
Abstract:
Theoretical evidence of a new five-membered divalent carbon (0) compound has been proposed. Calculations at different levels of density functional theory indicate that this compound is highly stable and a suitable candidate for synthesis. The proposed compound is highly basic and its basicity is almost equal or higher than that of related divalent carbon (0) compounds such as carbodicarbene and carbodiphosphorane.
Introduction
The seminal computational prediction of carbodicarbenes, C(NHC)2 (1) (), a compound with a central C (0) atom stabilized by two N-heterocyclic carbene (NHC) ligand by Tonner and Frenking1 and subsequent synthesis of its benzannulated derivative by Dyker et al,Citation2 has opened a new chapter in the knowledge book of carbon compounds.Citation3–Citation5 Early on, Ramirez et al synthesized the first carbodiphosphorane (CDP, 2) – C(PPh3)2 – in 1961,Citation6,Citation7 which was later on structurally characterized by an X-ray analysis in 1978.Citation6,Citation7 However, its carbon (0) character was not recognized at that time. It was only in 2006, when Tonner et al made an in-depth electronic structure analysis of this compound and established the carbon (0) character of the central carbon atom of this compound.Citation8 Recently, the bonding situation of carbon (0) compounds has received much theoretical attention and believed to take place through donor–acceptor interactions L→C←L.Citation9–Citation13 This bonding situation results in the retention of the four valence electrons of carbon as two lone pairs, one of σ symmetry and another of π symmetry.
Figure 1 Schematic representation of carbodicarbene, (1) C(NHC)2 and (2) CDP.
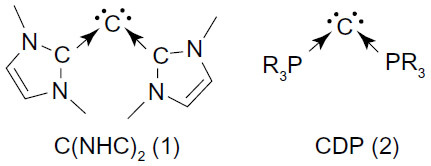
Hence, the name “carbone” was coined for these types of compounds. Various carbones have now been synthesized and structurally characterized.Citation2,Citation14–Citation16
In continuation of the search for suitable ligands to stabilize divalent carbon (0) compounds, we recently come across an interesting ligand viz, bis(imidazolin-2-iminium) dication (), which has been used to isolate a three-coordinate boron cation with boron–sulfur double bond, for the first time.Citation17 The use of this ligand in main-group element chemistryCitation18–Citation24 and transition metal chemistryCitation25–Citation32 has been recently reviewed.Citation33
Figure 2 Schematic representation of bis(imidazolin-2-iminium) cation (A) and the boron cation compound (B).
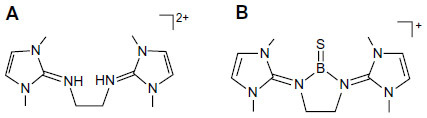
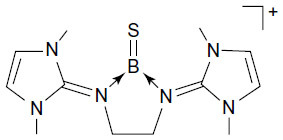
The central N–B bond in B is actually a donor–acceptor bond (), which resembles exactly the same as that of carbones, ie, L→C←L.Citation1
Figure 3 Donor–acceptor interaction in B.
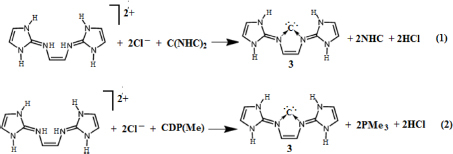
This has prompted us to investigate whether the ligand framework may stabilize a carbon atom to form a donor–acceptor complex. Quantum chemical calculations predict that the proposed compound (3) should be a synthetically accessible species. It is a neutral five-membered cyclic carbone (3) that contains a central divalent carbon (0) atom within the familiar N-heterocyclic framework. Moreover, the compound shows high basicity as that of other divalent carbon (0) compounds.Citation1–Citation16
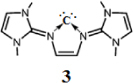
Computational details
All the structures were fully optimized without any symmetry constraints at BP86/TZVPCitation34–Citation37 level of theory. This level of theory has been widely used for this class of compounds.Citation1–Citation24 For Au atom, the relativistic small-core effective core potential basis set of Stuttgart/Dresden was used.Citation38 Stationary points were characterized as minima by calculating their Hessian matrix analytically at this level of theory. For the sake of analysis, the planar conformer of 3 is optimized using symmetry constraint. Proton affinities are calculated employing L + H+ → L–H+ equation. Zero-point corrections are also added in proton affinity calculations. For proton affinity calculations, single-point energies are calculated at MP2/aug-cc-PVDZCitation39,Citation40 level of theory on the BP86/TZVP optimized geometries. Solution phase model chemistry has been performed using the polarized continuum model (PCM)Citation41 employing water as a solvent (dielectric constant, ε =78.39). We have used the PCM calculation using integral equation formalism model with radii of the sphere defined by united atom topological model. The overlap index between two overlapping spheres is taken to be 0.89 (OFAC =0.89), and the minimum radii of the added sphere is 0.2 Å (RMIN =0.2 Å). All the calculations are performed using NWChem 6.1Citation42 and OrcaCitation43 suite of program.
