Here, we have reviewed several Plasmodium kinases investigated as potential drug targets (see for a graphical summary). PfPI4Kβ is the only clinically-validated kinase target so far; despite the termination of the first phase II clinical trial of MMV390048, we look forward to the clinical development of other PfPI4Kβ inhibitors, and the improved properties of UCT943 over MMV390048 gives us hope for this chemotype. Other promising Plasmodium kinase targets include PfPKG and PfCLK3; both kinases have had extensive characterization of their biological functions and their inhibitors. PfCDPK1 is also an extensively-characterized kinase, but unlike PfPKG and PfCLK3, these studies have instead led to questioning of its appropriateness as a drug target against the blood stages of P. falciparum. Other kinases, PfGSK3, PfPK6, and PfPK7 are shown to be potential targets for inhibition of the P. falciparum blood stage, but they are less well characterized; further understanding of their biological function and in-depth characterization of their inhibitors remains necessary. PfCDPK4 is an interesting kinase non-essential for the blood stage of P. falciparum but is a target for the development of transmission-blocking drugs. In this review, we have also highlighted known kinase inhibitor scaffolds that may be found within many large phenotypic screening datasets, and/or been investigated through separate SAR studies. Target identification for these compounds may prove fruitful for discovering novel kinase targets. HDT is a less-explored but nevertheless promising approach to discover novel antimalarials, considering that several host kinases have been implicated in the Plasmodium reproductive cycle, with one major benefit of HDT being reduced susceptibility to resistance development. Inhibition of Plasmodium and human kinases are therefore potential avenues toward novel antimalarial therapy.
Figure 4. Development stages of the Plasmodium life cycle. Kinase inhibitors that target parasites at the blood, liver and mosquito stages are indicated.
6. Expert opinion
The Plasmodium kinome, in general, is relatively understudied and we believe that further efforts toward improving our understanding of individual Plasmodium kinases would help to identify high quality targets. Significant efforts put into understanding the biological function of PfPI4Kβ, PfPKG and PfCLK3 have borne fruit in the development of potent and selective inhibitors with great potential at the clinical and preclinical stages of development. The inverse is also true; the development of their inhibitors have greatly enabled understanding of the roles and functions of these kinases. However, few other Plasmodium kinases have received as much attention and investment of resources, leading to scant information available. We hope that the availability of pioneering inhibitors would catalyze efforts toward further biological characterization of less-well studied kinases such as PfGSK3, PfPK6 and PfPK7. At the same time, it is also necessary to fully-characterize these inhibitors in terms of their kinome-wide inhibition profile, their activity against other life stages of the Plasmodium life cycle and in vivo activity in murine models, in order to better validate if the kinase targets are indeed of high potential. It is entirely possible that further biological characterization of these kinases would reveal that they are indeed suboptimal drug targets, as was the case for PfCDPK1, but this is nonetheless useful information which would be otherwise unattainable.
Polypharmacology is beneficial for drug discovery to combat resistance. Some scaffolds, such as the imidazopyrazines/pyridazine scaffold, demonstrate great potential for polypharmacology. The imidazopyrazines/pyridazines have been found to be inhibitors of multiple Plasmodium kinases in separate studies, for example PfPI4K [Citation13,Citation31,Citation74], PfPKG [Citation74,Citation75], PfCDPK1 [Citation75,Citation121–125], PfPK7 [Citation145,Citation146] and PfCLK1 [Citation180]. Furthermore, this scaffold is also identified from phenotypic screening hits from separate screening efforts [Citation90,Citation152]. However, as far as we are aware, no study has fully characterized these compounds against all identified targets of this series. Likewise, many other inhibitors are currently under-profiled against both human and Plasmodium kinomes and their true profile is currently uncharacterized. This demonstrates the necessity of further development and application of Plasmodium kinome-wide screening technologies. As the field of human protein kinase inhibition matured, the development of commercially-available human kinome-wide biochemical or binding assay panels has enabled a clearer picture of the pharmacological profile of each inhibitor. Such kinome-wide screens are now commonplace in both industry and academic drug discovery programs targeting human kinases. In the absence of similar biochemical or binding assay panels for the Plasmodium kinome, the closest alternative would be proteomic approaches such as Kinobeads, but this is less accessible for academic medicinal chemistry groups. As Kinobeads was optimized for human kinase binding, it is also of interest to characterize the binding profile of each individual immobilized inhibitor in Plasmodium lysate, and to optimize a set of inhibitors for improved Plasmodium kinome-wide capture. We believe that the development of Plasmodium kinome-wide biochemical or binding assays would greatly facilitate high-quality inhibitor development. If sufficient throughput in these assays is achieved, these assay panels could also serve to screen libraries of kinase inhibitor scaffolds enriched in large-scale phenotypic screens for hit discovery against novel Plasmodium kinases.
In parallel, identifying host cell targets and focusing on host-selective compounds with low toxicity may play an important role as co-administered drugs to protect classical, parasite-targeting drugs against the emergence and spread of resistance. We believe that understanding of the overall inhibition profile would be greatly beneficial to inform the malaria community of targets to prioritize, as well as to enable drug discovery. Finally, as integrated approaches to alleviate the increasingly concerning issue of anti-malarial drug resistance, we advocate for the development of drugs that kill asexual parasites through the inhibition of host cell kinases (host-directed therapy), as well as of transmission-blocking drugs targeting strictly sexual-stage specific kinases of the parasites such as PfCDPK4, so as to prevent selection of mutations during asexual proliferation.
Article highlights
Malaria remains a serious global health issue, and in view of the spread of resistance against all antimalarial drugs, new treatment strategies are urgently needed.
Protein kinases are attractive targets, in view of their essentiality for parasite survival and their established druggability.
Most drug discovery efforts have concentrated on a small number of Plasmodium kinases, and have identified promising lead compounds.
