
ABSTRACT
Rock-inhabiting fungi (RIF) are slow-growing microorganisms that inhabit rocks and exhibit exceptional stress tolerance owing to their thick melanised cell walls. This study reports the identification of a novel rock-inhabiting fungus, Cladophialophora brunneola sp. nov. which was isolated from a karst landform in Guizhou, China, using a combination of morphological and phylogenetic analyses. The genome of C. brunneola was sequenced and assembled, with a total size of approximately 33.8 Mb, encoding 14,168 proteins and yielding an N50 length of 1.88 Mb. C. brunneola possessed a larger proportion of species-specific genes, and phylogenomic analysis positioned it in an early diverged lineage within Chaetothyriales. In comparison to non-RIF, C. brunneola displayed reduction in carbohydrate-active enzyme families (CAZymes) and secondary metabolite biosynthetic gene clusters (BGCs). Transcriptome analysis conducted under PEG-induced drought stress revealed elevated expression levels of genes associated with melanin synthesis pathways, cell wall biosynthesis, and lipid metabolism. This study contributes to our understanding of the genomic evolution and polyextremotolerance exhibited by rock-inhabiting fungi.
1. Introduction
Fungi exhibit a global distribution, inhabiting diverse ecosystems such as tropical, polar, terrestrial, aquatic, and even endosymbiotic environments, acquiring nutrients through various ecological strategies, including saprophytic, parasitic, and predatory lifestyles (de Boer et al. Citation2005; Ohm et al. Citation2012; Yang et al. Citation2012, Citation2023; Pusztahelyi et al. Citation2015; Gostinčar et al. Citation2018; Coleine et al. Citation2022). Bare rock surface, characterised by limited moisture and nutrients, high UV radiation, and fluctuating temperatures, represents one of the most inhospitable habitats on earth (Gorbushina Citation2007). Rock-inhabiting fungi (RIF) have developed adaptations to thrive in this challenging environment, benefiting from the reduced competition with other microbes and their extremotolerance (Gorbushina et al. Citation1993; Wollenzien et al. Citation1995; Chertov et al. Citation2004). Despite their worldwide distribution, RIF have often been overlooked due to their small size, extremely slow growth, and lack of diagnostic morphological features. These fungi are characterised by the absence of sexual reproductive structures and form compact, melanised colonies on rocks. They are considered obligate rock dwellers with limited growth rates, often referred to as “lithophilic” fungi (Liu et al. Citation2021). RIF are a polyphyletic group mainly affiliated with Dothideomycetes, Eurotiomycetes, and Arthoniomycetes in Ascomycota, especially in the orders Dothideales, Capnodiales in Dothideomycetes, and Chaetothyriales in Eurotiomycetes (Gueidan et al. Citation2008; Ruibal et al. Citation2008, Citation2009; Egidi et al. Citation2014). Currently, limited genomic data is available for RIF. The first RIF genome to be sequenced was that of Cryomyces antarcticus MA 5682 (Sterflinger et al. Citation2014), followed by Coniosporium apollinis CBS 100218 (Teixeira et al. Citation2017) and Knufia petricola MA 5789 (Tesei et al. Citation2017). Proteomic data subsequently revealed changes in protein expression patterns under stress conditions, including a reduction in the number of expressed proteins and the upregulation of unidentified proteins (Tesei et al. Citation2012; Zakharova et al. Citation2013, Citation2014). It appears that RIF reduces metabolic levels as a mechanism to withstand adverse environmental factors. However, further transcriptomic and proteomic data are necessary to fully understand the mechanisms of stress adaption and metabolic activity during active growth and dormant phases.
Cladophialophora Borelli is classified as an asexual genus in the family Herpotrichiellaceae (Badali et al. Citation2008). It is composed of black yeast-like fungi species. The genus is considered to be simple hyphomycetes, characterised by brown hyphae that give rise to branched chains of pale brown conidia. Currently, Cladophialophora comprises over 40 species, which frequently act as opportunistic pathogens, causing a range of human infections from mild cutaneous lesions to fatal encephalitis (Horre and de Hoog Citation1999; Badali et al. Citation2008, Citation2009, Citation2010). These fungi can also be found as phytopathogens (Crous et al. Citation2007) and in environmental samples (Usui et al. Citation2016; Kiyuna et al. Citation2018; Das et al. Citation2019; Sun et al. Citation2020). Numerous molecular phylogenetic studies have shown that Cladophialophora is polyphyletic within the order Chaetothyriales. Additionally, this genus is closely related to various anamorphic genera, such as Exophiala, Cyphellophora, Fonsecaea, Knufia, Phialophora, and the teleomorphic genus Capronia (Kiyuna et al. Citation2018; Quan et al. Citation2020). Only five species of black yeast-like fungi have been sequenced in this genus, including three opportunistic pathogens (C. bantiana, C. carrionii, and C. yegresii) and two species from the environment (C. psammophila and C. immunda) with the ability to degrade polyaromatic hydrocarbons (Sterflinger et al. Citation2015; Teixeira et al. Citation2017).
In a survey of resources and diversity of RIF in Guizhou Province, a previously unrecognised species within the genus Cladophialophora was discovered. Through phylogenetic analysis of multiple gene sequences, including the internal transcribed spacer (ITS), small subunit of ribosomal RNA (SSU), ribosomal large subunit (LSU), translation elongation factor (TEF) exon genes, and β-tubulin (TUB) exon genes, the isolate was identified as Cladophialophora brunneola. Although RIF face numerous simultaneous stress factors in their natural habitat, the mechanisms underlying their adaptation to such niches remain poorly understood. Given that C. brunneola represents one of the typical lineages of RIF in the family Herpotrichiellaceae of Eurotiomycetes, we propose that analysing genomic and stress-response transcriptome data can provide new insights into the genome evolution and the development of polyextremotolerance in RIF.
2. Materials and methods
2.1. Sample collection on rocks and strain isolation
Rock samples were collected in a karst landform in Fanjingshan National Nature Reserve in Tongren City, Guizhou Province, China (27°55′ N, 108°42′ E, 2,220 m altitude). Vegetation was abundant around the circumstances of sampling sites (). Samples containing black colonies were obtained by using a sterile chisel to detach them from the stock rocks (). The collected samples were promptly sealed in plastic bags, stored in an ice box, and transported to the laboratory. Rock pieces (approximately 0.5–1 cm3) with black colonies were obtained from the rock samples using an industrial stone splitter (Model CM-10, Hydrasplit, Park Industries, Inc., St. Cloud, MN, USA). These rock pieces were subsequently surface-disinfected with 95% (v/v) ethanol for 3–5 seconds, transferred to physiological saline containing 0.001% (v/v) tween-20, washed with sterilised distilled water, and dried on sterilised filter papers. Approximately 1 gram of each sample was pulverised in a sterilised mortar, and the resulting powder was suspended in 2–3 mL of sterile water. Aliquots of suspensions (200 μL) were evenly spread onto Petri plates containing dichloran rose Bengal chlortetracycline (DRBC) agar with the addition of 100 mg/L streptomycin after autoclaving. The plates were then incubated at 10 °C for 4 weeks. Slow-developing colonies exhibiting dark pigmentation on the plates were transferred to MEA (malt extract 2%, peptone 2‰, and agar 1.5%) for further purification and identification. The type specimens were deposited in the Herbarium of Mycology at the Institute of Microbiology, Chinese Academy of Sciences in Beijing, China (HMAS), and ex-type living cultures were deposited in the China General Microbiological Culture Collection Center (CGMCC).
