
ABSTRACT
Prion disease is an infectious and fatal neurodegenerative disease. Western blotting (WB)-based identification of proteinase K (PK)-resistant prion protein (PrPres) is considered a definitive diagnosis of prion diseases. In this study, we aimed to detect PrPres using formalin-fixed paraffin-embedded (FFPE) specimens from cases of sporadic Creutzfeldt–Jakob disease (sCJD), Gerstmann–Sträussler–Scheinker disease (GSS), glycosylphosphatidylinositol-anchorless prion disease (GPIALP), and V180I CJD. FFPE samples were prepared after formic acid treatment to inactivate infectivity. After deparaffinization, PK digestion was performed, and the protein was extracted. In sCJD, a pronounced PrPres signal was observed, with antibodies specific for type 1 and type 2 PrPres exhibited a strong or weak signals depending on the case. Histological examination of serial sections revealed that the histological changes were compatible with the biochemical characteristics. In GSS and GPIALP, prion protein core-specific antibodies presented as PrPres bands at 8–9 kDa and smear bands, respectively. However, an antibody specific for the C-terminus presented as smears in GSS, with no PrPres detected in GPIALP. It was difficult to detect PrPres in V180I CJD. Collectively, our findings demonstrate the possibility of detecting PrPres in FFPE and classifying the prion disease types. This approach facilitates histopathological and biochemical evaluation in the same sample and is safe owing to the inactivation of infectivity. Therefore, it may be valuable for the diagnosis and research of prion diseases.
Introduction
Human prion diseases, also known as transmissible spongiform encephalopathies, are lethal neurodegenerative disorders that include sporadic Creutzfeldt–Jakob disease (sCJD), inherited prion diseases, and acquired human prion diseases. The pathogenesis of prion disease is characterized by the conversion of a normal cellular prion protein (PrP) isoform (PrPc) into an abnormal pathogenic form (scrapie prion protein: PrPSc) [Citation1]. PrPSc has a high beta-sheet content that renders it partially resistant to proteinase K (PK) digestion [Citation2].
The identification of PK-resistant PrP (PrPres) by western blotting (WB) is a critical diagnostic criterion for prion diseases [Citation3]. Prion proteins have two glycosylated sites near the C-terminus, and WB presents three PrP signals: unglycosylated, monoglycosylated, and diglycosylated PrP forms [Citation4]. The size of the unglycosylated PrPres fragment differs depending on the N-terminal PK digestion site and is distinguished into types 1 (21 kDa) and 2 (19 kDa).
Inherited prion diseases caused by mutations in the PrP gene (PRNP) account for approximately 10% of human prion diseases. In Japan, mutations in codon 180 (V180I) and 102 (P102L, Gerstmann–Sträussler–Scheinker disease [GSS]) are common [Citation5]. Diglycosylated PrPres do not appear in the WB of V180I CJD; conversely, two bands corresponding to the unglycosylated and monoglycosylated forms are identified [Citation6,Citation7]. GSS presents three PrPres bands between 21 and 35 kDa, consisting of unglycosylated, monoglycosylated, and diglycosylated PrPres, with a band pattern similar to that of sCJD type 1; furthermore, a low molecular weight of 8 kDa is also observed [Citation8,Citation9].
Glycosylphosphatidylinositol-anchorless prion disease (GPIALP) is an inherited prion disease that has recently been reported [Citation10–12]. A genetic mutation results in the lack of the C-terminal GPI anchor, resulting in PrPres smears and a 9 kDa signal detected by WB using a PrP core-specific antibody.
Traditionally, unfixed frozen samples are required for WB. In the event of suspected prion disease, a frozen sample is preserved during the autopsy. WB is performed to identify the presence of PrPres, which, together with the histological findings, is used to obtain a final diagnosis. Occasionally, prion disease may be suspected retrospectively after the autopsy; however, frozen samples are usually not preserved in such cases, and the diagnosis may remain unclear. Most preserved pathology specimens are formalin-fixed specimens, which complicates the evaluation of PrPres using conventional methods, such as WB.
Some reports have demonstrated the presence of PrPres in formalin-fixed paraffin-embedded (FFPE) samples, but all were animal samples, with no PrPc detection [Citation13–15]. Recently, Nakagaki et al. identified undiagnosed prion disease in a cadaver for anatomical practice by measuring prion seeding activity by real-time quaking-induced conversion (RT-QuIC) from formalin-fixed brains and histological findings from FFPE [Citation16]. However, this novel RT-QuIC method could only demonstrate PrPSc seeding activity, and the details regarding the PrPres type remained unknown. In this study, we employed novel modified protein extraction methods to facilitate the biochemical evaluation of PrP and PrPres in FFPE samples using WB. Furthermore, we evaluated whether the analysis of PrPres signal patterns using various PrP antibodies specific to different epitopes can be used for the detailed classification of prion diseases.
Materials and methods
Patient samples
This study was conducted using post-mortem brain specimens of 14 cases of prion diseases (seven sCJD, three GSS, two GPIALP, and two V180I CJD cases [one pentosan polysulfate [PPS]-treated and one untreated case]) and five cases of non-prion diseases (one liver cirrhosis, one spinocerebellar ataxia type 6, one Emery – Dreifuss muscular dystrophy, one chronic progressive external ophthalmoplegia, and one multiphasic disseminated encephalomyelitis). The examined cases are presented in . All cases were autopsied at the Department of Neuropathology, Graduate School of Medical Sciences, Kyushu University. We obtained informed written consent for the autopsy from the patients or their next of kin. All analyses were performed following the Declaration of Helsinki. This study was approved by the Ethics Committee of Omuta National Hospital (5–3) and the Ethics Committee of the Faculty of Medicine of Kyushu University (#2019–179).
Table 1. Case profiles.