Results and discussions
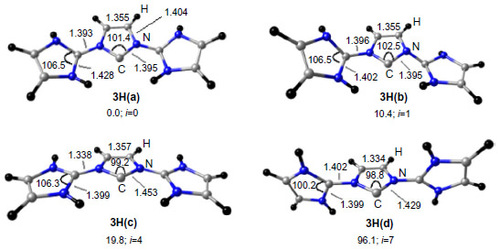
shows the optimized geometries of different conformers of 3 with H atom as the substituent at N atoms at BP86/TZVP level of theory. The equilibrium geometry 3H(a) has the two NHC units in the cis conformation with respect to the planar central five-membered ring. The C–N bond of the central five-membered ring in 3H(a) is 1.395 Å, which is very close to the exocyclic C–N bond (1.393 Å). This implies that the central C–N bond is as strong as the exocyclic one. The equilibrium conformer 3H(a) has an acute N–C–N angle of 101.4°. The trans conformer 3H(b) is 10.4 kcal/mol higher in energy with one imaginary frequency. The planar conformers 3H(c) and the 3H(d), where both the NHC units are perpendicular to the central ring, are higher in energy by 19.8 kcal/mol and 96.1 kcal/mol, respectively (). The C–C backbone length of the central ring in 3H(a), 3H(b), and 3H(c) is almost the same. It should be noted that the central C–N bond in 3H(a) (bond length 1.395 Å) may not reveal a donor–acceptor interaction. However, this donor–acceptor interaction becomes prominent when their reactivity is concerned.Citation9–Citation13 Moreover, the singlet–triplet energy separation of 3H(a) is 121.3 kcal/mol, a value quite higher than generally observed for carbenes.Citation44 The Mulliken atomic charge on the central carbon atom is −0.389e compared to −0.187e for the adjacent N atoms – suggesting a possible donor–acceptor interaction in 3. We would also like to emphasize here that the donor–acceptor bonding situation in 3 may not have any physical reality, however, they are quite useful while discussing their reactivity.Citation9–Citation13
Figure 4 Optimized geometries at BP86/TZVP level of theory and relative energies (kcal/mol at MP2/aug-cc-pVDZ//BP86/TZVP) of different conformers of 3H.
shows the frontier Kohn–Sham orbitals of 3H calculated at BP86/TZVP level of theory. The HOMO (highest occupied molecular orbital) is of π symmetry with an energy of −2.27 eV, whereas the HOMO-1 is of σ symmetry with an energy of −4.91 eV. These orbitals are of similar symmetry to that of C(NHC)2.Citation1 However, there is a contrasting feature. In C(NHC)2, the first protonation takes place at the σ symmetric orbital (HOMO-1) due to the smaller energy gap between the σ- and π-type molecular orbital. However, in case of 3, the first protonation takes place at the π symmetric lone pair orbital ().
Figure 5 Shape of frontier Kohn–Sham orbitals of 3H and orbital energies ε in eV at the BP86/TZVP level of theory.
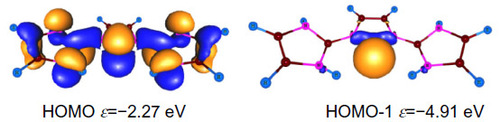
Figure 6 BP86/TZVP optimized first and second protonated derivatives of 3H.
