
ABSTRACT
A well-developed Target Product Profile (TPP) is central to successful in vitro diagnostic (IVD) device developments. A TPP is a key document that outlines the desired attributes of a target product that is aimed at a particular disease. The TPP framework can highlight technological gaps and focus developments on meeting specific needs that advance the field. A survey of various diagnostic technologies, such as those employed for respiratory disease diagnosis, reveals significant innovations that have been accomplished during the COVID-19 public health emergency. Nevertheless, the entire IVD diagnostic landscape continues to evolve, and it is important for device developers to identify the challenges and set appropriate targets to avoid pitfalls. While a TPP does not necessarily guarantee market success of an IVD, it is an important tool that can help developers define appropriate criteria for their device. A TPP can guide them through the multi-faceted development process to meet consumer demands, while ensuring clinical utility, safety, and effectiveness of their products. The following discussion highlights the TPP process for IVD device developments through the application of the basic tenets in various case studies. The aim is to help developers traverse the proverbial ‘valley of death’, where many great concepts fail to reach market success, and to encourage positive developments in the field beyond current IVD device limitations.
Introduction
When the COVID-19 pandemic was declared by the WHO in the Spring of 2020, the United States (US) was rapidly seeking diagnostic testing solutions to help address the public health crisis. In the US, the Food and Drug Administration (FDA) efforts helped to bolster testing capacity, especially during the early phase of the pandemic. This was in no small part due to the Emergency Use Authorization (EUA) mechanism. EUAs have been used by the FDA to expedite the availability of medical devices such as IVDs to protect public health. Since the start of the COVID-19 pandemic, the FDA has issued more than 650 individual EUAs. While a significant majority of these were for COVID-19 IVDs (Food and Drug Administration [FDA], Citation2020b; Ropes & Gray, Citation2023), EUAs have been granted for other product types including vaccines and therapeutics (FDA, Citation2020a, Citation2023c). Note that apart for the COVID-19 pandemic, the FDA has in the past issued EUAs for other public health crises (e.g. Ebola, Zika, and mpox outbreaks) to facilitate the availability and use of medical countermeasures (MCMs) needed during public health emergencies.
Following a public health emergency issued on 31 January 2020, a determination was made on 4 February 2020 by the HHS secretary that the emergency had a significant potential to affect national security or the health and security of United States citizens living abroad and that it involved the virus that causes COVID-19. These declarations laid the foundation for supporting the use of EUAs, allowing the FDA to quickly facilitate the authorisation of two primary types of diagnostic tests for SARS-CoV-2. These in Vitro Diagnostic (IVD) devices can be generally categorised based on their common technical principles (i.e. molecular and antigen tests). In brief, molecular devices are Nucleic Acid Amplification Tests (NAATs) such as what are commonly known as PCR-based tests. Alternatively, antigen tests are those that detect protein components associated with the SARS-CoV-2 virus and are sometimes referred to as either lateral flow or rapid tests.
The US National Institutes of Health (NIH) was instrumental to the rapidly evolving COVID-19 testing enterprise, in terms of the financial and research and development bootstrapping of IVD developers’ EUA submissions to the FDA. The novel RADx funding programme was launched by NIH, and the first awards were made in July 2020. According to published statistics (Johnson, Citation2022b, Citation2022c; National Institutes of Health [NIH], Citation2023; PATH, Citation2023), the cumulative production of 5 billion COVID-19 tests, and 46 FDA EUAs of COVID-19 tests, including one for the first over-the-counter (OTC) molecular diagnostic test for the disease, can be attributed to the RADx program.
Regarding the OTC designation, the COVID-19 EUA pathway opened an unprecedented diagnostic testing paradigm for infectious disease IVDs. In the past, this designation was only under remote consideration by both infectious disease IVD developers and regulators, and certainly not to the scale that was eventually implemented for the COVID pandemic. Generally, the COVID-19 EUA landscape began with initial authorisations of laboratory-based tests to mobilise high-throughput capacity and address the initial diagnostic demands. Subsequent waves included at-home testing such as mail-in sampling kits where specimens were collected safely by the individual and sent to test laboratories that would process and provide results. Eventually, authorisations included diagnostic tests where individuals could acquire the IVD devices without a prescription and perform the tests themselves. This paved a convenient path for performing diagnostic testing at-home and away from a physician’s office, and vastly improved the availability and accessibility of COVID IVDs.
Cumulatively, COVID-19 IVD EUAs represented the largest number of EUAs issued in history for a single disease diagnostic-intended use, and one of the greatest diagnostic market growth events in recent times. The aforementioned RADx initiative also established new methodologies for the industry to mobilise diagnostic products. As the IVD field transitions from the frantic, reactive response cycle of the early COVID-19 pandemic phase (APHL, Citation2023) to a normal, balanced regulatory and market landscape, there will be an anticipated reduction and consumer demand for EUA offerings. Successful products will include those devices that clear regulatory thresholds and meet market demands, while future developments will benefit from the lessons learned from the COVID-19 pandemic. One path to accomplish proper due diligence and to ensure that IVD developments address unmet needs while navigating past the proverbial ‘valley of death’, is to develop and incorporate a target product profile (TPP). The following sections will demonstrate the elements of a TPP to illustrate how it can be used to identify key regulatory hurdles, avoid potential developmental pitfalls, and guide innovation to meet critical clinical needs in disease diagnostics. In particular, the TPP framework will be used to help identify remaining gaps in IVD design while highlighting potential focal areas for innovation that are needed to better align diagnostic testing performance with the comprehensive disease status of the individual.
What a TPP is and is not
A TPP can be considered primarily as a planning tool that is not defined and executed once but may be continually refined and updated as additional relevant information becomes available during the development process. A TPP establishes key features and performance specifications that are aligned with a clinical need and by defining early-on the required validation metrics, they can guide developers to avoid late-stage development failures. Though a TPP is essentially a ‘living’ document, an organised approach to its development can help developers ensure it represents the right information and involves the appropriate stakeholders. Broadly, the TPP development process can be categorised into three phases – Scoping, Drafting, and Consensus Reviews (Cocco et al., Citation2020).
Conversely, before implementing a TPP to illustrate an IVD development process, it may be productive to highlight some of the pitfalls in an improperly defined profile. For example, a TPP is not a document to define a technical approach to solving a medical problem. The failure of many technologies to translate from bench to clinical practice is not necessarily due to the lack of an innovative approach (Cocco et al., Citation2020; Drucker & Krapfenbauer, Citation2013; Engel et al., Citation2011; P. Wang & Kricka, Citation2018). Many novel discoveries are centred around well-controlled laboratory conditions and fail to pass the crucible of practical and clinical needs. Therefore, a TPP should guide a developer through an extensive list of requirements to demonstrate the technology’s fitness for intended use, and ultimately its clinical utility. A test, for example, that successfully detects a marker of disease but requires invasive procedures to obtain a sample when simpler, safer diagnostic methods exist should quickly be identified by the TPP as unacceptable. A poor understanding of the true clinical need (not just the technical problem), positioning within the clinical workflow, and the overall risk vs benefit profile of a medical device often results in a high probability of the device failing validation and failing to be supported for clinical use. The consequences of missing such development outcomes can be devastatingly costly to both the developers and those in need of the product. Developers who use a TPP properly should steer clear of these developmental traps. The following summarises the general activities associated with each phase within the TPP decision-making framework.
Phase I: scoping
Scoping may be considered as a market analysis. During this phase, a developer should understand the disease area, the current technical limitations, and why a problem has not yet been solved. This also includes defining the clinical problem and unmet need. It may also involve identifying the competitive landscape and noting key competitors. The source of information for this analysis can be found in published literature and from interviewing stakeholders and consulting experts. For instance, published references can provide the background for developers to map the diagnostic ecosystem of interest, and help guide the corresponding solicitation of stakeholder input (Denkinger, Dolinger, et al., Citation2015). There are also published regulatory guidance documents provided by the FDA to guide IVD developers of certain device types, industry technical recommendations (e.g. for molecular assay development), and reference standards for appropriate verification and validation testing. Others have suggested that the TPP should include the main categories of scope, performance, operational characteristics, and pricing (FDA, Citation2023b), while some have ranked health needs as a scoping exercise to guide subsequent developments (Denkinger, Kik, et al., Citation2015, Forum for Collaborative Research, Citation2015; Gal et al., Citation2018; World Health Organization [WHO], Citation2014). Overall, there is no singular, correct methodology for conducting a survey to drive this phase. It is appropriate to apply the well-known Heilmeier Catechism (Defense Advanced Research Projects Agency DARPA, Citation2022) to this process. What appears to be universally important is to apply due diligence in understanding the needs and identifying the critical problems to solve.