Several kinase-inhibitor-like scaffolds with antimalarial activity have been identified that still require target deconvolution.
Kinase inhibition is suitable for strategies such as polypharmacology, host-directed therapy and transmission-blocking, all of which can contribute to alleviate the threat of drug resistance.
Declaration of interests
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- World Health Organization. World malaria report. Vol. 2021. Geneva: World Health Organization; 2021.
- World Health Organization. Global technical strategy for malaria 2016-2030, 2021 update. Geneva: World Health Organization; 2021.
- Dondorp AM, Nosten F, Yi P, et al. Artemisinin Resistance in Plasmodium falciparum Malaria. N Engl J Med. 2009;361(5):455–467.
- Uwimana A, Legrand E, Stokes BH, et al. Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat Med. 2020;26(10):1602–1608.
- Uwimana A, Umulisa N, Venkatesan M, et al. Association of Plasmodium falciparum kelch13 R561H genotypes with delayed parasite clearance in Rwanda: an open-label, single-arm, multicentre, therapeutic efficacy study. Lancet Infect Dis. 2021;21(8):1120–1128.
- Balikagala B, Fukuda N, Ikeda M, et al. Evidence of artemisinin-resistant Malaria in Africa. N Engl J Med. 2021;385(13):1163–1171.
- Moolman C, Sluis R, Beteck R, et al. An Update on Development of Small-Molecule Plasmodial Kinase Inhibitors. Molecules. 2020;25(21):5182.
- Mustière R, Vanelle P, Primas N. Plasmodial kinase inhibitors targeting malaria: recent developments. Molecules. 2020;25(24):5949.
- Adderley J, Doerig C. Comparative analysis of the kinomes of Plasmodium falciparum, Plasmodium vivax and their host Homo sapiens. BMC Genomics. 2022;23(1):237.
- Adderley J, Williamson T, Doerig C. Parasite and host erythrocyte kinomics of plasmodium infection. Trends Parasitol. 2021;37(6):508–524.
- Yang T, Ottilie S, Istvan ES, et al. MalDA, accelerating malaria drug discovery. Trends Parasitol. 2021;37(6):493–507.
- Forte B, Ottilie S, Plater A, et al. Prioritization of molecular targets for antimalarial drug discovery. ACS Infect Dis. 2021;7(10):2764–2776.
- McNamara CW, Lee MCS, Lim CS, et al. Targeting Plasmodium PI(4)K to eliminate malaria. Nature. 2013;504(7479):248–253.
- Paquet T, Le Manach C, Cabrera DG, et al. Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase. Sci Transl Med. 2017;9(387):eaad9735.
- Brunschwig C, Lawrence N, Taylor D, et al. UCT943, a Next-Generation Plasmodium falciparum PI4K inhibitor preclinical candidate for the treatment of malaria. antimicrob. Agents Chemother. 2018;62(9):e00012–18.
- Alam MM, Sanchez-Azqueta A, Janha O, et al. Validation of the protein kinase PfCLK3 as a multistage cross-species malarial drug target. Science. 2019;365(6456):eaau1682.
- Lu K, Mansfield CR, Fitzgerald MC, et al. Chemoproteomics for plasmodium parasite drug target discovery. ChemBioChem. 2021;22(16):2591–2599.
- Eberl HC, Werner T, Reinhard FB, et al. Chemical proteomics reveals target selectivity of clinical Jak inhibitors in human primary cells. Sci Rep. 2019;9(1):14159.
- Balestra AC, Koussis K, Klages N, et al. Ca2+ signals critical for egress and gametogenesis in malaria parasites depend on a multipass membrane protein that interacts with PKG. Sci Adv. 2021;7(13):eabe5396.
- Matralis AN, Malik A, Penzo M, et al. Development of chemical entities endowed with potent fast-killing properties against plasmodium falciparum malaria parasites. J Med Chem. 2019;62(20):9217–9235.
- Penzo M, de Las Heras-dueña L, Mata-Cantero, B L, et al. Ghidelli-disse et al. high-throughput screening of the plasmodium falciparum cGMP-dependent protein kinase identified a thiazole scaffold which kills erythrocytic and sexual stage parasites. Sci Rep. 2019;9(1):1–13.
- Vanaerschot M, Murithi JM, Pasaje CFA, et al. Inhibition of Resistance-Refractory P. falciparum Kinase PKG delivers prophylactic, blood stage, and transmission-blocking antiplasmodial activity. Cell Chem Biol. 2020;27(7):806–816.
- Kato N, Sakata T, Breton G, et al. Gene expression signatures and small-molecule compounds link a protein kinase to Plasmodium falciparum motility. Nat Chem Biol. 2008;4(6):347–356.
- Sinxadi P, Donini C, Johnstone H, et al. Safety, tolerability, pharmacokinetics, and antimalarial activity of the novel plasmodium phosphatidylinositol 4-Kinase Inhibitor MMV390048 in Healthy Volunteers. Antimicrob Agents Chemother. 2020;64(4):1–12.
- McCarthy JS, Donini C, Chalon S, et al. A phase 1, placebo-controlled, randomized, single ascending dose study and a volunteer infection study to characterize the safety, pharmacokinetics, and antimalarial activity of the plasmodium phosphatidylinositol 4-Kinase Inhibitor MMV390048. Clin Infect Dis. 2020;71(10):e657–e664.
- Hassett MR, Roepe PD. PIK-ing new malaria chemotherapy. Trends Parasitol. 2018;34(11):925–927.
- Balla T. Phosphoinositides: tiny Lipids With Giant Impact on Cell Regulation. Physiol Rev. 2013;93(3):1019–1137.
- Maurya R, Tripathi A, Kumar M, et al. PI4‐kinase and PfCDPK7 signaling regulate phospholipid biosynthesis in Plasmodium falciparum. EMBO Rep. 2022;23(2):1–15.