Protein extraction from FFPE
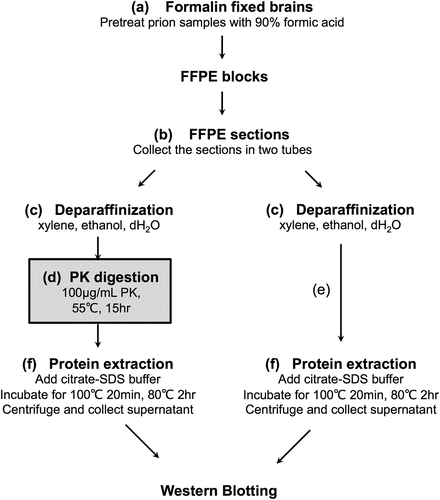
Brain specimens were fixed in formalin for at least two weeks. Prion disease samples were immersed in 90% formic acid for 1 h to inactivate infectivity (); FFPE blocks were prepared in the usual procedure. Detailed protein extraction methods have been previously described [Citation17,Citation18]. Briefly, each FFPE specimen was sectioned at 10-μm thickness and collected in two centrifuge tubes, with one or two sections each of 4–6 mm3 of brain volume (). Subsequently, they were deparaffinized with 100% xylene and 99% ethanol and hydrated (). Sections from one of the two tubes were incubated with PK (final concentration: 100 μg/mL) for 15 h at 55°C (). The conditions for PK digestion were modified from the paraffin-embedded tissue (PET)-blot technique [Citation19–21]. The other tube was used as a sample without PK treatment (). After quenching the PK reaction with Pefabloc SC (Roche, Indianapolis, IN), the samples were centrifuged at 15,000 × g for 10 min to discard the supernatant. Subsequently, the pellets were washed with citrate-sodium dodecyl sulphate (SDS) buffer (200 mM Tris-HCl at pH 7.5, 200 mM NaCl, 5% SDS, and 100 mM sodium citrate), and the supernatant was removed after centrifugation at 15,000 × g for 10 min at room temperature. Proteins were then extracted from the pellet by incubation at 100°C for 20 min and subsequently at 80°C for 2 h in 20 μL of citrate-SDS buffer with occasional vortexing. Finally, the supernatant was recovered after centrifugation at 15,000 × g for 15 min at room temperature and the sample buffer was added to the supernatant. This final solution was boiled at 95°C for 5 min and used as the sample for WB ().
Figure 1. Workflow of protein extraction from the formalin-fixed paraffin-embedded (FFPE) samples. The FFPE samples are sectioned into two tubes, one for proteinase K (PK) treatment (d) and the other without PK treatment (e), and the proteins are then extracted.
Western blotting
To detect PrPc and PrPres, samples were electrophoresed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) in 12% gels (BIO-RAD,-Hercules, CA, USA) and then transferred onto polyvinylidene difluoride membranes (BIO-RAD), and blocked with 5% skim milk. Four anti-prion antibodies (mouse monoclonal 3F4 [specific for PrP at 109–112], 1: 2000 (final concentration: 1 µg/mL); BioLegend, San Diego, CA, USA, rabbit monoclonal EP1802Y [specific for PrP at 212–230], 1: 2000 (final concentration: 0.33µg/mL); Abcam, Cambridge, UK, rabbit polyclonal Tohoku 1 [specific for type 1 PrPSc], 1: 5000 (protein concentration was not available), and rabbit polyclonal Tohoku 2 [specific for type 2 PrPSc], 1: 3000 (protein concentration was not available); Tohoku 1 and 2 were provided by Prof. Kitamoto of Tohoku University) were used [Citation22]. Peroxidase-conjugated anti-mouse immunoglobulin (Ig)G (1: 20000 (final concentration: 0.05 µg/mL); Vector, Burlingame, CA, USA) and peroxidase-conjugated anti-rabbit IgG (1: 20000 (final concentration: 0.05 µg/mL); Vector) were used as the secondary antibodies. Immunoreactivity was visualized using Immobilon Western Chemilum HRP substrate (Millipore, Billerica, MA, USA) and imaged on a Lumino Image Analyzer (Cytiva, Tokyo, Japan, ImageQuant 800). Western blot signals were measured using the ImageJ Fiji software (NIH). In each case group (sCJD, GSS, GPIALP, V180I CJD), the pattern of molecular weight of PrPres (high molecular weight PrPres (>30 kDa), three PrPres bands (19 or 21 ~ 30 kDa), and low molecular weight PrPres (<19 kDa)) was evaluated with Student’s T-test using Microsoft Excel software.
Immunohistochemistry
Sections with a thickness of 6 μm from the same FFPE block that was used for protein extraction were analysed. Hematoxylin and eosin (HE) staining was performed for histological examination. Immunohistochemistry (IHC) was performed using a primary antibody specific for anti-PrP: (mouse monoclonal 3F4 [1:400 (final concentration: 5 µg/mL)]). Envision system (Dako, Glostrup, Denmark) was used as the secondary antibody and visualized with 3,3′-diaminobenzidine (DOJINDO, Kumamoto, Japan).
Results
Western blotting
The results of WB using the 3F4 antibody are presented in . In control cases, PK-untreated samples (PK [−]) exhibited strong PrP signals from approximately 25–40 kDa (). In the PK-treated samples (PK [+]), no signal was detected in all control cases, and no PrPres. In the PK (−) samples of sCJD cases, PrP signals were observed mainly between 25 and 40 kDa with different intensities among cases (). PK (+) samples showed PrPres signals in all sCJD cases. The signal intensity and pattern varied among cases. In sCJD 1, 2, and 3, three PrPres bands were detected at 21–30 kDa (white arrowheads in ), and these were considered to correspond to unglycosylated, monoglycosylated, and diglycosylated PrPres, respectively. In addition, high molecular smear bands were also observed. sCJD 4 and 6 () also presented three PrPres bands, as well as high molecular smear bands; however, the bands corresponding to the unglycosylated PrPres were slightly wider at 19–21 kDa (black arrowheads in ). In sCJD 5, only faint PrPres was detected at 20–30 kDa. In sCJD 7, a broad band of unglycosylated PrPres at 19–21 kDa was detected (black arrowheads in ).