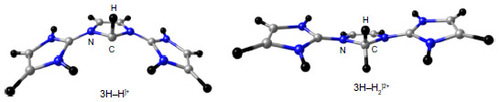
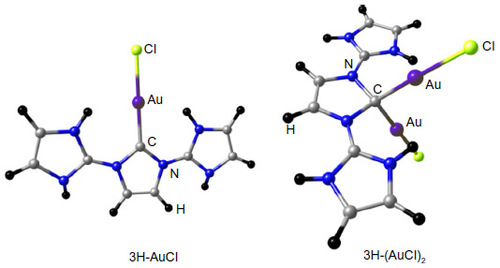
Visual inspection of these highest lying frontier molecular orbitals of 3 reveals that the largest coefficient is always at the central carbon atom (atomic orbital coefficient for the central carbon in HOMO is 0.73), which suggests that 3 may be considered as carbones.Citation9–Citation13 Thus, these two molecular orbitals may be considered as two lone pairs at the central carbon atom, and it should behave as double Lewis base. Hence, its first and second proton affinity has been calculated. contains the first and second proton affinities of 1–3. While the first proton affinity of 3 is close to 1 but higher than 2, the second proton affinity of 3 is much higher than 1 and 2. This indicates that the proposed compound is highly basic that might have wide application in transition metal catalysis.Citation44 Moreover, the calculated bond dissociation energies for the dissociation of one molecule of AuCl from 3 and 3-AuCl are very high (), further supporting their divalent carbon (0) character.Citation9–Citation13
Table 1 MP2/aug-cc-pVDZ//BP86/TZVP calculated proton affinities (in gas phase); PAs (kcal/mol) of 1–3
Figure 7 BP86/BSI (BSI = TZVP for H, C, N, Cl and SDD for Au) optimized protonated derivatives of 3H.
Abbreviation: SDD, Stuttgart/Dresden.
Calculations (The reaction energetics [including zero-point corrections] of reactions 1 and 2 are further calculated using the hybrid density functionals B3LYP [it is Becke’s three-parameter hybrid method using the Lee, Yang and Parr {LYP} correlation functional]Citation45–Citation47 and B3PW91Citation48,Citation49 using the TZVP basis sets. The calculated value of the reaction energetics are qualitatively similar to those obtained at BP86/TZVP level of theory) suggest that the reactions (1–2) are highly exergonic () suggesting the spontaneity of their formation. However, these equations may not be realistic as the reaction energetics is calculated in gas phase. To mimic the more realistic situation, we have calculated the reaction energetics in aqueous medium. In aqueous medium, both these reactions are exergonic albeit to a lesser extent than in gas phase – implying that the proposed compound may be a likely candidate for experimental realization.
Table 2 Calculated reaction Gibbs free energies (kcal/mol) of reactions 1 and 2 at T=298.15 K and P=1 atm
Conclusion
In summary, quantum chemical calculations provide a hint toward possible isolation of new five-membered neutral divalent carbon (0) compound within the familiar N-heterocyclic framework. The proposed compound contains two other NHC units whose electronic character can be modified in many ways,Citation51 and thus, the proposed compound may serve as an important perspective for transition metal catalysis.Citation44
Acknowledgments
The computational facility of Cotton College State University is greatly acknowledged. The authors declare no competing financial interest. Dedicated to my mentors Dr Ashwini K Phukan, Tezpur University, India, and Professor Shridhar R Gadre, IIT Kanpur, India.
Disclosure
The author reports no conflicts of interest in this work.
References
- Tonner R, Frenking G. C(NHC)2: divalent carbon(0) compounds with N-heterocyclic carbene ligands-theoretical evidence for a class of molecules with promising chemical properties. Angew Chem Int Ed Engl. 2007;46:8695.
- Dyker CA, Lavallo V, Donnadieu B, Bertrand G. Synthesis of an extremely bent acyclic allene (a “carbodicarbene”): a strong donor ligand. Angew Chem Int Ed Engl. 2008;47:3206.
- Alcarazo M, Lehmann CW, Anoop A, Thiel W, Fürstner A. Coordination chemistry at carbon. Nat Chem. 2009;1:295.
- Alcarazo M. On the metallic nature of carbon in allenes and heterocumulenes. Dalton Trans. 2011;40(9):1839–1845.