Figure 1. Sampling site and culture of Cladophialophora brunneola (CGMCC 3.18770). (a) Landscape of sampling site. (b) Rocks bearing black colonies. (c) Colony on 2% MEA after 4 weeks. (d–h) Conidial chains. (i, j) Conidia. Scale bars = 10 µm.

2.2. Morphological characterisation
The morphological characteristics of the strain were evaluated by incubating on 2% MEA for a duration of 4 weeks at 23 °C. After incubation, measurements were taken for colony diameter and colour. To examine the mycological characteristics, such as the presence of conidia, aerial mycelium, density, and pigment production, a light microscope (Nikon Eclipse 80i, Tokyo, Japan) was utilised.
2.3. DNA extraction, PCR amplification, and sequencing
The genomic DNA was extracted from fungal mycelia grown on MEA plates using the rapid “thermolysis” protocol (Zhang et al. Citation2010). The amplification of gene fragments, including ITS (internal transcribed spacer gene region), SSU (small subunit ribosomal RNA gene), LSU (large subunit ribosomal RNA gene), TEF (translation elongation factor), and TUB (the partial β-tubulin gene), was performed using the following primer pairs: ITS4/ITS5 for ITS (White et al. Citation1990), NS1/NS4 for partial SSU (White et al. Citation1990), LROR/LR5 for partial LSU (Rehner and Samuels Citation1994), 728 F/986 R for partial TEF (Carbone and Kohn Citation1999), and Bt2a/Bt2b for partial TUB (Glass and Donaldson Citation1995). The PCR programmes were performed as described by Su et al. (Citation2015). The PCR products were subsequently sequenced by Beijing Tianyihuiyuan Bioscience and Technology after evaluation through electrophoresis.
2.4. Phylogenetic analyses
The newly obtained sequences were queried against the nuclear database of NCBI. Related available sequences from GenBank were obtained and included in the analysis (). Multiple sequence alignments of five genes were generated using MAFFT v.7.407 with Q-INS-I strategy (Katoh and Standley Citation2013). Conserved blocks were selected from the initial alignments using Gblocks 0.91b (Castresana Citation2000). For phylogenetic reconstructions, both maximum likelihood (ML) and Bayesian inference (BI) methods were employed. ML analyses were conducted using RAxML v.8.2.12 (Stamatakis Citation2014) with the GTR + GAMMA model. Maximum likelihood bootstrap proportions (MLBP) were determined by performing 100 replicates. Models of evolution for BI analyses were estimated using PartitionFinder 2 (Lanfear et al. Citation2017). Bayesian inference posterior probabilities (BIPP) were calculated using Markov Chain Monte Carlo sampling (MCMC) in MrBayes v3.2.6 (Ronquist et al. Citation2012) under the estimated model of evolution. Two independent analyses with four Markov chains were run for 2,000,000 generations until the average standard deviation of the split frequencies dropped below 0.01. Trees were sampled every 100th generation. The initial 25% of MCMC sampling generations were discarded as burn-in. The trees were visualised using FigTree v1.4.4.
Table 1. Characteristics of genome assembly of Cladophialophora brunneola..
2.5. Genome sequencing, assembly, and annotation
We sequenced the genome of C. brunneola strain CGMCC 3.18770 using the PacBio Sequel and Illumina HiSeq platform at Allwegenes Tech (Tianjin, China). Library construction and genome sequencing were carried out using the company’s standardised pipeline. The PacBio Sequel reads were obtained and subjected to trimming, correction, and assembly using the Canu assembler version 1.8 (Koren et al. Citation2017). To minimise error rate, the read correction parameter “correctedErrorRate = 0.035” was employed. Moreover, Illumina short reads were used to further polish the assembly genome with Pilon v1.23 (Walker et al. Citation2014). The completeness of genome annotation was accessed using the ascomycota_odb10 database in BUSCO v5.2.2 (Seppey et al. Citation2019). Knufia petricola MA 5789, Coniosporium apollinis CBS 100218 from previous research were selected for RIF group. Other fungal genomes used for comparative analysis were downloaded from NCBI-GenBank database.
Tandem Repeat Finder v.4.04 (Benson Citation1999), LTR_FINDER (Xu and Wang Citation2007), RECON v.1.08, RepeatScout v.1.0.5, and RepeatModeler v.1.0.11 (http://www.repeatmasker.org; last accessed on 13 October 2022) were used to construct species-specific repeat sequence databases through identification of structural features and de novo prediction. Subsequently, these databases were merged with Repbase (Bao et al. Citation2015) to form a comprehensive reference repeat database. Finally, RepeatMasker v.4.0.8 (Chen Citation2004) was utilised to predict types and quantities of repeat sequences in the genome.
The GETA pipeline (https://github.com/chenlianfu/geta; last accessed on 13 October 2022) was used to generate a comprehensive set of protein-coding genes. RNA-seq reads obtained from different treatment tissues were mapped to the assembly using Hisat2 (Kim et al. Citation2019). Ab initio gene predictions were performed using Augustus (Stanke et al. Citation2004), which was trained with the RNA-seq data. The predicted gene sets were functionally annotated using eggNOG-mapper (Huerta-Cepas et al. Citation2019) and InterProScan (Jones et al. Citation2014) with default parameters. Noncoding RNA was predicted using Infernal v.1.1.2 (Nawrocki and Eddy Citation2013) with the Rfam 14.1 database (Kalvari et al. Citation2018). The presence of secondary metabolite BGCs was searched on the antiSMASH website (fungal version 5.2.0) (Blin et al. Citation2019). For the prediction of CAZymes, we employed blastp and HMMER v.2.2 (Finn et al. Citation2011) against the dbCAN2 (Zhang et al. Citation2018) database.
2.6. Phylogenomic reconstruction
Single-copy orthologs were identified in Orthofinder v.2.5.4 (Emms and Kelly Citation2015). Sequence alignments were performed using MAFFT v.7.407 (Katoh and Standley Citation2013), and gaps were eliminated using Gblocks 0.91b (Castresana Citation2000). Subsequently, the genes were concatenated to generate a supergene sequence. The best-fitting model was estimated with ModelTest-NG (Darriba et al. Citation2020). A maximum likelihood (ML) phylogenomic tree was built using RAxML v.8.2.12 (Stamatakis Citation2014) with 1,000 bootstraps to infer branch support.
2.7. Culture under PEG-induced drought stress and transcriptome analysis
Polyethylene-glycol (PEG), a non-permeable osmolyte known to induce significant water deficit (Lagerwerff et al. Citation1961), was used to assess the drought resistance of C. brunneola. Shake culture medium was prepared by adding 0%, 5%, 10%, 20%, and 40% PEG to 2% MEB solution, and was incubated at 23 °C for a month of observation. For RNA-seq analysis, samples treated with 0% (CK) and 20% (P) PEG for 7 days of cultivation were selected. Each sample was conducted in triplicate, and RNA was extracted using Trizol (InvitrogenTM, Carlsbad, CA, USA) (Meng and Feldman Citation2010). Sequencing was performed on the Illumina NovaSeq platform at Annoroad Gene Technology Co., Ltd. Library construction and genome sequencing followed the standardised pipeline of the company. TrimGalore-0.6.6 was used to remove adapters and low-quality bases, and the resulting clean reads were aligned to the genome of C. brunneola using hisat2 (Kim et al. Citation2015). Alignment files were then ordered using Samtools 1.6, and quantification of the mapped reads to exons was conducted with stringtie v1.3.5 (Pertea et al. Citation2015). The TPM (transcripts per million) method was used to calculate the expression level. Differentially expressed genes (DEGs) were identified using DEseq2 R package (Wang et al. Citation2010) with adjusted P-value < 0.05 and |log2Foldchange| > 1 as thresholds. Further Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of the DEGs was performed using the combination of Fisher’s exact test.