- Zhang M, Wang C, Otto TD, et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science. 2018;360(6388):7847.
- Plouffe D, Brinker A, McNamara C, et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc Natl Acad Sci. 2008;105(26):9059–9064.
- Zou B, Nagle A, Chatterjee AK, et al. Lead Optimization of Imidazopyrazines: a new class of antimalarial with activity on plasmodium liver stages. ACS Med Chem Lett. 2014;5(8):947–950.
- Zeeman A-M, Lakshminarayana SB, van der Werff N, et al. PI4 kinase is a prophylactic but not radical curative target in plasmodium vivax-type malaria parasites. Antimicrob Agents Chemother. 2016;60(5):2858–2863.
- Dembele L, Ang X, Chavchich M, et al. The Plasmodium PI(4)K inhibitor KDU691 selectively inhibits dihydroartemisinin-pretreated Plasmodium falciparum ring-stage parasites. Sci Rep. 2017;7(1):2325.
- Witkowski B, Lelièvre J, López Barragán MJ, et al. Increased tolerance to artemisinin in plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob Agents Chemother. 2010;54(5):1872–1877.
- Younis Y, Douelle F, Feng T-S, et al. 3,5-Diaryl-2-aminopyridines as a novel class of orally active antimalarials demonstrating single dose cure in mice and clinical candidate potential. J Med Chem. 2012;55(7):3479–3487.
- Fienberg S, Eyermann CJ, Arendse LB, et al. Structural basis for inhibitor potency and selectivity of plasmodium falciparum phosphatidylinositol 4-kinase inhibitors. ACS Infect Dis. 2020;6(11):3048–3063.
- MMV390048 POC in Patients With P. Vivax and P. Falciparum Malaria. ClinicalTrials.gov Identifier: NCT02880241. Available at https://clinicaltrials.gov/ct2/show/NCT02880241 cited 2022 Jul 21
- Mohammed R, Asres MS, Gudina EK, et al. Efficacy, safety, tolerability, and pharmacokinetics of MMV390048 in acute uncomplicated malaria. Am J Trop Med Hyg. 2023;108(1):81–84.
- González Cabrera D, Douelle F, Younis Y, et al. Structure–activity relationship studies of orally active antimalarial 3,5-substituted 2-aminopyridines. J Med Chem. 2012;55(24):11022–11030.
- Younis Y, Douelle F, González Cabrera D, et al. Structure–activity-relationship studies around the 2-amino group and pyridine core of antimalarial 3,5-diarylaminopyridines lead to a novel series of pyrazine analogues with oral in vivo activity. J Med Chem. 2013;56(21):8860–8871.
- Le Manach C, Nchinda AT, Paquet T, et al. Identification of a potential antimalarial drug candidate from a series of 2-aminopyrazines by optimization of aqueous solubility and potency across the parasite life cycle. J Med Chem. 2016;59(21):9890–9905.
- Gibhard L, Njoroge M, Paquet T, et al. Investigating sulfoxide-to-sulfone conversion as a prodrug strategy for a phosphatidylinositol 4-kinase inhibitor in a humanized mouse model of malaria. Antimicrob Agents Chemother. 2018;62(12):e00261–18.
- Liang X, Jiang Z, Huang Z, et al. Discovery of 6′-chloro-N-methyl-5’-(phenylsulfonamido)-[3,3′-bipyridine]-5-carboxamide (CHMFL-PI4K-127) as a novel Plasmodium falciparum PI(4)K inhibitor with potent antimalarial activity against both blood and liver stages of Plasmodium. Eur J Med Chem. 2020;188:112012.
- Kandepedu N, Gonzàlez Cabrera D, Eedubilli S, et al. Identification, characterization, and optimization of 2,8-disubstituted-1,5-naphthyridines as novel plasmodium falciparum phosphatidylinositol-4-kinase inhibitors with in vivo efficacy in a humanized mouse model of malaria. J Med Chem. 2018;61(13):5692–5703.
- Krishnan K, Ziniel P, Li H, et al. Torin 2 Derivative, NCATS-SM3710, Has Potent Multistage Antimalarial Activity through Inhibition of P. falciparum Phosphatidylinositol 4-Kinase (PfPI4KIIIβ). ACS Pharmacol Transl Sci. 2020;3(5):948–964.
- Ward P, Equinet L, Packer J, et al. Protein kinases of the human malaria parasite Plasmodium falciparum: the kinome of a divergent eukaryote. BMC Genomics. 2004;5(1):79.
- Deng W, Baker DA. A novel cyclic GMP-dependent protein kinase is expressed in the ring stage of the Plasmodium falciparum life cycle. Mol Microbiol. 2002;44(5):1141–1151.
- El Bakkouri M, Kouidmi I, Wernimont AK, et al. Structures of the cGMP-dependent protein kinase in malaria parasites reveal a unique structural relay mechanism for activation. Proc Natl Acad Sci. 2019;116(28):14164–14173.
- Hopp CS, Flueck C, Solyakov L, et al. Spatiotemporal and Functional Characterisation of the Plasmodium falciparum cGMP-Dependent Protein Kinase. PLoS One. 2012;7(11):e48206.
- Alam MM, Solyakov L, Bottrill AR, et al. Phosphoproteomics reveals malaria parasite Protein Kinase G as a signalling hub regulating egress and invasion. Nat Commun. 2015;6(1):7285.
- Govindasamy K, Khan R, Snyder M, et al. Plasmodium falciparum Cyclic GMP-Dependent Protein Kinase Interacts with a Subunit of the Parasite Proteasome. Infect Immun. 2019;87(1):e00523–18.
- Solyakov L, Halbert J, Alam MM, et al. Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat Commun. 2011;2(1):565.
