
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The acceleration of materials discovery has gained paramount importance due to its potential to overcome constraints in emerging technologies. Extensive exploration has been undertaken into three pivotal approaches: combinatorial synthesis, high-throughput characterization, and computational techniques, all aimed at unveiling new materials. This review article delves into recent progress in these domains. Combinatorial synthesis, especially in the development of thin-film materials libraries, emerges as a potent method for efficiently generating comprehensive multinary materials systems and composition gradients spanning the entire spectrum of required compositions. High-throughput characterization techniques play a pivotal role in assessing the compositional, structural, and functional attributes of materials within these libraries, yielding multidimensional datasets. Concurrently, recent advancements in computational materials science have notably expedited the discovery process by enabling high-throughput calculations and simulations of potential materials systems. These collective endeavors foster a more robust correlation between composition, processing, structure, and properties, facilitating the forecast and design of future materials through data-driven materials discovery. This approach allows for efficient optimization of newly identified materials. Furthermore, materials informatics, an integral element of this process, plays a crucial role in managing and extracting valuable insights from the vast data generated during materials discovery.
Graphical abstract
IMPACT STATEMENT
Combining high-throughput combinatorial methods, computational approaches, open-source libraries, and collaborative efforts leads to significant novelty, particularly in the discovery and understanding of new materials and their relationships between structure, properties, process, and composition.
1. Introduction
The discovery of new materials has been instrumental in driving significant technological advancements throughout history. Notable examples include the development of materials like bronze and steel in ancient times, as well as the advent of synthetic polymers in the twentieth century, all of which have had profound impacts on various industries and applications. However, it’s important to recognize the evolution of materials discovery over time. In Thomas Edison’s era, when he was searching for a suitable filament for the incandescent light bulb, the available options were limited, primarily relying on carbonized natural fibers. This historical context underscores the remarkable progress made in the field of materials science since then. In modern times, advanced materials frequently consist of complex combinations, including ternary, quaternary, or even more complex alloys. Furthermore, their properties can be finely tuned by manipulating factors like crystallinity, mesostructure, and layering arrangements. As a result, the process of screening and developing materials for contemporary applications often requires conducting a substantially greater number of experiments and necessitates a more advanced comprehension of material properties than what was sufficient a century ago. The ongoing quest for materials innovation remains a critical driving force for addressing urgent societal challenges, particularly in the realms of mitigating global climate change and securing a sustainable future energy supply. The development of novel materials with enhanced properties, such as heightened strength, improved energy efficiency, and superior environmental sustainability, is indispensable for advancing solutions in these vital areas. Thus, the relentless pursuit of materials discovery remains a foundational pillar for technological progress and is pivotal in tackling the most pressing challenges of our era [Citation1]. To make substantial advancements, it is crucial to carefully designate the materials systems for experimental exploration. This entails confining the selection of initial chemical elements to those that are abundant in the Earth, non-hazardous, and environmentally sustainable. Despite the immense potential in discovering new materials, a targeted approach is indispensable for achieving significant results. Despite the utilization of cutting-edge technologies, the process of materials discovery still relies heavily on extensive trial and error. Identifying a suitable material for a technological application can span decades of research, with additional time required for its optimization for commercial use. This prolonged journey primarily stems from the intricate and multifaceted nature of materials design, rendering it a formidable optimization challenge. Moreover, the absence of necessary data for making informed decisions regarding the prioritization of materials and experimental procedures further adds complexity to the materials discovery process [Citation2–9].
Materials used in engineering applications often have complex compositions and microstructures that consist of more than five elements. This is true for a variety of materials including steels, superalloys, bulk metallic glasses, and high-entropy alloys. In fact, the use of multinary materials is expected to increase in the future compared to materials composed of only one or two elements. However, the sheer number of possible combinations of chemical elements in multinary systems is vast, with over two million possible combinations for quinaries alone when using 50–60 starting elements. Achieving the exact composition, phase constitution, and microstructure required for specific applications is a difficult task [Citation10]. The vast and unexplored composition space is further expanded by the processing parameter space. Various manufacturing methods are employed to produce materials, leading to diverse material states, including metastable and stable forms, each characterized by distinct microstructures and properties that necessitate thorough investigation. Attaining the sought-after properties of multinary materials demands precise management of composition, processing techniques, and microstructural elements. To achieve these objectives, engineers and scientists must possess a profound comprehension of the fundamental physical and chemical principles governing these materials. This entails adopting an interdisciplinary approach that seamlessly integrates the fields of chemistry, physics, and materials science [Citation11–13]. Utilizing processing methods allows for the tailoring of materials’ microstructures and external attributes, including functional properties. The quest for novel materials involves a multidimensional exploration, encompassing intrinsic factors like composition and crystal structure, as well as extrinsic elements such as microstructure and functional properties. The identification of suitable properties for a particular application demands comprehensive investigation within this expansive and complex search domain [Citation3,Citation10,Citation14–23].
Quantum chemical approximations, like density functional theory (DFT), have proven highly effective in predicting a wide range of crucial materials properties with remarkable precision. Many scientific teams have harnessed high-throughput computational strategies to systematically evaluate thousands of compounds, assessing their potential suitability for emerging technological materials. This approach has significantly accelerated the exploration of novel materials with promising applications. This approach has been applied to a variety of fields, including solar water splitters, solar photovoltaics [Citation24], topological insulators [Citation25], scintillators, CO2 capture materials [Citation26], piezoelectrics, and thermoelectrics [Citation27]. The studies have suggested several new promising compounds for experimental follow-up [Citation28]. In the fields of catalysis, hydrogen storage materials [Citation29–31], and Li-ion batteries [Citation32], experimental ‘hits’ have already been reported from high-throughput computations [Citation2,Citation23,Citation33–37].
A promising trend that has emerged in recent years involves the integration of computational materials science with information technology. This integration encompasses the utilization of web-based dissemination, databases, and data-mining techniques to overcome the constraints typically associated with individual research groups [Citation2,Citation33]. Consequently, accessing computed materials datasets has become more accessible to new communities, fostering innovative collaborative approaches to materials discovery. Materials informatics, a field that utilizes prior knowledge stored in databases or extracted from electronic literature through computational methods, has the potential to enhance this filtering process. Theoretical and computational materials science can also formulate hypotheses regarding where to explore for new materials and provide predictions about novel materials. Computational techniques offer the advantage of assessing thousands of systems within a significantly shorter timeframe compared to experimental research. High-throughput computational materials science contributes invaluable insights by generating a practical inventory of prospective materials for subsequent combinatorial investigation. Utilizing computational methodologies for forecasting compositions serves as a means to refine the quest for new materials, transforming it from an extensive exploration to a curated selection of tens or hundreds of potential candidates. This streaterialsinformatics, a field that utilizes pamlined approach empowers researchers to channel their experimental endeavors towards a manageable roster of materials, thereby expediting the discovery of innovative materials tailored to meet specific application requirements [Citation35,Citation38–40].
The perspective delves into the possibilities of employing combinatorial synthesis, high-throughput calculations, and high-throughput characterization, coupled with computational techniques, to effectively uncover novel materials within intricate search spaces. This strategy seeks to shift away from chance-based discoveries and transition toward a data-centric and streamlined approach to materials discovery. The article also addresses broader considerations regarding materials discovery and examines potential experimental avenues utilizing thin-film combinatorial materials science (CMS) [Citation1,Citation41–44]. However, to achieve efficient materials discovery and design, the expansion of CMS with computational methods and materials informatics is necessary [Citation44–46]. Computational techniques offer the capability to predict and screen novel materials, enabling researchers to focus their experimental endeavors on a smaller subset of materials possessing the desired properties, thereby resulting in time and resource savings. Additionally, materials informatics plays a pivotal role in refining the quest for new materials by amalgamating prior knowledge from databases and literature. delineates the crucial stages within the realm of high-throughput material synthesis and characterization, seamlessly integrating theoretical material theory, modeling, and the development of material databases. Expediency in the fabrication and analysis cycle is paramount for expediting the introduction of new materials into the market. illustrates an instance of a thin-film fabrication system and candidate selection, wherein a single thin-film fabrication experiment can yield a 16 × 16 matrix comprising 256 distinct materials, each with a thickness measured in a few hundred nanometers [Citation47].
Figure 1. Illustration of the general process of high throughput material synthesis and characterization, which involves combining theoretical material theory, modeling, and material database development. The diagram shows that this process typically involves multiple stages, such as design and synthesis, characterization, and data analysis.
Figure 2. Illustrates two different approaches to high throughput material synthesis and screening. A combinatorial thin film material synthesis method is shown, where the thin film fabrication process is integrated in parallel using spatially addressable arrays of samples. A high throughput semiconductor thin film material screening process is depicted on the left side. Adapted from reference with permission [Citation47]. Copyright © 2015 The Chinese Ceramic Society.
![Figure 2. Illustrates two different approaches to high throughput material synthesis and screening. A combinatorial thin film material synthesis method is shown, where the thin film fabrication process is integrated in parallel using spatially addressable arrays of samples. A high throughput semiconductor thin film material screening process is depicted on the left side. Adapted from reference with permission [Citation47]. Copyright © 2015 The Chinese Ceramic Society.](/cms/asset/37ddd4b5-f076-4d0b-8b6d-47cd565153da/tstm_a_2292486_f0002_oc.jpg)
2. Exploring materials in multidimensional spaces: the quest for discovery
Materials discovery involves identifying new phases or combinations of phases with unique properties resulting from previously undiscovered combinations of composition, crystal structure, phase constitution, microstructure, and properties. This is different from materials development, which focuses on optimizing known materials through adjustments in composition or processing to enhance their existing properties. Materials design, on the other hand, begins with the desired properties and aims to create a material that exhibits these specific characteristics. Materials discovery, in contrast, entails the exploration of a wide range of materials to uncover various properties. While materials discovery can lead to the identification of exceptional properties in previously unknown materials, the vast and uncertain nature of the search space presents significant challenges [Citation10,Citation48].
Historically, materials discoveries often occurred serendipitously, where researchers stumbled upon unexpected findings while pursuing different research objectives. In such cases, the ability to distinguish between erroneous measurements and genuine discoveries played a vital role. Remarkable properties have also emerged in materials where they were not initially anticipated, such as the discovery of high-temperature superconductivity in oxides. More recently, innovative combinatorial synthesis and characterization methods led to the identification of a noble-metal-free nanoparticulate electrocatalyst. Additionally, exploring multinary systems for properties without prior reported results has yielded exciting discoveries. A noteworthy achievement in the field of resistive random-access memories is the successful control of conductive filament formation in mixed Hf-Ta anodic oxides (). This has enabled the identification of Hf-Ta anodic memristors with enhanced properties and made it possible to investigate the feasibility of forming-free Hf-Ta/HfO2-Ta2O5/Pt memristors. The recently discovered unipolar memristors exhibit a uniform chemical composition in their local regions, unlike bipolar memristors [Citation49]. Similarly, flexible electronics is an emerging field characterized by the development of electronic components that are both thin and flexible, suitable for integration into various applications like wearable devices, medical sensors, and electronic skins. A significant challenge in this field is the need for a cost-effective and efficient fabrication method for these components. High-throughput synthesis offers a solution by enabling the swift synthesis and screening of numerous materials to pinpoint their desirable properties. Recently, this approach has been applied to the production of thin metal oxide combinatorial films, which have applications in electronic component manufacturing. These films can be synthesized and screened for electronic properties like resistivity, conductivity, and dielectric constant in a high-throughput fashion, expediting the identification of materials suitable for specific electronic components. The successful implementation of high-throughput synthesis and anodic printing has paved the way for exciting advancements in the realm of flexible electronics (). This technology not only facilitates the development of flexible electronic components but also offers cost-effectiveness, efficiency, and compatibility with a diverse array of applications. Its potential extends to reshaping the dynamics of human interaction with electronic devices and fostering innovative possibilities across multiple domains, spanning from healthcare to consumer electronics [Citation50,Citation51]. Therefore, materials discoveries can occur in unexplored composition spaces, as well as in known composition spaces where special functionalities have not yet been investigated.
Figure 3. (a) Controlling formation of conductive filaments in hf-ta anodic oxides: a significant accomplishment in resistive random-access memories (b) revolutionizing flexible electronics: high throughput synthesis and anodic printing process, reproduced with permission [Citation49].
![Figure 3. (a) Controlling formation of conductive filaments in hf-ta anodic oxides: a significant accomplishment in resistive random-access memories (b) revolutionizing flexible electronics: high throughput synthesis and anodic printing process, reproduced with permission [Citation49].](/cms/asset/29b00040-d0f2-4154-9c9b-579c6b3a71f4/tstm_a_2292486_f0003_b.gif)
Given the vastness of the materials search space, achieving accurate predictions becomes paramount in identifying the most promising compositions or composition ranges for investigation. High-throughput computations offer an effective means of predicting potentially stable materials with intriguing properties from a pool of thousands of candidates. Subsequently, combinatorial materials science can step in to synthesize a limited number of compositions within materials libraries, strategically centered around the most promising predicted compositions. However, these predictions are often rooted solely in intrinsic properties and frequently lack precise and validated experimental data for validation. To overcome this limitation, high-throughput characterization of materials libraries becomes instrumental, serving the dual purpose of providing the missing datasets and validating the predictions. Through the generation of high-quality multidimensional data and its joint evaluation via high-throughput computations and experiments, researchers can enhance the accuracy of their predictions while also uncovering unexplored data points worthy of further exploration. In essence, this integrated approach holds significant potential for advancing materials discovery and expediting the development of cutting-edge technologies [Citation10,Citation52].
3. Synthesizing thin films through combinatorial approaches
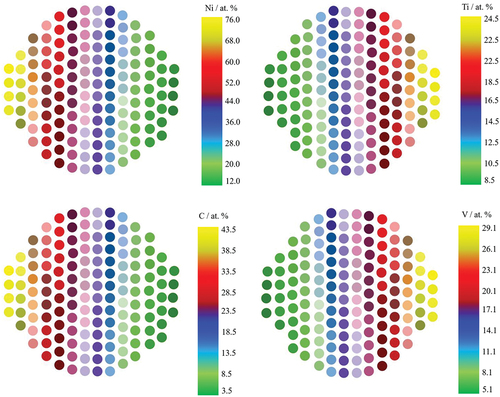
Combinatorial thin film libraries represent an approach for synthesizing and characterizing thin film materials with diverse compositions, thicknesses, and properties in a high-throughput manner. The primary objective behind creating such libraries is to expedite the discovery of new materials possessing sought-after characteristics applicable in various domains, including electronics, optics, and energy-related fields. This approach entails the precise deposition of a range of materials in a systematic and controlled manner. Each sample within the library comprises a specific combination of elements or compounds, resulting in the simultaneous and efficient synthesis of numerous distinct materials. This, in turn, facilitates the exploration of extensive regions within the materials phase diagram, opening up avenues for new material discoveries [Citation53]. Combinatorial thin film synthesis which was developed almost twenty years ago, has been used for screening materials for various applications, including high critical temperature superconductors and phosphors [Citation54,Citation55]. The method involves spatially selective deposition to create individual combinatorial material libraries using designed masks to delineate growth regions with different compositions. The most common method for depositing thin-film materials libraries is through (virtual) wedge-type films, which achieve well-defined composition gradients. This can be done by co-deposition from multiple sources or through multilayer deposition of nanoscale wedge-type layers. Combinatorial magnetron sputter processes are a useful method for fabricating materials libraries, as sputtering is a versatile process that can be applied in both scientific research and industrial applications. Findings from screening of sputtered materials libraries can be transferred to industrial thin-film applications. displays an example of how the composition of the four elements varies across the wafer in thermal co-deposition, with the different atomic percentages. These varying atomic percentages allow for the achievement of different target compositions, such as Ti12.2Ni58.5Cu25.2 V5.8. The distribution of these elements corresponds with the target gun positions for each element inside the sputtering chamber [Citation47]. Many examples regarding thin film combinatorial libraries can be found elsewhere [Citation14,Citation41,Citation45,Citation56].
Figure 4. The elemental distribution of Ni, Ti, Cu, and V on a si wafer, with each element’s composition indicated by a color scale ranging from high (yellow) to low (green) atomic percentage.
Another technique for creating thin-film materials libraries involves the development of ‘focused’ compositional gradient MLs (Materials Libraries). This method entails customizing the composition range within a limited region centered around a predicted composition of interest. To achieve this, co-deposition is employed, resulting in an atomic mixture within the deposited film. This approach is particularly suitable for producing metastable materials, especially when carried out at room temperature. Both the wedge-type and focused compositional gradient strategies guarantee comprehensive coverage of the composition space, ensuring that the compositions corresponding to any predicted phases are not overlooked, provided they are synthesizable. However, achieving synthesis requires variations in the conditions of both ML fabrication and processing, extending the combinatorial approach to encompass combinatorial processing libraries [Citation22].
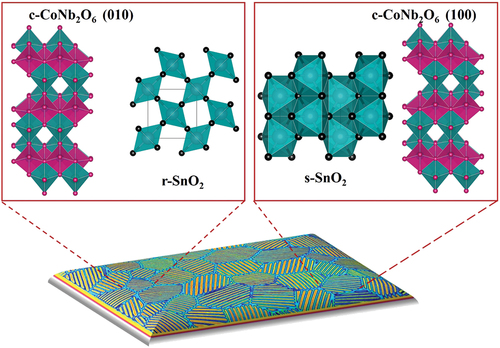
The orientation of the substrate’s crystal structure significantly influences the growth and crystallization of a thin film. When a substrate with a specific orientation structurally resembling the desired complex phase is employed, it can promote the formation of that desired polymorph or metastable phase. This approach enables the successful synthesis of phases that cannot be stabilized as bulk powders, as long as the appropriate growth template is provided for epitaxial stabilization. For example, illustrates the formation of the metastable scrutinyite-structured SnO2 rather than the stable rutile-structured SnO2 during epitaxial growth on polycrystalline CoNb2O6 substrates. The concept of combinatorial substrate epitaxy (CSE) was introduced as a potent method for determining the phase and orientation relationships between a substrate and a deposited film for all potential orientations. Polycrystalline substrates offer a broad spectrum of surface orientations, facilitating the investigation of the influence of orientation on film growth in a single experiment. The substrate’s orientation can be characterized using electron backscatter diffraction (EBSD) both before and after the deposition of a thin film onto the polycrystalline substrate [Citation57].
Figure 5. An illustration of a SnO2 film on a polycrystalline CoNb2O6 substrate, where each grain of the substrate allows the growth of SnO2 with a unique orientation relationship. The film consists of two types of SnO2: stable rutile (r-) SnO2 on some grains, and metastable scrutinyite-structured SnO2 on others.
4. Synthesizing nanoparticles through combinatorial approaches
The production of nanoparticles is a crucial technique for expediting material characterization and advancement. Nanoparticles possess distinctive physical and chemical characteristics owing to their minuscule dimensions, rendering them applicable in a wide array of fields such as catalysis, energy storage, and biomedical imaging. Among the prevailing techniques for crafting nanoparticles, chemical synthesis stands out as a widely employed method. This process entails the reduction of metal salts in the presence of both a reducing agent and a stabilizing agent. This method not only allows for precise control over nanoparticle size and shape but also facilitates the creation of tailored nanoparticles with properties optimized for specific applications [Citation58–60]. Another method for synthesizing nanoparticles is physical synthesis, which involves the use of high-energy sources such as laser ablation [Citation61], plasma sputtering [Citation62–64], and ball milling [Citation65,Citation66]. Physical methods for nanoparticle synthesis provide several advantages, including enhanced control over particle size and composition. These methods also offer the capability to create nanoparticles from a diverse array of materials. There are several reasons for the appeal of physical synthesis techniques in nanoparticle production [Citation67–69].
Precision in Process Control: Physical synthesis techniques offer the advantage of precise control over various process parameters, including energy input, temperature, pressure, and duration. These controllable factors allow for the production of nanoparticles with tailored properties, such as size-dependent optical, electronic, or magnetic characteristics. This customization capability is invaluable for applications like drug delivery, catalysis, and sensors.