2.8. Melanin coarse extraction and spectrophotometric measurements
Melanin was extracted following previously established methods with minor modifications (Pal et al. Citation2013; Liu et al. Citation2018; Pralea et al. Citation2019). Approximately 1 mg of fungal biomass from the culture filtrate was mixed with 1 mol/L NaOH and refluxed for 2 hours. Subsequently, the solution was centrifuged at 10,000 r/min for 5 minutes, and the resulting supernatant was acidified overnight at room temperature using concentrated HCl until pH reached to 2–3. The resulting precipitate was collected by centrifugation at 10,000 r/min for 5 minutes, and washed with 2 mL of ddH2O and 3 mL of organic solvents (chloroform:ethyl acetate:ethanol = 1:1:1). The precipitate was then dissolved in 1 mol/L NaOH, followed by acidification using concentrated HCl again as described above. The reacquired precipitate was freeze-dried to obtain crude melanin.
Extracted crude melanin samples were suspended in 1 mL of 1 mol/L NaOH to measure absorbance in the range of 200–800, utilising the ThermoGenesys-10S UV-Vis spectrophotometer. The blank control was prepared using 1 mol/L NaOH solution.
3. Results
3.1. Taxonomy
Cladophialophora brunneola W. Sun, L. Su, M.C. Xiang and X.Z. Liu, sp. nov. ()
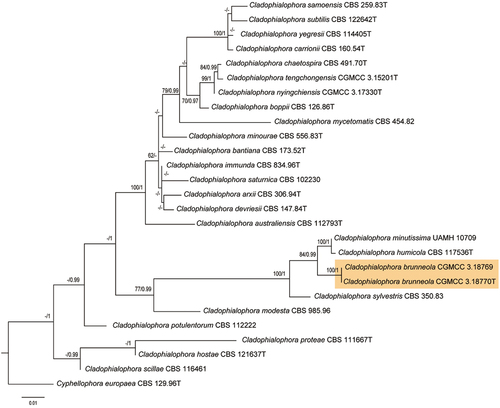
Figure 2. Phylogenetic tree of the Cladophialophora genus generated using maximum likelihood analysis with combined of ITS, SSU, and LSU sequences. Node values (ML/BI) indicating bootstrap support (left) and Bayesian posterior probability (right) are shown. Sequences generated in this study are highlighted in orange. The outgroup used in this analysis is Cyphellophora europaea strain CBS 129.96.
MycoBank: MB824202.
Etymology: brunneola (Lat., masc. adj.), referring to the colour of conidia and conidial chains.
Type: China, Guizhou Province, isolated from rock on 12 November 2014, by Meichun Xiang and Wenjiao Cai. The holotype is deposited at HMAS 247720 (dried culture), and the ex-type culture is preserved at CGMCC 3.18770. Another culture is also preserved at CGMCC 3.18769.
Description: Following a 4-week incubation period at 25 °C on MEA, the colonies reach a diameter of 12 mm. The central areas exhibit a light brown colouration and possess short aerial hyphae, whereas the periphery displays a dark brown, pale, and glossy appearance with pigment inside the medium, forming a zonate pattern (). The reverse of colonies appears olive brown. The mycelium is composed of less branched, smooth, thin-walled, septate hyphae that contain lipid droplets. These hyphae vary in colour from light to dark brown and exhibit a width of 1.1–1.9 μm. The conidia are unicellular, thick-walled, and range from hyaline to pale olivaceous in colour, from spherical to ellipsoid in shape. The dimensions of conidia are 3.5–5.5 × 4.2–4.9 μm, and they always form long chains (). No teleomorph form is observed.
Notes: Phylogenically, C. brunneola is closely related to C. humicola and C. minutissima. However, the conidia of C. brunneola are much smaller. Morphologically, C. humicola produces subcylindrical to narrowly ellipsoid conidia measuring 8–17 × 1.5–2.5 μm, characterised by an unthickened cell wall (Crous et al. Citation2007). C. minutissima produces cylindrical to fusiform conidia with truncated and thickened hila, measuring 8–22 × 1–2 μm (Davey and Currah Citation2007).
3.2. Phylogenetic analyses
The alignment results showed that the ITS sequence of C. brunneola was less than 97% similar to other Cladophialophora species, displaying 96.03% and 95.44% similarities to C. minutissima UAMH 10709T and C. humicola CBS 117536, respectively. Consequently, a 3-locus sequences alignment was analysed, incorporating ITS (1–300 characters), LSU (301–603 characters), and SSU (604–1512 characters), and involved a total of 27 taxa, with Cyphellophora europaea selected as the outgroup (Table S1). Bayesian inference (BI) and maximum likelihood (ML) generated concordant topologies. For the Bayesian analyses, the HKY + I + G model was the best fit for ITS and LSU regions, while the GTR + G model was suitable for SSU region. The maximum likelihood tree, presented in , depicted a strongly supported grouping (MLBP/BIPP = 100/1) of CGMCC 3.18770 and CGMCC 3.18769, distinct from C. minutissima and C. humicola ().
3.3. Genomic features of C. brunneola
The genome of C. brunneola (strain CGMCC 3.18770) was sequenced using Illumina paired-end and PacBio Sequel platforms, resulting in a total genome size of 33.8 Mb. The N50 length of assembly was 1.88 Mb. By searching approximately 100 bp in length with TTAGGG repeats, three contigs with telomeric repeats on both ends were identified. In addition, 21 scaffolds contained these characteristic telomeric repeats on either the 5′ or 3′ end (Table S2). To evaluate the completeness of the assembled genome, BUSCO analysis was performed, which detected 99.2% of the 758 expected genes, indicating a high-quality assembly.
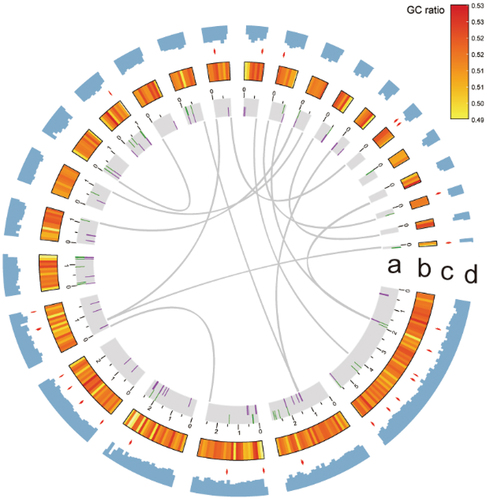
Approximately 1.53 Mb (representing 4.53% of the genome) in C. brunneola genome were repetitive DNA sequences and transposable elements (TEs). TEs comprised 0.97 Mb (2.87%) in total, consisting of 0.47 Mb (1.39%) retroelements, 0.09 Mb (0.28%) DNA transposons, and 0.41 Mb (1.21%) unknown transposons. The most prevalent transposable elements were LTRs, occupying 0.4 Mb (1.20%) of the genome (Table S3). Using the GETA pipeline, we obtained a final dataset comprising 14,168 protein-coding gene models. The average gene length was 2,299 bp, resulting in a gene density of 419 genes per Mb. The average CDS length was 2,183.07 bp, with an average of 2.27 exons and a mean exon length of 1,348.39 bp (, ).