- Tewari R, Straschil U, Bateman A, et al. The systematic functional analysis of plasmodium protein kinases identifies essential regulators of mosquito transmission. Cell Host Microbe. 2010;8(4):377–387.
- Taylor HM, McRobert L, Grainger M, et al. The Malaria Parasite Cyclic GMP-Dependent Protein Kinase Plays a Central Role in Blood-Stage Schizogony. Eukaryot Cell. 2010;9(1):37–45.
- McRobert L, Taylor CJ, Deng W, et al. Gametogenesis in Malaria Parasites Is Mediated by the cGMP-Dependent Protein Kinase. PLoS Biol. 2008;6(6):e139.
- Moon RW, Taylor CJ, Bex C, et al. A Cyclic GMP Signalling Module That Regulates Gliding Motility in a Malaria Parasite. PLoS Pathog. 2009;5(9):e1000599.
- Koussis K, Withers-Martinez C, Baker DA, et al. Simultaneous multiple allelic replacement in the malaria parasite enables dissection of PKG function. Life Sci. Alliance. 2020;3(4):e201900626.
- Collins CR, Hackett F, Strath M, et al. Malaria Parasite cGMP-dependent Protein Kinase Regulates Blood Stage Merozoite Secretory Organelle Discharge and Egress. PLoS Pathog. 2013;9(5):e1003344.
- Dvorin JD, Martyn DC, Patel SD, et al. A Plant-Like Kinase in Plasmodium falciparum Regulates Parasite Egress from Erythrocytes. Science. 2010;328(5980):910–912.
- Thomas JA, Tan MSY, Bisson C, et al. A protease cascade regulates release of the human malaria parasite Plasmodium falciparum from host red blood cells. Nat Microbiol. 2018;3(4):447–455.
- Brochet M, Collins MO, Smith TK, et al. Phosphoinositide Metabolism Links cGMP-Dependent Protein Kinase G to Essential Ca2+ Signals at Key Decision Points in the Life Cycle of Malaria Parasites. PLoS Biol. 2014;12(3):e1001806.
- Falae A, Combe A, Amaladoss A, et al. Role of Plasmodium berghei cGMP-dependent Protein Kinase in Late Liver Stage Development. J Biol Chem. 2010;285(5):3282–3288.
- Govindasamy K, Jebiwott S, Jaijyan DK, et al. Invasion of hepatocytes by Plasmodium sporozoites requires cGMP-dependent protein kinase and calcium dependent protein kinase 4. Mol Microbiol. 2016;102(2):349–363.
- Nare B, Allocco JJ, Liberator PA, et al. Evaluation of a Cyclic GMP-Dependent Protein Kinase Inhibitor in Treatment of Murine Toxoplasmosis: gamma Interferon Is Required for Efficacy. Antimicrob Agents Chemother. 2002;46(2):300–307.
- Donald RGK, Allocco J, Singh SB, et al. Toxoplasma gondii Cyclic GMP-Dependent Kinase: chemotherapeutic Targeting of an Essential Parasite Protein Kinase. Eukaryot Cell. 2002;1(3):317–328.
- Gurnett AM, Liberator PA, Dulski PM, et al. Purification and Molecular Characterization of cGMP-dependent Protein Kinase from Apicomplexan Parasites. J Biol Chem. 2002;277(18):15913–15922.
- Biftu T, Feng D, Fisher M, et al. Synthesis and SAR studies of very potent imidazopyridine antiprotozoal agents. Bioorganic Med. Chem. Lett. 2006;16(9):2479–2483.
- Baker DA, Stewart LB, Large JM, et al. A potent series targeting the malarial cGMP-dependent protein kinase clears infection and blocks transmission. Nat Commun. 2017;8(1):430.
- Large JM, Birchall K, Bouloc NS, et al. Potent inhibitors of malarial P. Falciparum protein kinase G: improving the cell activity of a series of imidazopyridines. Bioorganic Med. Chem. Lett. 2019;29(3):509–514.
- Large JM, Birchall K, Bouloc NS, et al. Potent bicyclic inhibitors of malarial cGMP-dependent protein kinase: approaches to combining improvements in cell potency, selectivity and structural novelty. Bioorg Med Chem Lett. 2019;29(19):126610.
- Tsagris DJ, Birchall K, Bouloc N, et al. Trisubstituted thiazoles as potent and selective inhibitors of Plasmodium falciparum protein kinase G (PfPKG). Bioorg Med Chem Lett. 2018;28(19):3168–3173.
- Mahmood SU, Cheng H, Tummalapalli SR, et al. Discovery of isoxazolyl-based inhibitors of Plasmodium falciparum cGMP-dependent protein kinase. RSC Med. Chem. 2020;11(1):98–101.
- Bheemanaboina RRY, de Souza ML, Gonzalez ML, et al. Discovery of Imidazole-Based Inhibitors of Plasmodium falciparum cGMP-Dependent Protein Kinase. ACS Med Chem Lett. 2021;12(12):1962–1967.
- Cheuka PM, Centani L, Arendse LB, et al. New Amidated 3,6-Diphenylated Imidazopyridazines with Potent Antiplasmodium Activity Are Dual Inhibitors of Plasmodium Phosphatidylinositol-4-kinase and cGMP-Dependent Protein Kinase. ACS Infect Dis. 2021;7(1):34–46.
- Green JL, Moon RW, Whalley D, et al. Imidazopyridazine inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 also target cyclic GMP-dependent protein kinase and heat shock protein 90 to kill the parasite at different stages of intracellular development. Antimicrob Agents Chemother. 2016;60(3):1464–1475.
- Baker DA, Matralis AN, Osborne SA, et al. Targeting the Malaria Parasite cGMP-Dependent Protein Kinase to Develop New Drugs. Front Microbiol. 2020;11:1–8.
- Rotella D, Siekierka J, Bhanot P. Plasmodium falciparum cGMP-Dependent Protein Kinase – a Novel Chemotherapeutic Target. Front Microbiol. 2021;11:1–7.