Figure 2. Western blot analyses of the formalin-fixed paraffin-embedded (FFPE) samples with the 3F4 anti-prion protein (PrP) antibody. (a) in control cases, proteinase K-untreated (PK [−]) samples show strong PrP signals at approximately 25–40kDa. Proteinase K-treated (PK [+]) samples show no signal. (b, c, d) in the sporadic Creutzfeldt–Jakob disease (sCJD) cases, PK-resistant PrP (PrPres) signals are observed with varied densities per case. In sCJD 1, 2, and 3, three PrPres bands are detected at 21–30 kDa (white arrowheads). sCJD 4, 6 and 7 also show three PrPres bands, but the unglycosylated PrPres are slightly wider at 19–21 kDa (black arrowheads). In sCJD 5, faint PrPres is detected at 21–30 kDa. In all sCJD cases, high molecular smear PrPres signals are observed with varied densities per case. (e) PK digestion reveals three bands of PrPres at 21–30 kDa (white arrowheads) in Gerstmann–Sträussler–Scheinker disease (GSS) 1 and 3, as well as low molecular weight bands at approximately 8 kDa (arrows) and strong high molecular weight smear bands. In GSS 2, a high molecular weight smear band and a band at approximately 8 kDa (arrow) are present. In glycosylphosphatidylinositol-anchorless prion disease (GPIALP) cases, PK digestion reveals smear bands from low to high molecular weight and low molecular bands at approximately 9 kDa (arrows). In V180I CJD cases, weak high molecular PrPres smear signals are observed in V180I CJD 1, PrPres signal are unclear in V180I CJD 2.
![Figure 2. Western blot analyses of the formalin-fixed paraffin-embedded (FFPE) samples with the 3F4 anti-prion protein (PrP) antibody. (a) in control cases, proteinase K-untreated (PK [−]) samples show strong PrP signals at approximately 25–40kDa. Proteinase K-treated (PK [+]) samples show no signal. (b, c, d) in the sporadic Creutzfeldt–Jakob disease (sCJD) cases, PK-resistant PrP (PrPres) signals are observed with varied densities per case. In sCJD 1, 2, and 3, three PrPres bands are detected at 21–30 kDa (white arrowheads). sCJD 4, 6 and 7 also show three PrPres bands, but the unglycosylated PrPres are slightly wider at 19–21 kDa (black arrowheads). In sCJD 5, faint PrPres is detected at 21–30 kDa. In all sCJD cases, high molecular smear PrPres signals are observed with varied densities per case. (e) PK digestion reveals three bands of PrPres at 21–30 kDa (white arrowheads) in Gerstmann–Sträussler–Scheinker disease (GSS) 1 and 3, as well as low molecular weight bands at approximately 8 kDa (arrows) and strong high molecular weight smear bands. In GSS 2, a high molecular weight smear band and a band at approximately 8 kDa (arrow) are present. In glycosylphosphatidylinositol-anchorless prion disease (GPIALP) cases, PK digestion reveals smear bands from low to high molecular weight and low molecular bands at approximately 9 kDa (arrows). In V180I CJD cases, weak high molecular PrPres smear signals are observed in V180I CJD 1, PrPres signal are unclear in V180I CJD 2.](/cms/asset/5ed6a7cc-4f00-4da1-ab91-999f14c2d9e2/kprn_a_2337981_f0002_b.gif)
In the PK (−) samples of the GSS cases, PrP signals were observed from 25 to 40 kDa (). PK digestion revealed low molecular PrPres signal at the molecular weight of 8 kDa in GSS 1, 2 and 3 (, arrow). Three PrPres bands were detected at 21–30 kDa in GSS 1 and 3 (, white arrowheads). High molecular weight strong PrPres smear signals were also observed in all three GSS cases. In GPIALP cases, PrP signals were also observed in PK (−) samples from 25 to 40 kDa; PK digestion revealed low molecular weight bands at approximately 9 kDa (, arrow) and smear bands from low to high molecular weight (). The PK (−) samples of V180I CJD cases also exhibited PrP signals from 25 to 40 kDa (). The PrPres signal was faint, with only faint high molecular PrPres seen in only V180I CJD case 1 ().
The results of WB using EP1802Y antibody are displayed in . In control cases, normal PrP signals were observed at 25–40 kDa in PK (−) samples; in PK (+), in all cases, a noticeable PrPres signal ranging from 21 to 30 kDa was not observed (). In the PK (−) samples of sCJD cases, as in 3F4, PrP signals were observed at 25–40 kDa with different intensities among cases (). The PK (+) samples of sCJD 1–5 and 7 showed moderate PrPres signals ranging from 21 to 30 kDa (, white arrowheads). In case 1–5 and 7, low molecular weight bands at approximately 16 kDa were also noted (arrows). In addition, in all cases, strong high molecular weight smear signal was observed (*). In sCJD 6, a band corresponding to the unglycosylated PrPres was observed at approximately 20 kDa (black arrowhead), which was lower in molecular weight than in sCJD 1–5 and 7. The 16 kDa band was not detected.