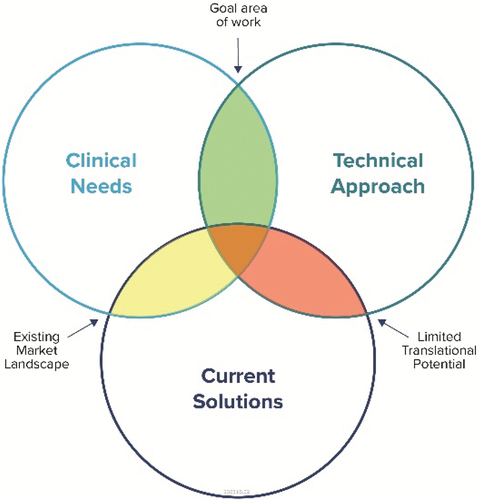
The IVD device development process can be visualised as a balance of competing attributes (). The target region, as denoted in (green), is where a candidate technology is focused on a clinical need that is not adequately addressed by current solutions.
Figure 1. IVD Development Competing Attributes. IVD device development can be viewed as a balance between meeting a clinical need, the technological innovation, and potential overlap with current offerings. Through this perspective, a developer can know where the product sits relative to competitors and in terms of prioritising the need. The green shaded region indicates the ideal goal of IVD development and where the innovation opportunity can be most impactful.
Overall, Phase I (scoping) should address questions such as those listed in .
Table 1. Questions (and sample responses) that guide developers in scoping their product development.
Phase II: drafting
Developers from the outset should remember that a TPP is a living document. Thus, the drafting phase may produce an initial draft for iteration, but this phase is never intended to be a one-time work process. Recommended approaches to this phase include forming a working group comprising multidisciplinary experts, performing iterative revisions and establishing a goal of creating a concise version that can be easily communicated to stakeholders for review in the next phase. Suggested categories and targets to include in a IVD development TPP are described in the corresponding section below.
Phase III: consensus reviews
With consensus reviews, a meeting with stakeholders and experts is held to survey their views towards the TPP draft. Discussion topics can include trade-offs between test attributes or rating of key parameters. The review can be approached via general questionnaires or through the Delphi method (Niederberger & Spranger, Citation2020). The Delphi method is a structured communication framework based on the results of multiple rounds of questionnaires sent to a panel of experts. After each round of questionnaires, the experts are presented with an aggregated summary of the previous round, allowing each expert to adjust their answers based on the group response. This process allows working towards a group opinion while benefiting from the incorporation of expert analysis.
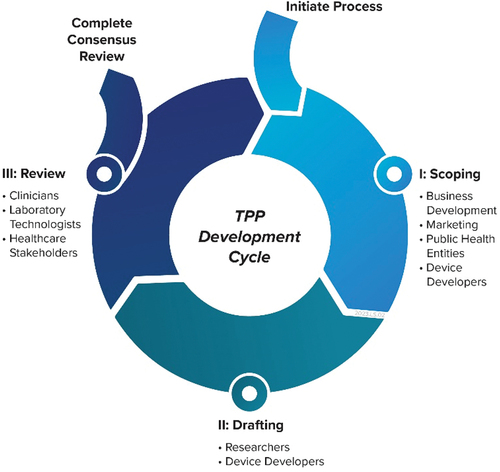
As stakeholders are integral to the successful execution of the above three phases, the following is a list of possible participant groups that are recommended for a developer’s own TPP development process ().
Figure 2. A notional TPP development and review cycle. Relevant participant groups are listed at the key stages for the TPP development process who may help in properly defining an IVD product roadmap.
Recommended IVD TPP categories and targets
Through a disciplined progression of the three phases described, an IVD device developer would prioritise and seek to balance several critical elements (Cocco et al., Citation2020). This would form what is commonly referred to as the trade space as it is near impossible to capture all needs. What is crucial is to define the development focus and identify the factors that drive the project. This can be framed within the TPP. A TPP should define the specific development criteria and corresponding targets as illustrated in the following notional developmental outline ().
Table 2. IVD development criteria. List of key criteria, corresponding description, and examples for framing a target product profile.
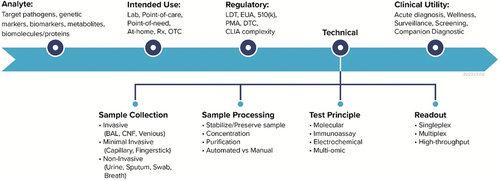
Among the factors listed above that are recommended for a TPP, a survey of published TPPs suggests that clinical utility is unfortunately a common oversight (FIND, Citation2022). Additionally, cost-effectiveness is an often neglected category, defined as the net benefit influenced by the device – including savings from medical interventions, weighed against direct device costs. Note that this isnot simply the outright pricing of the product. For IVD development, some of the common attributes and corresponding configurations that developers must choose to define for their device are shown in .
Figure 3. IVD attributes and corresponding configuration options that are commonly considered in the development process. The attributes along the critical development path (i.e. Analyte, Intended Use, etc.) relate to each other and define the development focus areas of an IVD product. The four elements under the Technical attribute outline the common, IVD operational workflow..
The following case studies help illustrate the impact of careful considerations for the various IVD device development criteria. A holistic approach with proper targets and balanced trade-offs for all TPP categories can lead to a more sustainable development process of game-changing innovations.
Case study I: gap analysis of IVD Dx “Valley of Death” and the role of a TPP to help identify potential pitfalls
The diagnostic field witnessed a large infusion of funding in 2020 where $200 M was allocated for COVID-19 devices (Johnson, Citation2022b). NIH through the RADx programme made significant investments and some participants were able to receive tens of millions of dollars for their developments (FDA, Citation2020b; Johnson, Citation2022c). Yet, not all funded projects were successful in receiving regulatory authorisations and reaching the market. Thus, funding alone was not sufficient to bridge from bench to bedside or what is commonly referred to as ‘the valley of death’. And while the failures were certainly not attributed to a lack of innovation, many of the technologies were novel.
As evident by the plethora of manuscripts highlighting a new technology that overcomes the limitations of a competing technique, the translation out of the lab is not a simple journey and many stumble on the hurdles of real-world concerns including manufacturing and distribution. Obtaining regulatory clearance from the FDA in terms of an Emergency Use Authorization (EUA) was one metric that RADx set for the majority of award recipients. Though it should be noted that obtaining an EUA is a lower regulatory bar than the typical full approval (PMA) or clearance (510(k)) from FDA, examination of public information in the form of posted Phase II RADx funding (National Institutes of Health [NIH], Citation2023) indicated that at least five (5) supported technical developments were still in progress towards the regulatory requirements as of April 2023.
According to a published analysis of these EUA non-issuances (Johnson, Citation2022c) one hypothesised reason relates to SARS-CoV-2 viral prevalence colliding with supply chain issues. In terms of the aforementioned TPP criteria, these issues could relate to failing to account for or missing appropriate targets for clinical performance, life cycle management, and cost-effectiveness. Further, as the pandemic evolved the prioritisation for different types of testing changed which impacted the funds available for development. This too relates to the clinical utility criteria, where a device that may have lagged in development and thus failed to meet a market need ultimately succumbed to missing the window of opportunity. Developers are advised to approach their IVD development with a wide aperture of the targeted clinical need or have the flexibility to pivot the technology to address multiple clinical applications (e.g. other diseases).