Mechanisms for Size Control: Physical synthesis methods directly influence nanoparticle size by adjusting specific parameters. For example, laser ablation can be fine-tuned by modifying laser parameters, while ball milling allows control over milling time, speed, and media, resulting in a well-defined size distribution of nanoparticles.
Versatility in Material Selection: Physical synthesis approaches are versatile and can be applied to a wide range of materials, including metals, ceramics, polymers, and composites. This versatility enables researchers to tailor nanoparticle properties to suit specific application requirements. For instance, refractory metals and alloys can be synthesized using methods like plasma sputtering, expanding the possibilities for various applications.
Minimized Chemical Reactions: Physical synthesis methods typically involve minimal chemical reactions, leading to reduced contamination and impurities in the final nanoparticle product. This results in nanoparticles characterized by higher purity and well-defined composition, which are particularly beneficial for electronics, catalysis, and biomedical applications.
Nonetheless, it is important to acknowledge that physical synthesis approaches may come with certain drawbacks. These can include increased complexity, higher costs in comparison to chemical synthesis methods, and the need for specialized equipment or facilities. The decision to opt for either physical or chemical synthesis methods for nanoparticles hinges on several considerations, including the targeted nanoparticle characteristics, the nature of the materials involved, available resources, and the precise demands of the intended application.
Combinatorial techniques, like matrix-assisted pulsed laser evaporation (MAPLE) and co-sputtering into ionic liquids, offer effective means of producing nanoparticle libraries in a high-throughput and efficient fashion, mirroring the principles employed in the creation of thin-film materials libraries. In the case of MAPLE, the procedure entails depositing a matrix material onto a substrate, followed by the utilization of a pulsed laser to ablate the matrix and generate nanoparticles [Citation68–71]. Control over the composition and characteristics of nanoparticles can be achieved by adjusting both the matrix material and the laser parameters, facilitating the combinatorial synthesis of a diverse array of nanoparticle compositions for high-throughput screening of nanoparticle properties. Additionally, another method called co-sputtering into ionic liquids utilizes sputtering as a physical synthesis technique to deposit nanoparticles onto an ionic liquid substrate [Citation72,Citation73]. A nanoparticle library is created when the ionic liquids are confined in wells across a plate. The composition and attributes of the nanoparticles can be finely controlled by altering the sputtering targets and the ionic liquids, enabling the synthesis of nanoparticles with diverse compositions in a combinatorial manner. However, employing combinatorial methods for nanoparticle synthesis presents a challenge in terms of requiring novel characterization techniques capable of handling the large number of nanoparticles with varying compositions produced in a single experiment. Conventional characterization methods like transmission electron microscopy (TEM) may not be suitable for the high-throughput characterization of combinatorial nanoparticle libraries. Hence, there is a pressing need to develop innovative characterization techniques capable of efficiently and accurately assessing the composition, size, shape, and other properties of nanoparticles in combinatorial libraries. This development would greatly facilitate rapid materials discovery and optimization. From a combinatorial standpoint, high-throughput screening (HTS) techniques can be employed to swiftly generate and analyze vast numbers of nanoparticles with diverse properties. For instance, HTS can be utilized to modify the size, shape, and surface chemistry of nanoparticles by adjusting synthesis conditions such as temperature, pressure, and reactant concentration. Subsequently, these nanoparticles can undergo screening using an array of techniques, including spectroscopic, microscopic, and analytical methods, to identify those exhibiting desired properties such as exceptional stability, biocompatibility, or catalytic activity [Citation72,Citation74–76].
5. High-throughput characterization of materials libraries
High-throughput characterization of multinary systems through the utilization of continuous composition spread thin-film multilayers serves as a potent technique for acquiring extensive and consistent datasets encompassing intrinsic and extrinsic properties. This method involves depositing thin films onto a substrate, incorporating composition gradients that continuously vary, thus enabling the comprehensive determination of material compositions, structures, and properties in a seamless manner. Notably, this approach surpasses conventional single-experiment methods in several ways. Firstly, the resultant datasets facilitate the efficient optimization of identified materials and expedite the discovery of new materials possessing the desired properties. Secondly, this methodology allows for the acquisition of expansive and reliable datasets pertaining to intrinsic and extrinsic properties, a feat unattainable through single-experiment procedures. These datasets contribute significantly to achieving a comprehensive understanding of the materials, encompassing their properties, microstructure, and processing parameters.
Multifunctional existence diagrams stand out as formidable tools for data visualization and decision support in the field of materials science. These diagrams offer a holistic perspective on the interplay among diverse material properties, processing variables, composition, and structure. By delineating regions within the materials domain where coveted properties manifest, they furnish valuable guidance for material enhancement and advancement. In the realm of high-throughput methods for materials exploration, it becomes paramount to prioritize data quality over measurement speed. Top-notch data holds the key to precise and meaningful analyses, ultimately expediting the materials discovery journey while curbing expenses. While this approach may demand time and resources, the ensuing datasets yield invaluable insights into material attributes and behaviors, thereby paving the path for the development of novel materials bearing the desired properties.
High-throughput techniques such as energy-dispersive X-ray analysis (EDX) [Citation77], wavelength-dispersive X-ray analysis (WDX) [Citation78] X-ray photoelectron spectroscopy (XPS) [Citation79], Rutherford backscatter spectroscopy (RBS) [Citation80,Citation81], and nuclear reaction analysis (NRA) [Citation82] can be used to measure the chemical composition of MLs. EDX is a fast method that can determine compositions of elements heavier than nitrogen with good accuracy while automated methods like WDX, XPS, RBS, and NRA can be used for heavier and lighter elements. These methods provide additional compositional information such as oxidation state [Citation79] and depth profiles [Citation81]. Combining techniques like XPS with RBS and NRA can allow determination of all elements in multinary systems, such as thin film Li-ion battery materials [Citation83,Citation84]. High-throughput X-ray diffraction (XRD) can be used to determine the phases present in a ML, with microfocus source and area detectors providing fast measurements with high good spatial resolution [Citation85–87].
shows the schematic principles of high throughput thin film composition and microstructure characterization, including the determination of the composition and microstructure using an integrated micro-beam X-ray fluorescence (XRF) technique and diffraction system (XRD) operated in a two-dimensional automatic scanning mode. An example of band-gap measurement for an oxide thin-film library and semiconductor transport property characterization approach based on ultrafast pulsed UV laser excitation can also be seen in the same library [Citation88–90].
Figure 6. Illustration of high throughput material characterization which shows a schematic of high throughput composition and microstructure characterization using micro-beam X-ray fluorescence and diffraction. High throughput band-gap characterization based on optical transmission and transport property characterization based on ultrafast pulsed laser excitation can also be seen.
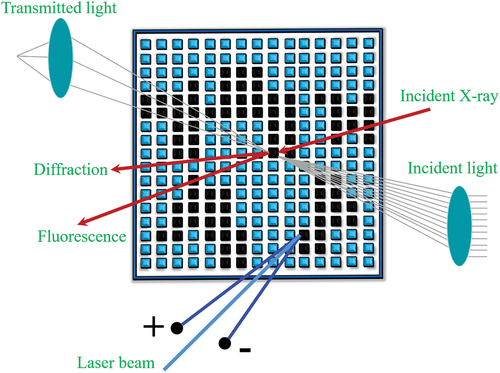
To gain a thorough understanding of materials, it’s imperative to assess a multitude of properties. High-throughput characterization techniques should be tailored to align with the particular objectives of material development and the targeted functional properties. It’s essential to establish appropriate screening parameters or descriptors corresponding to the properties of interest. In cases where a single parameter may prove inadequate, the design of machine learning models should encompass all the necessary parameters that require measurement. As an example, high-throughput electrical resistivity measurements serve as a valuable discovery tool and offer insights into phase zones and their demarcations [Citation91]. Temperature-dependent resistance measurements can also yield signatures for phase transformations, which can be analyzed using XRD for the few most interesting samples [Citation92,Citation93]. When it comes to determining magnetic properties, the magneto-optical Kerr effect is suitable for high-throughput measurements of magnetic hysteresis [Citation94,Citation95]. Basic optical properties can be determined through photography [Citation96,Citation97], while further properties require automated test-stands for optical transmission with a transparent substrate for MLs [Citation98]. Microstructure can be characterized using SEM [Citation99] and AFM [Citation100,Citation101], and mechanical properties can be measured using nanoindentation [Citation102,Citation103], taking into account microstructural variations over a ML that can influence measurement results. To uncover the crystal structure and properties of newly discovered materials, in-depth studies are necessary. One promising method, in addition to synchrotron measurements [Citation104–106], is advanced in situ transmission electron microscopy (TEM) [Citation107–109], such as automated diffraction tomography combined with precession electron diffraction [Citation109,Citation110]. Another approach involves studying the temperature and environment-dependent phase evolution of complex materials using combinatorial processing platforms [Citation10,Citation111]. These platforms are created by depositing multinary thin films on nanoscale Si-tip arrays, forming many identical nanoscale ‘reactor volumes’. This allows for fast diffusion and reaction, and immediate observation of the product phases on an atomic scale using atom-probe tomography (APT) and TEM [Citation111,Citation112]. With this approach, it is possible to rapidly map the phase space of multinary systems with regards to stability against decomposition into phases and reactions with the environment, such as oxidation of alloys [Citation113].
High-throughput techniques, like those employed in materials science, offer significant advantages in terms of efficiency and rapid assessment of material properties. However, it’s essential to recognize the limitations of current technology, such as Atomic Force Microscopy (AFM), Transmission Electron Microscopy (TEM), and radiation-based methods, and understand why they may not always meet unrealistic expectations for true high-throughput applications. These advanced techniques, while capable of providing incredibly high resolution and detailed insights, often come with complexities in terms of instrumentation, resolution versus speed trade-offs, sample size and preparation requirements, data handling and analysis challenges, resource intensity, experimental variability, safety concerns, and occasionally unrealistic expectations. While these techniques are indispensable for in-depth material characterization, achieving true high-throughput capabilities may necessitate the development of new tools and methodologies that can address these constraints effectively and align with the demands for automation and rapid data generation. Researchers should approach high-throughput characterization with a realistic understanding of the current technological limitations.
6. Data-intensive science: a paradigm shift in modern scientific exploration
In recent years, there has been a remarkable surge in the volume of data generated across various scientific disciplines. This data explosion owes much to the advancements in information technology, enabling scientists to collect and process vast datasets. Consequently, experimental, theoretical, and computational sciences are generating unprecedented volumes of data, presenting both a challenge and an opportunity. To harness this data effectively and extract valuable insights, scientists are required to adopt new methodologies, often drawing from the realm of data science. By embracing the principles of data science, scientists can analyze, interpret, and derive knowledge from extensive datasets, thereby advancing their research endeavors. This shift towards a data-driven approach holds the potential to transform the way we conduct scientific investigations, potentially leading to groundbreaking discoveries across diverse fields [Citation114,Citation115].
Traditionally, the scientific approach primarily focused on experimental and theoretical methods, yielding fundamental laws and equations. However, with the increasing complexity of theoretical models, the necessity for simulations emerged. These simulations have yielded copious amounts of data, supplemented by experimental observations obtained through sophisticated instrumentation. As a result, the scientific landscape has undergone a profound transformation. A new paradigm, often referred to as ‘data-intensive science’, or “Big Data’ has emerged, distinct from traditional computational science. () In this paradigm, the primary emphasis is placed on capturing and processing data, with scientists becoming more involved in the analytical stages during a later phase of the research process. This shift underscores the evolving nature of scientific exploration, marked by a heightened reliance on data acquisition, processing, and analysis, ultimately reshaping the scientific landscape [Citation116–118].
Figure 7. The left image illustrates the integrative role of data science, while the right image highlights the five defining characteristics of BigData.
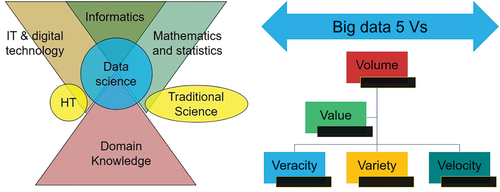
Data Science is a multidisciplinary field that integrates mathematical and statistical techniques, computer science, programming skills, and domain expertise to extract valuable insights and knowledge from data. Its primary objective is to oversee the entire data analysis process, encompassing data collection, data cleaning, thorough analysis, and the derivation of meaningful conclusions. The emergence of Data Science is a response to the challenges posed by the era of Big Data, which refers to massive and intricate datasets that transcend the capabilities of conventional data analysis methods. Data Science also encompasses data mining, an advanced analytical approach aimed at uncovering concealed patterns and insights within datasets. This process is an integral part of a broader framework recognized as Knowledge Discovery in Databases. To excel in the field of Data Science, individuals must possess a diverse skill set. This includes technical proficiencies in data manipulation, programming, and statistical modeling, as well as domain expertise to comprehend the specific context in which the data was generated. Consequently, Data Scientists hail from various backgrounds, including mathematics, statistics, computer science, engineering, and social sciences [Citation116,Citation119,Citation120].
The process of analysis in Data Science presents unique challenges, primarily attributed to the distinctive attributes of Big Data. In stark contrast to conventional datasets, Big Data distinguishes itself by its substantial volume, diverse variety, rapid velocity, and the crucial factor of veracity, signifying data quality and reliability (). These characteristics collectively contribute to the complexity associated with its analysis. Addressing this formidable challenge necessitates the employment of an array of advanced analytical techniques, with a notable emphasis on machine learning (ML) algorithms. Originally conceived in the domains of artificial intelligence (AI) and expert systems, ML algorithms have been adapted for the purpose of autonomously uncovering patterns within data, constructing predictive models, and optimizing outcomes. Their applicability in the context of Big Data is particularly noteworthy, as these algorithms possess the capability to learn from data and refine their performance iteratively. This facilitates the identification of patterns and insights that may elude conventional analytical methods [Citation121–123].
However, the effective implementation of these algorithms demands substantial computational resources. Recent advancements in high-performance computing have made this resource-intensive aspect more manageable. A key advantage of Data Science lies in its expansive repertoire of algorithms and techniques that can be applied to diverse datasets. This versatility empowers Data Scientists to compare the efficacy of various models and explanations, allowing for the selection of the most suitable approach, or even a combination of approaches through ensemble techniques. This departure from the traditional approach, where techniques are chosen based on a priori knowledge of the data and method, is a hallmark of Data Science. The integration of machine learning algorithms into Data Science signifies a shift in the epistemological approach. Rather than testing pre-existing theories against data to align the data with a theory, Data Scientists can derive insights directly from the data. This means that they can uncover previously unknown patterns and relationships, fundamentally altering our understanding of the data. The convergence of extensive and intricate datasets, advanced analytical techniques, and a novel epistemological standpoint represents a paradigm shift in data analysis. The realm of Big Data analytics holds transformative potential, capable of revolutionizing decision-making processes, problem-solving methodologies, and ultimately driving improved business outcomes, heightened efficiency, and innovation across various domains [Citation119,Citation124,Citation125].
6.1. Advances in computational materials science: from DFT to ML
Recent advancements in experimental and computational techniques have led to the generation of extensive and intricate datasets. Machine learning methods have emerged as valuable tools for extracting knowledge and insights from this data by uncovering patterns and correlations. In this section, we outline the fundamental approaches in a logical sequence, commencing with Density Functional Theory (DFT), a widely employed computational method in materials science. Subsequently, we delve into the High-Throughput (HT) approach, which can generate substantial volumes of data automatically through experimental or computational means. Regardless of the data’s origin, the Machine Learning (ML) approach plays a pivotal role in extracting knowledge from identified patterns [Citation123,Citation126,Citation127].
Computational materials science encompasses the utilization of computer simulations and calculations to investigate and comprehend the properties and behaviors of materials at the atomic and molecular levels. Over time, the methods employed in this field have evolved and can be categorized into three generations [Citation128].
First Generation: This generation centers on predicting material properties based on atomic structure. Local optimization algorithms, often reliant on Density Functional Theory (DFT) calculations, are used to forecast the properties of individual atoms and molecules. This approach remains influential, particularly for high-throughput calculations involving extensive material datasets.
Second Generation: The second generation focuses on predicting the crystal structure of a material while maintaining a fixed composition. Achieving this entails global optimization tasks, including genetic and evolutionary algorithms. This approach necessitates systematic execution of a substantial number of calculations and heavily relies on high-throughput methodologies.
Third Generation: The third generation of computational materials science embraces statistical learning. This approach harnesses the wealth of physical and chemical data available to expedite the discovery of novel compositions and the prediction of properties and crystalline structures. Machine learning (ML) algorithms are employed to analyze and learn from large datasets, enabling more precise predictions and discoveries at an accelerated pace.
These generational shifts underscore the dynamic evolution of computational materials science, with each stage representing a significant advancement in our ability to understand and manipulate materials for diverse applications [Citation128–130].
6.1.1. Density functional theory (DFT)
DFT is a prominent computational method in materials science employed to predict material properties based on their electronic structure. Operating on first-principles, it solves the Schrödinger equation to determine the electronic density of a material. DFT calculations offer insights into a material’s electronic structure, bonding, and can predict properties such as bandgap, electronic and magnetic attributes, as well as reaction energetics. Quantum Mechanics in the early 20th century revolutionized our understanding of material properties, with key concepts like Lewis model bonding stemming from Schrödinger equation solutions. However, this equation’s complexity for electron-electron interactions led to a computational shift. In the late 1920s, before computer availability, scientists devised approximations. In 1964, Hohenberg and Kohn introduced Density Functional Theory (DFT). Their theorems stated that the external potential V(r) for N electrons is uniquely tied to electronic density n(r) and that ground state energy E[n] minimizes for the exact density [Citation131,Citation132].
This equation signifies a many-electron system’s ground state energy as a functional of electronic density n(r). Its integration over space captures electron density variations. DFT, vital in condensed matter physics, materials science, and chemistry, offers versatile ab initio calculations. It assesses total energy, potential energy, and energy spectra in diverse systems, analyzes band structures, and determines structural properties. DFT’s accuracy has grown over time, becoming indispensable in materials research, drug development, and various applications [Citation133–135].
Density Functional Theory (DFT) has undergone substantial growth, emerging as a highly precise and predictive approach with notable contributions across various domains. DFT boasts versatile applications, enabling ab initio calculations in diverse systems. It encompasses assessments like total energy, potential energy, and energy spectra in crystalline structures, molecules, and organic complexes. DFT readily analyzes the band structures of metals, semiconductors, and insulators using plane-wave-based DFT equations, offering insights into electron and hole effective masses, band gaps, and optical transitions. Structural attributes, such as stress tensors, bulk modulus, and phonon spectra, are accessible via DFT, aiding material stability evaluations. While dispersion interaction isn’t inherently part of LSDA or GGA methods, many DFT codes now integrate parametrized models, enhancing its capability to describe non-covalent molecular bonding accurately. Overall, DFT’s evolution has rendered it increasingly precise and predictive, establishing it as a valuable tool spanning materials discovery, drug design, solar cell development, water splitting materials, and various other domains [Citation34,Citation136–142].
comprising commonly used DFT software tools for conducting electronic structure calculations in the realm of computational materials science.
Table 1. Compilation of software’s for conducting electronic structure calculations in the field of computational materials science.
6.1.2. The high-throughput (HT) approach
HT is a method for generating large amounts of data in an automated fashion. This can be done through either experimental or computational methods, or a combination of both. In materials science, HT experiments can involve synthesizing and characterizing large numbers of materials in a combinatorial fashion, while HT computational methods can involve running large numbers of simulations with varying input parameters to generate data on materials properties [Citation137,Citation158].
As computational power grows exponentially, the focus in materials science has shifted from lengthy calculations to streamlined simulation setup and analysis. Automation now allows for millions of parallel or sequential simulations, part of the high-throughput (HT) concept. HT entails generating vast electronic and thermodynamic data for real and hypothetical materials, enabling the discovery of desired materials. While HT doesn’t always include machine learning, the two are increasingly integrated. HT works with experimental, theoretical, and computational methods but faces the challenge of time-consuming individual calculations. The HT-DFT approach involves (i) calculations for many materials, (ii) systematic data storage, and (iii) data analysis for novel materials or insights. There’s been notable development of codes for managing simulations and repositories for sharing results, particularly in (i) and (ii). High-performance computers handle the calculations, and data management follows the FAIR concept. Material screening (iii) filters properties from repositories, but often researchers perform their own calculations to update databases. This has led to a surge in materials databases, including AFLOWLIB consortium, Materials Project, OQMD, NOMAD, and others () [Citation137,Citation139,Citation161,Citation169,Citation183–185].