Figure 3. Western blot analyses of the formalin-fixed paraffin-embedded (FFPE) samples with the EP1802Y anti-prion protein (PrP) antibody. (a) in control cases, proteinase K-untreated (PK [−]) samples show PrP signals at 25–40 kDa. In proteinase K-treated (PK [+]) samples, there is no PrPres signals. (b, c) the PK (+) samples of sporadic Creutzfeldt–Jakob disease (sCJD) 1–7 show unglycosylated PK-resistant PrP (PrPres) signals at 21 kDa (case 1–5 and 7: white arrowheads) and about 19 kDa (case 6: arrowhead). High molecular smear PrPres signals are observed in all sCJD cases (*). In addition, a low molecular weight band at approximately 16 kDa (arrows) are also found in sCJD cases 1–5 and 7. The 16 kDa PrPres signal is not observed in sCJD 6. (D) in the Gerstmann–Sträussler–Scheinker disease (GSS) cases, unglycosylated PrPres at 21 kDa (white arrowheads) and high molecular smear bands are present (*). The low molecular weight band at approximately 8 kDa is not detectable. In glycosylphosphatidylinositol-anchorless prion disease (GPIALP) cases, no PrPres signal is detected. In the PK (+) sample of V180I CJD 1 (pentosan polysulfate [PPS]-treated case), a high molecular smear band (*) is visible, whereas the PrPres signal is substantially weak in V180I CJD 2 (PPS-untreated case).
![Figure 3. Western blot analyses of the formalin-fixed paraffin-embedded (FFPE) samples with the EP1802Y anti-prion protein (PrP) antibody. (a) in control cases, proteinase K-untreated (PK [−]) samples show PrP signals at 25–40 kDa. In proteinase K-treated (PK [+]) samples, there is no PrPres signals. (b, c) the PK (+) samples of sporadic Creutzfeldt–Jakob disease (sCJD) 1–7 show unglycosylated PK-resistant PrP (PrPres) signals at 21 kDa (case 1–5 and 7: white arrowheads) and about 19 kDa (case 6: arrowhead). High molecular smear PrPres signals are observed in all sCJD cases (*). In addition, a low molecular weight band at approximately 16 kDa (arrows) are also found in sCJD cases 1–5 and 7. The 16 kDa PrPres signal is not observed in sCJD 6. (D) in the Gerstmann–Sträussler–Scheinker disease (GSS) cases, unglycosylated PrPres at 21 kDa (white arrowheads) and high molecular smear bands are present (*). The low molecular weight band at approximately 8 kDa is not detectable. In glycosylphosphatidylinositol-anchorless prion disease (GPIALP) cases, no PrPres signal is detected. In the PK (+) sample of V180I CJD 1 (pentosan polysulfate [PPS]-treated case), a high molecular smear band (*) is visible, whereas the PrPres signal is substantially weak in V180I CJD 2 (PPS-untreated case).](/cms/asset/9b14b386-50b5-4037-8717-900317a7757d/kprn_a_2337981_f0003_b.gif)
In the GSS cases, the PK (+) samples of GSS 1–3 showed bands corresponding to the unglycosylated PrPres at 21 kDa (, white arrowheads) and smear bands with a high molecular weight ( *). The low molecular weight band at approximately 8 kDa observed in 3F4 was not detected in EP1802Y. In GPIALP cases, PK (−) presented signals at approximately 25–40 kDa, whereas no signal was detected in PK (+) (). In the PK (+) sample of V180I CJD 1 (PPS-treated case), a high molecular smear band was observed (*). In contrast, the signal was substantially weak in the PK (+) sample of V180I CJD 2 (PPS-untreated case) ().
Relative percentage of PrPres by molecular weight is shown in . Using 3F4, in sCJD cases, the average density of the three PrPres bands was higher than that of the high molecular PrPres, but no significant difference was observed (p > 0.05). In GSS cases, the average density of the high molecular PrPres was significantly higher than three PrPres bands (p < 0.01) and low molecular PrPres (p < 0.01). The average density of the low molecular PrPres was significantly lower than three PrPres bands (p = 0.01). In GPIALP cases, the high molecular weight PrPres and three PrPres bands were summed because the signals were smear-like. Then, the low molecular weight PrPres was compared to that of the high molecular and three PrPres bands. The average density of low molecular weight PrPres of GPIALP cases was higher than that of GSS cases, but there was no significance (p = 0.15). In V180I CJD cases, only V180I CJD 1 case treated with PPS showed high molecular PrPres signal. Using EP1802Y, in sCJD cases, the average density of the high molecular PrPres was significantly higher than three PrPres bands (p < 0.01) and low molecular PrPres (p < 0.01). The average density of three PrPres bands was significantly higher than low molecular PrPres (p < 0.01). In GSS cases, the average density of the high molecular PrPres was significantly higher than three PrPres bands (p = 0.01). The low molecular PrPres signals were not detected. In GPIALP cases, there was no significant PrPres signal. In V180I CJD cases, only V180I CJD 1 case treated with PPS showed high molecular PrPres signal.
Table 2. Relative percentage of protease-resistant prion protein by molecular weight.
displays the WB results using Tohoku 1 and 2 as the primary antibodies against PK (+) samples of sCJD cases. Using Tohoku 1 antibody, sCJD 1–5 and 7 showed moderate to strong PrPres smear signals ranging from about 20 kDa to high molecular weight. Whereas, in sCJD 6, Tohoku 1 immunopositive PrPres smear signals was faint. Using Tohoku 2 antibody, strong Tohoku 2 immunopositive PrPres signals were observed in sCJD 6, with mild to moderate signals in sCJD 2,4 and 5. In summary, WB results showed a predominantly type 1 PrPres signal in sCJD 1 and 3, type 1 + 2 in sCJDs 2, 4, 5 and 7, and type 2 in sCJD 6.