Figure 3. Circos graph illustrating genome characteristics of Cladophialophora brunneola. (a) 25 scaffolds (>100 kb) representing over 93.83% of the assembled genome, with large fragment duplications marked. Transposons are shown in green and retrotransposons are shown in purple. (b) GC ratio per 100 kb. (c) Secondary metabolite BGCs indicated by red arrows. (d) Gene density per 100 kb.
3.4. Phylogenomic analysis and functional gene annotation
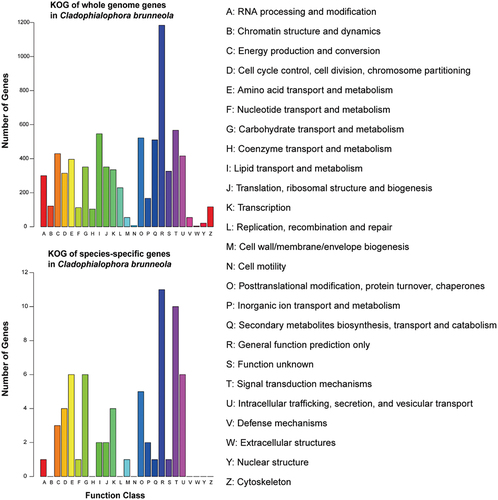
A maximum likelihood phylogenomic tree was conducted using 2,402 single-copy orthologs from 31 fungi within the order of Chaetothyriales (Eurotiomycetes), with representative species from Dothideomycetes serving as the outgroup (Table S4). The results showed that C. brunneola was distantly related to its relatives in Herpotrichiellaceae and represented an early lineage in the Chaetothyriales (). The large number of genes encoded in the C. brunneola genome can be attributed to the abundance of species-specific genes (2,137 genes). Similarly, the other two RIF, K. petricola and C. apollinis, which encode 9,308 and 10,469 genes respectively, exhibited a similar pattern in terms of orthologs. To determine the allocation of cellular functions within the proteome, protein functions were assigned based on sequence similarity using KOG classification. C. brunneola possessed a substantial number of genes categorised under “general function prediction only”. Additionally, in terms of the total count, C. brunneola had the highest number of proteins in the categories of “signal transduction mechanisms” (567 genes), followed by “lipid transport and metabolism” (542 genes) and “posttranslational modification, protein turnover, chaperones” (522 genes), as depicted in . Regarding the 4,906 annotated genes with KOG function, C. brunneola had the highest proportion of genes associated with “energy production and conversion” (429 genes, 5.69%), but the smallest proportion of genes related to “coenzyme transport and metabolism” (104 genes, 1.38%) compared to other fungal genomes in this study (Table S5). Of 2,137 species-specific genes in C. brunneola, only 45 were annotated with KOG, with “general function prediction only” and “signal transduction mechanisms” having the highest number of annotated genes ().
Figure 4. Phylogenomic relationships among representatives from Herpotrichiellaceae, trichomeriaceae, and cyphellophoraceae species. The tree was rooted using species in Dothideomycetes as outgroup. Three rock-inhabiting fungi (RIF) are highlighted in red. Genes were categorised as core (present in all species), shared (present in at least two species), and species-specific.
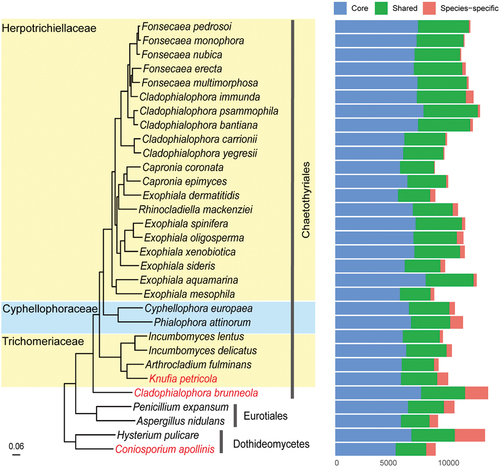
Figure 5. KOG functions of genes in whole genome and species-specific in Cladophialophora brunneola.
We performed a detailed analysis of the CAZymes in C. brunneola and compared them to those of other fungi, including two RIF species (K. petricola and C. apollinis) and three saprophytic fungi (Aspergillus nidulans, Hysterium pulicare, Penicillium expansum). C. brunneola exhibited a larger number of CAZymes (366 genes) (), primarily attributed to its glycoside hydrolases (GHs) (176 genes), which exceeded those of K. petricola and C. apollinis by 26 and 40 genes, respectively. In terms of proportion of total CAZymes, the profiles of 109 glycosyltransferases (GTs), 18 carbohydrate esterases (CEs), 15 carbohydrate-binding modules (CBMs), and 55 auxiliary activities (AAs) in C. brunneola resembled those of the other two RIF species. However, no protein with polysaccharide lyases (PLs) activity was identified in the genome of C. brunneola. In contrast to saprophytes, all three RIF species had significantly fewer CAZymes, particularly in GHs, PLs, CBMs, and AAs. Nonetheless, GTs constituted a higher proportion of these RIF species compared to saprophytes.
Table 2. Comparison of CAZymes between RIF and saprophytes.
A total of 26 putative BGCs responsible for secondary metabolite were identified in C. brunneola, encompassing 12 nonribosomal peptide synthetases (NRPSs), 6 polyketide synthases (PKSs), 3 terpene synthases, 2 beta lactone synthetases, 2 ribosomal synthesised and post-translational modified peptides (fungal RiPPs), and 1 NRPS-PKS hybrid enzyme (). In comparison to saprophytic species, the secondary metabolite biosynthetic gene clusters in RIF species exhibited a significant reduction, mainly due to the absence of PKS and NRPS clusters.
Table 3. Comparison of secondary metabolite BGCs between RIF and saprophytes.
3.5. Cultured features and transcriptome analysis of C. brunneola under PEG-induced drought stress
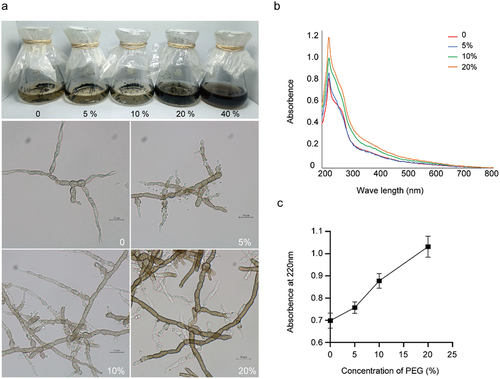
The accumulation of melanin in the culture filtrate and fungal biomass, as observed under microscopic examination (), showed a positive correlation with the increase in PEG concentration. Moreover, crude melanin extracts were obtained from the mycelia using an alkaline dissolution and acid precipitation procedure, with the exception of the 40% PEG culture due to its stickiness. UV absorption analysis of crude melanin extracts within the range of 200–800 nm further corroborated the increase in melanin accumulation with higher PEG concentrations, as depicted in .
Figure 6. (a) Melanin accumulation and hyphal morphology of Cladophialophora brunneola in liquid culture, scale bars = 10 µm. (b) UV-Vis absorption curve of crude melanin extraction (200–800 nm). (c) Absorption value at 220 nm of crude melanin extraction from samples treated with varying PEG concentrations.