The COVID-19 pandemic highlighted manufacturing bottlenecks in terms of supply chain issues. For example, the shortage of microprocessors had a widespread impact across many industries and had been called out as one of the reasons for Quidel’s failure to fully commercialise one of their products and to miss the current respiratory seasons (Johnson, Citation2022a). Other firms have adapted to this market situation, which continues to remain a key consideration in medical device development. For instance, Anavasi Diagnostics received a RADx award for their Ascencio device, and designed a system purpose-built to use only one simple chip while navigating the microprocessor supply chain issue. However, they still await for EUA authorisation as of the end of 2022. Supply chain considerations within the life cycle management category of a TPP, as described above (), should be a critical target for IVD device developers. Solutions to avoid such issues may facilitate overcoming regulatory hurdles as well as provide a market advantage over competitors that struggle with meeting device demands. After all, accessibility whether inherent in the technical design or due to market influences such as the supply chain bottlenecks still result in reduced clinical utility.
While the above case studies focus on those firms that failed the threshold of obtaining an EUA, it is by no means the only metric of success. Other devices did clear the EUA threshold and yet stumbled in their path to market. One example is the Meridian Biosciences RevoGene system. The device obtained an EUA in November of 2021. However, the market launch was delayed when it was discovered that the test could not detect the Omicron variant and was subsequently reauthorised by the FDA (Genomeweb, Citation2022). This illustrates the importance of careful consideration of clinical utility, assay configuration, and analytical performance of TPP targets. For an IVD, understanding the biological nature of the analyte that is intended such as anticipating the potential for variants and viral escape underscores potential costly development pitfalls. Similarly, many firms experienced constantly evolving market dynamics, perhaps amplified during the pandemic due to the extreme waves of public health measures (from lockdowns to masking to attempts to release restrictions) that impacted how people interacted with tests. COVID-19 admittedly continues to be cyclical where there are periods of great testing demand to times of decreased use or interest in only certain types of tests. The inability to accurately predict consumer demand far in advance presents additional pressures on manufacturers to adequately scale-up and respond. Again, trying to absorb these risk factors through creating a platform that developers can pivot to address multiple clinical needs may be more viable than pouring resources into a technology that is only useful for a specific disease application. These exemplify development issues on the business end, which are just as crucial as the technical approach.
Another case study of business struggles is illustrated with Ellume. The Australia-based firm reportedly entered into a procedure that is analogous to Chapter 11 bankruptcy proceedings in the US (Griffin, Citation2022). RADx awarded Ellume $30 million for its rapid antigen test, followed by $3.5 million for a combination flu/COVID test (Ellume, Citation2020). There was an additional significant influx of funds for the firm including major support from the US DoD and HHS on the order of $230 M for OTC, at-home COVID-19 test. At the end of 2020, Ellume received the first EUA for an over the counter COVID-19 test and opened a flagship US manufacturing plant in Frederick, Maryland, in March 2021. Yet, 8 months later it recalled 2.2 million tests due to the potential for false-positive results with certain device lots.
A deeper dive into Ellume’s assay configuration can elucidate perhaps where some of the pitfalls had occurred. The assay is based on quantum dot and optoelectronic detection technologies. Based on published reports, it appears to be an adaptation of the QIAreach system and uses fluorescent labelled antibodies to detect SARS-CoV-2 antigens as well as powered electronics to report out a result (Stieber et al., Citation2020). At its core, it is essentially a lateral flow assay. On the market, it was targeted at the $30 price point yet it competed with other rapid antigen tests that eventually came to market for a fraction of the cost at $3. A tear-down of the device showed excessive materials for accomplishing the same ‘control’ and ‘test’ readouts as simpler rapid antigen tests (The Signal Path, Citation2022). The higher price point due to the incorporation of relatively costly materials and the resultant generation of more material waste may be indicative of missing appropriate targets for cost-effectiveness and life cycle management TPP categories. While the technology was certainly innovative, the Ellume case study illustrates that a developer should ask, regardless of a novel approach (i.e. an electronic antigen test), whether the device is the most cost-effective method when there are cheaper and more efficient means to accomplish the same type of test.
A developer should also account for the market they are entering. In the Ellume case, rapid antigen tests are an established technology as exemplified by the prevalent pregnancy tests (i.e. antibody detection of human chorionic gonadotropin (hCG) in urine). Cost targets are more restrictive unless they can be offset by a demonstrated significance in performance, such as in comparison to molecular tests. Conversely, point-of-care molecular tests are known to be more complex, with more parts to accomplish the diagnostic assay and potentially more vulnerable to supply chain risks. A system that combines the clinical performance of a molecular test with the cost-effectiveness of a lateral flow assay may ultimately open additional opportunities to meet clinical utility in diagnostics for various diseases.
Case study II: applying TPP principles to guide innovation in IVD test configurations such as next generation rapid molecular diagnostics using CRISPR/Cas technologies
As indicated in the above case study, COVID-19 spurred many diagnostic innovations but one particular area that experienced critical growth was the acceleration of decentralised testing including point-of-care and home-use tests for infectious diseases (Satyanarayana, Citation2021). While there are no guarantees for sustaining this home-based testing modality, several TPP principles can guide more productive developments. For instance, a shift to multiplexed as opposed to single analyte at-home testing would expand clinical utility for other respiratory illnesses, reduce wasteful testing material, and facilitate more useful public health information. This may be the driving force behind several RADx funded performers addressing concurrent product applications from COVID-19 diagnostics to women's health and sexually transmitted infections (STI) (FDA, Citation2021). Rapid, portable detection at point-of-care (POC) or point-of-need (PON), if not at-home, is possible and critical for treatment decisions and transmission prevention for STIs (Renzoni et al., Citation2021). Further, at-home testing may encourage increased testing, and continued emphasis on addressing cost-effectiveness enables greater accessibility of high-performance diagnostics in low resource settings (Dean et al., Citation2021).
Towards greater accessibility of diagnostic testing, CRISPR/Cas technologies has the potential to accomplish the accuracy of PCR-based testing without the complexity and costs associated with elaborate instruments and reagent development. For example, dilution of samples collected in the specialised transport media to mitigate inhibitory effects of the media is also hypothesised as a possible cause of reduced sensitivity. Thus, apart from critical IVD development criteria that include addressing clinical utility, life cycle management, cost-effectiveness, and clinical performance, a CRISPR-based diagnostic should overcome the potentially complicated assay design cycle of a LAMP assay and may address some hardware and assay configuration limitations with an RT-PCR system (Kaminski et al., Citation2021).
CRISPR-based diagnostics uses a Cas protein targeted to a nucleic acid sequence-specific guide RNA (gRNA), the equivalent of primers in a PCR assay. One potential benefit of this mechanism over other NAATs is rapid assay reconfiguration against new targets due to relative gRNA simplicity (Chen et al., Citation2018; Slaymaker et al., Citation2019). Assuming the initial patient sample collection step is a fixed prerequisite for any diagnostic platform, CRISPR-based technologies are similar to other molecular diagnostic platforms that have extraction, amplification, and detection requirements. Besides potential assay reconfiguration, CRISPR-based diagnostic devices have two main differentiators – the potential to eliminate or incorporate simplified amplification and therefore no thermal cycling requirements and the possibility for high specificity in target recognition with fewer molecular target restrictions. By relying on an isothermal amplification protocol, CRISPR-based devices can be used in low resource settings using an ambient nucleic acid amplification technique (Harvard University, Citation2023). For PON applications, the inherent low-energy needs of the CRISPR assay chemistry mean that there is flexibility in the incorporation of power sources to support ancillary device elements such as microfluidic pumping. This includes using novel paper-based batteries such as those from Fuelium (Esquivel et al., Citation2019), which enable environmentally friendly system configurations and enable a holistic approach to life cycle management of the IVD product. In another configuration, different CRISPR-Cas enzyme types with an additional signal boosting enzyme called Csm6 allow for simultaneous detection of multiple nucleic acids at once in a bilateral flow strip made of paper for visual readouts without the need for instrumentation much like a pregnancy test (Rizk, Citation2019). In the case of CRISPR-based devices, key TPP characteristics that should be considered include – low-temperature requirements, low resource needs, easy interpretation, and generation of less waste. This helps drive towards accomplishing an ideal IVD platform over existing molecular assay counterparts.