Table 2. A comprehensive toolkit for high-throughput simulation results generation, manipulation, management, and analysis.
Materials screening is a vital step in high-throughput (HT) materials discovery workflows. This process entails filtering materials from a large database based on specific criteria or constraints. Typically performed after extensive data generation, materials screening involves using filters derived from various sources, including machine learning descriptors, theoretical models, or known materials properties. These filters are applied hierarchically to identify materials with desired attributes. Once materials pass through these filters, candidates with exceptional characteristics related to the desired properties are selected for further investigation, offering potential for novel applications or scientific insights. It’s important to note that the initial material selection plays a critical role in the materials screening process, whether it involves selecting materials with desired properties or excluding those with known unsuitable characteristics [Citation158].
6.1.3. Machine learning (ML)
The rapid expansion and achievements of machine learning (ML) have paved the way for its integration into various scientific fields, including material science. The continuous improvement of experimental methodologies has contributed to the accumulation of substantial data in material science, motivating researchers in this field to explore data-driven approaches for addressing scientific challenges. Despite the growing availability of resources for embarking on ML endeavors, there exists a scarcity of comprehensive guidance on navigating the intricate process of establishing a dependable and reliable ML solution. Machine learning (ML) techniques play a pivotal role in extracting knowledge and insights from extensive datasets by identifying correlations and patterns. In the field of materials science, ML serves as a fundamental tool for analyzing the large datasets generated by methods such as Density Functional Theory (DFT) or high-throughput (HT) approaches. These algorithms learn from data patterns, enabling predictions related to the discovery of novel materials with desired properties or the optimization of existing ones [Citation186].
Machine learning (ML) empowers computer programs to enhance their performance through three key elements: performance evaluation, task definition, and experience accumulation. In the context of materials science, the application of machine learning follows a structured framework referred to as Goal, Sample, Algorithm, and Model (GSAM). This framework involves the delineation of the problem, the selection of a relevant subset of data, the choice of an appropriate learning method, and the creation of a knowledge representation. However, before data can be effectively leveraged for machine learning, preprocessing steps are often essential. This preprocessing phase entails data cleansing, which involves identifying and eliminating incomplete, inaccurate, or extraneous information. While high-throughput (HT) methods have become indispensable for generating extensive datasets, the primary challenge lies in distilling valuable knowledge and insights from this abundance of information. Machine learning techniques excel in uncovering relationships and patterns within the data, even in high-dimensional spaces that surpass human reasoning capabilities [Citation187].
The process of classifying materials based on their performance benefits greatly from the use of filters. These filters help systematically categorize materials, identifying those that exhibit the desired behavior at an exceptional level as potential candidates for further exploration. This approach has proven highly effective in uncovering new technological and scientific applications. The fifth ‘V’ of big data, which revolves around extracting value from data, plays a crucial role in the success of this method. Machine learning (ML) has played a pivotal role in this process by providing automated tools for data analysis capable of identifying patterns within the data. In simple terms, ML is a technology that empowers computers to learn and improve their performance on tasks without requiring explicit programming. This technology is rooted in artificial intelligence (AI), a branch of computer science dedicated to creating intelligent machines capable of performing tasks typically associated with human intelligence. AI encompasses various subfields, including machine learning and deep learning. Since its inception in the 1950s, AI has made significant advancements in statistics, computer science, technology, and neuroscience. Today, ML has reached a more advanced level, offering a wide range of algorithms and techniques, including supervised learning, unsupervised learning, semi-supervised learning, and reinforcement learning [Citation188].
Machine learning (ML) algorithms have found diverse applications across various domains, including finance, navigation control, speech processing, computer vision, and bioinformatics, among others. Artificial intelligence (AI) encompasses a range of techniques that enable computers to mimic human intelligence. This emulation can be achieved not only through ML but also by employing less flexible, rule-based strategies like decision trees, if-then rules, knowledge bases, and computer logic. In recent years, deep learning (DL) has gained significant prominence within the ML field due to its remarkable achievements in various domains. DL involves representation learning and is loosely inspired by biological neural networks, featuring multiple layers between input and output layers. Data science is inherently intertwined with machine learning as it provides the essential data required for algorithms to learn. Data science encompasses a wide spectrum of techniques, including data mining, statistical analysis, and machine learning, all aimed at extracting knowledge and insights from complex datasets. By acquiring, processing, and analyzing large datasets, data science empowers organizations to make informed, data-driven decisions that were previously challenging with traditional methods. Therefore, data science is an indispensable component of machine learning, playing a pivotal role in enabling organizations to extract valuable insights from intricate data collections [Citation209]. For a list of libraries and platforms dedicated to materials prediction and machine learning, please refer to .
Table 3. Compilation of libraries and platforms for machine learning and materials prediction.
Creating machine learning (ML) models for materials involves a structured series of essential steps. Initially, data collection is paramount, where a diverse dataset containing various materials and their corresponding properties is collected from multiple sources. This dataset serves as the basis for training the ML model. Next, comes feature extraction, a crucial process for identifying pertinent material characteristics essential for determining material properties. These features typically encompass aspects like the material’s structure, composition, symmetry, and the properties of its constituent elements. The selection of the appropriate ML model is a pivotal step, contingent on the specific problem being addressed, whether it involves regression or classification, and the inherent characteristics of the data. Once the model is chosen, the training phase begins. During training, the selected model is fine-tuned using the extracted features and corresponding material properties from the training dataset. The objective is to optimize the model’s parameters to minimize the difference between predicted and actual properties. Subsequent to training, model validation takes center stage, where an evaluation of the trained model’s performance occurs. This evaluation is conducted on a validation set, which is a subset of the original dataset that was not used during training. Various metrics like accuracy, precision, recall, and F1-score are employed to assess the model’s performance. If the model fails to meet the desired performance criteria, optimization steps are implemented. These might involve adjusting hyperparameters, modifying the model architecture, or incorporating additional data. Once the model is well-tuned and successfully validated, it can be deployed to predict the properties of new materials. This is accomplished by inputting the features of these new materials into the model, which then generates predictions about their properties as output. Finally, the results produced by the model are meticulously analyzed to extract valuable insights into the underlying relationships between material features and properties. These insights are instrumental in guiding the design of new materials customized to possess specific desired properties [Citation209–213].
represents a typical machine learning project. This process begins with data loading and preprocessing, followed by data splitting, feature engineering, the application of various machine learning models, performance assessment of these models, comparison of performance among different models, and finally, visualization of the results [Citation214].
Figure 8. A visual representation depicting the application of machine learning in materials science. Modified with permission from reference [Citation214].
![Figure 8. A visual representation depicting the application of machine learning in materials science. Modified with permission from reference [Citation214].](/cms/asset/a3907606-8cd0-4b65-a146-da0dda4643ee/tstm_a_2292486_f0008_oc.jpg)
It’s crucial to emphasize that the choice of a machine learning algorithm should be tailored to the specific problem and dataset under analysis. Each algorithm comes with its own set of strengths and weaknesses, making it imperative to select the most suitable one to yield optimal results. Probability estimation algorithms like Bayesian networks and support vector machines find common application, particularly in the quest for new materials. These algorithms excel in estimating the likelihood of a material exhibiting specific properties based on existing data. Regression algorithms, such as linear regression and neural networks, are harnessed to predict material properties at both macro and micro levels. They leverage the intricate relationship between input features and output values to make precise predictions. Clustering algorithms like k-means and hierarchical clustering are employed to group materials based on their resemblances and distinctions, facilitating the identification of trends and patterns within extensive datasets. Classification algorithms, including decision trees and random forests, prove invaluable for categorizing materials according to their features. This is instrumental in tasks like identifying materials with particular properties or classifying new materials based on their inherent characteristics. Furthermore, the fusion of machine learning methodologies with intelligent optimization algorithms like genetic algorithms and particle swarm optimization enhances the accuracy and efficiency of material property prediction and optimization endeavors [Citation215].
As a tangible illustration of the practical application of machine learning (ML), the work conducted by D. Xue and colleagues exemplifies the fusion of ML techniques and experimental testing to uncover novel materials with outstanding thermal properties [Citation216]. Their approach commenced with a dataset featuring established materials alongside their thermal characteristics. Utilizing a machine learning model, they made predictions regarding the performance of various material compositions (see ). These predictive insights steered the selection of materials for real-world testing. Following each round of experiments, the outcomes were integrated back into the dataset, progressively refining the accuracy of their predictions. In this iterative process, tailored to address their specific design challenge, they meticulously followed a systematic sequence: (i) They initiated with an initial dataset that encompassed experimental data on diverse alloys, including known thermal dissipation (DT) values and essential material descriptors. This dataset served as the model’s input. (ii) Subsequently, the model underwent training and cross-validation using this initial alloy dataset. (iii) They introduced a separate dataset comprising unexplored alloys, thereby defining an extensive search space teeming with potential candidates. (iv) Leveraging the knowledge imparted by the trained model from step (ii), they applied it to predict DT values for all alloys within this uncharted dataset (iii). (v) The design phase encompassed the discerning selection of the ‘best’ four candidates for subsequent synthesis and characterization. Through this iterative approach, they successfully identified the most optimal combination for thermal behavior among the novel materials they discovered, surpassing the previously established benchmarks. This methodology brilliantly underscores the immense potential of amalgamating artificial intelligence with laboratory experiments, propelling the horizons of materials science and ushering in exciting prospects for material design and discovery [Citation215,Citation217].
Figure 9. Feedback derived from the experiments included the augmentation of the dataset with the addition of four newly discovered alloys. Modified with permission from reference [Citation216].
![Figure 9. Feedback derived from the experiments included the augmentation of the dataset with the addition of four newly discovered alloys. Modified with permission from reference [Citation216].](/cms/asset/be8c2204-69b9-4091-941b-89fa2208ed3c/tstm_a_2292486_f0009_oc.jpg)
6.1.4. Materials informatics
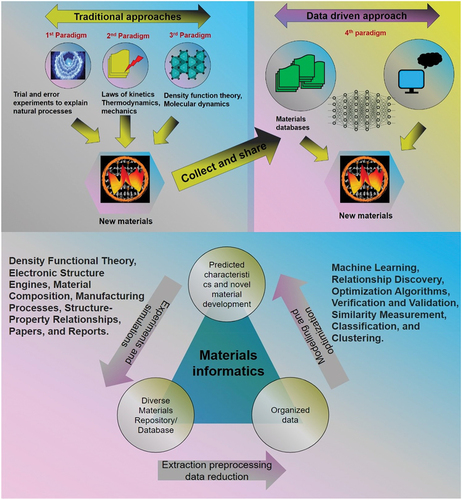
Materials Informatics is a specialized subfield within materials science that employs machine learning and data-driven techniques to uncover relationships between known material attributes and their properties. These attributes typically encompass a material’s structure, composition, symmetry, and the properties of its constituent elements. In Materials Informatics, the primary objective revolves around determining a material’s properties based on its inherent attributes. This process involves training machine learning models using existing datasets of materials and their respective properties. These models are then used to predict the properties of new materials not present in the original dataset. Furthermore, Materials Informatics can address the reverse query of identifying materials with specific desired properties. The overarching goal of Materials Informatics, also referred to as data-driven materials science, is to apply powerful tools from data mining, machine learning, and mathematical optimization to systematically unveil relationships between materials processing, structure, properties, and performance (PSPP) [Citation218,Citation219]. Once these PSPP relationships are unveiled, they can serve as a driving force behind predictive discovery and the design of innovative materials, as well as the optimization of manufacturing processes [Citation220–221]. This shift toward data-driven discovery aligns with the broader trend in modern research known as the fourth paradigm of science [Citation118,Citation122,Citation222]. Coined by Jim Gray in 2007, this term signifies the evolution of scientific methods from empirical science (the first paradigm) to theoretical science (the second paradigm), then computational science (the third paradigm), and now data-driven science (the fourth paradigm) [Citation223,Citation224] (). The rapid rise of materials informatics coincided with the introduction of the US Materials Genome Initiative (MGI) in 2011. This initiative explicitly emphasized the importance of digital data and associated software tools in materials research. Following the launch of MGI, materials informatics has played a pivotal role in facilitating laboratory discoveries related to materials and manufacturing processes. These discoveries have spanned diverse fields, including thermoelectrics and hydrothermal synthesis. Furthermore, there has been a notable surge in research publications featuring the development of various materials informatics-based models to elucidate relationships within the processing-structure-properties-performance (PSPP) framework.
Figure 10. Materials informatics workflow depicting the progression through traditional paradigms to the fourth paradigm shift.
Illustrated in is a generic materials informatics workflow. This analysis commences with the extraction and preprocessing of data, a phase where the essential elements of the dataset are identified and selected. The refined dataset is then subject to further scrutiny to unveil relationships among the components of interest. These discovered relationships serve as the foundation for creating what are known as inverse and forward models. The former enables the design of materials with specific desired properties, while the latter supports predictive analytics. To complete the cycle, experiments and computer simulations, guided by theoretical models, are employed to generate fresh data for the materials databases. This process effectively closes the loop, ensuring that the materials informatics workflow remains dynamic and continuously evolves.
In materials informatics, ML models serve as approximate functions that take inputs (materials features) and generate outputs (material properties). These models can be considered as phenomenological or empirical, as they offer heuristic functions that describe available data. The primary objective of machine learning in this context is to unveil feature-property relationships that might elude human researchers. It’s important to distinguish these machine learning models from theoretical models used in traditional materials science, which aim to comprehend the fundamental physics underlying material properties. ML models in materials informatics are not focused on understanding physics intricacies but rather on predicting properties based on material features. Nonetheless, the insights gleaned from materials informatics can eventually contribute to the development of theories and a deeper understanding of the fundamental physics governing material properties [Citation225].
The materials informatics workflow closely mirrors the general machine learning workflow, albeit with some specialized components. This workflow includes problem definition, data collection, representation of materials, and the selection, evaluation, and optimization of machine learning algorithms. Problem definition entails specifying the desired machine learning outcome and identifying the necessary input variables. Adequate data collection is crucial to comprehensively address the problem. The representation of materials is a critical step, as it significantly influences the performance of the machine learning algorithm. Machine learning algorithms must be chosen and assessed based on factors such as accuracy, training time, and model complexity or interpretability [Citation226–228].
Expanding on our previous discussion from materials perspective, illustrates the intricate interplay between materials informatics and the machine learning workflow employed for the prediction of mechanical deformation properties. This diagram delineates the various stages integral to this process. Starting from the left-hand side of the illustration, we observe the establishment of databases stemming from microscopy and (nano)mechanics experiments. These databases serve as invaluable repositories of data pertaining to the mechanical behavior of materials when subjected to diverse conditions, encompassing factors such as stress, strain, and temperature. In the subsequent stage, multiscale modeling simulations assume a pivotal role, providing essential descriptors and predictors that elucidate the underlying physical mechanisms governing the behavior of these materials [Citation229].
Figure 11. Materials informatics and the workflow involving machine learning for mechanical deformation strategy. Adopted with permission [Citation229].
![Figure 11. Materials informatics and the workflow involving machine learning for mechanical deformation strategy. Adopted with permission [Citation229].](/cms/asset/bdf6f7c1-79bd-47f5-91b2-971d2c6161c7/tstm_a_2292486_f0011_oc.jpg)
These descriptors encapsulate information about the material’s microstructure, crystal arrangement, defects, and other characteristics that exert influence on its mechanical properties. Transitioning to the center of the figure, these generated descriptors and predictors become indispensable for the training and validation of machine learning algorithms. These algorithms harness the extensive data derived from both the databases and simulations to formulate predictions concerning the mechanical properties of materials. These predictions offer crucial insights into various facets of material deformation, including critical parameters like strength, ductility, and toughness. It’s important to note that these predictions are not merely abstract outcomes; they find practical applications in structural characterization, materials design, and discovery. By elucidating how materials respond under diverse conditions, this approach equips materials scientists and engineers with a rapid and effective means to evaluate potential materials, identifying those with promise for specific applications. In essence, outlines a streamlined pathway that expedites the process of materials screening and the identification of optimal candidates tailored to meet specific mechanical requirements [Citation229].
provides an illustration of the implementation of a machine learning (ML) approach, highlighting two pivotal steps: the detection of patterns within input experimental or simulation data and subsequent predictions. These steps are of utmost importance in gaining a comprehensive understanding of the fundamental properties of materials. To effectively detect patterns in input data, a substantial volume of data is essential, often sourced from diverse origins. These sources encompass data generated through various means, including high-throughput simulations (such as ab initio, atomistic, and continuum-based simulations), extensive experimentation (such as mechanical testing and microscopy), and pre-existing materials databases. These repositories of data yield valuable insights into the properties and behaviors of materials under various conditions. In the quest to uncover structure-property relationships, a pertinent material dataset is defined by a feature space that comprises a set of atomic and/or local structural descriptors. These descriptors play a pivotal role in capturing essential physical mechanisms and microstructural intricacies relevant to a specific group of materials. This wealth of features serves to characterize the materials and unveil latent patterns within the data, potentially revealing underlying connections between these features and material properties. With these identified patterns at hand, machine learning algorithms come to the forefront, facilitating predictions related to uncharted materials. The ultimate goal is to harness these discerned patterns for the precise prediction of properties associated with new or unexplored materials. This methodology empowers materials scientists by streamlining the screening process for a wide range of potential materials, significantly reducing the time and resources required for the development of novel materials tailored to specific properties.
When deciding whether to apply machine learning (ML) to a research problem, it’s paramount to assess the availability of sufficient, consistent, validated, and representative data related to the specific behavior of interest. It’s worth noting that data generation is often better suited to traditional or high-throughput methods, particularly in the initial stages of research. Additionally, ML techniques excel at navigating high-dimensional spaces to uncover patterns in data, and they have the capacity to explicitly encode these discovered patterns, creating computational models that can be manipulated. Therefore, ML methods are most valuable when tackling problems that prove challenging for traditional approaches, where human intuition may not suffice to develop a physical model. In general, ML methods can offer significant utility in problems that fall within one of the following categories, arranged in increasing order of added value and complexity:
Replacing the collection of challenging, intricate, or expensive properties or data.
Generalizing a pattern observed in a dataset to a similar class of data.
Revealing relationships between correlated variables, particularly when the links are unknown or indirect, surpassing the scope of intuition or domain knowledge.
Establishing a comprehensive approximate model for a complex, poorly understood property or phenomenon that lacks fundamental theories or equations.
Historically, ML methods have proven successful in various domains, including automation, image and language processing, social sciences, chemistry, biology, and many more. Moreover, recent times have witnessed the emergence of numerous new applications of ML in diverse fields. To recap, data-driven approaches in materials science prove exceptionally valuable in scenarios where fundamental theories or equations are absent, or where traditional methods encounter complexity, difficulty, or high costs. These applications encompass tasks like creating models for enigmatic phenomena, substituting resource-intensive calculations with streamlined ML models, and implementing feature selection techniques to uncover approximate models and descriptors. Among the prevalent challenges in materials science, where machine learning has demonstrated success, are instances where it replaces resource-intensive Density Functional Theory (DFT) calculations with more efficient models, such as atomistic potentials for Molecular Dynamics (MD) simulations, or the prediction of various material properties.