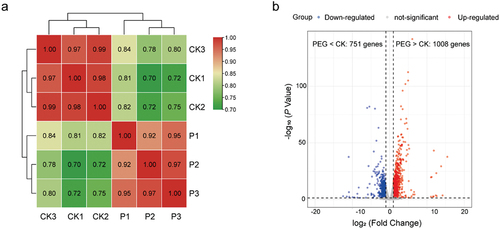
Simultaneously, we conducted RNA-seq analysis on mycelia harvested from liquid cultures supplemented with 0% and 20% PEG over a period of 7 days. Alignment of clean reads to the C. brunneola genome revealed expression in 80.24% (11,368 genes) of the predicted genes across at least one sample (Table S6). Prominent distinctions were observed between PEG-treated and control groups (), with the identification of 1,759 differentially expressed genes (DEGs). Among these DEGs, 1,008 genes were upregulated, while 751 genes showed downregulation in the PEG-treated samples compared to control samples without PEG-induced stress ().
Figure 7. (a) Pairwise Pearson correlation coefficients comparing the transcriptome data of control (CK) and PEG-treated (P) samples in Cladophialophora brunneola. (b) Volcano plots illustrating differentially expressed genes (DEGs) between control and PEG-treated samples.
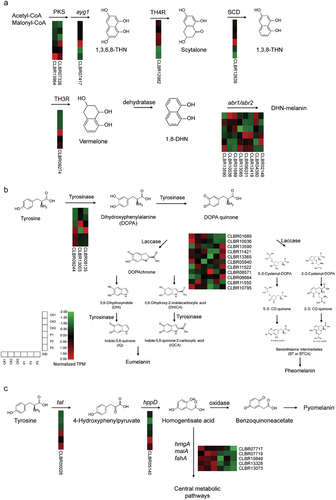
In the genome of C. brunneola, we identified homologs participating in three different melanin biosynthesis pathways (Table S7) as well as upregulated genes that may contribute to melanin production (). Under PEG-induced stress, 8 out of the 14 genes in the DHN-melanin pathway were up-regulated in three replicates (). In the DOPA-melanin pathway, tyrosinases and laccases, which play vital roles, were activated and expressed (). CLBR09244, a homolog of tyrosinase, exhibited more than a 4-fold increase in expression compared to the control condition (TPM 783.8 vs. TPM 193.7), significantly surpassing the average expression level of all genes (TPM 71.2). Additionally, C. brunneola conserves the alternative pathway of producing a third type of melanin through the utilisation of L-tyrosine degradation pathway, which results in the production of pyomelanin as a side-product (Schmaler-Ripcke et al. Citation2009; Keller et al. Citation2011) (). In response to drought stress, tyrosine aminotransferase (Tat) and hydroxyphenylpyruvate dioxygenase (hppD) exhibited slight upregulation, while hmgA, maiA, and fahA were downregulated, leading to pyomelanin accumulation.
Figure 8. Gene expression levels in melanin synthesis pathways of Cladophialophora brunneola. (a) DHN-melanin pathway. (b) DOPA-melanin pathway. (c) L-tyrosine degradation pathway.
Under PEG-induced stress, the upregulated genes (z-score > 0) showed enrichment in GO terms related to organisation or biogenesis of the cell wall (GO:0071554; P-value = 5.4e-10), metabolic process involving macromolecules in the cell wall (GO:0044036; P-value = 1.1e-08), metabolic process involving polysaccharides in the cell wall (GO:0010383; P-value = 2.34e-08), and metabolic process involving chitin in the cell wall (GO:0006037; P-value = 3.20e-07) in biology process (BP) category. Additionally, in the cellular component (CC) category, the enriched GO terms include the cell periphery (GO:0071944; P-value = 3.41e-05), the cell septum (GO:0030428; P-value = 2.31E–04), cell tip (GO:0051286, P-value = 2.81E–04) and the cell wall (GO:0005618, P-value = 9.43E–04) (see and Table S8).
Figure 9. (a) GO enrichment plot of different expression genes (DEGs) in Cladophialophora brunneola, with z-score calculated as (number of upregulated genes – number of downregulated genes) divided by the square root of the number of genes in a specific GO term. (b) KEGG pathway enrichment of DEGs. (c) Expression level of genes involved in fatty acid degradation and biosynthesis.
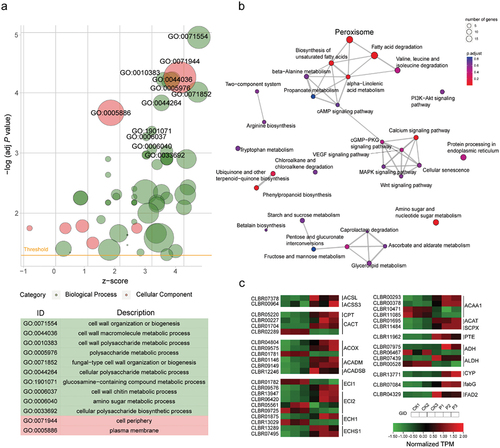
The results of KEGG pathway enrichment analysis indicated the upregulation of genes associated with lipid metabolism (Table S9). These included fatty acid degradation (ko00071, P-value = 1.85E–08), biosynthesis of unsaturated fatty acids (ko01040, P-value = 1.46E–03), metabolism of alpha-linolenic acid (ko00592, P-value = 4.55E–03), and degradation of valine, leucine and isoleucine (ko00280, P-value = 1.18E–02) and transport and catabolism in peroxisome (ko04146, P-value = 2.55E–04) (). Additionally, the upregulated genes were enriched in amino sugar and nucleotide sugar metabolism (ko00520, P-value = 1.36E–03), pentose and glucuronate interconversions (ko00040, P-value = 3.63E–03) related to carbohydrate metabolism, as well as tyrosine metabolism (ko00350, P-value = 1.44E–02) and arginine and proline metabolism (ko00330, P-value = 3.32E–02) related to amino acid metabolism.
Twenty-six genes involved in mitochondrial and peroxisomal fatty acid degradation were upregulated in DEGs of lipid metabolism (Table S10, ), likely providing energy for cellular maintenance. Two acyl-CoA synthetases (ACSL and ACSS3) among these DEGs play a critical role in catalysing the activation of free fatty acids (FAs) by forming acyl-CoA thioesters. Carnitine O-acetyltransferase (CPT, CLBR05220) and mitochondrial carnitine/acylcarnitine transporter (CACT, CLBR00227, and CLBR01704), which are components of the carnitine system responsible for transferring long chains of fatty acyl-CoA across the mitochondrial inner membrane, were also upregulated. Five acyl-CoA oxidases or dehydrogenases (ACOX, ACADM, and ACADSB), six enoyl-CoA hydratases (ECI1, ECI2, ECH1, and ECHS1), thiolases (ACAA1, ACAT, and SCPX), and thioesterases (PTE, CLBR11962) were also upregulated in the subsequent cycle of β-oxidation. Alcohol dehydrogenase (CLBR07439), the first and rate-limiting enzyme in the oxidation of fatty acids in peroxisomes, was also slightly upregulated. Additionally, the production of unsaturated fatty acids by C. brunneola as a means to maintain membrane fluidity was indicated by the upregulation of 3-oxoacyl-[acyl-carrier-protein] reductase (FabG, CLBR07084) and fatty acid desaturase (FAD2, CLBR04329).
Compatible osmolytes, such as trehalose, polyols, proline, and malondialdehyde are essential for cells to resist osmotic stress (Jennings Citation1984; Jennings and Burke Citation1990; Pfyffer et al. Citation1990; Hounsa et al. Citation1998). CLBR10229 and CLBR08760, which encode pyrroline-5-carboxylate reductase and trehalose 6-phosphate synthase, respectively, were significantly upregulated after PEG treatment (Table S6), resulting in the accumulation of pyrroline and trehalose.