From a regulatory perspective, SHERLOCK was the first CRISPR assay to obtain an FDA EUA for COVID-19. Mammoth’s DETECTR also received an FDA EUA for COVID-19. Sense Detection, another firm seeking to develop a CRISPR assay, claims that it has advanced a device that does not require amplification for a molecular assay and received a CE mark (FDA, Citation2020b; Genomeweb, Citation2023). Subsequently, SHERLOCK acquired Sense Detection and thus collectively provided a portfolio of CRISPR-based detection chemistries and assay platforms to bring forth into the commercial market (Johnson, Citation2023). However, at present a rapid, PON CRISPR-based IVD has not yet received an FDA EUA.
Ultimately, as with any new technological principle, there are many development details for CRISPR-based diagnostics that remain to be solved and could be guided by proper TPP establishment for the IVD product. For example, as illustrated in the above of potential design criteria within the TPP framework, sensitivity and specificity are key performance metrics for molecular tests. Recent studies have shown that CRISPR-based detection has less sensitivity than previously reported (Chen et al., Citation2018, Citation2021; Ramachandran & Santiago, Citation2021; Slaymaker et al., Citation2019, Citation2021). Thus, further verification and validation testing and refinements in properly characterising CRISPR technologies are needed to truly assess their utility and place in the IVD market. Moreover, a developer of a CRISPR-based device may define their TPP to include key factors such as guide RNA requirements, amplification-free sensitivity to avoid potential assay inhibition, and a defined supply chain that minimises reagent costs and considers potential material waste.
Ideally, with a clear TPP roadmap, the developer would identify their differentiating characteristics to create an IVD product that is successfully marketed, enhances diagnostic accessibility to low resource or underserved communities, and that ultimately has clinical utility.
Case study III: applying TPP principles to guide the design of microbiological IVD devices using novel sample types
The move towards more accessible diagnostics, such as point-of-need or at-home test devices, has elevated the need to consider appropriate sample types that are conducive to such testing algorithms (IDSA, Citation2020; Tran & Albahra, Citation2021; Vandenberg et al., Citation2021; Zhou et al., Citation2021). This often neglected IVD development criterion is critically linked to device performance and cannot necessarily be resolved by innovating new technical approaches, as exemplified with the infamous Theranos technology (Clerici et al., Citation2021; Kidd et al., Citation2016). Factors such as sampling methods, sampling site restrictions, and potential interferents are integral to the overall test performance and must be considered before proposing a novel diagnostic modality (Diamandis, Citation2015; L. Wang et al., Citation2019). The following deeper dive into potential specimen types and how they relate to infectious diseases in terms of analyte concentration and other clinical matrix factors will help elucidate more productive IVD development processes.
First and foremost, decentralised testing, particularly devices designed for the lay user to perform at-home necessarily restricts the type of samples that can be collected. Specifically, samples that are minimally or non-invasive would likely be approved for such IVD products, as how the specimen is collected is part of the overall safety profile of the device that would be reviewed by the FDA for regulatory clearances. Within the TPP framework, the use of novel sample types (i.e. specimens not traditionally used for a disease diagnosis) relate to user type, intended use environment, human factors, indication, analytical/clinical performance, assay configuration, quality control, life cycle management, clinical utility, and even stability criteria. Clearly, selection of sample type influences multiple factors in IVD device development (Charlton et al., Citation2018; Spencer et al., Citation2019).
Saliva specimen type
As an example, COVID IVDs opened the possibility of using saliva as a sample type for respiratory disease diagnosis. Saliva is a non-invasive sample type and is easily collected. Further, although nasopharyngeal (NP) swabs are widely considered to be the preferred specimen for SARS-CoV-2 testing (Loens et al., Citation2009), they are not a viable option for at-home testing because of the need for skilled staff to preclude harm to the patient during collection and difficulty in proper use by non-healthcare professionals. Hence, saliva is an attractive option; however, variability in collection methods greatly impact the viability of this sample type. Therefore, detailed validation studies are needed to determine if saliva is a viable sample type, at least for SARS-CoV-2 detection (Hanson et al., Citation2021). Prospective studies with paired NP/saliva samples have been performed by researchers and indicate that overall there can be a slight performance decrease with saliva samples (i.e. positive NP swabs vs missed detections with saliva) (Azzi et al., Citation2020, Citation2021; Landry et al., Citation2020; Williams et al., Citation2020). A possible contribution to variable saliva performance is the consistency and viscosity of the sample itself, which can lead to variability in how the sample can be processed and handled. It has been reported that thick saliva may be more frequently encountered in potentially dehydrated symptomatic outpatients, but researchers have also observed similar sample characteristics in about 10% of the asymptomatic volunteers (Hanson et al., Citation2021).
In another study that evaluated the diagnostic validity, specimen collection time and cost associated with saliva for use with automated molecular Flu/RSV assays, there was again a decrease in sensitivity performance of the sample type when compared to the recommended NP samples (K. K. To et al., Citation2020). This appeared to indicate a lower viral load in saliva for most patients, even those hospitalised with severe disease as were the primary participants in the study. However, the saliva samples still had an observed sensitivity of >90% versus 96% for NP samples. When compared to other saliva-based studies, the use of the automated multiplex PCR system – namely the Cepheid Xpress Flu/RSV was suggested as a contributor to higher performance with this sample type because of the integrated sample processing. It was hypothesised that systems that used a separate extraction can lose sensitivity due to loss of extracted sample whereas the automated system used all extracted nucleic acid and thus circumvented the problem of low viral load for some patients. Reducing collection time and thus overall testing time while facilitating lower costs are also important for resource-limited settings.
But can the relative success in detecting analytes for COVID-19 and Flu/RSV in saliva sample type be translated to other respiratory infections? One study evaluated sputum samples, NP swab specimens, tracheal aspirates (TA), and bronchoalveolar lavage (BAL) specimens for PCR detection performance of MERS-CoV (K. K. W. To et al., Citation2019). The researchers observed that samples from deeper in the respiratory tract (i.e. TA and BAL specimens) yielded significantly higher MERS-CoV genome loads than samples from other respiratory tract anatomical sites. This translates into the need for a more sensitive detection modality to account for potentially reduced analyte from the upper respiratory specimens. It should be noted that TA and BAL specimens are invasive and therefore are typically only performed on patients admitted into intensive care unit where the sample is more accessible.
Overall, the analyte load in a clinical sample at any given time regardless of the infection status reflects the dynamic interaction between the aetiological agent and its interaction with the host, specifically the immune system (Memish et al., Citation2014). Since the natural history of infection can vary among diseases (e.g., peak viral load at 10 days post onset of symptoms (dpos) for SARS-CoV vs. 5 dpos for SARS-CoV-2)(Hung et al., Citation2004, Puhach et al., Citation2023), it is recommended that the amount of analyte in an intended sample type be properly evaluated to understand the appropriate TPP targets to be defined for device development criteria (Ahmadzadeh et al., Citation2021).
Case study IV: applying TPP principles to guide the design of next generation diagnostic paradigms
Diagnostic testing technology and needs are changing rapidly. Classically, IVD device developments need to be medically supported in terms of safety and efficacy for regulatory approval. Secondly, the diagnostic must provide clinical utility to the health care professional and patient – the device should provide as much information to aid in diagnosis and guide the appropriate treatment and intervention(s). This approach has enabled clinicians in traditional laboratory settings and even through at-home testing to diagnose disease and metabolic states that inform the clinical algorithms of treatment options. However, current diagnostic capabilities are limited, and it is evident that diagnostic paradigms need to evolve to aid in facilitating more comprehensive clinical decision-making for effecting better care of patients. As these clinical diagnostic needs change, proper application of a TPP helps outline the desired characteristics and performance criteria of a medical product to ensure that it meets the needs of the intended user.
One major gap that still challenges the current diagnostics field is the lack of accurate and rapid point-of-care tests. Rapid diagnostic tests that can be performed at the point-of-care are essential for timely and effective management of infectious diseases, especially in low-resource settings. However, many existing point-of-care tests have limitations in terms of accuracy, sensitivity, and specificity, which can lead to false-positive or false-negative results.