Current and forthcoming challenges in the field of materials informatics encompass several critical aspects. Firstly, the issue of data heterogeneity and siloing hampers systematic data mining due to the diverse nature of materials datasets originating from various sources and stored in multiple formats across numerous isolated repositories, complicating seamless access and analysis. Secondly, the absence of consistent metadata poses challenges in assessing data quality, exacerbated by uncertainties and errors associated with materials data generation. Thirdly, the development of inverse materials Property-Structure-Processing-Performance (PSPP) models presents a more complex problem than forward models, yet it holds greater relevance for materials discovery by determining optimal material design parameters to achieve desired properties and performance. Fourthly, the representation of materials concepts in computational forms, such as networks, is crucial for materials informatics, potentially revealing relationships between materials based on various criteria. Lastly, scientific and technological advancements are needed to explicitly integrate experimental data, computational data, and materials theory for effective multiscale modeling and to establish similarity metrics for materials, facilitating systematic assessment of material similarities and enabling network analysis techniques. Addressing these challenges is essential for the progress of materials informatics and its potential to revolutionize materials science [Citation227,Citation230].
6.2. Applications in materials science
6.2.1. Applications of DFT
DFT finds extensive applications across various material systems, encompassing atomic, molecular, and solid-state structures, including surfaces, defects, and low-dimensional systems. It serves as a versatile tool for probing a multitude of material properties, including structural, electronic, thermal, optical, catalytic, magnetic, and topological aspects [Citation231–233]. Structurally, DFT aids in determining geometries, bond characteristics, and lattice parameters while predicting material stability and phase transitions. It accurately elucidates electronic properties like band structures, density of states, and charge carrier mobility [Citation234–237]. Additionally, DFT provides insights into thermodynamic characteristics such as entropy and heat capacity, facilitating stability assessments. Moreover, DFT explores vital aspects like electron-phonon coupling and optical behavior, including absorption spectra. It delves into magnetic properties, topological features, and catalytic activity, thereby offering indispensable insights into material design and a wide range of applications [Citation238–241].
6.2.2. Applications of HT
High-throughput methods, such as density functional theory (DFT) calculations, find extensive applications in materials science, spanning properties like structural, electronic, thermal, optical, catalytic, magnetic, and topological. While these methods are invaluable for material screening, some corrections may be needed to enhance accuracy. In the realm of high entropy alloys (HEAs), predicting phase transitions poses challenges due to complex compositions. The LTVC model addresses this by amalgamating HT DFT calculations with cluster expansion and statistical analysis, improving predictive capabilities [Citation242–244]. HT calculations also play a crucial role in identifying materials for energy recovery in thermoelectrics, enabling ZT calculations across numerous materials via advanced interpolation schemes. These methods are similarly useful in identifying optoelectronic materials, solar absorbers, and characterizing elastic properties in inorganic materials [Citation12,Citation39,Citation139,Citation139,Citation184,Citation245]. Furthermore, HT calculations hold potential in the discovery of topological insulators (TIs) with applications in spintronics and quantum computing, aided by descriptors for TI selection [Citation246–249]. Within the 2D materials domain, databases like the Materials Project and 2D Materials Database compile data, streamlining material screening, and expediting the discovery of materials with distinctive properties, thus propelling materials research [Citation22,Citation235,Citation250–253].
6.2.3. Applications of ML
This section provides an overview of the extensive applications of machine learning (ML) techniques in the field of materials science, highlighting the adaptability of materials informatics (MI). These applications in materials science can be broadly divided into two primary categories: predicting material properties and discovering new materials. Property prediction, whether at the macroscopic or microscopic level, often involves regression analysis. In contrast, the discovery of novel materials leverages ML to screen various combinations of structures and components, guided by probabilistic models, with subsequent validation through density functional theory (DFT) analysis. Beyond these functions, ML proves beneficial for process optimization and the approximation of density functional theory (DFT) calculations, resulting in computational cost reduction without compromising accuracy [Citation232,Citation254–256].
Understanding material properties, whether on macroscopic or microscopic scales, frequently necessitates intricate computational simulations or experimental measurements. However, constructing precise simulations capable of capturing complex material-property relationships can be daunting, often involving undiscovered interactions. Moreover, experimental measurements typically occur late in the materials selection process, leading to inefficiencies in time and costs when results are unsatisfactory. In certain scenarios, studying material properties remains exceptionally challenging or nearly impossible despite extensive computational or experimental efforts. Consequently, there is a growing demand for intelligent, efficient, and cost-effective ML prediction models capable of extracting knowledge from existing empirical data, identifying nonlinear relationships between material properties and factors, and delivering accurate predictions with minimal computational resources. In the realm of discovering new materials, the ML process consists of two crucial components: a learning system and a prediction system. The learning system encompasses data preprocessing, relevant feature selection, feature extraction, and the training and evaluation of ML models. Following this, the prediction system employs these trained models to suggest novel material compositions and structures with improved properties. This suggestion-and-validation approach significantly expedites the discovery of novel materials, reducing the time and resources needed for testing and development [Citation257–259].
Various ML techniques, including supervised, unsupervised, and reinforcement learning, are deployed for these tasks, often with a focus on crystal structure prediction and composition-based strategies. One significant challenge in ML for materials research is feature selection, which involves identifying the most critical variables in a dataset relevant to the property of interest. Feature selection can be particularly intricate, especially when dealing with extensive datasets. To tackle this challenge, researchers have adopted diverse methods such as sure independence screening and sparsifying operator (SISSO), as well as the least absolute shrinkage and selection operator (LASSO). Additionally, innovative techniques like the Δ-approach to ML, subgroup discovery, and multi-fidelity learning aim to enhance the accuracy and efficiency of ML models tailored for materials science research. ML algorithms have proven invaluable in predicting a wide array of material properties, including the conductivity of thermoelectric materials and the occurrence of superconductivity. When coupled with appropriate descriptors, these algorithms excel at accurately predicting material properties, thus diminishing the need for costly and time-consuming experimental tests. Furthermore, ML has played a pivotal role in the discovery of novel magnetic materials, offering promising advancements with applications in science and technology. Additionally, ML has been instrumental in predicting critical properties of superconductors, providing insights into novel materials and their potential applications [Citation260,Citation261]. ML-driven predictions of superconductors’ critical temperatures (TC) and the discovery of new non-cuprate and non-iron-based oxide superconductors hold the promise of revolutionizing various technological sectors [Citation260,Citation261]. These materials find applications in medical imaging, particle accelerators, power transmission, and beyond, potentially leading to even more innovative and efficient technologies [Citation262,Citation263]. Furthermore, ML has been applied to explore topological phases in materials, a field that necessitates calculating topological invariants related to the Berry curvature of occupied states [Citation264–267]. ML algorithms have been effectively employed to classify topological phase transitions and predict topological materials, capitalizing on extensive datasets within this domain. ML approaches offer indispensable tools for investigating and identifying materials with unique properties and diverse potential applications [Citation268–270].
7. Open-source libraries
To extract meaningful insights from the extensive and intricate datasets generated in materials science research, it is imperative to possess accessible analytical tools capable of interpreting and comprehending the data. One illustrative instance of this capability is the creation of phase diagrams, which serves as a means to evaluate material stability through energy assessments derived from fundamental electronic structure calculations. The scrutiny of these phase diagrams enables the identification of materials with enduring stability and provides valuable insights into their performance across various conditions [Citation52,Citation271]. Analysis tools play a pivotal role in the design of functional electronic materials, including thermoelectrics and transparent conducting oxides. A prime example is the examination of materials’ computed band structures, which yields crucial insights into properties like band gaps, the character of optical transitions (be it indirect or direct), and the effective masses of charge carriers. This process ensures the creation of original content without plagiarism. Numerous open-source tools are readily available for conducting calculations and analyzing datasets in materials science. A notable example is the Python Materials Genomics library [Citation52], offering a collection of core Python elements for representing materials data. Another invaluable open-source resource is the Materials Genome Project [Citation2], a freely accessible database of materials properties accessible to researchers globally. Supported by government funding, the Material Genome Initiative (MGI) strives to expedite the development and commercialization of novel materials for emerging technologies. Recent advancements in resources and algorithms have empowered the computational materials community to significantly contribute to materials discovery and development. The MGI primarily funds research and development endeavors aimed at accelerating materials discovery, synthesis, and characterization, ultimately reducing the time and costs associated with bringing new materials-based technologies to market. Additionally, the Materials Project provides comprehensive data on the electronic, structural, and thermodynamic properties of a broad array of materials, serving as a valuable asset for materials design and discovery. In this context, Pymatgen, another open-source tool, stands as a widely adopted Python library that equips materials scientists with a diverse array of tools for conducting calculations on materials datasets. These tools encompass structure analysis, electronic structure analysis, and phase diagram construction. The Pymatgen community boasts over 100 global collaborators dedicated to enhancing and expanding its functionality continuously [Citation33,Citation139,Citation272].
presents an illustrative workflow for the computational design of materials, delineating the sequential stages within the process of discovering novel materials. The workflow commences with the inception of an initial concept, which subsequently evolves into a set of design criteria predicated on the intended material properties and functionalities. These design criteria then serve as the basis for a computational screening process encompassing a vast array of compounds, with the aim of pinpointing prospective candidates warranting in-depth scrutiny. Following the identification of a subset of potential materials, further comprehensive analyses are conducted utilizing a spectrum of computational tools, including electronic structure calculations and the construction of phase diagrams. This analytical phase facilitates the assessment of material stability, electronic characteristics, and other pivotal attributes, ultimately discerning the most promising candidates meriting subsequent intensive investigation.
Figure 12. Illustration of the computational material design process, which involves investigating a large number of compounds to identify stable materials.
The scenario depicted in underscores the intricate challenge of striking a balance between the desired properties of a material and its commercial feasibility. In this context, the scientist initiates the quest by formulating strategies for identifying stable materials, leveraging a repertoire of software tools, prior knowledge, intuition, and databases housing materials properties and structures to pinpoint promising compounds aligning with the desired property criteria. Nevertheless, even if a compound aligns with the sought-after property criteria, its commercial viability may be in question if it proves too costly to produce or utilize. Factors like raw material expenses, manufacturing processes, and market demand can collectively influence the economic viability of a material. In the scenario outlined, the scientist grapples with this dilemma as a potential compound emerges as a front-runner but is ultimately deemed prohibitively expensive due to the presence of elements like ruthenium, known for its rarity and high cost. This situation prompts the scientist to explore alternative materials that fulfill the desired property criteria without incorporating exorbitant elements. Potential strategies could encompass substitution approaches, such as substituting ruthenium with a more cost-effective transition metal, or delving into entirely novel material categories that obviate the reliance on rare or expensive constituents. Overall, this scenario underscores how computational material design serves as an accelerant in the quest for new materials, endowing researchers with a methodical and efficient framework for screening extensive compound libraries and singling out promising contenders for deeper investigation. It also highlights the importance of validating computational predictions through suitable experimental techniques like thin film combinatorial synthesis, which enables cost-effective and streamlined screening processes with minimal material wastage. By adopting this approach, researchers can expedite the exploration of diverse material compositions and combinations, hastening the discovery of new materials exhibiting desired properties, all while mitigating resource consumption. Furthermore, the integration of high-throughput techniques in thin film combinatorial materials synthesis allows for the rapid analysis of extensive datasets, unveiling correlations between material properties and composition, thereby guiding the development of new materials tailored to specific requirements [Citation10,Citation273].
8. Collaboration between computational methods and experimental data
The synergy between experimental and computational materials science teams is gaining paramount significance in expediting the exploration of novel material phases. Computational materials science has witnessed substantial progress in recent times, enabling high-throughput computations and simulations for potential material systems. This progress has notably expanded the scope and efficiency of materials discovery [Citation10,Citation274]. Nevertheless, for computational materials science to further enhance its accuracy and applicability, the availability of extensive databases containing high-throughput experimental data is imperative. At present, there are databases primarily focused on calculated materials data, which serve as valuable resources for researchers engaged in computational materials science. One prominent example of such a database is the Materials Project, an open-access repository of computed materials properties designed to expedite materials discovery and design. This repository encompasses data on more than 100,000 materials, encompassing a wide range of information, including crystal structures, electronic properties, thermodynamic characteristics, and more. These details are derived from various computational methodologies. The Materials Project empowers researchers to investigate diverse materials, forecast the emergence of new materials featuring specific properties, and even simulate material performance within real-world applications. This integration of computational and experimental data sources stands as a pivotal step in advancing the capabilities and accuracy of computational materials science [Citation52]. Furthermore, the Open Quantum Materials Database (OQMD) represents another notable instance of a calculated materials data repository. The OQMD has been meticulously crafted to archive the outcomes of extensive high-throughput assessments of the electronic structure of various materials. Within its extensive dataset, the OQMD offers comprehensive information regarding thermodynamic characteristics, crystal configurations, electronic properties, and magnetic attributes for an impressive compilation of over 400,000 materials. The OQMD database is thoughtfully accessible via a user-friendly web interface, with an additional application programming interface (API) available for those who wish to programmatically harness its wealth of data. Importantly, the OQMD database is freely accessible to researchers, serving as a valuable and easily accessible resource for the computational materials science community. This kind of data repository is instrumental in advancing the capabilities of computational materials science by providing a wealth of structured information for analysis and modeling [Citation168,Citation275]. These databases contain information on the physical and chemical properties of materials, which allows to select potential materials systems for further investigation [Citation52]. The potential advantages of establishing a joint database or harmonizing existing databases for both experimental and computational materials science are undeniably substantial. Such a collaborative effort can bridge the gap between the two domains, leading to mutual benefits. Experimental data can serve as a vital means to validate and enhance the accuracy of computational models, while computed data can guide experimental investigations by pinpointing materials systems worthy of exploration. This synergy between experimental and computational methodologies holds immense promise for expediting the discovery of novel materials boasting desired properties. A compelling example of this synergy lies in the realm of catalyst development. The amalgamation of experimental and computational data within a shared catalyst database has the potential to revolutionize the field. It not only aids researchers in identifying promising new materials for catalytic applications but also offers a robust platform for rigorously verifying the precision of computational models in predicting material properties. Consequently, this collaborative effort can streamline the discovery of more efficient catalysts, opening doors to transformative industrial applications such as clean energy production and greenhouse gas emissions reduction. In summary, the harmonization of experimental and computational data through shared databases represents a pivotal step toward unlocking innovative materials with groundbreaking properties and application [Citation276]. Materials informatics is an exponentially expanding field with the capacity to bridge the divide between experimental and computational materials science cohorts. Employing machine learning algorithms to scrutinize extensive datasets comprising both experimental and calculated materials information, materials informatics emerges as a potent tool for unearthing fresh materials systems endowed with coveted properties. Furthermore, it empowers predictions regarding how materials will respond in diverse environmental conditions, providing a holistic perspective on material behavior. This convergence of data-driven insights from experimentation and computation holds the promise of expediting the discovery and optimization of materials with groundbreaking attributes [Citation227]. Effectively handling digital research data can pose considerable challenges for high-throughput experimentation teams. To surmount these hurdles, the establishment of a structured database housing experimental datasets obtained through uniform protocols emerges as a valuable solution. This approach guarantees the comparability of all materials within a dataset while upholding stringent standards for data quality and consistency, primarily through continuous composition gradients. Consequently, researchers gain streamlined access to high-caliber experimental data, which, in turn, can substantially enhance the precision and dependability of their computational models, ultimately facilitating more robust and reliable scientific outcomes [Citation34,Citation46].
Machine learning (ML) in materials science can greatly benefit from the inclusion of reference materials, which can contribute to the generation of high-quality datasets. It’s crucial to incorporate all research outcomes, including negative findings, to maximize the utility of the data for machine learning purposes. Nevertheless, challenges pertaining to data organization, quality assurance, and data accessibility persist, underscoring the need for consensus on metadata standards and data formats to facilitate seamless data exchange between experimental and computational research groups. The semi-automation of data input and retrieval from databases is recommended to streamline the combinatorial research workflow, thereby enhancing the management and analysis of extensive materials datasets. To bolster the reliability of data within a collaborative database, it is advisable to produce and measure nominally identical materials libraries of newly discovered systems in diverse laboratories using different methodologies to validate their properties. In the exploration of multinary materials systems, the sheer volume of data generated by computational materials science (CMS) exceeds the capacity for manual management, necessitating the development and deployment of advanced computational tools for data processing and interpretation. Effectively visualizing compositions and properties within intricate multinary materials systems presents a formidable yet essential challenge for comprehensive data analysis. As a result, there is a demand for the creation of novel algorithms and the implementation of corresponding software to construct functional phase diagrams or existence diagrams, encompassing metastable phases, for multinary alloys. These endeavors are poised to lay the foundation for future materials design and innovation.
Significant advancements in software development have greatly accelerated phase analysis through high-throughput X-ray diffraction (XRD) by incorporating machine learning techniques. Notably, POLYSNAP is a cluster analysis software designed for the efficient sorting of X-ray spectra acquired from thin film composition spreads. Its primary purpose is to rapidly unveil the distribution of phases within ternary diagrams. Cluster analysis, a method that involves categorizing data points into clusters based on their similarity, is harnessed by POLYSNAP to group X-ray spectra according to their resemblance. This enables the identification of various structural phases present in composition spreads. The utilization of POLYSNAP offers several advantages, including its capability to handle a substantial volume of data points concurrently, enabling the analysis of numerous spectra from composition spreads within a relatively short timeframe. Additionally, POLYSNAP boasts a user-friendly interface that simplifies the visualization and interpretation of analysis outcomes [Citation87]. An additional noteworthy example is the creation of the AgileFD artificial intelligence algorithm, specifically developed to tackle the complexities of interpreting X-ray diffraction data. This algorithm excels in swiftly mapping phases within a combinatorial library of diffraction patterns, enhancing the identification of component phases, and mapping their concentration and lattice parameter variations based on composition. AgileFD leverages Gibbs’ phase rule to generate physically meaningful phase maps autonomously, with the added flexibility of refining solutions with expert input when necessary. Moreover, as an open-source algorithm, AgileFD is adaptable for integration into a wide array of high-throughput workflows, significantly expediting the process of materials discovery [Citation277]. Rather than manually analyzing hundreds of diffractograms, non-negative matrix factorization algorithms can obtain sets of tens of similar patterns to initiate further in-depth analysis [Citation86]. In addition, concepts have been introduced for using machine learning to guide data-driven experimentation, which can be adapted and further developed to improve high-throughput experimentation [Citation86]. Traditionally, measurements across all areas within a material library are conducted sequentially, resulting in a substantial volume of data. To address this issue, an innovative approach involves the initial use of fast, high-throughput measurements to identify areas of particular interest. Subsequently, more time-consuming methods can be selectively applied to these identified regions. This strategy aligns with the screening and confirmatory approach commonly employed in various scientific disciplines. An alternative technique entails the integration of design of experiment principles with machine learning to minimize the necessary number of measuring areas. However, there is potential for further optimization, reducing the number of required measuring areas while still achieving comparable results. As an illustration, instead of measuring the entire area within a region of interest, it might prove more efficient to focus solely on the boundaries delineating these regions [Citation278,Citation279]. A potential approach involves initiating the analysis by examining a set of random measuring areas and subsequently measuring areas that are farthest from this initial dataset. This method effectively extends exploration within the search space within the same timeframe. Furthermore, rather than opting for arbitrary measuring areas, a more efficient strategy might involve selecting areas based on prior knowledge or scientific insight. In combinatorial studies, one example of this tailored approach is directing attention to specific regions within a material phase diagram that are anticipated to exhibit noteworthy properties or characteristics. For instance, when the objective is to identify novel materials with enhanced catalytic capabilities, researchers can opt to concentrate on measuring areas with a high probability of containing materials possessing the desired composition and crystalline structure for catalysis. This targeted selection can streamline the measuring process while simultaneously increasing the likelihood of discovering valuable materials [Citation55,Citation280,Citation281]. Finally, predictions of synthesizability could be highly useful in further reducing the number of necessary experiments.