- Maier AG, Rug M, O’Neill MT, et al. Exported Proteins Required for Virulence and Rigidity of Plasmodium falciparum-Infected Human Erythrocytes. Cell. 2008;134(1):48–61.
- Droucheau E, Primot A, Thomas V, et al. Plasmodium falciparum glycogen synthase kinase-3: molecular model, expression, intracellular localisation and selective inhibitors. Biochim Biophys Acta - Proteins Proteomics. 2004;1697(1–2):181–196.
- Mundwiler-Pachlatko E, Beck H-P. Maurer’s clefts, the enigma of Plasmodium falciparum. Proc Natl Acad Sci. 2013;110(50):19987–19994.
- Prinz B, Harvey KL, Wilcke L, et al. Hierarchical phosphorylation of apical membrane antigen 1 is required for efficient red blood cell invasion by malaria parasites. Sci Rep. 2016;6(1):34479.
- Triglia T, Healer J, Caruana SR, et al. Apical membrane antigen 1 plays a central role in erythrocyte invasion by Plasmodium species. Mol Microbiol. 2000;38(4):706–718.
- Fugel W, Oberholzer AE, Gschloessl B, et al. 3,6-Diamino-4-(2-halophenyl)-2-benzoylthieno[2,3-b]pyridine-5-carbonitriles Are Selective Inhibitors of Plasmodium falciparum Glycogen Synthase Kinase-3. J Med Chem. 2013;56(1):264–275.
- Masch A, Kunick C. Selective inhibitors of Plasmodium falciparum glycogen synthase-3 (PfGSK-3): new antimalarial agents? Biochim Biophys Acta - Proteins Proteomics. 2015;1854(10):1644–1649.
- Masch A, Nasereddin A, Alder A, et al. Structure-activity relationships in a series of antiplasmodial thieno[2,3-b]pyridines. Malar J. 2019;18(1):1–10.
- Schweda SI, Alder A, Gilberger T, et al. 4-Arylthieno[2,3-b]pyridine-2-carboxamides Are a New Class of Antiplasmodial Agents. Molecules. 2020;25(14):3187.
- Moolman C, van der Sluis R, Beteck RM, et al. Exploration of benzofuran-based compounds as potent and selective Plasmodium falciparum glycogen synthase kinase-3 (PfGSK-3) inhibitors. Bioorg Chem. 2021;112:104839.
- Galal KA, Truong A, Kwarcinski F, et al. Identification of Novel 2,4,5-Trisubstituted Pyrimidines as Potent Dual Inhibitors of Plasmodial PfGSK3/PfPK6 with Activity against Blood Stage Parasites In Vitro. J Med Chem. 2022;65(19):13172–13197.
- Bracchi-Ricard V, Barik S, DelVecchio C, et al. PfPK6, a novel cyclin-dependent kinase/mitogen-activated protein kinase-related protein kinase from Plasmodium falciparum. Biochem J. 2000;347(1):255.
- Gamo F-J, Sanz LM, Vidal J, et al. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465(7296):305–310.
- Crowther GJ, Hillesland HK, Keyloun KR, et al. Biochemical Screening of Five Protein Kinases from Plasmodium falciparum against 14,000 Cell-Active Compounds. PLoS One. 2016;11(3):e0149996.
- Ong HW, Truong A, Kwarcinski F, et al. Discovery of potent Plasmodium falciparum protein kinase 6 (PfPK6) inhibitors with a type II inhibitor pharmacophore. Eur J Med Chem. 2023;249:115043.
- Moyano PM, Němec V, Paruch K. Cdc-Like Kinases (CLKs): biology, Chemical Probes, and Therapeutic Potential. Int J Mol Sci. 2020;21(20):7549.
- Lindberg MF, Meijer L. Dual-Specificity, Tyrosine Phosphorylation-Regulated Kinases (DYRKs) and cdc2-Like Kinases (CLKs) in Human Disease, an Overview. Int J Mol Sci. 2021;22(11):6047.
- Agarwal S, Kern S, Halbert J, et al. Two nucleus-localized CDK-like kinases with crucial roles for malaria parasite erythrocytic replication are involved in phosphorylation of splicing factor. J Cell Biochem. 2011;112(5):1295–1310.
- Kern S, Agarwal S, Huber K, et al. Inhibition of the SR Protein-Phosphorylating CLK Kinases of Plasmodium falciparum Impairs Blood Stage Replication and Malaria Transmission. PLoS One. 2014;9(9):e105732.
- Bourgeois CF, Lejeune F, Stévenin J. Broad Specificity of SR (Serine/Arginine) Proteins in the Regulation of Alternative Splicing of Pre-Messenger RNA, in. Nucleic Acid Res Mol Biol. 2004;78:37–88.
- Stamm S. Regulation of Alternative Splicing by Reversible Protein Phosphorylation. J Biol Chem. 2008;283(3):1223–1227.
- Zhou Z, Fu X-D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma. 2013;122(3):191–207.
- Talevich E, Mirza A, Kannan N. Structural and evolutionary divergence of eukaryotic protein kinases in Apicomplexa. BMC Evol. Biol. 2011;11(1):321.
- Dellaire G, Makarov EM, Cowger JM, et al. Mammalian PRP4 Kinase Copurifies and Interacts with Components of Both the U5 snRNP and the N-CoR Deacetylase Complexes. Mol Cell Biol. 2002;22(14):5141–5156.
- Schneider M, Hsiao -H-H, Will CL, et al. Human PRP4 kinase is required for stable tri-snRNP association during spliceosomal B complex formation. Nat Struct Mol Biol. 2010;17(2):216–221.