Figure 4. Western blotting analyses of the formalin-fixed paraffin-embedded (FFPE) samples with the Tohoku 1 (specific for type 1 scrapie prion protein [PrPSc]) and Tohoku 2 (specific for type 2 PrPSc) anti-proteinase K-resistant PrP (PrPres) antibodies. (a, b) Sporadic Creutzfeldt–Jakob disease (sCJD) 1 and 3 show bands of PrPres at 21–30 kDa and high molecular smears with Tohoku 1, whereas Tohoku 2 shows no signal. In sCJD 2, 4, and 5, PrPres signals are observed both with Tohoku 1 and 2. sCJD 6 shows a weak signal with Tohoku 1 but a strong signal with Tohoku 2. (c, d) sCJD 7 shows a smear band with Tohoku 1 and a weak smear band at approximately 20 kDa with Tohoku 2.
![Figure 4. Western blotting analyses of the formalin-fixed paraffin-embedded (FFPE) samples with the Tohoku 1 (specific for type 1 scrapie prion protein [PrPSc]) and Tohoku 2 (specific for type 2 PrPSc) anti-proteinase K-resistant PrP (PrPres) antibodies. (a, b) Sporadic Creutzfeldt–Jakob disease (sCJD) 1 and 3 show bands of PrPres at 21–30 kDa and high molecular smears with Tohoku 1, whereas Tohoku 2 shows no signal. In sCJD 2, 4, and 5, PrPres signals are observed both with Tohoku 1 and 2. sCJD 6 shows a weak signal with Tohoku 1 but a strong signal with Tohoku 2. (c, d) sCJD 7 shows a smear band with Tohoku 1 and a weak smear band at approximately 20 kDa with Tohoku 2.](/cms/asset/a65bb028-86a5-443f-88c7-c9d06ae3b71f/kprn_a_2337981_f0004_b.gif)
Histopathological findings
In sCJD 1, severe neuronal loss and gliosis with coarse neuropil degeneration in the cerebral cortex were observed (). IHC revealed synaptic PrP deposits (). In sCJD 2, neuronal loss, gliosis, spongiform changes (), and some large confluent vacuoles (, inset) were observed. IHC revealed fine granular synaptic deposits (). Irregularly shaped PrP around the large confluent vacuoles were also observed (, inset). Severe neuronal loss and gliosis were also observed in sCJD3 (). IHC indicated diffuse, frequent fine granular synaptic PrP deposits throughout the cortex (). In sCJD 4, spongiform changes were observed in the cortex. However, neuronal cell loss and hypertrophic astrocyte proliferation were relatively mild, although some large confluent vacuoles were also observed (). IHC revealed synaptic PrP deposits and irregularly shaped PrP around the large confluent vacuoles (). Perineuronal PrP deposits (, inset) were also observed. In sCJD 5, spongiform changes and occasional large confluent vacuoles were observed (). IHC revealed synaptic PrP deposits and PrP deposits around the large confluent vacuoles (). Spongiform changes and reactive astrocytes were observed in sCJD 6 (). IHC revealed synaptic, perineuronal, and plaque-type deposits (). In sCJD 7, extensive spongiform changes and gliosis were observed, along with large confluent vacuoles (). IHC revealed perivacuolar deposition and diffuse synaptic deposits (). In summary, IHC showed large confluent vacuoles and perivacuolar PrP deposits, a histological feature of sCJD type 2, in sCJD 2, 4, 5 and 7.
Figure 5. Histological findings of the prion disease cases. (A, C, E, G, I, K, M, O, Q, S, U, W: Hematoxylin and eosin staining (HE), B, D, F, H, J, L, N, P, R, T, V, X: Immunohistochemistry (IHC) with 3F4), (A, B: sporadic Creutzfeldt–Jakob disease case 1 (sCJD 1), C, D: sCJD 2, E, F: sCJD 3, G, H: sCJD 4, I, J: sCJD 5, K, L: sCJD 6, M, N: sCJD 7, O, P: Gerstmann–Sträussler–Scheinker disease case 1 (GSS 1), Q, R: glycosylphosphatidylinositol-anchorless prion disease case 1 (GPIALP 1), S, T: V180I CJD 1 (treated with pentosan polysulfate (PPS)), U, V: V180I CJD 2 without PPS, W, X: non prion case: control 3). (A, B: sCJD 1) HE staining indicating neuronal loss, gliosis and spongiform changes (a). IHC with 3F4 displaying synaptic prion protein (PrP) deposits in the cerebral cortex (b). (C, D: sCJD 2) HE staining shows spongiform changes and some large confluent vacuoles (inset) (c). 3F4 reveals synaptic PrP and perivacuolar PrP deposits (inset) (d). (E, F: sCJD 3) HE staining indicating neuronal loss, gliosis and spongiform changes (e). 3F4 displaying synaptic PrP deposits in the cerebral cortex (f). (G, H: sCJD 4) HE staining shows spongiform changes and some large confluent vacuoles (g). 3F4 reveals synaptic PrP deposits, perivacuolar PrP deposits and perineuronal PrP deposits (inset) (h). (I, J: sCJD 5) HE staining shows spongiform changes and some large confluent vacuoles (I). 3F4 reveals synaptic PrP and perivacuolar PrP deposits (j). (K, L: sCJD 6) HE staining shows spongiform changes (k). 3F4 reveals synaptic, perineuronal (inset), and plaque-type deposits (inset) (l). (M, N: sCJD 7) HE staining shows spongiform changes, gliosis, and large confluent vacuoles (m). 3F4 reveals PrP perivacuolar and diffuse synaptic deposits (n). (O, P: GSS 1) HE staining shows spongiform changes and PrP plaques (inset) (o). 3F4 reveals numerous PrP plaques (inset) and synaptic deposits (p). (Q, R: GPIALP 1) HE staining shows minimal spongiform changes and only mild neuronal loss (q). 3F4 reveals numerous PrP coarse granular deposits in the neuropil and around the vessels (r). (S, T: V180I CJD 1 treated with PPS) HE staining shows spongiform changes and gliosis (s). 3F4 reveals synaptic deposits and many PrP coarse deposits (t). (U, V: V180I CJD 2 without PPS) HE staining shows extensive and severe spongiform changes (u). 3F4 shows virtually no abnormal PrP deposits (v). (W, X: non prion case: control 3) HE shows that the neurons and neuropil are preserved, and there is no obvious neurodegenerative pathology (w). 3F4 shows a weak positive PrP staining in the cortex (x). (Scale bars: A-V: 50 µm, inset: C, D, H, O, P: 25 µm, L: 20 µm).