- Kaufhold O, Hahn FE. Carbodicarbenes: divalent carbon(0) compounds. Angew Chem Int Ed Engl. 2008;47:4057.
- Ramirez F, Desai NB, Hansen B, McKelvie N. Hexaphenylcarbodiphosphorane, (C6H5)3PCP(C6H5)3. J Am Chem Soc. 1961;83:3539.
- Hardy GE, Zink JI, Kaska WC, Baldwin JC. Structure and triboluminescence of polymorphs of hexaphenylcarbodiphosphorane. J Am Chem Soc. 1978;100:8001.
- Tonner R, öxler F, Neumüller B, Petz W, Frenking G. Carbodiphosphoranes: the chemistry of divalent carbon(0). Angew Chem Int Ed Engl. 2006;45:8038.
- Tonner R, Frenking G. Divalent carbon(0) chemistry, part 1: parent compounds. Chemistry. 2008;14:3260.
- Klein S, Frenking G. Carbodiylides C(ECp*)2 (E=B-Tl): another class of theoretically predicted divalent carbon(0) compounds. Angew Chem Int Ed Engl. 2010;49:7106.
- Tonner R, Frenking G. Divalent carbon(0) compounds. Pure Appl Chem. 2009;81:597.
- Tonner R, Heydenrych G, Frenking G. First and second proton affinities of carbon bases. ChemPhysChem. 2008;9:1474.
- Esterhuysen C, Frenking G. Distinguishing carbones from allenes by complexation to AuCl. Chemistry. 2011;17:9944.
- Marrot S, Kato T, Gornitzka H, Baceiredo A. Cyclic carbodiphosphoranes: strongly nucleophilic sigma-donor ligands. Angew Chem Int Ed. 2006;45:2598.
- Fernandez I, Dyker CA, DeHope A, Donnadieu B, Frenking G, Bertrand G. Exocyclic delocalization at the expense of aromaticity in 3,5-bis(pi-donor) substituted pyrazolium ions and corresponding cyclic bent allenes. J Am Chem Soc. 2009;131:11875.
- Schmidbaur H; Schier A. Coordination chemistry at carbon: the patchwork family comprising (Ph3P)2C, (Ph3P)C(C2H4), and (C2H4)2C. Angew Chem Int Ed Engl. 2013;52:176.
- Franz D, Irran E, Inoue S. Isolation of a three-coordinate boron cation with a boron-sulfur double bond. Angew Chem Int Ed. 2014;53:14264.
- Kuhn N, Fawzi R, Steimann M, Wiethoff J. Derivate des Imidazols. XXIII. 2-Iminoimidazolin-Derivate des Magnesiums und Aluminiums. Z Anorg Allg Chem. 1997;623:554.
- Kuhn N, Abram U, Maichle-Mössmer, C, Wiethoff J. Derivate des Imidazols. XXIV. Li12O2Cl2(ImN)8(THF)4] 8THF: Ein Peroxo-Komplex des Lithiums mit neuartiger Käfigstruktur. Z Anorg Allg Chem. 1997;623:1121.
- Kuhn N, Grathwohl M, Steimann M, Henkel G. 1,2-Bis(1′,3′- dimethylimidazolin-2′-iminato)ethanein neuer Chelatligand [1]. Z Naturforsch B. 1998;53:997.
- Kuhn N, Göhner M, Grathwohl M, Wiethoff J, Frenking G, Chen Y. 2-Iminoimidazoline – starke Stickstoffbasen als Koordinationspartner in der Anorganischen Chemie. Z Anorg Allg Chem. 2003;629:793.
- Kinjo R, Donnadieu B, Bertrand G. Isolation of a carbene-stabilized phosphorus mononitride and its radical cation (PN+*). Angew Chem Int Ed Engl. 2010;49:5930.
- Inoue S, Leszczynska K. An acyclic imino-substituted silylene: synthesis, isolation, and its facile conversion into a zwitterionic silaimine. Angew Chem Int Ed Engl. 2012;51:8589.
- Dielmann F, Back O, Henry-Ellinger M, Jerabek P, Frenking G, Bertrand G. A crystalline singlet phosphinonitrene: a nitrogen atom-transfer agent. Science. 2012;337:1526.