- Mahindra A, Janha O, Mapesa K, et al. , A. Amambua-Ngwa et al. Development of Potent Pf CLK3 Inhibitors Based on TCMDC-135051 as a New Class of Antimalarials. J Med Chem. 2020;63(17):9300–9315.
- Swale C, Bellini V, Bowler MW, et al. Altiratinib blocks Toxoplasma gondii and Plasmodium falciparum development by selectively targeting a spliceosome kinase. Sci Transl Med. 2022;14(656):eabn3231.
- Hui R, El Bakkouri M, Sibley LD. Designing selective inhibitors for calcium-dependent protein kinases in apicomplexans. Trends Pharmacol Sci. 2015;36(7):452–460.
- Kadian K, Gupta Y, Kempaiah P, et al. Calcium Dependent Protein Kinases (CDPKs): key to Malaria Eradication. Curr Top Med Chem. 2017;17(19):2215–2220.
- Zhao Y, Kappes B, Franklin RM. Gene structure and expression of an unusual protein kinase from Plasmodium falciparum homologous at its carboxyl terminus with the EF hand calcium-binding proteins. J Biol Chem. 1993;268(6):4347–4354.
- Zhao Y, Franklin RM, Kappes B. Plasmodium falciparum calcium-dependent protein kinase phosphorylates proteins of the host erythrocytic membrane. Mol Biochem Parasitol. 1994;66(2):329–343.
- Möskes C, Burghaus PA, Wernli B, et al. Export of Plasmodium falciparum calcium-dependent protein kinase 1 to the parasitophorous vacuole is dependent on three N-terminal membrane anchor motifs. Mol Microbiol. 2004;54(3):676–691.
- Azevedo MF, Sanders PR, Krejany E, et al. Inhibition of Plasmodium falciparum CDPK1 by conditional expression of its J-domain demonstrates a key role in schizont development. Biochem J. 2013;452(3):433–441.
- Bansal A, Singh S, More KR, et al. Characterization of Plasmodium falciparum Calcium-dependent Protein Kinase 1 (PfCDPK1) and Its Role in Microneme Secretion during Erythrocyte Invasion. J Biol Chem. 2013;288(3):1590–1602.
- Green JL, Rees-Channer RR, Howell SA, et al. The Motor Complex of Plasmodium falciparum: phosphorylation by a Calcium-Dependent Protein Kinase. J Biol Chem. 2008;283(45):30980–30989.
- Thomas DC, Ahmed A, Gilberger TW, et al. Regulation of Plasmodium falciparum Glideosome Associated Protein 45 (PfGAP45) Phosphorylation. PLoS One. 2012;7(4):e35855.
- Ridzuan MAM, Moon RW, Knuepfer E, et al. Subcellular location, phosphorylation and assembly into the motor complex of GAP45 during Plasmodium falciparum schizont development. PLoS One. 2012;7(3):e33845.
- Frénal K, Dubremetz J-F, Lebrun M, et al. Gliding motility powers invasion and egress in Apicomplexa. Nat Rev Microbiol. 2017;15(11):645–660.
- Kumar S, Kumar M, Ekka R, et al. PfCDPK1 mediated signaling in erythrocytic stages of Plasmodium falciparum. Nat Commun. 2017;8(1):63.
- Bansal A, Molina-Cruz A, Brzostowski J, et al. Pf CDPK1 is critical for malaria parasite gametogenesis and mosquito infection. Proc Natl Acad Sci. 2018;115(4):774–779.
- Sebastian S, Brochet M, Collins MO, et al. A Plasmodium Calcium-Dependent Protein Kinase Controls Zygote Development and Transmission by Translationally Activating Repressed mRNAs. Cell Host Microbe. 2012;12(1):9–19.
- Govindasamy K, Bhanot P. Overlapping and distinct roles of CDPK family members in the pre-erythrocytic stages of the rodent malaria parasite, Plasmodium berghei. PLOS Pathog. 2020;16(8):e1008131.
- Jebiwott S, Govindaswamy K, Mbugua A, et al. Plasmodium berghei Calcium Dependent Protein Kinase 1 Is Not Required for Host Cell Invasion. PLoS One. 2013;8(11):e79171.
- Lemercier G, Fernandez-Montalvan A, Shaw JP, et al. Identification and Characterization of Novel Small Molecules as Potent Inhibitors of the Plasmodial Calcium-Dependent Protein Kinase 1. Biochemistry. 2009;48(27):6379–6389.
- Chapman TM, Osborne SA, Bouloc N, et al. Substituted imidazopyridazines are potent and selective inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1). Bioorg Med Chem Lett. 2013;23(10):3064–3069.
- Ansell KH, Jones HM, Whalley D, et al. Biochemical and Antiparasitic Properties of Inhibitors of the Plasmodium falciparum Calcium-Dependent Protein Kinase PfCDPK1. Antimicrob Agents Chemother. 2014;58(10):6032–6043.
- Large JM, Osborne SA, Smiljanic-Hurley E, et al. Imidazopyridazines as potent inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1): preparation and evaluation of pyrazole linked analogues. Bioorganic Med. Chem. Lett. 2013;23(21):6019–6024.
- Chapman TM, Osborne SA, Wallace C, et al. Optimization of an Imidazopyridazine Series of Inhibitors of Plasmodium falciparum Calcium-Dependent Protein Kinase 1 (PfCDPK1). J Med Chem. 2014;57(8):3570–3587.
- Bansal A, Ojo KK, Mu J, et al. Reduced Activity of Mutant Calcium-Dependent Protein Kinase 1 Is Compensated in Plasmodium falciparum through the Action of Protein Kinase G. MBio. 2016;7(6):e02011–16.
- Kumar S, Haile MT, Hoopmann MR, et al. Plasmodium falciparum Calcium-Dependent Protein Kinase 4 is Critical for Male Gametogenesis and Transmission to the Mosquito Vector. MBio. 2021;12(6):605.