The use of computational methods such as quantum mechanics, molecular dynamics, and machine learning can provide valuable insights into the properties and behavior of materials [Citation34]. While first-principles calculations stand as a potent instrument for materials design and discovery, they come with a computational cost and time-consuming nature. Despite enhancements in the reliability and accessibility of first-principles software, the automation and management of numerous calculations remain a formidable challenge. This is primarily due to the resource-intensive nature of first-principles calculations, often demanding several hundred central processing unit (CPU) hours merely to extract fundamental properties for a single material [Citation282,Citation283]. Furthermore, these calculations frequently involve sequential steps, where the optimization of a crystal structure must precede property calculations. This sequential nature can lead to time inefficiencies and contribute to a deceleration of the overall workflow. Additionally, first-principles calculations often necessitate human intervention to attain a converged and dependable outcome [Citation283,Citation284]. This sensitivity to input parameters often necessitates manual adjustments to ensure accuracy and consistency in the results. High-throughput computation can pose challenges related to data accuracy due to several factors. One prevalent factor is the utilization of approximations or simplifications within computational models, which can introduce errors, especially in complex systems with numerous variables. Furthermore, data accuracy can be affected by the quality of input data and the calibration of computational models [Citation285–287].
Despite encountering these obstacles, the advantages offered by first-principles calculations in materials design and discovery are substantial. These calculations offer in-depth understanding of materials’ electronic, structural, and thermodynamic characteristics, equipping researchers with the knowledge needed to make well-founded choices regarding materials worth pursuing for further exploration. Furthermore, with the continual advancements in computational hardware and software, the utilization of first-principles calculations in materials science is expected to experience ongoing growth and broadening horizons in the years ahead.
8.1. Elevating battery technology: combinatorial synthesis, high-throughput techniques, and machine learning – a case study
Advanced battery technology has become a crucial catalyst in the pursuit of a mobile, environmentally friendly, and sustainable society in the coming decades. Lithium batteries, celebrated for their attributes such as high voltage, impressive specific energy density, swift recharging capabilities, and wide operational temperature range, have found extensive applications in an array of devices. These devices include portable electronic gadgets, electric vehicles, and energy storage solutions for harnessing renewable energy sources like wind and solar power. The evolution of battery technology is intrinsically intertwined with the innovation of new materials. For instance, lithium-rich layered oxide materials have garnered significant attention due to their suitability as positive electrodes in high-energy-density lithium-ion batteries. Additionally, nano silicon-based anodes have emerged as promising alternatives, displaying reversible capacities ranging from 380 to 2000 milliampere-hours per gram (mAh g−1). It is indisputable that the exploration of advanced materials and the deliberate practice of rational design are central to the field of battery research [Citation288–290].
To expedite the progress of chemical materials in lithium batteries, a range of high-throughput techniques has been deployed. These encompass high-throughput simulations, synthesis methods, and precise measurements, all aimed at exploring novel battery materials. In parallel, the integration of data mining and machine learning has emerged as a powerful tool for extracting valuable insights from the wealth of information generated by high-throughput techniques. These technologies open up new avenues for in-depth exploration of the intricate relationships between the structure and properties of battery materials, often leading to the discovery of materials with enhanced characteristics. Additionally, the comparison of theoretical predictions or model outcomes with extensive experimental data serves to pinpoint sources of error and uncertainty in battery research. This critical analysis, in turn, facilitates the refinement of theoretical models and the optimization of research equipment, thereby advancing our comprehension of battery technology. The collaborative advancement of these elements, as illustrated in , holds the promise of accelerating the discovery of potential compounds in the future. This synergistic approach is poised to reduce both the time and financial investments involved in research endeavors, benefiting not only lithium batteries but also emerging energy storage technologies such as sodium (Na), zinc (Zn), magnesium (Mg), and aluminum (Al) batteries, among others [Citation291].
Figure 13. The collaborative approach of combining combinatorial synthesis, high-throughput calculations, and data sciences in the development of new materials for lithium batteries.
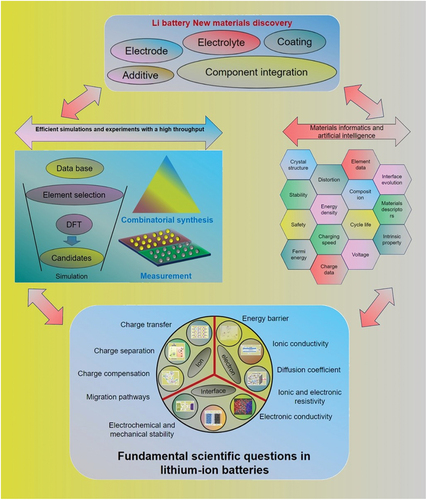
8.1.1. Innovations and challenges
The establishment of a high-throughput calculation framework has its foundations in density functional theory simulations. Furthermore, an inventive strategy involving the amalgamation of calculation techniques at varying accuracy levels has been proposed to expedite the exploration of novel materials. The former approach has been successfully applied to survey the inorganic crystal structure database, with the aim of identifying potential electrode materials characterized by high voltage and capacitance properties. Taking inspiration from the latter approach, a groundbreaking superionic conductor has been conceptualized. As the pursuit of battery systems with improved energy density and safety gains momentum, there is a growing expectation surrounding the adoption of inorganic solid electrolytes to replace liquid electrolytes in the next generation of lithium batteries. The utilization of solid electrolytes holds the potential to mitigate challenges such as leakage, vaporization, decomposition, and undesired side reactions commonly observed in conventional lithium-ion batteries. Nonetheless, the quest for solid electrolytes demonstrating exceptional performance remains a formidable endeavor, primarily due to the intricate relationship between material structures and ionic conductivity. In a holistic battery system, performance is not solely dependent on the attributes of individual components; it is also significantly shaped by the intricate interactions among these components. A prime example of this is the interface between the electrode and electrolyte, which exerts a profound influence on the battery’s stability, charging/discharging rate, and longevity. Consequently, the quest for harmonious combinations of battery components assumes paramount significance. Expanding our comprehension of the fundamental scientific challenges inherent in battery systems remains the primary research focus on the journey to uncover innovative lithium battery materials [Citation291,Citation292].
To address the challenges outlined above, it is essential to make progress in both scientific understanding and technological development. provides an overview of the goals in battery technology and the necessary methodologies for the near future. Firstly, one approach involves the creation of intricate prototypes to uncover the fundamental scientific phenomena in batteries through high-throughput experiments and simulations. Employing advanced measurement and analytical tools allows for a deeper exploration of complex microstructures and evolutionary processes. This approach aids in gaining insights into the failure mechanisms in lithium batteries and directs the quest for innovative battery materials. Secondly, it is crucial to develop an automated screening and prediction workflow that offers the required precision and efficiency. Each component of the battery, including the electrode, electrolyte, additive, and collector, must meet multiple criteria to ensure outstanding overall device performance. For example, a high-voltage cathode should demonstrate both high capacitance and exceptional conductivity. Similarly, electrolyte materials must possess swift ionic conductivity and a broad electrochemical window. Therefore, tools for screening and predicting materials based on multiple objectives must be devised. Moreover, substantial progress is needed in the field of data science and technology to support material design. Machine learning techniques and big data methods are expected to play a pivotal role in statistically unraveling the relationships between material properties and intricate physical factors. This statistical approach underpins material design and, conversely, material design informs statistical analyses. Nevertheless, it is essential to acknowledge that material informatics is still a developing field with challenges such as data standardization, the diversity of material types, and potential conflicts in research culture. To address these issues, tailored data management approaches specific to battery materials should be developed, along with the exploration of suitable descriptors for this particular domain [Citation293].
In summary, through the collaborative integration of combinatorial synthesis, high-throughput calculations, simulations, machine learning, and materials informatics, the prospects for advancing battery technology are highly promising. Combinatorial synthesis expedites the discovery of new materials, while high-throughput calculations and simulations enable efficient material testing and modeling. Machine learning and informatics harness data-driven insights to uncover intricate relationships between material properties. This synergistic approach not only accelerates the identification of novel battery materials but also enhances the understanding of fundamental scientific phenomena. The convergence of these methodologies offers the potential to revolutionize battery technology, leading to improved energy storage solutions for a sustainable future [Citation294].
The presented below outlines the future trajectory for advancing lithium battery technology, emphasizing a collaborative approach that incorporates high-throughput simulations, calculations, data mining, and artificial intelligence (AI) design. This comprehensive strategy explores various critical factors, including thermal stability, electronic structure, ionic transport, ionic conductivity, mechanical modulus, electronic conductivity, and phonon interfaces. The intricate interplay between electrode and electrolyte materials assumes a central role in this forward-looking strategy. Through the application of high-throughput simulations and calculations, researchers aim to gain profound insights into these essential properties that shape battery performance and safety. Uncovering the intricate relationships between these properties and material structures is poised to transform the design of lithium battery systems, making them more efficient, durable, and sustainable to meet the evolving demands of a mobile and eco-conscious society. High-throughput experiments play a vital role in streamlining the integration of battery material synthesis, property measurements, and battery device production. The synergy between experimentation and synthesis enables rapid advancements in battery design and production, allowing researchers to efficiently explore a vast array of materials and configurations. Ultimately, this approach contributes to the creation of more effective and sustainable energy storage solutions. Furthermore, data mining serves as a powerful tool for revealing the intricate connections between voltage, conductivity, and volume change concerning atomic structure. Data mining also illuminates how microstructure and atomic arrangements influence material properties. Additionally, the integration of artificial intelligence (AI) into the rational design of new electrode materials and electrolytes for batteries significantly enhances the efficiency and precision of the design process. AI-driven approaches facilitate the exploration of extensive material design spaces, predicting optimal compositions and structures while considering performance characteristics, safety, and sustainability. This synergy between human expertise and AI-driven insights accelerates the development of innovative battery materials, leading to more efficient and sustainable energy storage solutions for the future. The incorporation of data science further enhances our understanding of material properties, enabling more informed and data-driven decisions in the pursuit of advanced battery technology.
Figure 14. The evolving trajectory of lithium batteries in the near future, incorporating high-throughput simulations, high-throughput experiments, data mining, and artificial intelligence.
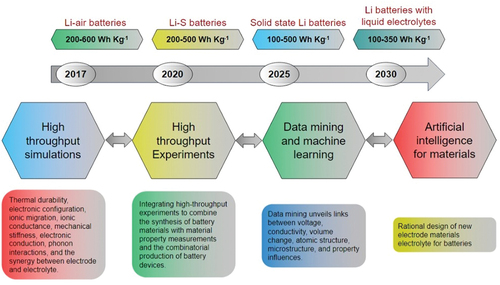
The collaborative efforts among high-throughput simulations, calculations, data mining, and artificial intelligence are poised to reshape the landscape of battery research in the future. This collaborative synergy streamlines the process of material discovery, optimization, and design, ultimately accelerating the development of next-generation battery technologies. High-throughput simulations, experiments, and calculations provide in-depth insights into material properties, while data mining uncovers valuable relationships within vast datasets. Artificial intelligence augments human expertise, facilitating the exploration of complex material design spaces. Together, these collaborative endeavors hold the potential to revolutionize energy storage solutions, creating batteries that are not only more efficient but also more sustainable and eco-friendlier, meeting the growing demands of a mobile and environmentally conscious society. One primary approach to incorporating combinatorial synthesis into experimental workflows is through benchtop solution-based synthesis instruments. These instruments allow for the robotic coupling of synthesis and metrology and involve using robotic systems to dispense and mix different materials and solutions onto a substrate. The resulting material compositions are then characterized and analyzed to determine their properties. One early example of such an instrument is “Ada,” as shown in . [Citation295] The use of robotic systems in the synthesis and characterization process enables the rapid screening of a large number of material compositions. In the case of Ada, the instrument was capable of synthesizing and analyzing up to 300 different material compositions per day. This high-throughput screening approach allows for the efficient exploration of the thermal processing space and the discovery of new materials with desirable properties.
Figure 15. The Ada self-driving laboratory. Adopted with permission from reference [Citation295].
![Figure 15. The Ada self-driving laboratory. Adopted with permission from reference [Citation295].](/cms/asset/a717d963-060d-4df7-8808-68c6eb99430b/tstm_a_2292486_f0015_oc.jpg)
9. Summary and future perspective
The quest for discovering new materials with coveted properties has long been a formidable challenge in the realm of materials science. Traditionally, this endeavor was fraught with a slow and costly trial-and-error approach, demanding substantial investments of time, energy, and resources. However, the contemporary landscape of materials discovery has been profoundly reshaped by recent advancements in various domains, including combinatorial synthesis, high-throughput characterization, computational methodologies, web-based collaboration, and open-source analytical tools. Combinatorial synthesis, a dynamic technique, has emerged as a potent catalyst for rapid material exploration. It empowers researchers to synthesize an extensive array of materials in parallel, systematically varying their chemical compositions, structures, and properties. This method, characterized by its efficiency, enables the creation of material libraries that can be swiftly screened for desired attributes. By concurrently producing a multitude of materials, combinatorial synthesis not only expedites the discovery process but also widens the scope of materials available for investigation. High-throughput characterization, an indispensable counterpart to combinatorial synthesis, facilitates the expeditious and efficient assessment of material properties. Leveraging automated instrumentation, this methodology swiftly evaluates a broad spectrum of material characteristics, spanning electrical conductivity, magnetic behavior, hardness, and more. By rapidly appraising numerous materials, high-throughput techniques amass a wealth of data, which serves as a valuable resource for identifying promising candidates warranting further investigation. In parallel, computational techniques have ascended to prominence, revolutionizing materials discovery. Employing quantum mechanical calculations, Density Functional Theory (DFT), and machine learning algorithms, researchers can simulate material properties, forecast novel materials boasting desired attributes, and even blueprint virtual prototypes. By synergizing computational tools with experimental methods, material scientists significantly curtail the temporal and financial investments traditionally associated with material discovery. The advent of web-based sharing platforms and open-source analysis tools further underpins this transformative shift in materials science. Online platforms facilitate data dissemination, global collaboration, and the accessibility of vast material datasets. Simultaneously, open-source analytical tools empower researchers to dissect and visualize data, construct predictive models, and exchange code and algorithms, fostering transparency, reproducibility, and an environment conducive to collaborative innovation. In summation, the amalgamation of combinatorial synthesis, high-throughput characterization, computational methodologies, web-based collaboration, and open-source analytical tools represents a paradigm shift in the approach to material discovery. These innovations collectively serve to expedite the exploration of new materials, broaden the spectrum of materials subject to study, and foster collaboration, transparency, and replicability in the realm of materials science research.
The future of high-throughput materials discovery and design could be built on several actionable plans, including:
9.1. Collaboration between computational and experimental research groups
Collaboration between computational and experimental research groups plays a pivotal role in expediting the materials discovery process. This synergistic partnership, initiated at the inception of materials development, yields substantial reductions in both time and cost. The computational group offers invaluable insights into material properties, aiding the experimental group in the judicious selection of materials for synthesis and testing. Reciprocally, the experimental team provides feedback on the applicability of the computational approach and contributes to the refinement of computational models. This collaboration serves as a bridge between theoretical predictions and empirical observations. While computational models generate predictions about material properties, experimental measurements validate these predictions. The amalgamation of these two methodologies facilitates model enhancement and refinement, resulting in more precise predictions and accelerated discovery timelines. Furthermore, interdisciplinary collaboration empowers researchers to tackle intricate challenges, such as designing materials with specific properties. For instance, computational techniques can anticipate the properties of a material with desired functionality, subsequently enabling the experimental group to synthesize and validate these properties through testing and analysis. This collaborative synergy propels the materials discovery process, fostering innovation and scientific advancement.
9.2. Addressing materials data science challenges
Materials data science presents a distinct array of challenges that demand attention to expedite the materials discovery process. One of the foremost challenges revolves around data-guided experimentation within expansive search spaces. The burgeoning volume of materials data accentuates the difficulty in singling out promising candidates for experimental evaluation. Hence, the imperative arises to pioneer novel data-driven methodologies that empower researchers in making judicious decisions regarding the synthesis and testing of materials. Moreover, grappling with the analysis of high-dimensional data presents another hurdle. Materials data typically encompasses multiple variables, complicating the discernment of intricate relationships among them. To surmount these hurdles, there exists a pressing need to cultivate a new breed of materials data scientists equipped with the acumen to formulate and deploy sophisticated data analysis techniques.
9.3. Open access publishing results and visualizing data
Disseminating research outcomes and data constitutes a pivotal facet of expediting the materials discovery process. Through the act of sharing their discoveries, researchers can circumvent redundant endeavors and construct upon the groundwork laid by their peers. Furthermore, the act of sharing data and findings serves as a mechanism for corroborating experimental results and safeguarding their reproducibility. To foster a culture of data and result-sharing, researchers must document their findings comprehensively, encompassing the methodologies employed, the materials synthesized, and the empirical data amassed. This information dissemination can be accomplished through various avenues such as scientific journals, conferences, and online repositories. Moreover, researchers ought to ensure that their data and results are readily accessible and intelligible to their counterparts. An efficacious means to facilitate data-informed decision-making and encourage the sharing of findings is through interactive multifunctional existence diagrams. These diagrams furnish an all-encompassing perspective of the data, empowering researchers to discern patterns and trends expeditiously. By visualizing the entirety of pertinent data within a system, scientists can judiciously chart their course for future research endeavors.
9.4. Automating experiments towards autonomous experimentation
One of the significant challenges within materials informatics has been the absence of a dependable experimental materials database, mainly because manually compiling such a resource proves impractical. Addressing this challenge has led to the emergence of autonomous experimental systems, representing an innovative solution. Autonomous experimentation stands at the forefront of materials discovery and design, capitalizing on the collaborative power of artificial intelligence and robotics to streamline the synthesis and testing of materials. This groundbreaking approach harnesses machine learning algorithms for generating hypotheses regarding promising materials, all while employing robotic systems to autonomously execute the synthesis and testing procedures, markedly reducing the necessity for human intervention. The primary objectives of autonomous experimentation are threefold: to expedite the materials discovery process, significantly reduce associated costs, and mitigate the errors frequently encountered in manual experimentation. Through the automation of these processes, researchers gain the capability to efficiently screen a vast array of potential materials and promptly pinpoint the most promising candidates for further in-depth investigation. Autonomous experimental systems excel in comprehensive data storage, encompassing essential information such as synthesis conditions, experimental parameters, structural characteristics, compositional data, and even instances where desired values were not achieved (commonly referred to as negative results). This holistic approach substantially amplifies the volume of data available, enhancing the precision of predictions through machine learning. Moreover, given that data collection transpires without human intervention, it inherently becomes more reproducible and less susceptible to human errors. Consequently, this approach facilitates comprehensive analysis, unveiling correlations between process parameters and material properties, ultimately culminating in the development of predictive technologies for synthetic processes, often termed process informatics. The remarkable enhancement in experimental efficiency, courtesy of autonomous experimentation, directly translates into a substantial increase in materials data. Autonomous experiments are estimated to outperform conventional methods by at least an order of magnitude in terms of speed. When synergistically combined with materials checkup systems, the acquisition of measurement data can be further expedited by yet another order of magnitude.
An early illustration of such an apparatus is ‘Ada’, originally devised for the synthesis of organic transport layers in solar cell applications. The instrument ingeniously employed robotic pipetting to precisely dispense organic hole transport material, dopants, and plasticizers onto microscope slides. These coated slides were subsequently subjected to annealing within a forced convection furnace, yielding a diverse array of material compositions. The integration of robotic systems into the synthesis and characterization processes proved instrumental in swiftly screening numerous material compositions. Remarkably, in the case of Ada, the instrument demonstrated the capability to synthesize and analyze up to 300 distinct material compositions within a single day. This high-throughput screening methodology has proven to be exceptionally efficient for systematically exploring the thermal processing parameter space and unearthing novel materials possessing coveted properties [Citation295].
Acknowledgements
This research was funded in whole, or in part, by the Austrian Science Fund (FWF) [P32847-N]. For the purpose of open access, the authors have applied a CC BY public copyright license to the accepted manuscript version arising from this submission. The financial support received from the European Union Horizon 2020 through the project Medical Device Obligation Taskforce (MDOT) is gratefully acknowledged. MDOT received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement number 814654.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Notes on contributors

Khurram Shahzad
Khurram Shahzad is a materials engineer and served as a Material Engineer/faculty member in Pakistan Institute of Engineering and Applied Sciences (PIEAS) for more than 10 years. He received his Ph.D. from Hokkaido University, Japan with the support of MEXT scholarship. He expanded his research expertise at JKU, Austria, where he specialized in combinatorial oxide films growth and EIS characterization. Dr. Shahzad’s research approach is interdisciplinary, combining simulation, experimentation, and characterization. His research focuses on advancements in materials science, with a particular emphasis on combining computational and experimental techniques for material design and characterization.