Another area for general improvement in IVD developments is to address the current limited detection capability of diagnostic devices. Many infectious diseases have overlapping clinical symptoms, making it difficult to diagnose them accurately with a single test, especially with home-use tests. Multiplexed assays that can detect multiple pathogens simultaneously are therefore needed to improve diagnosis and treatment. However, the availability of such assays is currently limited, and they can be expensive and complex to use. The ability to detect analytes must also consider the natural history of the disease course and where they appear in a sampling site. This is illustrated in showing for instance where a nucleic acid and antibody test may be most reliable in their respective application of a typical disease course from viral infection (UKRI, Citation2023).
Figure 4. Graphical representation showing generalised natural history of infection and notional patient condition during respiratory disease course. The illustration helps visualise how current diagnostic algorithms involving Nucleic Acid Amplification Tests (NAATs) and serological IVDs are implemented. Actual levels of analyte (i.e. pathogen nucleic acids or antibodies) could vary depending on disease.
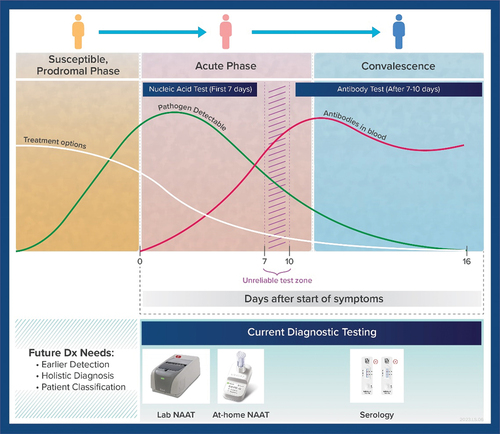
For acute infection, depending on the aetiological agent it may be possible to capture the diagnostic window based on symptomology. However, a factor to consider is whether testing based on symptoms would necessarily effect a change that matters from a clinical intervention perspective (e.g. would it be prudent to wait for a test result before starting a critical treatment when a broad spectrum of antibiotics could be immediately administered and could be life-saving). Alternatively, as observed with COVID-19, infected individuals could be asymptomatic and thus testing cannot necessarily be based on symptoms alone. Others have proposed a diagnostic algorithm where, based on time after exposure (FDA, Citation2023b), an individual could repeat testing over a course of days as opposed to discrete ‘one-time’ testing to preclude risk of false-negative results (FDA, Citation2023a). In such cases, the cost of testing is a serious consideration to enable such a diagnostic approach.
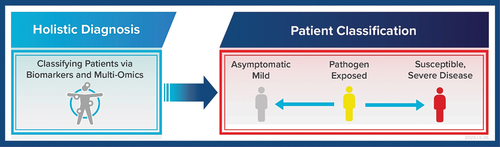
Another significant limitation with current approaches in diagnostic testing is the inability to effectively classify patients based on potential disease severity. This is a critical need to be efficient with resources and focus treatments and critical care on individuals who are more prone to developing severe disease. Thus, an IVD that can accomplish holistic diagnosis such as via biomarker detection and/or multi-omic testing would revolutionise current diagnostic algorithms ().
Figure 5. Holistic diagnostic approach in future IVD developments is needed to classify patients and thus focus treatments and care management on individuals susceptible to developing severe disease. This can be considered part of a future precision medicine approach to addressing infectious diseases through novel IVDs that for instance can accomplish multi-omics testing.
Finally, with the advent of at-home testing, a challenge is how to enable lay users to properly interpret test results and facilitate their proper incorporation into their clinical care. In the traditional diagnostic paradigm, patients would visit their health care provider and consult with their physician to help guide the interpretation of any test results. While the value of clinicians and their years of professional expertise cannot be trivialised, there is also a recognition of the need for greater accessibility of testing, especially in low-resource environments or the underserved communities while simultaneously ensuring safe and effective IVD developments. Some efforts include WHO’s prequalification program to ensure access to safe, appropriate, and affordable IVDs especially in low- and middle-income (LMICs) countries (Morin et al., Citation2018). Digital health initiatives are also being developed to harness modern information technology (IT) for better outcomes, particularly in meeting the needs of underserved populations such as US Health and Human Services (HHS) health equity telehealth program (HHS, Citation2023). Other innovative approaches include implementing the burgeoning developments in artificial intelligence (AI) and machine learning (ML) tools to develop personal health assistants that can provide some of the guidance that had in the past been provided by health care practitioners (Mikhail, Citation2022). Developers are also incorporating AI/ML algorithms to facilitate the interpretation of point-of-care diagnostics to enable simplified workflows without compromising the depth and quality of information that can be used to safely and effectively obtain a clinically useful result (Frontiers, Citation2023).
Applying TPP principles to the design of next-generation diagnostic assays can help to ensure that the assays are fit-for-purpose, accurate, and appropriate for their intended use. By defining the intended use, diagnostic accuracy, sample types, required instrumentation, turnaround time, and other key factors, the TPP can guide the development of assays that meet the needs of patients, healthcare providers, and public health agencies.
Conclusion
As described above, a well-developed TPP is central to successful IVD device developments. The TPP framework can highlight technological gaps and focus developments on meeting specific needs that advance the field. A survey of various diagnostic technologies, such as those employed for respiratory disease diagnosis, reveals significant innovations that have been accomplished in recent years, and have revolutionised the treatment of infections (Das et al., Citation2018). Yet, many of the significant technological strides have been largely laboratory-based, with relatively complex instruments and requirement for large amounts of resources. There is a growing need for greater accessibility and clinical utility of next-generation diagnostic capabilities.
One phenomenon that evolved from the COVID-19 diagnostic landscape was promoting a consumerist bent on testing – people sought as much personal health information (i.e. infection status) as possible that was facilitated with at-home testing products. This unprecedented increase in IVD test availability highlighted highly desired device attributes including – broad accessibility, cost-effectiveness, usability and convenience, and direct access to results and medical information. As the entire IVD diagnostic landscape continues to evolve, where appropriate and feasible away from a centralised lab testing model, it is important for device developers to identify the challenges and set appropriate targets to avoid pitfalls. As demonstrated in the above Case Study I/II, careful consideration of the relevant TPP criteria can guide developers through the process of meeting these consumer demands, while ensuring clinical utility and safety and effectiveness of their products.
The TPP framework also helps identify new development opportunities to address this trend in decentralised testing model. As examined in the above Case Study III, it can guide developers in assessing the appropriateness of novel sample types that meet usability requirements while addressing clinical unity among other factors. Further, the TPP can guide IVD developers to innovate novel technical principles beyond just mechanisms for detecting aetiological agents. As discussed in Case Study IV, current infectious disease diagnostic paradigms are focused solely on the presence of an active infection. IVD devices do not directly provide other critical diagnostic information such as whether the test subject is at higher risk for severe infection or whether a different preventative course (e.g. vaccine boosting) would lead to better clinical outcomes.
Information from IVD devices is just one piece of the comprehensive clinical case definition. Disease diagnosis has always been in the context of other information and facilitated by experienced, licenced health professionals. The limited availability of relevant medical information on a patient can lead to great subjectivity in interpretation. Having a diagnostic device that provides more information relating to the natural history of disease (broader coverage of disease states) can have greater clinical utility but requires the appropriate developmental targets. Moreover, with more complex data streams becoming available, AI/ML algorithms are assuming greater roles to help interpret diagnostic information beyond just a singular test result. Digital health technologies help bridge the gap between heterogeneous data that is becoming more accessible with greater diagnostic accessibility and limited availability of trained healthcare practitioners who can properly interpret the information. As we look to the future of diagnostics, many interrelated challenges remain with no one-size-fits-all solutions. Should next-generation IVD devices be developed that analyse aetiological agents and biomarkers simultaneously in a multi-omics configuration? What features would enable personalised (i.e. individual based) versus precision (i.e. customised, sub-grouping or population based) diagnostic and treatment outcomes? These and other pertinent questions may be addressed through the TPP process.