- Ranjan R, Ahmed A, Gourinath S, et al. Dissection of Mechanisms Involved in the Regulation of Plasmodium falciparum Calcium-dependent Protein Kinase 4. J Biol Chem. 2009;284(22):15267–15276.
- Kato K, Sudo A, Kobayashi K, et al. Characterization of Plasmodium falciparum calcium-dependent protein kinase 4. Parasitol Int. 2009;58(4):394–400.
- Billker O, Dechamps S, Tewari R, et al. Calcium and a Calcium-Dependent Protein Kinase Regulate Gamete Formation and Mosquito Transmission in a Malaria Parasite. Cell. 2004;117(4):503–514.
- Fang H, Klages N, Baechler B, et al. Multiple short windows of calcium-dependent protein kinase 4 activity coordinate distinct cell cycle events during Plasmodium gametogenesis. Elife. 2017;6:1–23.
- Fang H, Gomes AR, Klages N, et al. E.M. Walker et al. Epistasis studies reveal redundancy among calcium-dependent protein kinases in motility and invasion of malaria parasites. Nat Commun. 2018;9(1):4248.
- Ojo KK, Pfander C, Mueller NR, et al. Transmission of malaria to mosquitoes blocked by bumped kinase inhibitors. J Clin Invest. 2012;122(6):2301–2305.
- Ojo KK, Eastman RT, Vidadala R, et al. A Specific Inhibitor of PfCDPK4 Blocks Malaria Transmission: chemical-genetic Validation. J Infect Dis. 2014;209(2):275–284.
- Vidadala RSR, Ojo KK, Johnson SM, et al. A. Mitra et al. Development of potent and selective Plasmodium falciparum calcium-dependent protein kinase 4 (PfCDPK4) inhibitors that block the transmission of malaria to mosquitoes. Eur J Med Chem. 2014;74:562–573.
- Huang W, Hulverson MA, Zhang Z, et al. 5-Aminopyrazole-4-carboxamide analogues are selective inhibitors of Plasmodium falciparum microgametocyte exflagellation and potential malaria transmission blocking agents. Bioorganic Med. Chem. Lett. 2016;26(22):5487–5491.
- Van Voorhis WC, Doggett JS, Parsons M, et al. Extended-spectrum antiprotozoal bumped kinase inhibitors: a review. Exp Parasitol. 2017;180(12):71–83.
- Reininger L, Tewari R, Fennell C, et al. An Essential Role for the Plasmodium Nek-2 Nima-related Protein Kinase in the Sexual Development of Malaria Parasites. J Biol Chem. 2009;284(31):20858–20868.
- Reininger L, Garcia M, Tomlins A, et al. The Plasmodium falciparum, Nima-related kinase Pfnek-4: a marker for asexual parasites committed to sexual differentiation. Malar J. 2012;11(1):250.
- Dorin D, Semblat J-P, Poullet P, et al. PfPK7, an atypical MEK-related protein kinase, reflects the absence of classical three-component MAPK pathways in the human malaria parasite Plasmodium falciparum. Mol Microbiol. 2005;55(1):184–186.
- Dorin-Semblat D, Sicard A, Doerig C, et al. Disruption of the PfPK7 Gene Impairs Schizogony and Sporogony in the Human Malaria Parasite Plasmodium falciparum. Eukaryot Cell. 2008;7(2):279–285.
- Pease BN, Huttlin EL, Jedrychowski MP, et al. Characterization of Plasmodium falciparum Atypical Kinase PfPK7 – dependent Phosphoproteome. J Proteome Res. 2018;17(6):2112–2123.
- Koyama FC, Ribeiro RY, Garcia JL, et al. Ubiquitin proteasome system and the atypical kinase PfPK7 are involved in melatonin signaling in Plasmodium falciparum. J. Pineal Res. 2012;53(2):147–153.
- Rezende Lima W, Tessarin-Almeida G, Rozanski A, et al. Signaling transcript profile of the asexual intraerythrocytic development cycle of Plasmodium falciparum induced by melatonin and cAMP. Genes Cancer. 2016;7(9–10):323–339.
- Merckx A, Echalier A, Langford K, et al. Structures of P. falciparum Protein Kinase 7 Identify an Activation Motif and Leads for Inhibitor Design. Structure. 2008;16(2):228–238.
- Bouloc N, Large JM, Smiljanic E, et al. Synthesis and in vitro evaluation of imidazopyridazines as novel inhibitors of the malarial kinase PfPK7. Bioorg Med Chem Lett. 2008;18(19):5294–5298.
- Lima MNN, Borba JVB, Cassiano GC, et al. Artificial Intelligence Applied to the Rapid Identification of New Antimalarial Candidates with Dual‐Stage Activity. ChemMedChem. 2021;16(7):1093–1103.
- Crowther GJ, Napuli AJ, Gilligan JH, et al. Identification of inhibitors for putative malaria drug targets among novel antimalarial compounds. Mol Biochem Parasitol. 2011;175(1):21–29.
- Guiguemde WA, Shelat AA, Bouck D, et al. Chemical genetics of Plasmodium falciparum. Nature. 2010;465(7296):311–315.
- Avery VM, Bashyam S, Burrows JN, et al. Screening and hit evaluation of a chemical library against blood-stage Plasmodium falciparum. Malar J. 2014;13(1):1–12.
- Dorjsuren D, Eastman RT, Wicht KJ, et al. Chemoprotective antimalarials identified through quantitative high-throughput screening of Plasmodium blood and liver stage parasites. Sci Rep. 2021;11(1):2121.
- Hallyburton I, Grimaldi R, Woodland A, et al. Screening a protein kinase inhibitor library against Plasmodium falciparum. Malar J. 2017;16(1):446.
- Harmse L, van Zyl R, Gray N, et al. Structure-activity relationships and inhibitory effects of various purine derivatives on the in vitro growth of Plasmodium falciparum. Biochem Pharmacol. 2001;62(3):341–348.