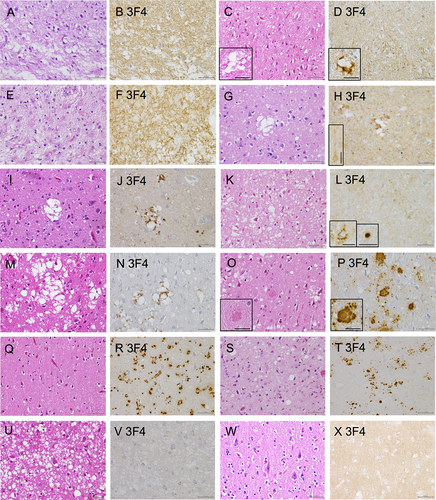
In GSS 1, neuronal loss, spongiform changes, and gliosis (), as well as PrP plaques, were also observed (, inset). IHC revealed numerous PrP plaques and synaptic PrP deposits (). GPIALP 2 exhibited minimal spongiform changes and only mild neuronal loss (). IHC revealed numerous PrP coarse granular deposits in the neuropil and around the vessels (). In V180I CJD 1 (PPS-treated case), neuronal loss, spongiform changes, and gliosis were evident in the cerebral cortex; however, coarsening of the tissue was relatively mild (). IHC revealed synaptic deposits and many PrP coarse deposits (). V180I CJD 2 (PPS-untreated case) showed extensive and severe spongiform changes in the cerebral cortex with relatively mild neuronal loss (). IHC showed virtually no abnormal PrP deposits (). In the control case, neurons are preserved and there are no spongiform changes of neuropil (). IHC revealed a uniform and weak positive PrP staining in the neuropil, suggesting the presence of normal PrP in the cerebral cortex ().
Discussion
Our new method clearly proved PrPres signal from FFPE samples. The use of PK before the protein extraction stage from FFPE was important for a high efficiency of PrPres extraction. Furthermore, the characteristic signal could be identified and differentiated in sCJD types 1 and 2, as well as in inherited prion diseases such as GSS, GPIALP, and V180I CJD, using multiple antibodies. Collectively, our method facilitates the biochemical diagnosis of prion disease from FFPE samples.
The sCJD cases 1 and 3, in which the unglycosylated PrPres signal was observed mainly at 21 kDa, were predominantly PrPres type 1 with a strong Tohoku 1 signal, and these findings were consistent with the histological findings of 3F4, which showed fine granular synaptic PrP deposition. Whereas, in cases 2, 4, 5 and 7, PrPres was detected in both Tohoku 1 and Tohoku 2; these results were compatible with the histology findings of type 2 sCJD, such as perivacuolar and perineuronal PrP depositions in addition to synaptic deposition characteristic of type 1. In contrast, case 6 showed predominantly Tohoku 2 PrPres signals with histological findings of confluent large vacuole and perivacuolar PrP deposits, consistent with sCJD type 2. In summary, cases 1 and 3 were sCJD type 1, cases 2, 4, 5 and 7 were sCJD type 1 + 2 and case 6 was sCJD type 2, consistent with WB and histological results. Furthermore, our new method used the EP1802Y antibody and revealed a 16 kDa C-terminal PrPres fragment suggestive of type 1 PrPres [Citation23]. The 16 kDa C-terminal PrPres fragment was noted in cases 1–5 and 7, whereas not in case 6.
The PrPres signals by 3F4 in GSS and GPIALP cases were 8/9 kDa low molecular PrPres signals and high molecular smear PrPres signals, which are considerably different from the signal pattern of sCJD and can be distinguished from it. Conversely, GSS and GPIALP are difficult to distinguish because of their similarity. However, the results of EP1802Y are dramatically different between these cases, and the PrPres signal was undetected in GPIALP. This is because GPIALP does not include the epitope recognized by EP1802Y owing to the stop codon at the C-terminus.
In V180I CJD, it was difficult to detect the PrPres signal in WB using 3F4. Identifying PrPres deposition in tissues and WB using frozen samples for V180I is challenging. We previously reported a case of V180I CJD treated with PPS [Citation7]. This case had relatively high histological deposition of PrP with easy identification of PrPres in WB using frozen samples. In the present study, the PrPres signal using 3F4 was slightly stronger in the PPS-treated case than in the PPS-untreated case. Using EP1802Y, a strong smear signal was observed in the PPS-treated case, whereas the smear signal was weak in the PPS-untreated case. In V180I CJD, EP1802Y, which recognizes the C-terminus, may be able to identify PrPres more easily than 3F4, which recognizes the prion protein core.
In our method, high molecular smear PrPres signals were evident using 3F4 and EP1802Y in almost all prion disease cases. On the other hand, no high smear PrP signals were found in non-prion cases. The high molecular smear PrPres signal is rarely observed in frozen samples. The aggregation of PrPres may have become stronger during the FFPE sample preparation process, which may have made it difficult to isolate PrP during thermal boiling, resulting in a high molecular smear PrPres signals. In GPIALP, 3F4 revealed marked high molecular smear PrPres signals, but no signals using EP1802Y. As GPIALP does not have an epitope for EP1802Y, the result that it is only recognized by 3F4 suggests that PrPres without the C-terminus is accurately extracted. Furthermore, even in V180I CJD, where PrPres signals are usually difficult to obtain, high molecular smear PrPres signals were relatively easy to obtain when EP1802Y was used. In view of these, high molecular smear PrPres signals seem to be a very powerful PrPres detection signal in this method using FFPE samples.