Acknowledgments
We wish to thank Garrett Dalton for his assistance with the graphical illustrations. We also wish to thank Joe Bogan for his critical review and comments.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Notes on contributors
Ricky Soong
Ricky Soong is a Senior Advisor at MRIGlobal. In this role, he advises clients on effective medical device designs, appropriate analytical and clinical study designs towards FDA premarket submissions, and applicable technical solutions to accomplish program requirements. Dr. Soong’s background is in Biomedical Engineering with multidisciplinary expertise including molecular biology, protein engineering, microfluidics, and nanotechnology. His experience includes a previous role as Scientific Reviewer of infectious disease IVD devices at the US FDA. At the FDA he addressed COVID-19 EUA IVD issues such as LDT testing policies, responding to inquiries from test developers, and leading the market authorisation of the first COVID-19 IVD De Novo. He also worked with stakeholders to establish Digital Health regulatory review policies by achieving consensus among internal and external stakeholders on mobile medical apps, cybersecurity, and AI/ML medical algorithms. Dr. Soong has also served as a Subject Matter Expert (SME) advisor to the Department of Defense Joint Program Executive Office to evaluate the technical feasibility of products from advanced developers to address CBRN defense needs of the Joint Forces. During his career spanning both academia and industry, he has authored industry guidances and numerous peer reviewed publications in bioengineering, biomedical engineering, and nanotechnology.
Elaine Bradford, is a Staff Scientist in the Infectious Disease, Surveillance, and Diagnostics Group at MRIGlobal. She is a Technical Lead across multiple projects and has experience across industry Research & Development teams. Her background spans molecular biology, microbiology, and host-pathogen interactions.
Kenneth B. Yeh
Kenneth B. Yeh is a senior director at MRIGlobal. He has created partnerships and reinforced best practices to develop technical solutions in the areas of biodefense, biotechnology, and global health security. Kenny’s work spans industry experience for detecting and identifying biological agents in the field and fielding cooperative biological research studies among global partners. His related publications include panel topics on end users and first movers in biotechnology, sharing of data and sample material, and research in high-containment facilities. Kenny has a background in microbiology.
Gene Olinger
Gene Olinger has actively collaborated with government agencies, academic institutions, industry partners, the WHO, the UN, and NGOs to deliver solutions for global protection with a strong focus on One Health. Dr. Olinger’s expertise extends to diagnostics, outbreak monitoring, and biodefense committees, and he serves as an associate director for maximum containment laboratory training at Boston University’s NEIDL. He has been an adjunct instructor at Hood College for the past 2 years supporting the Biology, Ecology, and Sustainability Studies programs.
References
- Ahmadzadeh, M., Vahidi, H., Mahboubi, A., Hajifathaliha, F., Nematollahi, L., & Mohit, E. (2021). Different respiratory samples for COVID-19 detection by standard and direct quantitative RT-PCR: A literature review. Iranian Journal of Pharmaceutical Research, 20(3), 285–16. https://doi.org/10.22037/ijpr.2021.115458.15383
- Association of Public Health Laboratories. (2023). APHL Response Efforts. Retrieved August 4,2023, from https://www.aphl.org/programs/infectious_disease/SARS-CoV-2/Pages/APHL-response.aspx.
- Azzi, L., Carcano, G., Dalla Gasperina, D., Sessa, F., Maurino, V., & Baj, A. (2021). Two cases of COVID-19 with positive salivary and negative pharyngeal or respiratory swabs at hospital discharge: A rising concern. Oral Diseases, 27(Suppl S3), 707–709. https://doi.org/10.1111/odi.13368
- Azzi, L., Carcano, G., Gianfagna, F., Grossi, P., Gasperina, D. D., Genoni, A., Fasano, M., Sessa, F., Tettamanti, L., Carinci, F., Maurino, V., Rossi, A., Tagliabue, A., & Baj, A. (2020). Saliva is a reliable tool to detect SARS-CoV-2. Journal of Infection, 81(1), e45–e50. https://doi.org/10.1016/j.jinf.2020.04.005
- Charlton, C. L., Babady, E., Ginocchio, C. C., Hatchette, T. F., Jerris, R. C., Li, Y., Loeffelholz, M., McCarter, Y. S., Miller, M. B., Novak-Weekley, S., Schuetz, A. N., Tang, Y.-W., Widen, R., & Drews, S. J. (2018). Practical guidance for clinical microbiology laboratories: Viruses causing acute respiratory tract infections. Clinical Microbiology Reviews, 32(1), e00042–18. https://doi.org/10.1128/CMR.00042-18
- Chen, J. S., Ma, E., Harrington, L. B., Da Costa, M., Tian, X., Palefsky, J. M., & Doudna, J. A. (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science: Advanced Materials and Devices, 360(6387), 436–439. https://doi.org/10.1126/science.aar6245
- Chen, J. S., Ma, E., Harrington, L. B., Da Costa, M., Tian, X., Plafesky, J. M., & Doudna, J. A. (2021). Erratum for the report “CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity”. Science: Advanced Materials and Devices, 371(6531). https://doi.org/10.1126/science.abh0317
- Clerici, B., Muscatello, A., Bai, F., Pavanello, D., Orlandi, M., Marchetti, G. C., Castelli, V., Casazza, G., Costantino, G., & Podda, G. M. (2021). Sensitivity of SARS-CoV-2 detection with nasopharyngeal swabs. Frontiers in Public Health, 8, 593491. https://doi.org/10.3389/fpubh.2020.593491
- Cocco, P., Ayaz-Shah, A., Messenger, M. P., West, R. M., & Shinkins, B. (2020). Target product profiles for medical tests: A systematic review of current methods. BMC Medicine, 18(1), 119. https://doi.org/10.1186/s12916-020-01582-1
- Das, S., Dunbar, S., & Tang, Y. W. (2018). Laboratory diagnosis of respiratory tract infections in children – the state of the art. Frontiers in Microbiology, 9(9), 2478. https://doi.org/10.3389/fmicb.2018.02478
- Dean, D., Swaminathan, S., Kama, M., Goemans, S., Faktaufon, D., Alnabelseya, N., Spelke, D., Kahrizi, K., Black, M., Mitra, D., & Munson, E. (2021). Development and evaluation of a point-of-care test in a low-resource setting with high rates of Chlamydia trachomatis urogenital infections in Fiji. Journal of Clinical Microbiology, 59(7), e0018221. https://doi.org/10.1128/JCM.00182-21
- Defense Advanced Research Projects Agency. (2022). The heilmeier catechism. Retrieved December 21, 2022, from https://www.darpa.mil/work-with-us/heilmeier-catechism:.
- Denkinger, C., Dolinger, D., Schito, M., Wells, W., Cobelens, F., Pai, M., Zignol, M., Cirillo, D. M., Alland, D., Casenghi, M., Gallarda, J., Boehme, C. C., & Perkins, M. D. Target product profile of a molecular drug-susceptibility test for use in microscopy centers. (2015). The Journal of Infectious Diseases, 211(Suppl suppl_2), S39–S49. https://doi.org/10.1093/infdis/jiu682
- Denkinger, C., Kik, S., Cirillo, D., Casenghi, M., Shinnick, T., Weyer, K., Gilpin, C., Boehme, C. C., Schito, M., Kimerling, M., & Pai, M. (2015). Defining the needs for next generation assays for tuberculosis. Journal of Infectious Diseases, 211(Suppl suppl_2), S29–38. https://doi.org/10.10.1093/infdis/jiu821
- Diamandis, E. P. (2015). Theranos phenomenon: Promises and fallacies. Clinical Chemistry and Laboratory Medicine, 53(7), 989–993. https://doi.org/10.1515/cclm-2015-0356
- Drucker, E., & Krapfenbauer, K. (2013). Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. EPMA Journal, 4(1), 4–7. https://doi.org/10.1186/1878-5085-4-7
- Ellume. (2020). Ellume awarded US$30M from U.S. National Institutes of health RADx initiative to accelerate development of rapid COVID-19 diagnostics. Retrieved December 7, 2022, from https://www.ellumehealth.com/news/ellume-awarded-us-30m-from-us-national-institutes-of-health-radx-initiative-to-accelerate-development-of-rapid-covid-19-diagnostics-7-october-2020:.