4. Discussion
CGMCC 3.18770 and CGMCC 3.18769 are two isolates that represent the novel fungus, C. brunneola sp. nov., from the genus Cladophialophora. Phylogenetic comparisons suggest that C. minutissima, C. humicola, C. sylvestris, and C. modesta are the closest relatives of C. brunneola. However, extensive phylogenetic analyses demonstrate that C. brunneola is a distinct species, separate from these related fungi in the family Herpotrichiellaceae, and it appears in the early lineage of the order Chaetothyriales. These results suggest that C. brunneola exhibits significant differences in single-copy ortholog sequence compared to its relatives and has undergone specific environmental selection during genome evolution.
The influence of natural selection in the evolution of RIF genomes may be much deeper than phylogeny. Comparative genomic analysis of C. brunneola and other two published RIF species reveals genomic characteristics consistent with a rock-inhabiting lifestyle. Despite having a relatively small genome size, RIF displayed higher gene density (374.4 vs. 339.9 genes per Mb) compared to saprophytic fungi. Genes encoding enzymes involved in the degradation of glucans, polysaccharides, and chitin were reduced. For example, chitinase (GH18), chitosanase (GH75), CBM50, and CBM18, which bind to chitinous components, as well as gluco/chitooligosaccharide oxidase (AA7) and family GT31, which encode a diverse set of transferases, were less abundant in RIF. Fungal secondary metabolites (SMs) are known to play a role in self-protection, defence, competition, and communication with other organisms (Keller Citation2015). The average number of SMs biosynthetic gene clusters in RIF was almost one-third of saprophytes fungi (17.3 vs. 48). Clearly, RIF have reduced primary and secondary metabolic requirements, enabling themselves to be compatible with oligotrophic rock surfaces with fewer competitors.
The rock surface and its surroundings are inherently stressful environments, characterised by extreme temperature, UV exposure, and osmotic pressure. In this study, drought and osmotic stress were simulated using PEG treatment. We observed that C. brunneola produces significant quantities of melanin, either excreted into surroundings or deposited on the cell wall. Melanin, a pigment widely distributed in both Prokaryotes and Eukaryotes, serves as an ultraviolet absorber (Plonka and Grabacka Citation2006), provides structural rigidity in cell walls, and can store water and ions, making it advantageous in preventing desiccation (Bell and Wheeler Citation1986; Butler and Day Citation1998). Furthermore, genes involved in cell wall biogenesis and melanin synthesis pathways were found to be upregulated under PEG-induced stress, suggesting that the thickening cell wall creates more space for the increased deposition of melanin. The phenomenon of upregulated genes significantly enriching lipid metabolism is intriguing. It is speculated that the abundance of lipid droplets possessed in the cells of RIF (Gorbushina Citation2003; Onofri et al. Citation2007) is critical for desiccation tolerance, as the oxidation of fatty acids can supply cells with energy and water.
In conclusion, this study identified a novel rock-inhabiting fungus, Cladophialophora brunneola sp. nov., which was isolated from a karst landform in Guizhou, China, using a combination of morphological and phylogenetic analyses. The genome characteristics and transcriptional response of C. brunneola CGMCC 3.18770 clearly illustrated its adaptation to diverse stresses and harsh environments. The integrity and rigidity of the cell wall play a crucial role in the survival of C. brunneola in its niche. The constitutive production and upregulation of melanin biosynthesis throughout its life cycle, during drought conditions, enable melanised C. brunneola to survive and adapt to a multi-stress environment. Furthermore, C. brunneola shares genomic characteristics in common with rock-inhabiting fungi from various taxa, indicating a convergence in genome evolution. Therefore, the availability of genome sequences for other rock-inhabiting fungal species will facilitate more comprehensive investigations of the convergent mechanisms underlying their adaptation to extreme environments.
Supplemental Material
Download Zip (1.2 MB)Acknowledgments
The author thank Dr. Baosong Chen (State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China) for kindly help in melanin extraction.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The sequencing data are available at NCBI BioProject ID PRJNA952342.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21501203.2023.2256764
Additional information
Funding
References
- Badali H, Carvalho VO, Vicente V, Attili-Angelis D, Kwiatkowski IB, Van den Ende AHGG, De Hoog GS. 2009. Cladophialophora saturnica sp. nov., a new opportunistic species of Chaetothyriales revealed using molecular data. Med Mycol. 47(1):51–62. doi: 10.1080/13693780802291452.
- Badali H, de Hoog GS, Curfs-Breuker I, Klaassen CHW, Meis JF. 2010. Use of amplified fragment length polymorphism to identify 42 Cladophialophora strains related to cerebral phaeohyphomycosis with in vitro antifungal susceptibility. J Clin Microbiol. 48(7):2350–2356. doi: 10.1128/jcm.00653-10.
- Badali H, Gueidan C, Najafzadeh MJ, Bonifaz A, van den Ende AH, de Hoog GS. 2008. Biodiversity of the genus Cladophialophora. Stud Mycol. 61:175–191. doi: 10.3114/sim.2008.61.18.
- Bao W, Kojima KK, Kohany O. 2015. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 6(1):11. doi: 10.1186/s13100-015-0041-9.
- Bell AA, Wheeler MH. 1986. Biosynthesis and functions of fungal melanins. Annu Rev Phytopathol. 24(1):411–451. doi: 10.1146/annurev.py.24.090186.002211.
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27(2):573–580. doi: 10.1093/nar/27.2.573.
- Blin K, Shaw S, Steinke K, Villebro R, Ziemert N, Lee SY, Medema MH, Weber T. 2019. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47(W1):W81–W87. doi: 10.1093/nar/gkz310.
- Butler MJ, Day AW. 1998. Fungal melanins: a review. Can J Microbiol. 44(12):1115–1136. doi: 10.1139/w98-119.
- Carbone I, Kohn LM. 1999. A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia. 91(3):553–556. doi: 10.1080/00275514.1999.12061051.
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17(4):540–552. doi: 10.1093/oxfordjournals.molbev.a026334.
- Chen N. 2004. Using RepeatMasker to identify repetitive elements in genomic sequences. CP In Bioinformatics. 5(1). Chapter 4:Unit 4.10. doi: 10.1002/0471250953.bi0410s05.
- Chertov O, Gorbushina A, Deventer B. 2004. A model for microcolonial fungi growth on rock surfaces. Ecol Modell. 177(3):415–426. doi: 10.1016/j.ecolmodel.2004.02.011.
- Coleine C, Stajich JE, Selbmann L. 2022. Fungi are key players in extreme ecosystems. Trends Ecol Evol. 37(6):517–528. doi: 10.1016/j.tree.2022.02.002.
- Crous PW, Schubert K, Braun U, de Hoog GS, Hocking AD, Shin HD, Groenewald JZ. 2007. Opportunistic, human-pathogenic species in the Herpotrichiellaceae are phenotypically similar to saprobic or phytopathogenic species in the venturiaceae. Stud Mycol. 58:185–234. doi: 10.3114/sim.2007.58.07.
- Darriba D, Posada D, Kozlov AM, Stamatakis A, Morel B, Flouri T, Crandall K. 2020. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol. 37(1):291–294. doi: 10.1093/molbev/msz189.