- Petrovic D, Glöge T, Bannenberg T, et al. Synthesis and reactivity of 16-electron pentamethylcyclopentadienyl–ruthenium(II) complexes with Bis(imidazolin-2-imine) ligands. Eur J Inorg Chem. 2007;2007:3472.
- Petrovic D, Hill LMR, Jones PG, Tolman WB, Tamm M. Synthesis and reactivity of copper(I) complexes with an ethylene-bridged bis(imidazolin-2-imine) ligand. Dalton Trans. 2008;7:887–894.
- Beer S, Brandhorst K, Hrib CG, et al. Experimental and theoretical investigations of catalytic alkyne cross-metathesis with imidazolin-2-iminato tungsten alkylidyne complexes. Organometallics. 2009;28:1534.
- Glöge T, Petrovic D, Hrib CG, et al. Synthesis and structural characterization of an isomorphous series of Bis(imidazolin-2-imine) metal dichlorides containing the first row transition metals Mn, Fe, Co, Ni, Cu, and Zn. Z Anorg Allg Chem. 2010;636:2303.
- Trambitas AG, Yang J, Melcher D, et al. Synthesis and structure of rare earth dicarbollide complexes with an imidazolin-2-iminato ligand featuring very short metal-nitrogen bonds. Organometallics. 2011;30:1122.
- Shoken D, Sharma M, Botoshansky M, Tamm M, Eisen MS. Mono(imidazolin-2-iminato) titanium complexes for ethylene polymerization at low amounts of methylaluminoxane. J Am Chem Soc. 2013;135:12592.
- Nomura K, Bahuleyan BK, Zhang S, et al. Synthesis and structural analysis of (imido)vanadium(V) dichloride complexes containing imidazolin-2-iminato- and imidazolidin-2-iminato ligands, and their use as catalyst precursors for ethylene (co)polymerization. Inorg Chem. 2014;53:607.
- Karmel ISR, Botoshansky M, Tamm M, Eisen MS. Uranium(IV) imidazolin-2-iminato complexes: a new class of actinide complexes. Inorg Chem. 2014;53:694.
- Wu X, Tamm M. Transition metal complexes supported by highly basic imidazolin-2-iminato and imidazolin-2-imine N-donor ligands. Coord Chem Rev. 2014;260:116.
- Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A. 1988;38:3098.
- Perdew JP. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B Condens Matter. 1986;33:8822.
- Schäefer A, Horn H, Ahlrichs R. Fully optimized contracted gaussian basis sets for atoms Li to Kr. J Chem Phys. 1992;97:2571.
- Schäefer A, Horn H, Ahlrichs R. Fully optimized contracted gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J Chem Phys. 1994;100:5829.
- Dolg M, Wedig U, Stoll H, Preuss H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J Chem Phys. 1987;86:866.
- Møller C, Plesset MS. Note on an approximation treatment for many-electron systems. Phys Rev. 1934;46:618.
- Binkley JS, Pople JA. Møller–Plesset theory for atomic ground state energies. Int J Quantum Chem. 1975;9:229.
- Tomasi J, Persico M. Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem Rev. 1994;94:2027.
- Valiev M, Bylaska EJ, Govind N, et al. NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations. Comput Phys Commun. 2010;181:1477.
- Neese F. ORCA – An ab Initio, Density Functional and Semiempirical Program Package, Version 3.0.2. Bonn: University of Bonn; 2008.
- Herrmann WA. N-heterocyclic carbenes: a new concept in organometallic catalysis. Angew Chem Int Ed Engl. 2002;41:1290.
- Becke AD. Densityfunctional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648.
- Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B Condens Matter. 1988;37:785.
- Vosko SH, Wilk L, Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can J Phys. 1980;58:1200.
- Perdew JP. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B Condens Matter. 1986;33:8822.
- Perdew JP, Burke K, Wang Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys Rev B Condens Matter. 1996;54:16533.
- Schmidbaur H. The aurophilicity phenomenon: a decade of experimental findings, theoretical concepts and emerging applications. Gold Bull. 2000;33:3.
- Guha AK, Sarmah S, Phukan AK. Effect of substituents at the heteroatom on the structure and ligating properties of heterocyclic carbene, silylene, germylene and abnormal carbene: a theoretical study. Dalton Trans. 2010;39:7374.