- Engel, N., Wachter, K., Pai, M., Gallarda, J., Boehme, C., Celentano, I., & Weintraub, R. (2016). Addressing the challenges of diagnostics demand and supply: Insights from an online global health discussion platform. BMJ Global Health, 1(4), e000132. https://doi.org/10.1136/bmjgh-2016-000132
- Esquivel, J. P., Castellarnau, M., Gasso, S., & Sabate, N. (2019). Paper-based batteries as sustainable power source for disposable electronic devices, Smart systems integration. 13th International Conference and Exhibition on Integration Issues of Miniaturized Systems, Barcelona, Spain, pp. 1–4.
- FIND. (2022). A need for novel diagnostics: Meeting the moment. Retrieved December 15, 2022, from https://www.finddx.org/wp-content/uploads/2022/12/20221209_rep_novel_diagnostics_FV_EN.pdf:.
- Food and Drug Administration. (2020a). 85 FR 18250 – for a complete overview of FDA’s COVID-19 EUA efforts related to vaccines. https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#vaccines.
- Food and Drug Administration. (2020b). 85 FR 7316 – for a complete overview of FDA’s COVID-19 EUA efforts related to IVDs. https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas.
- Food and Drug Administration. (2021). FDA decision summary, K200748. Visby medical sexual health click test. Retrieved December 14, 2022, from https://www.accessdata.fda.gov/cdrh_docs/reviews/K200748.pdf.
- Food and Drug Administration. (2023a). At-homeCOVID-19 antigen tests-take steps to reduce your risk of false negative results: FDA safety communication. Retrieved March 22, 2023, from https://www.fda.gov/medical-devices/safety-communications/home-covid-19-antigen-tests-take-steps-reduce-your-risk-false-negative-results-fda-safety:.
- Food and Drug Administration. (2023b). At-homeCOVID-19 diagnostic tests: frequently asked questions. Retrieved March 22, 2023, from https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/home-covid-19-diagnostic-tests-frequently-asked-questions:.
- Food and Drug Administration. (2023c). Drugs and non-vaccine biological products. Retrieved March 22, 2023, from https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#coviddrugs:.
- The Forum for Collaborative Research. (2015). High-priority target product profile for hepatitis C diagnosis in decentralized settings: Report of a consensus meeting. Retrieved December 8, 2022, from https://forumresearch.org/storage/documents/2015/hcv/hcv-tpp-report17july2015final.pdf.
- Frontiers. (2023). Retrieved March 22, 2023, from https://www.frontiersin.org/research-topics/33459/artificial-intelligence-in-point-of-care-diagnostics:.
- Gal, M., Francis, N., Hood, K., Villacian, J., Goossens, H., Watkins, A., Butler, C. C., & Gupta, V. Matching diagnostics development to clinical need: Target product profile development for a point of care test for community-acquired lower respiratory tract infection. (2018). PloS One, 13(8), e0200531. 2018. https://doi.org/10.1371/journal.pone.0200531
- Genomeweb 360 dx. (2022). FDA reissues EUA for meridian bioscience COVID-19 test after omicron variant trouble. 360Dx. Retrieved December 13, 2022, from https://www.360dx.com/molecular-diagnostics/fda-reissues-eua-meridian-bioscience-covid-19-test-after-omicron-variant#.Y6MhhtXMI2x.
- Genomeweb 360 dx. (2023). Coronavirus test tracker: Commercially available COVID-19 diagnostic tests. 360Dx. Retrieved April 5, 2023, from https://www.360dx.com/coronavirus-test-tracker-launched-covid-19-tests:.
- Griffin, R. (2022). Covid test maker central to US response appoints administrators. Bloomberg Law. Retrieved December 7, 2022, from https://news.bloomberglaw.com/bankruptcy-law/covid-test-maker-ellume-appoints-administrators-in-australia:.
- Hanson, K. E., Caliendo, A. M., Arias, C. A., Hayden, M. K., Englund, J. A., Lee, M. J., Loeb, M., Patel, R., El Alayli, A., Altayar, O., Patel, P., Falck-Ytter, Y., Lavergne, V., Morgan, R. L., Murad, M. H., Sultan, S., Bhimraj, A., & Mustafa, R. A. (2021). The infectious diseases society of America guidelines on the diagnosis of COVID-19: Molecular diagnostic testing. Clinical Infectious Diseases, ciab048. https://doi.org/10.1093/cid/ciab048
- Harvard University. (2023). Sherlock biosciences licenses Wyss Institute’s ambient nucleic acid amplification technology from Harvard to develop highly accurate, low-cost diagnostics for point-of-need. Wyss Institute. Retrieved March 21, 2023, from https://wyss.harvard.edu/news/sherlock-biosciences-licenses-wyss-institutes-ambient-nucleic-acid-amplification-technology-from-harvard-to-develop-highly-accurate-low-cost-diagnostics-for-point-of-need/.
- Health and Human Services. (2023). Health equity in telehealth. Retrieved March 22, 2023, from https://telehealth.hhs.gov/providers/health-equity-in-telehealth?gclid=EAIaIQobChMIsPat-bDw_QIVYxatBh3h1w-PEAAYAyAAEgKLpvD_BwE:.
- Hung, I. F., Cheng, V. C., Wu, A. K., Tang, B. S., Chan, K. H., Chu, C. M., Wong, M. M., Hui, W. T., Poon, L. L., Tse, D. M., Chan, K. S., Woo, P. C., Lau, S. K., Peiris, J. S., & Yuen, K. Y. (2004). Viral loads in clinical specimens and SARS manifestations. Emerging Infectious Diseases, 10(9), 1550–1557. https://doi.org/10.3201/eid1009.040058
- Infectious Diseases Society of America. (2020). IDSA guidelines on the diagnosis of COVID-19: Molecular diagnostic testing. Retrieved December 8, 2022.
- Johnson, M. (2022a). QuidelOrtho continues to see supply chain impacts; Combined Q3 revenues up 54 percent. Genomeweb 360 dx.
- Johnson, M. (2022b). Radx revved up COVID testing development. Will it also change the US Dx ecosystem? Part 1. Genomeweb 360 dx.
- Johnson, M. (2022c). Radx revved up COVID testing development. Will it also change the US Dx ecosystem? Part 2. Genomeweb 360 dx.
- Johnson, M. (2023). Sherlock biosciences expects sense biodetection buy to accelerate commercialization plans. Genomeweb 360Dx.
- Kaminski, M. M., Abudayyeh, O. O., Gootenberg, J. S., Zhang, F., & Collins, J. J. (2021). CRISPR-based diagnostics. Nature Biomedical Engineering, 5(7), 643–656. https://doi.org/10.1038/s41551-021-00760-7
- Kidd, B. A., Hoffman, G., Zimmerman, N., Li, L., Morgan, J. W., Glowe, P. K., Botwin, G. J., Parekh, S., Babic, N., Doust, M. W., Stock, G. B., Schadt, E. E., & Dudley, J. T. (2016). Evaluation of direct-to-consumer low-volume lab tests in healthy adults. Journal of Clinical Investigation, 126(5), 1734–1744. https://doi.org/10.1172/JCI86318
- Landry, M. L., Criscuolo, J., & Peaper, D. R. (2020). Challenges in use of saliva for detection of SARS CoV-2 RNA in symptomatic outpatients. Journal of Clinical Virology, 130, 104567. https://doi.org/10.1016/j.jcv.2020.104567
- Loens, K., Van Heirstraeten, L., Malhotra-Kumar, S., Goossens, H., & Ieven, M. (2009). Optimal sampling sites and methods for detection of pathogens possibly causing community-acquired lower respiratory tract infections. Journal of Clinical Microbiology, 47(1), 21–31. https://doi.org/10.1128/JCM.02037-08
- Memish, Z. A., Al-Tawfiq, J. A., Makhdoom, H. Q., Assiri, A., Alhakeem, R. F., Albarrak, A., Alsubaie, S., Al-Rabeeah, A. A., Hajomar, W. H., Hussain, R., Kheyami, A. M., Almutairi, A., Azhar, E. I., Drosten, C., Watson, S. J., Kellam, P., Cotten, M., & Zumla, A. (2014). Respiratory tract samples, viral load, and genome fraction yield in patients with Middle East respiratory syndrome. The Journal of infectious diseases, 210(10), 1590–1594. https://doi.org/10.1093/infdis/jiu292
- Mikhail, A. (2022). It’s estimated that the majority of seniors don’t take their medications as prescribed. This AI driven personal health assistant could help solve that. Fortune. Retrieved March 22, 2023, from https://fortune.com/well/2022/10/07/renee-ai-driven-personal-health-assistant/.