- Das K, Lee SY, Jung HY. 2019. Cladophialophora lanosa sp. nov., a new species isolated from soil. Mycobiology. 47(2):173–179. doi: 10.1080/12298093.2019.1611242.
- Davey ML, Currah RS. 2007. A new species of Cladophialophora (hyphomycetes) from boreal and montane bryophytes. Mycol Res. 111(Pt 1):106–116. doi: 10.1016/j.mycres.2006.10.004.
- de Boer W, Folman LB, Summerbell RC, Boddy L. 2005. Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol Rev. 29(4):795–811. doi: 10.1016/j.femsre.2004.11.005.
- Egidi E, de Hoog GS, Isola D, Onofri S, Quaedvlieg W, de Vries M, Verkley GJM, Stielow JB, Zucconi L, Selbmann L. 2014. Phylogeny and taxonomy of meristematic rock-inhabiting black fungi in the Dothideomycetes based on multi-locus phylogenies. Fungal Divers. 65(1):127–165. doi: 10.1007/s13225-013-0277-y.
- Emms DM, Kelly S. 2015. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16(1):157. doi: 10.1186/s13059-015-0721-2.
- Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39(Web Server issue):W29–37. doi: 10.1093/nar/gkr367.
- Glass NL, Donaldson GC. 1995. Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl Environ Microbiol. 61(4):1323–1330. doi: 10.1128/aem.61.4.1323-1330.1995.
- Gorbushina A. 2003. Microcolonial fungi: survival potential of terrestrial vegetative structures. Astrobiology. 3(3):543–554. doi: 10.1089/153110703322610636.
- Gorbushina A. 2007. Life on the rocks. Environ Microbiol. 9(7):1613–1631. doi: 10.1111/j.1462-2920.2007.01301.x.
- Gorbushina AA, Krumbein WE, Hamman CH, Panina L, Soukharjevski S, Wollenzien U. 1993. Role of black fungi in color change and biodeterioration of antique marbles. Geomicrobiol J. 11(3–4):205–221. doi: 10.1080/01490459309377952.
- Gostinčar C, Zajc J, Lenassi M, Plemenitaš A, Gunde-Cimerman N, Al-Hatmi AMS, Gunde-Cimerman N. 2018. Fungi between extremotolerance and opportunistic pathogenicity on humans. Fungal Divers. 93(1):195–213. doi: 10.1007/s13225-018-0414-8.
- Gueidan C, Villasenor CR, de Hoog GS, Gorbushina AA, Untereiner WA, Lutzoni F. 2008. A rock-inhabiting ancestor for mutualistic and pathogen-rich fungal lineages. Stud Mycol. 61:111–119. doi: 10.3114/sim.2008.61.11.
- Horre R, de Hoog GS. 1999. Primary cerebral infections by melanized fungi: a review. Stud Mycol. 43:176–193. doi: 10.1055/s-0039-3400957.
- Hounsa CG, Brandt EV, Thevelein J, Hohmann S, Prior BA. 1998. Role of trehalose in survival of Saccharomyces cerevisiae under osmotic stress. Microbiol. 144(3):671–680. doi: 10.1099/00221287-144-3-671.
- Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, et al. 2019. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47(D1):D309–D314. doi:10.1093/nar/gky1085.
- Jennings DH. 1984. Polyol metabolism in fungi. Adv Microb Physiol. 25:149–193. doi: 10.1016/S0065-2911(08)60292-1.
- Jennings DH, Burke RM. 1990. Compatible solutes - the mycological dimension and their role as physiological buffering agents. New Phytol. 116(2):277–283. doi: 10.1111/j.1469-8137.1990.tb04715.x.
- Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics. 30(9):1236–1240. doi: 10.1093/bioinformatics/btu031.
- Kalvari I, Nawrocki EP, Argasinska J, Quinones-Olvera N, Finn RD, Bateman A, Petrov AI. 2018. Non-coding RNA analysis using the RFAM database. Curr Protoc Bioinformatics. 62(1):e51. doi: 10.1002/cpbi.51.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. doi: 10.1093/molbev/mst010.
- Keller NP. 2015. Translating biosynthetic gene clusters into fungal armor and weaponry. Nat Chem Biol. 11(9):671–677. doi: 10.1038/nchembio.1897.
- Keller S, Macheleidt J, Scherlach K, Schmaler-Ripcke J, Jacobsen ID, Heinekamp T, Brakhage AA, Chauhan N. 2011. Pyomelanin formation in Aspergillus fumigatus requires HmgX and the transcriptional activator HmgR but is dispensable for virulence. PLoS One. 6(10):e26604. doi: 10.1371/journal.pone.0026604.
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 12(4):357–U121. doi: 10.1038/nmeth.3317.
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 37(8):907–915. doi: 10.1038/s41587-019-0201-4.
- Kiyuna T, An KD, Kigawa R, Sano C, Sugiyama J. 2018. Two new Cladophialophora species, C. tumbae sp. nov. and C. tumulicola sp. nov., and chaetothyrialean fungi from biodeteriorated samples in the Takamatsuzuka and Kitora Tumuli. Mycoscience. 59(1):75–84. doi: 10.1016/j.myc.2017.08.008.
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27(5):722–736. doi: 10.1101/gr.215087.116.
- Lagerwerff JV, Ogata G, Eagle HE. 1961. Control of osmotic pressure of culture solutions with polyethylene glycol. Sci. 133(3463):1486–1487. doi: 10.1126/science.133.3463.1486.
- Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. 2017. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 34(3):772–773. doi: 10.1093/molbev/msw260.
- Liu B, Fu R, Wu B, Liu X, Xiang M. 2021. Rock-inhabiting fungi: terminology, diversity, evolution and adaptation mechanisms. Mycology. 13(1):1–31. doi: 10.1080/21501203.2021.2002452.
- Liu Q, Xiao J, Liu B, Zhuang Y, Sun L. 2018. Study on the preparation and chemical structure characterization of melanin from boletus griseus. Int J Mol Sci. 19(12):3736. doi: 10.3390/ijms19123736.
- Meng L, Feldman L. 2010. A rapid TRIzol-based two-step method for DNA-free RNA extraction from Arabidopsis siliques and dry seeds. Biotechnol J. 5(2):183–186. doi: 10.1002/biot.200900211.
- Nawrocki EP, Eddy SR. 2013. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29(22):2933–2935. doi: 10.1093/bioinformatics/btt509.
- Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, Barry KW, Condon BJ, Copeland AC, Dhillon B, Glaser F, et al. 2012. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen dothideomycetes fungi. PLoS Pathog. 8(12):e1003037. doi:10.1371/journal.ppat.1003037.
- Onofri S, Seltimann L, de Hoog GS, Grube M, Barreca D, Ruisi S, Zucconi L. 2007. Evolution and adaptation of fungi at boundaries of life. Adv Space Res. 40(11):1657–1664. doi: 10.1016/j.asr.2007.06.004.
- Pal AK, Gajjar DU, Vasavada AR. 2013. DOPA and DHN pathway orchestrate melanin synthesis in Aspergillus species. Med Mycol. 52(1):10–18. doi: 10.3109/13693786.2013.826879.
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 33(3):290–295. doi: 10.1038/nbt.3122.
- Pfyffer GE, Boraschigaia C, Weber B, Hoesch L, Orpin CG, Rast DM. 1990. A further report on the occurrence of acyclic sugar alcohols in fungi. Mycol Res. 94:219–222. doi:10.1016/s0953-7562(09)80617-5.