These results are summarized in . The prion pathotype can be diagnosed from FFPE samples if tested according to this chart. The representative band patterns are also summarized in .
Figure 6. Schema for classing prion diseases using western blotting (WB) from formalin-fixed paraffin-embedded (FFPE) samples. In proteinase K-treated FFPE samples by western blotting with 3F4 antibody, the presence of 21–30 kDa three PrPres bands, high molecular smear PrPres signals, or low molecular PrPres signals strongly suggests prion disease. In 3F4, three PrPres bands + moderate high molecular smear PrPres signals suggest sCJD, strong high molecular smear PrPres signals + moderate three PrPres bands + moderate low molecular PrPres signals suggests GSS or GPIALP. In V180I CJD, it is difficult to detect, and only V180I CJD 1 PPS-treated case show weak high molecular smear PrPres signals. In sCJD cases, western blotting using Tohoku 1 (TH1) and Tohoku 2 (TH2) antibodies specific for type 1 and type 2 PrPres, respectively, can also distinguish between type 1 and type 2 PrPres with this method. In the case of V180I CJD, western blotting with EP1802Y antibody enhanced the high molecular smear PrPres signal, which was weak in 3F4. However, this was also only in PPS-treated case (V180I CJD 1), and it was difficult to identify the PrPres signal in untreated V180I CJD (V180I CJD 2).
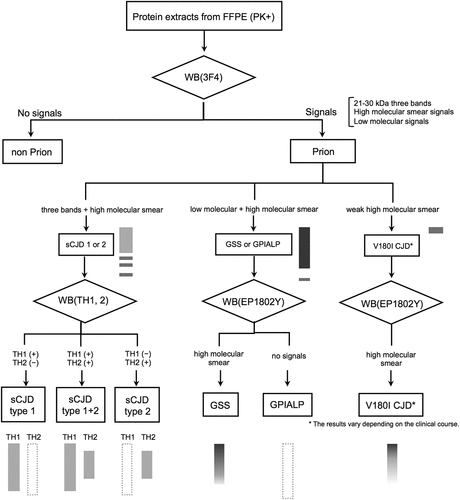
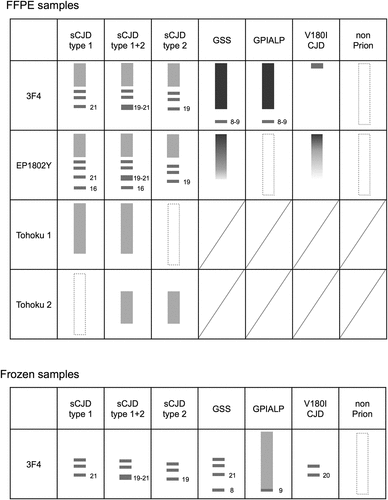
Figure 7. Representative band patterns of western blotting (WB) of formalin-fixed paraffin-embedded (FFPE) samples using multiple antibodies specific for different epitopes. Band patterns for the conventional method using frozen samples are displayed at the bottom of the table.
Since the sections from the same FFPE block are used as samples, pathological and biochemical evaluations can be performed at the same site. Our new PrPres detection method using FFPE showed biochemical PrPres characteristics of sCJD and hereditary prion diseases, which were compatible with the histological features of each disease type. Many samples, such as the brain and spinal cord, are commonly processed as FFPE, and biochemical evaluations such as type 1 and type 2 can also be performed on them. Therefore, this method can theoretically be used to evaluate the biochemical status of PrPres in all autopsy cases of prion diseases where FFPE is available. Even for areas, such as the hypothalamus, where it is challenging to obtain frozen samples during the autopsy, FFPE samples can be used first for observation and subsequent biochemical characterization. However, the limitation of this method is that if formalin fixation is prolonged, the PrP band will not be detectable [Citation24].
FFPE samples are inactivated by formic acid treatment; therefore, no special infection control measures are required, and the samples can be assessed in a general laboratory. Furthermore, WB can be performed without frozen samples, and biochemical evaluation is possible even if prion diseases are suspected after the autopsy; therefore, a definitive diagnosis can be obtained. Prion disease-derived infection is a risk factor during cadaveric surgical training [Citation25]; however, this risk may be mitigated by screening using our proposed methodology.
Author Contribution
Sachiko Koyama and Hiroyuki Honda conceptualized the study, conducted examinations, analysed the data, and wrote manuscript. Kaoru Yagita, Hideomi Hamasaki, Hideko Noguchi and Masahiro Shijo, contributed to the data analysis and examinations. Kosuke Matsuzono, Kei-Ichiro Takase, Keita Kai, Shin-Ichi Aishima, Kyoko Itoh Toshiharu Ninomiya and Naokazu Sasagasako, provided the autopsy samples and clinical data.