Andrei Ionut Mardare
Andrei Ionut Mardare is a physicist working in the field of thin film combinatorial development of inorganic materials. He studied Physics at University of Bucharest, Romania and conducted his doctoral work at Max-Planck Institute for Iron Research in Düsseldorf, Germany. He received his PhD degree in Physics in 2009 from Ruhr University Bochum (RUB) Germany, then he joined the Institute of Chemical Technology of Inorganic Materials at Johannes Kepler University Linz, Austria. His current research interests include ultra-thin anodic oxides on valve metals and their dynamic processes at atomic scale.

Achim Walter Hassel
Achim Walter Hassel received his Ph.D. in 1997 from University of Düsseldorf, Germany. After that, he was an Alexander von Humboldt and JSPS-fellow until 1999 at Hokkaido University (Sapporo, Japan). 2000–2009 he was head of the Electrochemistry and Corrosion group at the Max-Planck-Institute for Iron Research and scientific director of the IMPRS Surmat. Since 2009 he holds a chair in Chemistry at the Johannes Kepler University Linz, Austria. He was project leader of the Christian Doppler Laboratory for Combinatorial Oxide Chemistry and several EU projects. His research interests are in the field of combinatorial and electrochemical materials science.
References
- Moskowitz SL. The advanced materials revolution: technology and economic growth in the age of globalization. Hoboken (NJ): John Wiley & Sons; 2008.
- Jain A, Ong SP, Hautier G, et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater. 2013;1(1):1–41. doi: 10.1063/1.4812323
- Potyrailo RA, Amis EJ. High-throughput analysis: a tool for combinatorial materials science. New York: Kluwer Academic/Plenum Publishers; 2003.
- Tabor DP, Roch LM, Saikin SK, et al. Accelerating the discovery of materials for clean energy in the era of smart automation. Nat Rev Mater. 2018;3:5–20. doi: 10.1038/s41578-018-0005-z
- Maier WF, Stöwe K, Sieg S. Combinatorial and high-throughput materials science. Angew Chem Int Ed. 2007;46(32):6016–6067. doi: 10.1002/anie.200603675
- Terakura K, Takeuchi I. Focus on materials genome and informatics. Sci Technol Adv Mater. 2017;18(1):1–2. doi: 10.1080/14686996.2016.1246226
- Wang Y, Zhang W, Chen L, et al. Quantitative description on structure–property relationships of Li-ion battery materials for high-throughput computations. Sci Technol Adv Mater. 2017;18(1):134–146. doi: 10.1080/14686996.2016.1277503
- Grasset F, Pic L. Focus on overview of innovative materials for energy. Sci Technol Adv Mater. 2017;18(1):704. doi: 10.1080/14686996.2017.1379729
- Lam Pham T, Kino H, Terakura K, et al. Machine learning reveals orbital interaction in materials. Sci Technol Adv Mater. 2017;18(1):756–765. doi: 10.1080/14686996.2017.1378060
- Ludwig A. Discovery of new materials using combinatorial synthesis and high-throughput characterization of thin-film materials libraries combined with computational methods. Npj Comput Mater. 2019;5(1):70. doi: 10.1038/s41524-019-0205-0
- Chen Y, Chen C, Zheng C, et al. Database of ab initio L-edge X-ray absorption near edge structure. Sci Data. 2021;8(1):153. doi: 10.1038/s41597-021-00936-5
- Horton MK, Dwaraknath S, Persson KA. Promises and perils of computational materials databases. Nat Comput Sci. 2021;1(1):3–5. doi: 10.1038/s43588-020-00016-5
- Gregoire JM, Zhou L, Haber JA. Combinatorial synthesis for AI-driven materials discovery. Nat Synth. 2023;2(6):493–504. doi: 10.1038/s44160-023-00251-4
- Ogale SB. Thin films and heterostructures for oxide electronics. New York: Springer; 2005.
- Cabo MJ, NP M, Song JI. Synthesis of non-phosphorylated epoxidised corn oil as a novel green flame retardant thermoset resin. Sci Rep. 2021;11(1):24140. doi: 10.1038/s41598-021-03274-z
- Li P, Materials research Society. Fall Meeting. Mineralization in natural and synthetic biomaterials: symposium held November 29-December 1 1999, Boston, MA. 2000.
- Fasolka MJ, Materials Research Society. Fall Meeting. Combinatorial methods and informatics in materials science : symposium held November 28-December 1, 2005, Boston, MA, Warrendale, PA: Materials Research Society; 2006.
- Park JC, Heo GS, Lee JH, et al. Synthesis of TaZnO thin films using combinatorial magnetron sputtering and its electrical, structural and optical properties. J Nanosci Nanotechnol. 2012;12(7):5303–5306. doi: 10.1166/jnn.2012.6303
- McGinn PJ. Thin-film processing routes for combinatorial materials investigations—a review. ACS Comb Sci. 2019;21(7):501–515. doi: 10.1021/acscombsci.9b00032
- Potyrailo RA, Materials Research Society. Fall Meeting. Combinatorial and artificial intelligence methods in materials science II : symposium held December 1–4 2003, Boston, MA, Warrendale, PA: Materials Research Society; 2004.
- Al Hasan NM, Hou H, Sarkar S, et al. Combinatorial synthesis and high-throughput characterization of microstructure and phase transformation in ni–ti–cu–V quaternary thin-film library. Eng. 2020;6(6):637–643. doi: 10.1016/j.eng.2020.05.003
- Green ML, Takeuchi I, Hattrick-Simpers JR. Applications of high throughput (combinatorial) methodologies to electronic, magnetic, optical, and energy-related materials. J Appl Phys. 2013;113(23):231101. doi: 10.1063/1.4803530
- Potyrailo RA, Amis EJ. Battery materials for ultrafast charging and discharging. Nature. 2009;458(7235):190–193. doi: 10.1038/nature07853
- Yu L, Zunger A. Identification of potential photovoltaic absorbers based on first-principles spectroscopic screening of materials. Phys Rev Lett. 2012;108(6):68701. doi: 10.1103/PhysRevLett.108.068701
- Yang K, Setyawan W, Wang S, et al. A search model for topological insulators with high-throughput robustness descriptors. Nat Mater. 2012;11(7):614–619. doi: 10.1038/nmat3332
- Hack J, Maeda N, Meier DM. Review on CO2 capture using Amine-functionalized materials. ACS Omega. 2022;7(44):39520–39530. doi: 10.1021/acsomega.2c03385
- Armiento R, Kozinsky B, Fornari M, et al. Screening for high-performance piezoelectrics using high-throughput density functional theory. Phys Rev B. 2011;84(1):14103. doi: 10.1103/PhysRevB.84.014103
- Chen H, Hautier G, Ceder CG. Synthesis, computed stability, and crystal structure of a new family of inorganic compounds: carbonophosphates. J Am Chem Soc. 2012;134(48):19619–19627. doi: 10.1021/ja3040834
- Alapati SV, Johnson JK, Sholl DS. Identification of destabilized metal hydrides for hydrogen storage using first principles calculations. J Phys Chem B. 2006;110(17):8769–8776. doi: 10.1021/jp060482m
- Lu J, Fang ZZ, Choi YJ, et al. Potential of binary lithium magnesium nitride for hydrogen storage applications. J Phys Chem C. 2007;111(32):12129–12134. doi: 10.1021/jp0733724
- Yang J, Sudik A, Wolverton C, et al. High capacity hydrogen storage materials: attributes for automotive applications and techniques for materials discovery. Chem Soc Rev. 2010;39(2):656–675. doi: 10.1039/B802882F
- Jain A, Hautier G, Moore C, et al. A computational investigation of Li9M3(P2O7)3(PO4)2 (M = V, Mo) as cathodes for li ion batteries. J Electrochem Soc. 2012;159(5):A622. doi: 10.1149/2.080205jes
- Mathew K, Montoya JH, Faghaninia A, et al. Atomate: a high-level interface to generate, execute, and analyze computational materials science workflows. Comput Mater Sci. 2017;139:140–152. doi: 10.1016/j.commatsci.2017.07.030
- Jain A, Hautier G, Moore CJ, et al. A high-throughput infrastructure for density functional theory calculations. Comput Mater Sci. 2011;50(8):2295–2310. doi: 10.1016/j.commatsci.2011.02.023
- Jain A, Shin Y, Persson KA. Computational predictions of energy materials using density functional theory. Nat Rev Mater. 2016;1(1):1–13. doi: 10.1038/natrevmats.2015.4
- Castelli IE, Olsen T, Datta S, et al. Computational screening of perovskite metal oxides for optimal solar light capture. Energy Environ Sci. 2012;5(2):5814–5819. doi: 10.1039/C1EE02717D
- Kim JC, Moore CJ, Kang B, et al. Synthesis and electrochemical properties of monoclinic LiMnBO[sub 3] as a li intercalation material. J Electrochem Soc. 2011;158(3):A309. doi: 10.1149/1.3536532
- Hou T, Fong KD, Wang J, et al. Correction: the solvation structure, transport properties and reduction behavior of carbonate-based electrolytes of lithium-ion batteries. Chem Sci. 2022;13(27):8205. doi: 10.1039/D2SC90129C
- Rutt A, Shen J-X, Horton M, et al. Expanding the material search space for multivalent cathodes. ACS Appl Mater Interfaces. 2022;14(39):44367–44376. doi: 10.1021/acsami.2c11733
- Siron M, Andriuc O, Persson KA. Data-driven investigation of tellurium-containing semiconductors for CO2 reduction: trends in adsorption and scaling relations. J Phys Chem C. 2022;126(31):13224–13236. doi: 10.1021/acs.jpcc.2c04810
- Ionut A, Ludwig A, Savan A, et al. Scanning droplet cell microscopy on a wide range Hafnium – Niobium thin film combinatorial library. Electrochim Acta. 2013;110:539–549. doi: 10.1016/j.electacta.2013.03.065
- Alexandrakis V, Wallisch W, Hamann S, et al. Combinatorial development of Fe–Co–Nb thin film magnetic nanocomposites. ACS Comb Sci. 2015;17(11):698–703. doi: 10.1021/acscombsci.5b00116
- Fackler SW, Alexandrakis V, König D, et al. Combinatorial study of Fe-Co-V hard magnetic thin films. Sci Technol Adv Mater. 2017;18(1):231–238. doi: 10.1080/14686996.2017.1287520
- Kawashima K, Okamoto Y, Annayev O, et al. Combinatorial screening of halide perovskite thin films and solar cells by mask-defined IR laser molecular beam epitaxy. Sci Technol Adv Mater. 2017;18(1):307–315. doi: 10.1080/14686996.2017.1314172
- Mardare AI, Savan A, Ludwig A, et al. A combinatorial passivation study of Ta–Ti alloys. Corros Sci. 2009;51(7):1519–1527. doi: 10.1016/j.corsci.2008.12.003
- Curtarolo S, Hart GLW, Nardelli MB, et al. The high-throughput highway to computational materials design. Nat Mater. 2013;12(3):191–201. doi: 10.1038/nmat3568
- Mao SS, Burrows PE. Combinatorial screening of thin film materials: an overview. J Mater. 2015;1(2):85–91. doi: 10.1016/j.jmat.2015.04.002
- Potyrailo R, Rajan K, Stoewe K, et al. Combinatorial and high-throughput screening of materials libraries: review of state of the art. ACS Comb Sci. 2011;13(6):579–633. doi: 10.1021/co200007w
- Zrinski I, Minenkov A, Cancellieri C, et al. Mixed anodic oxides for forming-free memristors revealed by combinatorial screening of hafnium-tantalum system. Appl Mater Today. 2022;26:101270. doi: 10.1016/j.apmt.2021.101270
- Siket CM, Tillner N, Mardare AI, et al. Direct writing of anodic oxides for plastic electronics. Npj Flex Electron. 2018;2(1):23. doi: 10.1038/s41528-018-0036-y
- Mardare AI, Kaltenbrunner M, Sariciftci NS. Ultra-thin anodic alumina capacitor films for plastic electronics. Phys Status Solidi Appl Mater Sci. 2012;209(5):813–818. doi: 10.1002/pssa.201100785
- Ong SP, Richards WD, Jain A, et al. Python materials Genomics (pymatgen): a robust, open-source python library for materials analysis. Comput Mater Sci. 2013;68:314–319. doi: 10.1016/j.commatsci.2012.10.028
- Shahzad K, Mardare CC, Mardare AI, et al. Growth of mixed anodic films on combinatorial Al-Gd alloys and their superimposed potential-pH diagrams. J Electroanal Chem. 2022;911:116227. doi: 10.1016/j.jelechem.2022.116227
- Xiang XD, Sun X, Briceño G, et al. A combinatorial approach to materials discovery. Science. 1995;268(5218):1738–1740. ited: in: PMID: 17834993. doi: 10.1126/science.268.5218.1738
- Koinuma H, Takeuchi I. Combinatorial solid-state chemistry of inorganic materials. Nat Mater. 2004;3(7):429–438. doi: 10.1038/nmat1157
- Dover RB, Schneemeyer LF, Fleming RM. Discovery of a useful thin-film dielectric using a composition-spread approach. Nature. 1998;392(6672):162–164. doi: 10.1038/32381
- Zhang Y, Schultz AM, Li L, et al. Combinatorial substrate epitaxy: a high-throughput method for determining phase and orientation relationships and its application to BiFeO3/TiO2 heterostructures. Acta Mater. 2012;60(19):6486–6493. doi: 10.1016/j.actamat.2012.07.060
- Hyeon T. Chemical synthesis of magnetic nanoparticles. Chem Commun. 2003;8:927–934. doi: 10.1039/b207789b
- Saldan I, Semenyuk Y, Marchuk I, et al. Chemical synthesis and application of palladium nanoparticles. J Mater Sci. 2015;50(6):2337–2354. doi: 10.1007/s10853-014-8802-2
- Talapin DV, Chevchanko EV. Introduction: nanoparticle chemistry. Chem Rev. 2016;116(18):10343–10345. doi: 10.1021/acs.chemrev.6b00566
- Ghorbani HR. A review of methods for synthesis of Al nanoparticles. Orient J Chem. 2014;30(4):1941–1949. doi: 10.13005/ojc/300456
- Hu X, Takai O, Saito N. Simple synthesis of platinum nanoparticles by plasma sputtering in water. Jpn J Appl Phys. 2013;52(1S):01AN05. doi: 10.7567/JJAP.52.01AN05
- Orozco-Montes V, Caillard A, Brault P, et al. Synthesis of platinum nanoparticles by plasma sputtering onto glycerol: effect of argon pressure on their physicochemical properties. J Phys Chem C. 2021;125(5):3169–3179. doi: 10.1021/acs.jpcc.0c09746
- Liang J, Liu Q, Li T, et al. Magnetron sputtering enabled sustainable synthesis of nanomaterials for energy electrocatalysis. Green Chem. 2021;23(8):2834–2867. doi: 10.1039/D0GC03994B
- Giri PK, Bhattacharyya S, Singh DK, et al. Correlation between microstructure and optical properties of ZnO nanoparticles synthesized by ball milling. J Appl Phys. 2007;102(9):93515. doi: 10.1063/1.2804012
- Li L, Pu S, Liu Y, et al. High-purity disperse α-Al2O3 nanoparticles synthesized by high-energy ball milling. Adv Powder Technol. 2018;29(9):2194–2203. doi: 10.1016/j.apt.2018.06.003
- Abid N, Khan AM, Shujait S, et al. Synthesis of nanomaterials using various top-down and bottom-up approaches, influencing factors, advantages, and disadvantages: a review. Adv Colloid Interface Sci. 2022;300:102597. doi: 10.1016/j.cis.2021.102597
- Ristoscu C, Mihailescu IN. Thin films and nanoparticles by pulsed laser deposition: wetting, adherence, and nanostructuring. Pulsed laser ablation. New York: Jenny Stanford Publishing; 2018. p. 245–276.