- Morin, S., Bazarova, N., Jacon, P., & Vella, S. (2018). The manufacturers’ perspective on world health organization prequalification of in vitro diagnostics. Clinical Infectious Diseases, 66(2), 301–305. https://doi.org/10.1093/cid/cix719
- National Institutes of Health. (2023). Radx® tech and ATP programs: Phase 2 awards. Retrieved April 14, 2023, from https://www.nibib.nih.gov/covid-19/radx-tech-program/radx-tech-phase2-awards:.
- Niederberger, M., & Spranger, J. (2020). Delphi technique in health sciences: A map. Frontiers in Public Health, 8, 457. https://doi.org/10.3389/fpubh.2020.00457
- PATH. (2023). Global availability of COVID-19 diagnostic tests. Retrieved April 5, 2023, from https://www.path.org/programs/diagnostics/covid-dashboard-global-availability-covid-19-diagnostic-tests/:.
- Puhach, O., Meyer, B., & Eckerle, I. (2023). SARS-CoV-2 viral load and shedding kinetics. Nature reviews. Microbiology, 21(3), 147–161. https://doi.org/10.1038/s41579-022-00822-w
- Ramachandran, A., & Santiago, J. G. (2021). Enzyme kinetics of CRISPR molecular diagnostics. bioRxiv Analytical Chemistry, 93(20), 7456–7464. https://doi.org/10.1021/acs.analchem.1c00525
- Renzoni, A., Perez, F., Ngo Nsoga, M. T., Yerly, S., Boehm, E., Gayet-Ageron, A., Kaiser, L., & Schibler, M. (2021). Analytical evaluation of Visby medical RT-PCR portable device for rapid detection of SARS-CoV-2. Diagnostics (Basel), 11(5), 813. https://doi.org/10.3390/diagnostics11050813
- Rizk, C. (2019). Sherlock biosciences aims to create Dx Tool for ‘Where testing should be done, but isn’t. Genomeweb.
- Ropes & Gray. (2023). The end of an era: How will terminating the COVID-19 public health emergency affect life sciences. Retrieved March 21, 2023, from https://www.ropesgray.com/en/newsroom/alerts/2023/02/the-end-of-an-era-how-will-terminating-the-covid-19-public-health-emergency-affect-life-sciences:.
- Satyanarayana, M. (2021). Over-the-counter COVID-19 tests make big promises. Do they deliver? Chemical & Engineering News, 99(20), 28–35. https://doi.org/10.1021/cen-09920-cover
- The Signal Path. (2022). TNP #9 - teardown & analysis of an electronic COVID-19 home test kit (ellume). Youtube. Retrieved December 16, 2022.
- Slaymaker, I. M., Mesa, P., Kellner, M. J., Kannan, S., Brignole, E., Koob, J., Feliciano, P. R., Stella, S., Abudayyeh, O. O., Gootenberg, J. S., Strecker, J., Montoya, G., & Zhang, F. (2019). High-resolution structure of Cas13b and biochemical characterization of RNA targeting and cleavage. Cell Reports, 26(13), 3741–3751. https://doi.org/10.1016/j.celrep.2019.02.094
- Slaymaker, I. M., Mesa, P., Kellner, M. J., Kannan, S., Brignole, E., Koob, J., Feliciano, P. R., Stella, S., Abudayyeh, O. O., Gootenberg, J. S., Strecker, J., Montoya, G., & Zhang, F. (2021). Correction: High-resolution structure of Cas13b and biochemical characterization of RNA targeting and cleavage. Cell Reports, 26(13), 3741–3751. https://doi.org/10.1016/j.celrep.2021.108865
- Spencer, S., Thompson, M. G., Flannery, B., Fry, A., & Kraft, C. S. (2019). Comparison of respiratory specimen collection methods for detection of influenza virus infection by reverse transcription-PCR: A literature review. Journal of Clinical Microbiology, 57(9), e00027–19. https://doi.org/10.1128/JCM.00027-19
- Stieber, F., Howard, J., Rao, S. N., Kawamura, L. M., Manissero, D., Love, J., Yang, M., Uchiyama, R., Parsons, S., Miller, C., Douwes, H., McDonald, A., Fairburn, L., & Boyle, J. (2020). First performance report of QIAreach™ anti-SARS-CoV-2 total test, an innovative nanoparticle fluorescence digital detection platform. Journal of Clinical Virology, 133, 104681. https://doi.org/10.1016/j.jcv.2020.104681
- To, K. K., Tsang, O. T., Leung, W. S., Tam, A. R., Wu, T. C., Lung, D. C., Yip, C. C., Cai, J. P., Chan, J. M., Chik, T. S., Lau, D. P., Choi, C. Y., Chen, L. L., Chan, W. M., Chan, K. H., Ip, J. D., Ng, A. C., Poon, R. W. … Yuen, K. Y. (2020). Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: An observational cohort study. Lancet Infectious Disease, 20(5), 565–574. https://doi.org/10.1016/S1473-3099(20)30196-1
- To, K. K. W., Yip, C. C. Y., Lai, C. Y. W., Wong, C. K. H., Ho, D. T. Y., Pang, P. K. P., Ng, A. C. K., Leung, K. H., Poon, R. W. S., Chan, K. H., Cheng, V. C. C., Hung, I. F. N., & Yuen, K. Y. (2019). Saliva as a diagnostic specimen for testing respiratory virus by a point-of-care molecular assay: A diagnostic validity study. Clinical Microbiology and Infection, 25(3), 372–378. https://doi.org/10.1016/j.cmi.2018.06.009
- Tran, N., & Albahra, S. (2021). Comparison of SARS-CoV-2 specimen types: Nasal Swabs, Nasopharyngeal Swabs and beyond. Retrieved August 4, 2023.https://health.ucdavis.edu/blog/lab-best-practice/comparison-of-sars-cov-2-specimen-types-nasal-swabs-nasopharyngeal-swabs-and-beyond/2021/09
- UK Research and Innovation. (2023). What is the purpose of testing for COVID-19? Retrieved March 22, 2023, from https://coronavirusexplained.ukri.org/en/article/vdt0006:.
- Vandenberg, O., Martiny, D., Rochas, O., Vvan Belkum, A., & Kozlakidis, Z. (2021). Considerations for diagnostic COVID-19 tests. Nature Reviews Microbiology, 19(3), 171–183. https://doi.org/10.1038/s41579-020-00461-z
- Wang, P., & Kricka, L. (2018). Current and emerging trends in point-of-care technology and strategies for clinical validation and implementation. Clinical Chemistry, 64(10), 1439–1452. https://doi.org/10.1373/clinchem.2018.287052
- Wang, L., Yang, S., Yan, X., Liu, T., Feng, Z., & Li, G. (2019). Comparing the yield of oropharyngeal swabs and sputum for detection of 11 common pathogens in hospitalized children with lower respiratory tract infection. Virology Journal, 16(1), 84. https://doi.org/10.1186/s12985-019-1177-x
- Williams, E., Bond, K., Zhang, B., Putland, M., Williamson, D. A., & McAdam, A. J. (2020). Saliva as a Noninvasive Specimen for detection of SARS-CoV-2. Journal of Clinical Microbiology, 58(8), e00776–20. https://doi.org/10.1128/JCM.00776-20
- World Health Organization. (2014). High-priority target product profiles for new tuberculosis diagnostics: Report of a consensus meeting. Retrieved December 21, 2022, from https://apps.who.int/iris/bitstream/handle/10665/135617/WHO_HTM_TB_2014.18_eng.pdf:.
- Zhou, Y., O’Leary, T. J., & Hozbor, D. F. (2021). Relative sensitivity of anterior nares and nasopharyngeal swabs for initial detection of SARS-CoV-2 in ambulatory patients: Rapid review and meta-analysis. PLoS One, 16(7), e0254559. https://doi.org/10.1371/journal.pone.0254559