- Mallari JP, Guiguemde WA, Guy RK. Antimalarial activity of thiosemicarbazones and purine derived nitriles. Bioorg Med Chem Lett. 2009;19(13):3546–3549.
- Houzé S, Hoang N-T, Lozach O, et al. Several human cyclin-dependent kinase inhibitors, structurally related to roscovitine, as new anti-malarial agents. Molecules. 2014;19(9):15237–15257.
- Le Roch K, Sestier C, Dorin D, et al. Activation of a Plasmodium falciparum cdc2-related kinase by heterologous p25 and Cyclin H. J. Biol Chem. 2000;275(12):8952–8958.
- Phuangsawai O, Beswick P, Ratanabunyong S, et al. Evaluation of the anti-malarial activity and cytotoxicity of 2,4-diamino-pyrimidine-based kinase inhibitors. Eur J Med Chem. 2016;124:896–905.
- Toviwek B, Phuangsawai O, Konsue A, et al. Preparation, biological & cheminformatics-based assessment of N2,N4-diphenylpyrimidine-2,4-diamine as potential Kinase-targeted antimalarials. Bioorg Med Chem. 2021;46:116348.
- Lebar MD, Hahn KN, Mutka T, et al. CNS and antimalarial activity of synthetic meridianin and psammopemmin analogs. Bioorg Med Chem. 2011;19(19):5756–5762.
- Bharate SB, Yadav RR, Khan SI, et al. Meridianin G and its analogs as antimalarial agents. Medchemcomm. 2013;4(6):1042.
- Yadav RR, Khan SI, Singh S, et al. Synthesis, antimalarial and antitubercular activities of meridianin derivatives. Eur J Med Chem. 2015;98:160–169.
- Bharate SB, Yadav RR, Battula S, et al. Meridianins: marine-derived potent kinase inhibitors. mini-reviews med. Chem. 2012;12(7):618–631.
- Moy B, Kirkpatrick P, Kar S, et al. Lapatinib. Nat Rev Drug Discov. 2007;6(6):431–432.
- Patel G, Karver CE, Behera R, et al. Kinase scaffold repurposing for neglected disease drug discovery: discovery of an efficacious, lapatanib-derived lead compound for trypanosomiasis. J Med Chem. 2013;56(10):3820–3832.
- Devine W, Woodring JL, Swaminathan U, et al. Protozoan parasite growth inhibitors discovered by cross-screening yield potent scaffolds for lead discovery. J Med Chem. 2015;58(14):5522–5537.
- Woodring JL, Patel G, Erath J, et al. Evaluation of aromatic 6-substituted thienopyrimidines as scaffolds against parasites that cause trypanosomiasis, leishmaniasis, and malaria. Medchemcomm. 2015;6(2):339–346.
- Woodring JL, Bachovchin KA, Brady KG, et al. Optimization of physicochemical properties for 4-anilinoquinazoline inhibitors of trypanosome proliferation. Eur J Med Chem. 2017;141:446–459.
- Woodring JL, Behera R, Sharma A, et al. Series of Alkynyl-Substituted Thienopyrimidines as Inhibitors of Protozoan Parasite Proliferation. ACS Med Chem Lett. 2018;9(10):996–1001.
- Devine W, Thomas SM, Erath J, et al. Antiparasitic lead discovery: toward optimization of a chemotype with activity against multiple protozoan parasites. ACS Med Chem Lett. 2017;8(3):350–354.
- Mehta N, Ferrins L, Leed SE, et al. Optimization of Physicochemical Properties for 4-Anilinoquinoline Inhibitors of Plasmodium falciparum Proliferation. ACS Infect Dis. 2018;4(4):577–591.
- Ferrins L, Sharma A, Thomas SM, et al. Anilinoquinoline based inhibitors of trypanosomatid proliferation. PLoS Negl Trop Dis. 2018;12(11):e0006834.
- Bachovchin KA, Sharma A, Bag S, et al. Improvement of aqueous solubility of lapatinib-derived analogues: identification of a quinolinimine lead for human African trypanosomiasis drug development. J Med Chem. 2019;62(2):665–687.
- Singh B, Bernatchez JA, McCall L-I, et al. Scaffold and Parasite Hopping: discovery of New Protozoal Proliferation Inhibitors. ACS Med Chem Lett. 2020;11(3):249–257.
- Katiyar S, Kufareva I, Behera R, et al. Lapatinib-Binding protein kinases in the African trypanosome: identification of cellular targets for kinase-directed chemical scaffolds. PLoS One. 2013;8(2):e56150.
- Adderley JD, John von Freyend S, Jackson SA, et al. Analysis of erythrocyte signalling pathways during Plasmodium falciparum infection identifies targets for host-directed antimalarial intervention. Nat Commun. 2020;11(1):1–13.
- Prudêncio M, Rodrigues CD, Hannus M, et al. Kinome-Wide RNAi screen implicates at least 5 host hepatocyte kinases in plasmodium sporozoite infection. PLoS Pathog. 2008;4(11):e1000201.
- Chien HD, Pantaleo A, Kesely KR, et al. Imatinib augments standard malaria combination therapy without added toxicity. J Exp Med. 2021;218(10):1–9.
- Wei L, Adderley J, Leroy D, et al. Host-directed therapy, an untapped opportunity for antimalarial intervention. Cell Reports Med. 2021;2(10):100423.
- Adderley J, Grau GE. Host-directed therapies for malaria: possible applications and lessons from other indications. Curr Opin Microbiol. 2023;71:102228.
- Bendjeddou LZ, Loaëc N, Villiers B, et al. Exploration of the imidazo[1,2-b]pyridazine scaffold as a protein kinase inhibitor. Eur J Med Chem. 2017;125:696–709.