- Serbezov V. Pulsed laser deposition: the road to hybrid nanocomposites coatings and novel pulsed laser adaptive technique. Recent Pat Nanotechnol. 2013;7(1):26–40. doi: 10.2174/187221013804484863
- Narayanan KB, Sakthivel N. Biological synthesis of metal nanoparticles by microbes. Adv Colloid Interface Sci. 2010;156(1–2):1–13. doi: 10.1016/j.cis.2010.02.001
- Siegwart DJ, Whitehead KA, Nuhn L, et al. Combinatorial synthesis of chemically diverse core-shell nanoparticles for intracellular delivery. Proc Natl Acad Sci. 2011;108(32):12996–13001. doi: 10.1073/pnas.1106379108
- König D, Richter K, Siegel A, et al. High-throughput fabrication of Au–Cu nanoparticle libraries by combinatorial sputtering in ionic liquids. Adv Funct Mater. 2014;24(14):2049–2056. doi: 10.1002/adfm.201303140
- Richter K, Campbell PS, Baecker T, et al. Ionic liquids for the synthesis of metal nanoparticles. Phys Status Solidi. 2013;250(6):1152–1164. doi: 10.1002/pssb.201248547
- Fan Y, Yen C-W, Lin H-C, et al. Automated high-throughput preparation and characterization of oligonucleotide-loaded lipid nanoparticles. Int J Pharm. 2021;599:120392. doi: 10.1016/j.ijpharm.2021.120392
- Damoiseaux R, George S, Li M, et al. No time to lose—high throughput screening to assess nanomaterial safety. Nanoscale. 2011;3(4):1345–1360. doi: 10.1039/c0nr00618a
- Ramishetti S, Hazan-Halevy I, Palakuri R, et al. A combinatorial library of lipid nanoparticles for RNA delivery to leukocytes. Adv Mater. 2020;32(12):1906128. doi: 10.1002/adma.201906128
- Shi Y, Yang B, Rack PD, et al. High-throughput synthesis and corrosion behavior of sputter-deposited nanocrystalline Alx(CoCrFeNi)100-x combinatorial high-entropy alloys. Mater Des. 2020;195:109018. doi: 10.1016/j.matdes.2020.109018
- Stock N. High-throughput investigations employing solvothermal syntheses. Microporous Mesoporous Mater. 2010;129(3):287–295. doi: 10.1016/j.micromeso.2009.06.007
- Bodenstein-Dresler LCW, Kama A, Frisch J, et al. Prospect of making XPS a high-throughput analytical method illustrated for a CuxNi1−xOy combinatorial material library. RSC Adv. 2022;12(13):7996–8002. doi: 10.1039/D1RA09208A
- Stremy M, Horvath D, Vana D, et al. RBS channeling MATLAB application for automated measurement control and evaluation for 6MV tandetron accelerator. Appl Sci. 2021;11(9):11. doi: 10.3390/app11093817
- Möller S, Höschen D, Kurth S, et al. A new high-throughput focused MeV ion-beam analysis setup. Instruments. 2021;5(1):10. doi: 10.3390/instruments5010010
- Möller S, Ding R, Xie H, et al. 13C tracer deposition in EAST D and He plasmas investigated by high-throughput deuteron nuclear reaction analysis mapping. Nucl Mater Energy. 2020;25:100805. doi: 10.1016/j.nme.2020.100805
- Światowska-Mrowiecka J, de Diesbach S, Maurice V, et al. Li-ion intercalation in thermal oxide thin films of MoO3 as studied by XPS, RBS, and NRA. J Phys Chem C. 2008;112(29):11050–11058. doi: 10.1021/jp800147f
- Barfoot KM. Ion beam analysis. Phys Bull. 1984;35(12):511. doi: 10.1088/0031-9112/35/12/022
- Wendelbo R, Akporiaye DE, Karlsson A, et al. Combinatorial hydrothermal synthesis and characterisation of perovskites. J Eur Ceram Soc. 2006;26(6):849–859. doi: 10.1016/j.jeurceramsoc.2004.12.031
- Kusne AG, Gao T, Mehta A, et al. On-the-fly machine-learning for high-throughput experiments: search for rare-earth-free permanent magnets. Sci Rep. 2014;4(1):6367. doi: 10.1038/srep06367
- Long CJ, Hattrick-Simpers J, Murakami M, et al. Rapid structural mapping of ternary metallic alloy systems using the combinatorial approach and cluster analysis. Rev Sci Instrum. 2007;78(7):72217. doi: 10.1063/1.2755487
- EAG Laboratories. The global leader in materials testing services [Internet]. [cited 2023 Mar 14]. Available from: https://www.eag.com/
- Zarnetta R, Kneip S, Somsen C, et al. High-throughput characterization of mechanical properties of Ti–Ni–Cu shape memory thin films at elevated temperature. Mater Sci Eng A. 2011;528(21):6552–6557. doi: 10.1016/j.msea.2011.05.006
- Naujoks D, Eggeler YM, Hallensleben P, et al. Identification of a ternary μ-phase in the Co–Ti–W system – an advanced correlative thin-film and bulk combinatorial materials investigation. Acta Mater. 2017;138:100–110. doi: 10.1016/j.actamat.2017.07.037
- Curtarolo S, Setyawan W, Wang S, et al. AFLOWLIB.ORG: a distributed materials properties repository from high-throughput ab initio calculations. Comput Mater Sci. 2012;58:227–235. doi: 10.1016/j.commatsci.2012.02.002
- Shinde PA, Lokhande AC, Chodankar NR, et al. Temperature dependent surface morphological modifications of hexagonal WO3 thin films for high performance supercapacitor application. Electrochim Acta. 2017;224:397–404. doi: 10.1016/j.electacta.2016.12.066
- Bunn JK, Fang RL, Albing MR. A high-throughput investigation of Fe–Cr–Al as a novel high-temperature coating for nuclear cladding materials. Nanotechnology. 2015;26(27):274003. doi: 10.1088/0957-4484/26/27/274003
- Hattrick-Simpers JR, Jun C, Murakami M, et al. High-throughput screening of magnetic properties of quenched metallic-alloy thin-film composition spreads. Appl Surf Sci. 2007;254(3):734–737. doi: 10.1016/j.apsusc.2007.07.104
- Takeuchi I, Famodu OO, Read JC, et al. Identification of novel compositions of ferromagnetic shape-memory alloys using composition spreads. Nat Mater. 2003;2(3):180–184. doi: 10.1038/nmat829
- Decker P, Naujoks D, Langenkämper D, et al. High-throughput structural and functional characterization of the thin film materials system Ni–Co–Al. ACS Comb Sci. [Internet] 2017;19:618–624. doi: 10.1021/acscombsci.6b00176
- Xia R, Brabec CJ, Yip H-L, et al. High-throughput optical screening for efficient semitransparent organic solar cells. Joule. 2019;3(9):2241–2254. doi: 10.1016/j.joule.2019.06.016
- Kashiwagi T, Sue K, Takebayashi Y, et al. High-throughput synthesis of silver nanoplates and optimization of optical properties by machine learning. Chem Eng Sci. 2022;262:118009. doi: 10.1016/j.ces.2022.118009
- Shen C, Wang C, Huang M, et al. A generic high-throughput microstructure classification and quantification method for regular SEM images of complex steel microstructures combining EBSD labeling and deep learning. J Mater Sci Technol. 2021;93:191–204. doi: 10.1016/j.jmst.2021.04.009
- Yablon D, Chakraborty I, Passino H, et al. Deep learning to establish structure property relationships of impact copolymers from AFM phase images. MRS Commun. 2021;11(6):962–968. doi: 10.1557/s43579-021-00103-2
- Sormana J-L, Meredith JC. High-throughput discovery of structure−mechanical property relationships for segmented poly(urethane−urea)s. Macromolecules. 2004;37(6):2186–2195. doi: 10.1021/ma035385v
- Li Y, Hu X, Liu H, et al. High-throughput assessment of local mechanical properties of a selective-laser-melted non-weldable Ni-based superalloy by spherical nanoindentation. Mater Sci Eng A. 2022;844:143207. doi: 10.1016/j.msea.2022.143207
- Tong Y, Zhang H, Huang H, et al. Strengthening mechanism of CoCrNiMox high entropy alloys by high-throughput nanoindentation mapping technique. Intermetallics. 2021;135:107209. doi: 10.1016/j.intermet.2021.107209
- Gregoire JM, Van Campen DG, Miller CE, et al. A. High-throughput synchrotron X-ray diffraction for combinatorial phase mapping. J Synchrotron Radiat. 2014;21(6):1262–1268. doi: 10.1107/S1600577514016488
- Tanaka M, Katsuya Y, Yamamoto A. A new large radius imaging plate camera for high-resolution and high-throughput synchrotron x-ray powder diffraction by multiexposure method. Rev Sci Instrum. 2008;79(7):75106. doi: 10.1063/1.2956972
- Caskey CM, Richards RM, Ginley DS, et al. Thin film synthesis and properties of copper nitride, a metastable semiconductor. Mater Horizons. 2014;1(4):424–430. doi: 10.1039/C4MH00049H
- Graham BJ, Hildebrand DGC, Kuan AT, et al. High-throughput transmission electron microscopy with automated serial sectioning. bioRxiv. 2019;657346. doi:10.1101/657346
- Sáfrán G. “One-sample concept” micro-combinatory for high throughput TEM of binary films. Ultramicroscopy. 2018;187:50–55. doi: 10.1016/j.ultramic.2018.01.001
- Young R, Carleson PD, Da X, et al. High-yield and high-throughput TEM sample preparation using focused ion beam automation. Istfa’98 Proc 24th Int Symp Test Fail Anal. ASM International. 1998;332.
- Ornelas IM, Unwin PR, Bentley CL. High-throughput correlative electrochemistry–microscopy at a transmission electron microscopy grid electrode. Anal Chem. 2019;91(23):14854–14859. doi: 10.1021/acs.analchem.9b04028
- Li YJ, Savan A, Kostka A, et al. Accelerated atomic-scale exploration of phase evolution in compositionally complex materials. Mater Horizons. 2018;5(1):86–92. doi: 10.1039/C7MH00486A
- Mugnaioli E, Gorelik T, Kolb U. “Ab initio” structure solution from electron diffraction data obtained by a combination of automated diffraction tomography and precession technique. Ultramicroscopy. 2009;109(6):758–765. doi: 10.1016/j.ultramic.2009.01.011
- Li YJ, Kostka A, Savan A, et al. Atomic-scale investigation of fast oxidation kinetics of nanocrystalline CrMnFeCoNi thin films. J Alloys Compd. 2018;766:1080–1085. doi: 10.1016/j.jallcom.2018.07.048
- Tolle KM, Tansley DSW, Hey AJ. The fourth paradigm: data-intensive scientific discovery. 2011;99(8):1334–1337.
- Bell G, Hey T, Szalay A. Beyond the data deluge. Science. 2009;323(5919):1297–1298. doi: 10.1126/science.1170411
- Kitchin R. Big data, new epistemologies and paradigm shifts. Big Data Soc. 2014;1(1):1–12. doi: 10.1177/2053951714528481
- Kuhn TS. The structure of scientific revolutions. Chicago: University of Chicago Press; 1962.
- Agrawal A, Choudhary A. Perspective: materials informatics and big data: realization of the “fourth paradigm” of science in materials science. APL Mater. 2016;4(5):053208–10. doi: 10.1063/1.4946894
- Jain A, Persson KA, Ceder G. Research update: the materials genome initiative: data sharing and the impact of collaborative ab initio databases. APL Mater. 2016;4(5):53102. doi: 10.1063/1.4944683
- Szalay A. Extreme data-intensive scientific computing. Comput Sci Eng. 2011;13(6):34–41. doi: 10.1109/MCSE.2011.74
- Faris J, Kolker E, Szalay A, et al. Communication and data-intensive science in the beginning of the 21st century. Omi A J Integr Biol. 2011;15(4):213–215. doi: 10.1089/omi.2011.0008
- Szabo C, Sheng QZ, Kroeger T, et al. Science in the cloud: allocation and execution of data-intensive scientific workflows. J Grid Comput. 2014;12(2):245–264. doi: 10.1007/s10723-013-9282-3
- Burns R, Vogelstein JT, Szalay AS. From cosmos to connectomes: the evolution of data-intensive science. Neuron. 2014;83(6):1249–1252. doi: 10.1016/j.neuron.2014.08.045
- Nukarapu D, Tang B, Wang L, et al. Data replication in data intensive scientific applications with performance guarantee. IEEE Trans Parallel Distrib Syst. 2010;22(8):1299–1306. doi: 10.1109/TPDS.2010.207
- Oliveira SF, Fürlinger K, Kranzlmüller D Trends in computation, communication and storage and the consequences for data-intensive science. 2012 IEEE 14th Int Conf High Perform Comput Commun 2012 IEEE 9th International Conference on Embed Softw Syst. Liverpool (UK): IEEE; 2012. p. 572–579.
- Tian Y, Yuan R, Xue D, et al. Role of uncertainty estimation in accelerating materials development via active learning. J Appl Phys. 2020;128(1):14103. doi: 10.1063/5.0012405
- Shen C, Wang C, Wei X, et al. Physical metallurgy-guided machine learning and artificial intelligent design of ultrahigh-strength stainless steel. Acta Mater. 2019;179:201–214. doi: 10.1016/j.actamat.2019.08.033
- Lee JG. Computational materials science: an introduction. Boca Raton: CRC press; 2016.
- Raabe D. Computational materials science: the simulation of materials microstructures and properties. Germany: Wiley-Vch; 1998.
- LeSar R. Introduction to computational materials science: fundamentals to applications. New York: Cambridge University Press; 2013.
- Schrödinger E. An undulatory theory of the mechanics of atoms and molecules. Phys Rev. 1926;28(6):1049–1070. doi: 10.1103/PhysRev.28.1049
- Dirac PAM, Fowler RH. Quantum mechanics of many-electron systems. Proc R Soc London Ser A, Contain Pap a Math Phys Character. 1997;123:714–733.
- Hartree DR. The wave mechanics of an atom with a non-Coulomb central field. Part I. Theory and methods. Proc Camb Philol Soc. 1928;24:89. doi: 10.1017/S0305004100011919
- Thomas LH. The calculation of atomic fields. Math Proc Cambridge philos Soc. Cambridge: Cambridge University Press; 1927.
- Kohn W, Sham LJ. Self-consistent equations including exchange and correlation effects. Phys Rev. 1965;140(4A):A1133–A1138. doi: 10.1103/PhysRev.140.A1133
- Schleder GR, Padilha ACM, Acosta CM, et al. From DFT to machine learning: recent approaches to materials science – a review. J Phys Mater. 2019;2(3):032001. doi: 10.1088/2515-7639/ab084b
- Persson KA, Waldwick B, Lazic P, et al. Prediction of solid-aqueous equilibria: Scheme to combine first-principles calculations of solids with experimental aqueous states. Phys Rev B – Condens Matter Mater Phys. 2012;85:1–12. doi: 10.1103/PhysRevB.85.235438
- Cohen AJ, Mori-Sánchez P, Yang W. Challenges for density functional theory. Chem Rev. 2012;112(1):289–320. doi: 10.1021/cr200107z
- Doe RE, Persson KA, Meng YS, et al. First-principles investigation of the Li−Fe−F phase diagram and equilibrium and nonequilibrium conversion reactions of Iron fluorides with lithium. Chem Mater. 2008;20(16):5274–5283. doi: 10.1021/cm801105p
- Ping S, Davidson W, Jain A, et al. Python materials genomics (pymatgen): a robust, open-source python library for materials analysis. Comput Mater Sci. 2013;68:314–319. doi: 10.1016/j.commatsci.2012.10.028
- Grimme S, Antony J, Ehrlich S, et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. 2010;132(15):154104. doi: 10.1063/1.3382344
- Cai T, Han H, Yu Y, et al. Study on the ground state of NiO: the LSDA (GGA)+ U method. Phys B Condens Matter. 2009;404(1):89–94. doi: 10.1016/j.physb.2008.10.009
- Kresse G, Hafner J. Ab initio molecular dynamics for liquid metals. Phys Rev B. 1993;47(1):558–561. doi: 10.1103/PhysRevB.47.558
- Goedecker S, Teter M, Hutter J. Separable dual-space Gaussian pseudopotentials. Phys Rev B. 1996;54(3):1703–1710. doi: 10.1103/PhysRevB.54.1703
- Giannozzi P, Baroni S, Bonini N, et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J Phys Condens Matter. 2009;21(39):395502. doi: 10.1088/0953-8984/21/39/395502
- Segall MD, Lindan PJD, Probert MJ, et al. First-principles simulation: ideas, illustrations and the CASTEP code. J Phys Condens Matter. 2002;14(11):2717. doi: 10.1088/0953-8984/14/11/301
- Iftimie R, Minary P, Tuckerman ME. Ab initio molecular dynamics: concepts, recent developments, and future trends. Proc Natl Acad Sci. 2005;102(19):6654–6659. doi: 10.1073/pnas.0500193102
- Skylaris C-K, Haynes PD, Mostofi AA, et al. Introducing ONETEP: linear-scaling density functional simulations on parallel computers. J Chem Phys. 2005;122(8):84119. doi: 10.1063/1.1839852
- Schwarz K, Blaha P. Solid state calculations using WIEN2k. Comput Mater Sci. 2003;28(2):259–273. doi: 10.1016/S0927-0256(03)00112-5
- Gulans A, Kontur S, Meisenbichler C, et al. Exciting: a full-potential all-electron package implementing density-functional theory and many-body perturbation theory. J Phys Condens Matter. 2014;26(36):363202. PMID: 25135665. doi: 10.1088/0953-8984/26/36/363202
- Andrade X, Strubbe D, De Giovannini U, et al. Real-space grids and the octopus code as tools for the development of new simulation approaches for electronic systems. Phys Chem Chem Phys. 2015;17(47):31371–31396. doi: 10.1039/C5CP00351B
- Mortensen JJ, Hansen LB, Jacobsen KW. Real-space grid implementation of the projector augmented wave method. Phys Rev B. 2005;71(3):35109. doi: 10.1103/PhysRevB.71.035109
- Neese F. The ORCA program system. Wiley Interdiscip Rev Comput Mol Sci. 2012;2(1):73–78. doi: 10.1002/wcms.81
- Ahlrichs R, Bär M, Häser M, et al. Electronic structure calculations on workstation computers: the program system turbomole. Chem Phys Lett. 1989;162(3):165–169. doi: 10.1016/0009-2614(89)85118-8
- Werner H-J, Knowles PJ, Knizia G, et al. Molpro: a general-purpose quantum chemistry program package. Wiley Interdiscip Rev Comput Mol Sci. 2012;2(2):242–253. doi: 10.1002/wcms.82
- García A, Papior N, Akhtar A, et al. Siesta: recent developments and applications. J Chem Phys. 2020;152(20):204108. doi: 10.1063/5.0005077
- Blum V, Gehrke R, Hanke F. Ab initio molecular simulations with numeric atom-centered orbitals. Comput Phys Commun. 2009;180(11):2175–2196. doi: 10.1016/j.cpc.2009.06.022
- Setyawan W, Curtarolo S. High-throughput electronic band structure calculations: challenges and tools. Comput Mater Sci. 2010;49(2):299–312. doi: 10.1016/j.commatsci.2010.05.010
- Aflow - Automatic FLOW for materials discovery [Internet]. Available from: http://aflowlib.org/
- Gražulis S, Chateigner D, Downs RT, et al. Crystallography open database – an open-access collection of crystal structures. J Appl Crystallogr. 2009;42(4):726–729. Cited: in: PMID: 22477773. doi: 10.1107/S0021889809016690
- Shishkin M, Sato H. Self-consistent parametrization of DFT + U framework using linear response approach: application to evaluation of redox potentials of battery cathodes. Phys Rev B. 2016;93(8). doi: 10.1103/PhysRevB.93.085135
- AFLOWπ [Internet]. Available from: http://aflowlib.org/src/aflowpi/index.html
- Materials Project [Internet]. Available from: https://materialsproject.org/
- Automated interactive infrastructure and database for computational science — AiiDA documentation [Internet]. Available from: http://aiida.net/
- Pizzi G, Cepellotti A, Sabatini R, et al. AiiDA: automated interactive infrastructure and database for computational science. Comput Mater Sci. 2016;111:218–230. doi: 10.1016/j.commatsci.2015.09.013
- Crystallography Open Database [Internet]. Available from: http://crystallography.net/cod/
- Mathew K, Singh AK, Gabriel JJ, et al. Mpinterfaces: A materials project based python tool for high-throughput computational screening of interfacial systems. Comput Mater Sci. 2016;122:183–190. doi: 10.1016/j.commatsci.2016.05.020
- OQMD [Internet]. Available from: https://oqmd.org/
- Jain A, Ong SP, Chen W, et al. FireWorks: a dynamic workflow system designed for high-throughput applications. Concurr Comput Pract Exp. 2015;27(17):5037–5059. doi: 10.1002/cpe.3505
- ICSD [Internet]. Available from: https://icsd.fiz-karlsruhe.de/
- Lambert H, Fekete A, Kermode JR, et al. Imeall: a computational framework for the calculation of the atomistic properties of grain boundaries. Comput Phys Commun. 2018;232:256–263. doi: 10.1016/j.cpc.2018.04.029
- Haastrup S, Strange M, Pandey M, et al. The computational 2D materials database: high-throughput modeling and discovery of atomically thin crystals. 2D Mater. 2018;5(4):042002. doi: 10.1088/2053-1583/aacfc1
- Pylada documentation [Internet]. Available from: http://pylada.github.io/pylada/
- Ashton M, Paul J, Sinnott SB, et al. Topology-scaling identification of layered solids and stable exfoliated 2D materials. Phys Rev Lett. 2017;118(10):106101. doi: 10.1103/PhysRevLett.118.106101
- Hjorth Larsen A, Mortensen J J, Blomqvist J, et al. The atomic simulation environment—a python library for working with atoms. J Phys Condens Matter. 2017;29(27):273002. Cited: in: PMID: 28323250. doi: 10.1088/1361-648X/aa680e
- Atomic Simulation Environment [Internet]. Available from: https://wiki.fysik.dtu.dk/ase/
- NOMAD [Internet]. Available from: https://nomad-lab.eu/nomad-lab/
- Mathew K, Montoya JH, Faghaninia A, et al. Atomate: A high-level interface to generate, execute, and analyze computational materials science workflows. Comput Mater Sci. 2017;139:140–152. doi: 10.1016/j.commatsci.2017.07.030
- Atomate (materials science workflows) — atomate 1.0.3 documentation [Internet]. Available from: https://atomate.org/
- Talirz L, Kumbhar S, Passaro E, et al. Materials Cloud, a platform for open computational science. Sci Data. [Internet] 2020;7:299. doi: 10.1038/s41597-020-00637-5
- Choudhary K, Cheon G, Reed E, et al. Elastic properties of bulk and low-dimensional materials using van der Waals density functional. Phys Rev B. 2018;98(1):14107. doi: 10.1103/PhysRevB.98.014107
- Choudhary K, Cheon G, Reed E, et al. Computational materials chemistry [Internet]. Available from: https://cmr.fysik.dtu.dk/
- Agapito LA, Curtarolo S, Nardelli MB. Reformulation of DFT + U as a pseudohybrid hubbard density functional for accelerated materials discovery. Phys Rev X. 2015;5(1). doi: 10.1103/PhysRevX.5.011006
- Jain A, Hautier G, Ong SP, et al. Formation enthalpies by mixing GGA and GGA + U calculations. Phys Rev B - Condens Matter Mater Phys. 2011;84:1–10. doi: 10.1103/PhysRevB.84.045115
- Calderon CE, Plata JJ, Toher C, et al. The AFLOW standard for high-throughput materials science calculations. Comput Mater Sci. 2015;108:233–238. doi: 10.1016/j.commatsci.2015.07.019
- Zhou Z-H. Machine learning. Singapore Pte Ltd; 2021.