- Plonka PM, Grabacka M. 2006. Melanin synthesis in microorganisms - biotechnological and medical aspects. Acta Biochim Pol. 53(3):429–443. doi: 10.18388/abp.2006_3314.
- Pralea IE, Moldovan RC, Petrache AM, Ilieș M, Hegheș SC, Ielciu I, Nicoară R, Moldovan M, Ene M, Radu M, et al. 2019. From extraction to advanced analytical methods: the challenges of melanin analysis. Int J Mol Sci. 20(16):3943. doi:10.3390/ijms20163943.
- Pusztahelyi T, Holb IJ, Pocsi I. 2015. Secondary metabolites in fungus-plant interactions. Front Plant Sci. 6:573. doi:10.3389/fpls.2015.00573.
- Quan Y, Muggia L, Moreno LF, Wang M, Al-Hatmi AMS, da Silva Menezes N, Shi D, Deng S, Ahmed S, Hyde KD, et al. 2020. A re-evaluation of the Chaetothyriales using criteria of comparative biology. Fungal Divers. 103(1):47–85. doi: 10.1007/s13225-020-00452-8.
- Rehner SA, Samuels GJ. 1994. Taxonomy and phylogeny of Gliocladium analysed from nuclear large subunit ribosomal DNA sequences. Fungal Biol. 98:625–634. doi:10.1016/S0953-7562(09)80409-7.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542. doi: 10.1093/sysbio/sys029.
- Ruibal C, Gueidan C, Selbmann L, Gorbushina AA, Crous PW, Groenewald JZ, Muggia L, Grube M, Isola D, Schoch CL, et al. 2009. Phylogeny of rock-inhabiting fungi related to Dothideomycetes. Stud Mycol. 64:123–133. doi: 10.3114/sim.2009.64.06.
- Ruibal C, Platas G, Bills GF. 2008. High diversity and morphological convergence among melanised fungi from rock formations in the central mountain system of Spain. Persoonia. 21(6):93–110. doi: 10.3767/003158508x371379.
- Schmaler-Ripcke J, Sugareva V, Gebhardt P, Winkler R, Kniemeyer O, Heinekamp T, Brakhage AA. 2009. Production of pyomelanin, a second type of melanin, via the tyrosine degradation pathway in Aspergillus fumigatus. Appl Environ Microbiol. 75(2):493–503. doi: 10.1128/aem.02077-08.
- Seppey M, Manni M, Zdobnov EM. 2019. BUSCO: assessing genome assembly and annotation completeness. In: Kollmar M, editor. Gene prediction: methods and protocols. New York, NY: Springer New York; pp. 227–245.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313. doi: 10.1093/bioinformatics/btu033.
- Stanke M, Steinkamp R, Waack S, Morgenstern B. 2004. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32(Suppl_2):W309–312. doi: 10.1093/nar/gkh379.
- Sterflinger K, Lopandic K, Blasi B, Poynter C, de Hoog S, Tafer H. 2015. Draft genome of Cladophialophora immunda, a black yeast and efficient degrader of polyaromatic hydrocarbons. Genome Announc. 3(1):e01283–01214. doi: 10.1128/genomeA.01283-14.
- Sterflinger K, Lopandic K, Pandey R, Blasi B, Kriegner A. 2014. Nothing special in the specialist? Draft genome sequence of Cryomyces antarcticus, the most extremophilic fungus from Antarctica. PLoS One. 9(10):e109908. doi: 10.1371/journal.pone.0109908.
- Su L, Guo LY, Hao Y, Xiang MC, Cai L, Liu XZ. 2015. Rupestriomyces and Spissiomyces, two new genera of rock-inhabiting fungi from China. Mycologia. 107(4):831–844. doi: 10.3852/14-305.
- Sun W, Su L, Yang S, Sun J, Liu B, Fu R, Wu B, Liu X, Cai L, Guo L, et al. 2020. Unveiling the hidden diversity of rock-inhabiting fungi: chaetothyriales from China. J Fungi (Basel). 6(4):187. doi:10.3390/jof6040187.
- Teixeira MM, Moreno LF, Stielow BJ, Muszewska A, Hainaut M, Gonzaga L, Abouelleil A, Patané JSL, Priest M, Souza R. 2017. Exploring the genomic diversity of black yeasts and relatives (Chaetothyriales, Ascomycota). Stud Mycol. 86:1–28. doi:10.1016/j.simyco.2017.01.001.
- Tesei D, Marzban G, Zakharova K, Isola D, Selbmann L, Sterflinger K. 2012. Alteration of protein patterns in black rock inhabiting fungi as a response to different temperatures. Fungal Biol. 116(8):932–940. doi: 10.1016/j.funbio.2012.06.004.
- Tesei D, Tafer H, Poyntner C, Piñar G, Lopandic K, Sterflinger K. 2017. Draft genome sequences of the black rock fungus Knufia petricola and its spontaneous nonmelanized mutant. Genome Announc. 5(44):e01242–01217. doi: 10.1128/genomea.01242-17.
- Usui E, Takashima Y, Narisawa K. 2016. Cladophialophora inabaensis sp nov., a new species among the dark septate endophytes from a secondary forest in Tottori, Japan. Microbes Environ. 31(3):357–360. doi: 10.1264/jsme2.ME16016.
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9(11):e112963. doi:10.1371/journal.pone.0112963.
- Wang L, Feng Z, Wang X, Wang X, Zhang X. 2010. Degseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 26(1):136–138. doi: 10.1093/bioinformatics/btp612.
- White TJ, Bruns T, Lee S, Taylor J. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis M, Gelfand D Sninsky J, editors. PCR protocols. San Diego: Academic Press; pp. 315–322. doi: 10.1016/B978-0-12-372180-8.50042-1.
- Wollenzien U, de Hoog GS, Krumbein WE, Urzi C. 1995. On the isolation of microcolonial fungi occurring on and in marble and other calcareous rocks. Sci Total Environ. 167(1–3):287–294. doi: 10.1016/0048-9697(95)04589-S.
- Xu Z, Wang H. 2007. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35(suppl_2):W265–W268. doi: 10.1093/nar/gkm286.
- Yang EC, Xu LL, Yang Y, Zhang XY, Xiang MC, Wang CS, An ZQ, Liu XZ. 2012. Origin and evolution of carnivorism in the Ascomycota (fungi). Proc Natl Acad Sci USA. 109(27):10960–10965. doi: 10.1073/pnas.1120915109.
- Yang J, Yun J, Liu X, Du W, Xiang M. 2023. Niche and ecosystem preference of earliest diverging fungi in soils. Mycology. 14(3):239–255. doi: 10.1080/21501203.2023.2237047.
- Zakharova K, Marzban G, de Vera JP, Lorek A, Sterflinger K. 2014. Protein patterns of black fungi under simulated mars-like conditions. Sci Rep. 4(1):5114. doi: 10.1038/srep05114.
- Zakharova K, Tesei D, Marzban G, Dijksterhuis J, Wyatt T, Sterflinger K. 2013. Microcolonial fungi on rocks: a life in constant drought? Mycopathologia. 175(5–6):537–547. doi: 10.1007/s11046-012-9592-1.
- Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. 2018. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46(W1):W95–w101. doi: 10.1093/nar/gky418.
- Zhang YJ, Zhang S, Liu XZ, Wen HA, Wang M. 2010. A simple method of genomic DNA extraction suitable for analysis of bulk fungal strains. Lett Appl Microbiol. 51(1):114–118. doi: 10.1111/j.1472-765X.2010.02867.x.