Acknowledgments
This work was funded by a Grant of The Clinical Research Promotion Foundation (2021). The authors greatly appreciate the patients and their families for their collaboration with this study. We would also like to thank Tetsuyuki Kitamoto (Department of Neurological Science, Tohoku University Graduate School of Medicine, Miyagi, Japan) for genetic test, and Kyoko Iinuma (Department of Anatomy and Neuroscience, Graduate School of Medical Sciences, Kyushu University, Fukuoka, Japan) and Toru Iwaki (Kyushu University, Fukuoka, Japan) for technical and conceptual assistance, and Editage (www.editage.com) for the English language editing.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data supporting the findings of this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Prusiner SB. The prion diseases. Brain Pathology. 1998;8(3):499–513. doi: 10.1111/j.1750-3639.1998.tb00171.x
- Pan KM, Baldwin M, Nguyen J, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A. 1993;90(23):10962–10966. doi: 10.1073/pnas.90.23.10962
- Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46(2):224–233. doi: 10.1002/1531-8249(199908)46:2<224:AID-ANA12>3.0.CO;2-W
- Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39(6):767–778. doi: 10.1002/ana.410390613
- Nozaki I, Hamaguchi T, Sanjo N, et al. Prospective 10-year surveillance of human prion diseases in Japan. Brain. 2010;133(10):3043–3057. doi: 10.1093/brain/awq216
- Iwasaki Y, Mori K, Ito M, et al. An autopsied case of V180I Creutzfeldt-Jakob disease presenting with panencephalopathic-type pathology and a characteristic prion protein type. Neuropathology. 2011;31(5):540–548. doi: 10.1111/j.1440-1789.2010.01192.x
- Honda H, Ishii R, Hamano A, et al. Microsphere formation in a subtype of Creutzfeldt–Jakob disease with a V180I mutation and codon 129 MM polymorphism. Neuropathol Appl Neurobiol. 2013;39(7):844–848. doi: 10.1111/nan.12047
- Piccardo P, Dlouhy SR, Lievens PM, et al. Phenotypic variability of Gerstmann-Sträussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol. 1998;57(10):979–988. doi: 10.1097/00005072-199810000-00010
- Parchi P, Chen SG, Brown P, et al. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann–Sträussler–Scheinker disease. Proc Natl Acad Sci U S A. 1998;95(14):8322–8327. doi: 10.1073/pnas.95.14.8322
- Mead S, Gandhi S, Beck J, et al. A novel prion disease associated with diarrhea and autonomic neuropathy. N Engl J Med. 2013;369(20):1904–1914. doi: 10.1056/NEJMoa1214747
- Matsuzono K, Ikeda Y, Liu W, et al. A novel familial prion disease causing pan-autonomic-sensory neuropathy and cognitive impairment. Eur J Neurol. 2013;20(5):e67–9. doi: 10.1111/ene.12089
- Honda H, Matsuzono K, Fushimi S, et al. C-Terminal-deleted prion protein fragment is a Major Accumulated Component of Systemic PrP deposits in hereditary prion disease with a 2-bp (CT) deletion in PRNP codon 178. J Neuropathol Exp Neurol. 2016;75(11):1008–1019. doi: 10.1093/jnen/nlw077
- Kunkle RA, Nicholson EM, Lebepe-Mazur S, et al. Western blot detection of PrP Sc in archived paraffin-embedded brainstem from scrapie-affected sheep. J Vet Diagn Invest. 2008;20(4):522–526. doi: 10.1177/104063870802000421
- Nicholson EM. Enrichment of PrPSc in formalin-fixed, paraffin-embedded tissues prior to analysis by Western blot. J Vet Diagn Invest. 2011;23(4):790–792. doi: 10.1177/1040638711407886
- Dorj G, Okada H, Miyazawa K, et al. Retrospective analysis of sheep scrapie by western blotting with formalin-fixed paraffin-embedded (FFPE) tissues. J Vet Med Sci. 2012;74(9):1207–1210. doi: 10.1292/jvms.12-0037
- Nakagaki T, Kaneko M, Satoh K, et al. Detection of prions in a cadaver for anatomical practice. N Engl J Med. 2022;386(23):2245–2246. doi: 10.1056/NEJMc2204116
- Rodríguez-Rigueiro T, Valladares-Ayerbes M, Haz-Conde M, et al. A novel procedure for protein extraction from formalin-fixed paraffin-embedded tissues. Proteomics. 2011;11(12):2555–2559. doi: 10.1002/pmic.201000809
- Inoue T, Hagiyama M, Yoneshige A, et al. Increased ectodomain shedding of cell adhesion molecule 1 from pancreatic islets in type 2 diabetic pancreata: correlation with hemoglobin A1c levels. PloS One. 2014;9(6):e100988. doi: 10.1371/journal.pone.0100988
- Schulz-Schaeffer WJ, Tschöke S, Kranefuss N, et al. The paraffin-embedded tissue blot detects PrP(Sc) early in the incubation time in prion diseases. Am J Pathol. 2000;156(1):51–56. doi: 10.1016/S0002-9440(10)64705-0
- Schulz-Schaeffer WJ, Fatzer R, Vandevelde M, et al. Detection of PrP(Sc) in subclinical BSE with the paraffin-embedded tissue (PET) blot. Arch Virol Suppl. 2000;16:173–180. doi: 10.1007/978-3-7091-6308-5_16
- Hofmann A, Wrede A, Jürgens-Wemheuer WM, et al. Prion type 2 selection in sporadic Creutzfeldt–Jakob disease affecting peripheral ganglia. Acta Neuropathol Commun. 2021;9(1):187. doi: 10.1186/s40478-021-01286-4
- Kobayashi A, Sakuma N, Matsuura Y, et al. Experimental verification of a traceback phenomenon in prion infection. J Virol. 2010;84(7):3230–3238. doi: 10.1128/JVI.02387-09
- Notari S, Strammiello R, Capellari S, et al. Characterization of truncated forms of abnormal prion protein in Creutzfeldt-Jakob disease. J Biol Chem. 2008;283(45):30557–30565. doi: 10.1074/jbc.M801877200
- Nicholson EM, Kunkle RA, Hamir AN, et al. Detection of the disease-associated isoform of the prion protein in formalin-fixed tissues by Western blot. J Vet Diagn Invest. 2007;19(5):548–552. doi: 10.1177/104063870701900515
- Ogami-Takamura K, Saiki K, Endo D, et al. The risk of Creutzfeldt–Jakob disease infection in cadaveric surgical training. Anat Sci Int. 2022;97(3):297–302. doi: 10.1007/s12565-022-00662-x