- El Naqa I, Murphy MJ. What is machine learning? Berlin: Springer; 2015.
- What is machine learning course| its importance and types-FORE [Internet]. Available from: https://www.fsm.ac.in/blog/an-introduction-to-machine-learning-its-importance-types-and-applications/
- Phoenics: phoenics: bayesian optimization for efficient experiment planning [Internet]. Available from: https://github.com/aspuru-guzik-group/phoenics
- Häse F, Roch LM, Kreisbeck C, et al. Phoenics: a Bayesian optimizer for chemistry. ACS Cent Sci. 2018;4(9):1134–1145. doi: 10.1021/acscentsci.8b00307
- Schütt KT, Kessel P, Gastegger M, et al. SchNetPack: A deep learning toolbox for atomistic systems. J Chem Theory Comput. 2019;15(1):448–455. Cited: in: PMID: 30481453. doi: 10.1021/acs.jctc.8b00908
- Atomistic-machine-learning/schnetpack: SchNetPack - deep neural networks for atomistic systems.
- Bartók AP, Payne MC, Kondor R, et al. Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons. Phys Rev Lett. 2010;104(13):136403. doi: 10.1103/PhysRevLett.104.136403
- TensorFlow [Internet]. Available from: https://www.tensorflow.org/
- Smith JS, Isayev O, Roitberg AE. ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost. Chem Sci. 2017;8(4):3192–3203. doi: 10.1039/C6SC05720A
- ASE_ANI: ANI-1 neural net potential with python interface (ASE) [Internet]. Available from: https://github.com/isayev/ASE_ANI
- amp [Internet]. Available from: https://bitbucket.org/andrewpeterson/amp/src/master/
- Scikit-learn: machine learning in python — scikit-learn 1.2.2 documentation [Internet]. Available from: https://scikit-learn.org/stable/
- Weka 3 - data mining with open source machine learning software in java [Internet]. Available from: https://www.cs.waikato.ac.nz/ml/weka/
- SISSO: a data-driven method combining symbolic regression and compressed sensing for accurate & interpretable models. [Internet]. Available from: https://github.com/rouyang2017/SISSO
- AFLOW ML [Internet]. Available from: http://aflowlib.org/aflow-ml/
- Matminer (materials data mining) — matminer 0.8.0 documentation [Internet]. Available from: https://hackingmaterials.lbl.gov/matminer/
- PROPhet [Internet]. Available from: https://biklooost.github.io/PROPhet/
- Wolverton/Magpie — bitbucket. [Internet] Available from: https://bitbucket.org/wolverton/magpie/src/master/
- Wang H, Zhang L, Han J, et al. DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics. Comput Phys Commun. 2018;228:178–184. doi: 10.1016/j.cpc.2018.03.016
- Deepmodeling/deepmd-kit: a deep learning package for many-body potential energy representation and molecular dynamics [Internet]. [cited 2023 Mar 19]. Available from: https://github.com/deepmodeling/deepmd-kit
- Combo: COMmon Bayesian optimization [Internet]. Available from: https://github.com/tsudalab/combo
- Geilhufe RM, Olsthoorn B, Balatsky AV. Shifting computational boundaries for complex organic materials. Nat Phys. 2021;17(2):152–154. doi: 10.1038/s41567-020-01135-6
- Alpaydin E. Machine learning. Cambridge (USA): Mit Press; 2021.
- Awad M, Khanna R. Machine learning BT - efficient learning machines: theories, concepts, and applications for engineers and system designers. Berkeley, CA: Apress; 2015.
- Machine learning a probabilistic perspective by Kevin P. Murphy – Bridget market [Internet]. Available from: https://bridgetmarket.us/product/machine-learning-a-probabilistic-perspective-by-kevin-p-murphy/?gclid=Cj0KCQjwwtWgBhDhARIsAEMcxeDbvFRioGBCRTKU5no7ClDW8En05BHyw9v_I_wCKLOTE6wXewVvvaAaAjndEALw_wcB
- Pilania G, Wang C, Jiang X, et al. Accelerating materials property predictions using machine learning. Sci Rep. 2013;3(1):2810. doi: 10.1038/srep02810
- Mannodi-Kanakkithodi A, Pilania G, Huan TD, et al. Machine learning strategy for accelerated design of polymer dielectrics. Sci Rep. 2016;6(1):20952. doi: 10.1038/srep20952
- Wang A-T, Murdock RJ, Kauwe SK, et al. Machine learning for materials scientists: an Introductory guide toward best practices. Chem Mater. 2020;32(12):4954–4965. doi: 10.1021/acs.chemmater.0c01907
- Kohavi R, Quinlan R. Decision-tree discovery handbook of data mining and knowledge discovery. Oxford: Oxford University Press; 2002.
- Xue D, Balachandran PV, Hogden J, et al. Accelerated search for materials with targeted properties by adaptive design. Nat Commun. 2016;7: doi: 10.1038/ncomms11241
- Mitchell TM. Machine learning. New York: McGraw-Hill Science; 1997.
- Hill J, Mulholland G, Persson K, et al. Materials science with large-scale data and informatics: unlocking new opportunities. MRS Bull. 2016;41(5):399–409. doi: 10.1557/mrs.2016.93
- Agrawal A, Choudhary A. Perspective: materials informatics and big data: realization of the “fourth paradigm” of science in materials science. APL Mater. 2016;4(5):053208. doi: 10.1063/1.4946894
- Ward L, Wolverton C. Atomistic calculations and materials informatics: a review. Curr Opin Solid State Mater Sci. 2017;21(3):167–176. doi: 10.1016/j.cossms.2016.07.002
- Agrawal A, Choudhary A. Deep materials informatics: applications of deep learning in materials science. MRS Commun. 2019;9(3):779–792. doi: 10.1557/mrc.2019.73
- Hey T. The fourth paradigm, data-intensive scientific discovery, microsoft research. Berlin (Heidelberg): Springer-Verlag; 2009.
- Hey T, Tansley S, Tolle KM. Jim Gray on eScience: a transformed scientific method. In: The fourth paradigm; 2009.
- Hey T, Trefethen A. The fourth paradigm 10 years on. Inform Spektrum. 2020;42(6):441–447. doi: 10.1007/s00287-019-01215-9
- Dimiduk DM, Holm EA, Niezgoda SR. Perspectives on the impact of machine learning, deep learning, and artificial intelligence on materials, processes, and structures engineering. Integr Mater Manuf Innov. 2018;7:157–172. Internet. doi: 10.1007/s40192-018-0117-8
- Isayev O, Tropsha A, Curtarolo S. Materials informatics: methods, tools, and applications. New York (USA): John Wiley & Sons; 2019.
- Ramprasad R, Batra R, Pilania G, et al. Machine learning in materials informatics: recent applications and prospects. Npj Comput Mater. 2017;3(1):54. doi: 10.1038/s41524-017-0056-5
- Takahashi K, Tanaka Y. Materials informatics: a journey towards material design and synthesis. Dalton Trans. 2016;45:10497–10499. Internet. doi: 10.1039/C6DT01501H
- Frydrych K, Karimi K, Pecelerowicz M, et al. Materials informatics for mechanical deformation: a review of applications and challenges. Materials. 2021;14(19):5764.
- Alberi K, Nardelli MB, Zakutayev A, et al. The 2019 materials by design roadmap. J Phys D Appl Phys. 2019;52(1):013001. doi: 10.1088/1361-6463/aad926
- Allam O, Cho BW, Kim KC, et al. Application of DFT-based machine learning for developing molecular electrode materials in li-ion batteries. RSC Adv. 2018;8(69):39414–39420. doi: 10.1039/C8RA07112H
- Cai J, Chu X, Xu K, et al. Machine learning-driven new material discovery. Nanoscale Adv. 2020;2(8):3115–3130. doi: 10.1039/D0NA00388C
- Hafner J, Wolverton C, Ceder G, et al. Toward computational materials design: The impact of density functional theory on materials research. MRS Bull. 2006;31(9):659–668. doi: 10.1557/mrs2006.174
- Butler KT, Frost JM, Skelton JM, et al. Computational materials design of crystalline solids. Chem Soc Rev. 2016;45(22):6138–6146. doi: 10.1039/C5CS00841G
- Hammer B, Nørskov JK. Theoretical surface science and catalysis—calculations and concepts. Adv Catal. 2000;45:71–129.
- Perdew JP, Chevary JA, Vosko SH, et al. Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation. Phys Rev B. 1992;46(11):6671–6687. doi: 10.1103/PhysRevB.46.6671
- Kohn W, Becke AD, Parr RG. Density functional theory of electronic structure. J Phys Chem. 1996;100(31):12974–12980. doi: 10.1021/jp960669l
- Mazurek AH, Ł S, Pisklak DM. Periodic DFT calculations—review of applications in the pharmaceutical sciences. Pharmaceutics. 2020;12(5):415. doi: 10.3390/pharmaceutics12050415
- Paul A, Birol T. Applications of DFT + DMFT in materials science. Ann Rev Mater Res. 2019;49(1):31–52. doi: 10.1146/annurev-matsci-070218-121825
- Smith SW. Digital Signal Processing. San Diego (CA): California Technical Publishing; 1997.
- Tolba SA, Gameel KM, Ali BA, et al. The DFT+U: Approaches, accuracy, and applications. In: Density functional calculations: recent progresses of theory and application; 2018. p. 30–35.
- Lederer Y, Toher C, Vecchio KS, et al. The search for high entropy alloys: a high-throughput ab-initio approach. Acta Mater. 2018;159:364–383. doi: 10.1016/j.actamat.2018.07.042
- Tomczak JM. Thermoelectricity in correlated narrow-gap semiconductors. J Phys Condens Matter. 2018;30(18):183001. doi: 10.1088/1361-648X/aab284
- Lin L. Materials databases infrastructure constructed by first principles calculations: a review. Mater Perform Charact. 2015;4. doi: 10.1520/MPC20150014
- Montoya JH, Persson KA. A high-throughput framework for determining adsorption energies on solid surfaces. Npj Comput Mater. 2017;3(1):1–3. doi: 10.1038/s41524-017-0017-z
- Armitage NP, Mele EJ, Vishwanath A. Weyl and Dirac semimetals in three-dimensional solids. Rev Mod Phys. 2018;90(1):15001. doi: 10.1103/RevModPhys.90.015001
- Ando Y, Fu L. Topological crystalline insulators and topological superconductors: from concepts to materials. Annu Rev Condens Matter Phys. 2015;6(1):361–381. doi: 10.1146/annurev-conmatphys-031214-014501
- Cano J, Bradlyn B, Wang Z, et al. Building blocks of topological quantum chemistry: elementary band representations. Phys Rev B. 2018;97(3):35139. doi: 10.1103/PhysRevB.97.035139
- Bradlyn B, Elcoro L, Vergniory MG, et al. Band connectivity for topological quantum chemistry: band structures as a graph theory problem. Phys Rev B. 2018;97(3):35138. doi: 10.1103/PhysRevB.97.035138
- Zhang C, Gao M, Yeh J, et al. High-entropy alloys: fundamentals and applications. Switzerland: Springer; 2016.
- Kuisma M, Ojanen J, Enkovaara J, et al. Kohn-Sham potential with discontinuity for band gap materials. Phys Rev B. 2010;82(11):115106. doi: 10.1103/PhysRevB.82.115106
- Hasan MZ, Kane CL. Colloquium: topological insulators. Rev Mod Phys. 2010;82(4):3045–3067. doi: 10.1103/RevModPhys.82.3045
- Yang K. High-throughput design of functional materials using materials genome approach. Chinese Phys B. 2018;27(12):128103. doi: 10.1088/1674-1056/27/12/128103
- Schmidt J, Marques MRG, Botti S, et al. Recent advances and applications of machine learning in solid-state materials science. Npj Comput Mater. 2019;5(1):83. doi: 10.1038/s41524-019-0221-0
- Jain A, Ong SP, Hautier G, et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater. 2013;1(1):11002. doi: 10.1063/1.4812323
- Blaiszik B, Chard K, Pruyne J, et al. The materials data facility: data services to advance materials science research. JOM. Internet 2016;68:2045–2052. doi: 10.1007/s11837-016-2001-3
- Wen C, Zhang Y, Wang C, et al. Machine learning assisted design of high entropy alloys with desired property. Acta Mater. 2019;170:109–117. doi: 10.1016/j.actamat.2019.03.010
- Curtarolo S, Morgan D, Persson K, et al. Predicting crystal structures with data mining of quantum calculations. Phys Rev Lett. 2003;91(13):1–4. doi: 10.1103/PhysRevLett.91.135503
- Fischer CC, Tibbetts KJ, Morgan D, et al. Predicting crystal structure by merging data mining with quantum mechanics. Nat Mater. 2006;5(8):641–646. Cited: in: PMID: 16845417. doi: 10.1038/nmat1691
- Coey JM. Magnetism and magnetic materials. Cambridge (UK): Cambridge University Press; 2010.
- Andreoni W, Yip S. Handbook of materials modeling: applications: current and emerging materials. Switzerland: Springer Nature; 2020.
- Lee J, Seko A, Shitara K, et al. Prediction model of band gap for inorganic compounds by combination of density functional theory calculations and machine learning techniques. Phys Rev B. 2016;93(11):115104. doi: 10.1103/PhysRevB.93.115104
- Yamawaki M, Ohnishi M, Ju S, et al. Multifunctional structural design of graphene thermoelectrics by Bayesian optimization. Sci Adv. 2023;4(6):eaar4192. doi: 10.1126/sciadv.aar4192
- Faber FA, Lindmaa A, von Lilienfeld OA, et al. Machine learning energies of 2 million elpasolite (ABCD6) crystals. Phys Rev Lett. 2016;117(13):135502. doi: 10.1103/PhysRevLett.117.135502
- V BP, Young J, Lookman T, et al. Learning from data to design functional materials without inversion symmetry. Nat Commun. 2017;8(1):14282. doi: 10.1038/ncomms14282
- Jain A, Bligaard T. Atomic-position independent descriptor for machine learning of material properties. Phys Rev B. 2018;98(21):214112. doi: 10.1103/PhysRevB.98.214112
- Davies DW, Butler KT, Walsh A. Data-driven discovery of photoactive quaternary oxides using first-principles machine learning. Chem Mater. 2019;31(18):7221–7230. doi: 10.1021/acs.chemmater.9b01519
- Saad Y, Gao D, Ngo T, et al. Data mining for materials: computational experiments with AB compounds. Phys Rev B. 2012;85(10):104104. doi: 10.1103/PhysRevB.85.104104
- Houhou R, Bocklitz T. Trends in artificial intelligence, machine learning, and chemometrics applied to chemical data. Anal Sci Adv. 2021;2(3–4):128–141. doi: 10.1002/ansa.202000162
- Martin TB, Audus DJ. Emerging trends in machine learning: a polymer perspective. ACS Polym Au. 2023;3(3):239–258. doi: 10.1021/acspolymersau.2c00053
- Zhao J-C. Combinatorial approaches as effective tools in the study of phase diagrams and composition–structure–property relationships. Prog Mater Sci. 2006;51(5):557–631. doi: 10.1016/j.pmatsci.2005.10.001
- Introduction — pymatgen 2023.3.23 documentation [Internet]. Available from: https://pymatgen.org/
- Hassel AW, Lohrengel MM. The scanning droplet cell and its application to structured nanometer oxide films on aluminium. Electrochim Acta. 1997;42(20–22):3327–3333. doi: 10.1016/S0013-4686(97)00184-9
- Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2012;64:4–17. doi: 10.1016/j.addr.2012.09.019
- Kirklin S, Saal JE, Hegde VI, et al. High-throughput computational search for strengthening precipitates in alloys. Acta Mater. Internet 2016;102:125–135. doi: 10.1016/j.actamat.2015.09.016
- Studt F, Sharafutdinov I, Abild-Pedersen F, et al. Discovery of a Ni-Ga catalyst for carbon dioxide reduction to methanol. Nat Chem. 2014;6(4):320–324. doi: 10.1038/nchem.1873
- Suram SK, Xue Y, Bai J, et al. Automated phase mapping with AgileFD and its application to light absorber discovery in the V–Mn–Nb oxide system. ACS Comb Sci. 2017;19(1):37–46. doi: 10.1021/acscombsci.6b00153
- Huber L, Hadian R, Grabowski B, et al. A machine learning approach to model solute grain boundary segregation. Npj Comput Mater. 2018;4(1):64. doi: 10.1038/s41524-018-0122-7
- Aykol M, Dwaraknath SS, Sun W, et al. Thermodynamic limit for synthesis of metastable inorganic materials. Sci Adv. 2018;4:1–8. doi: 10.1126/sciadv.aaq0148
- Zhao J-C. A combinatorial approach for efficient mapping of phase diagrams and properties. J Mater Res. 2001;16(6):1565–1578. doi: 10.1557/JMR.2001.0218
- Takahashi R, Kubota H, Murakami M, et al. Design of combinatorial shadow masks for complete ternary-phase diagramming of solid state materials. J Comb Chem. 2004;6(1):50–53. doi: 10.1021/cc030038i
- Hasegawa Y, Iwata J-I, Tsuji M, et al. First-principles calculations of electron states of a silicon nanowire with 100,000 atoms on the K computer. Proc 2011 Int Conf High Perform Comput Networking, Storage Anal. 2011. p. 1–11.
- Jinnouchi R, Karsai F, Kresse G. Making free-energy calculations routine: combining first principles with machine learning. Phys Rev B. 2020;101(6):60201. doi: 10.1103/PhysRevB.101.060201
- Duan C, Janet JP, Liu F, et al. Learning from failure: predicting electronic structure calculation outcomes with machine learning models. J Chem Theory Comput. 2019;15(4):2331–2345. doi: 10.1021/acs.jctc.9b00057
- McAndrew CC, Victory JJ. Accuracy of approximations in MOSFET charge models. IEEE Trans Electron Devices. 2002;49(1):72–81. doi: 10.1109/16.974752
- Van Gunsteren WF, Berendsen HJC. Computer simulation of molecular dynamics: methodology, applications, and perspectives in chemistry. Angew Chemie Int Ed English. 1990;29(9):992–1023. doi: 10.1002/anie.199009921
- Argaman N, Makov G. Density functional theory: an introduction. Am J Phys. 2000;68(1):69–79. doi: 10.1119/1.19375
- Tarascon J-M, Armand M. Issues and challenges facing rechargeable lithium batteries. Nature. 2001;414(6861):359–367 [Internet]. doi: 10.1038/35104644
- Thackeray MM, Kang S-H, Johnson CS, et al. Li2MnO3-stabilized LiMO2 (M = Mn, Ni, Co) electrodes for lithium-ion batteries. J Mater Chem. 2007;17(30):3112–3125. Internet. doi: 10.1039/b702425h
- Li H, Huang X, Chen L, et al. A high capacity nano Si composite anode material for lithium rechargeable batteries. Electrochem Solid-State Lett. 1999;2(11):547. Internet. doi: 10.1149/1.1390899
- Xiao R, Li H, Chen L. High-throughput design and optimization of fast lithium ion conductors by the combination of bond-valence method and density functional theory. Sci Rep. 2015;5(1):14227. Internet. doi: 10.1038/srep14227
- Hautier G, Jain A, Chen H, et al. Novel mixed polyanions lithium-ion battery cathode materials predicted by high-throughput ab initio computations. J Mater Chem. 2011;21(43):17147–17153. Internet. doi: 10.1039/c1jm12216a
- Knauth P. Inorganic solid li ion conductors: an overview. Solid State Ion. 2009;180(14–16):911–916. Internet. doi: 10.1016/j.ssi.2009.03.022
- Wang X, Xiao R, Li H, et al. Quantitative structure-property relationship study of cathode volume changes in lithium ion batteries using ab-initio and partial least squares analysis. J Mater [Internet]. 2017;3(3):178–183. doi: 10.1016/j.jmat.2017.02.002
- MacLeod BP, Parlane FGL, Morrissey TD, et al. Self-driving laboratory for accelerated discovery of thin-film materials. Sci Adv. 2020;6(20):eaaz8867. doi: 10.1126/sciadv.aaz8867